

Abstract
Protein arginine methyltransferases (PRMTs) catalyze the transfer of methyl groups to arginine residues in proteins. PRMT inhibitors are novel, promising drugs against cancer that are currently in clinical trials, which include oral administration of the drugs. However, off-target activities of systemically available PRMT inhibitors have not yet been investigated. In this work, we study the relevance of arginine methylation in platelets and investigate the effect of PRMT inhibitors on platelet function and on the expression of relevant platelet receptors. We show that (1) key platelet proteins are modified by arginine methylation; (2) incubation of human platelets with PRMT inhibitors for 4 h results in impaired capacity of platelets to aggregate in response to thrombin and collagen, with IC50 values in the μM range; and (3) treatment with PRMT inhibitors leads to decreased membrane expression and reduced activation of the critical platelet integrin αIIbβ3. Our contribution opens new avenues for research on arginine methylation in platelets, including the repurposing of arginine methylation inhibitors as novel antiplatelet drugs. We also recommend that current and future clinical trials with PRMT inhibitors consider any adverse effects associated with platelet inhibition of these emerging anticancer drugs.
Keywords: antiplatelet, arginine methylation, cancer, inhibitor, platelet, protein arginine methyltransferase
Platelets, or thrombocytes, are small anucleate discoid cells of 3 μm in diameter and key components of normal hemostasis.1 In the event of injury, platelets adhere to the injured vessel wall, aggregate with other platelets, and combine with soluble fibrinogen and insoluble fibrin to form a platelet plug. Platelets are the second most numerous cells in circulating blood, typically present in 150–440 × 106 cells/ml and have an average life span of 5–10 days.2
Platelets can be activated by agonists such as thrombin, collagen, adenosine diphosphate (ADP), and von Willebrand factor (vWF). Platelet membranes contain glycoprotein receptors that induce signaling mechanisms inside the cell upon ligand binding to trigger platelet activation. Activated platelets undergo actin cytoskeletal rearrangements to form membrane extensions, called lamellipodia, which facilitate platelet adhesion to vascular wall structures by increasing the surface area of the platelet.3 Platelets can also adhere to each other through the formation of interplatelet bridges, in a process known as platelet aggregation.4 Platelet aggregation is dependent on the activity of the αIIbβ3 receptor, the most abundant integrin in human platelets.5 Activation of the αIIbβ3 fibrinogen receptor is the final step in the activation process and the first step toward aggregation, which is amplified by secretion of platelet dense and alpha granules.6
Although platelet function is vital to hemostasis, pharmaceutical interventions to decrease the activity of platelets can be very useful in the clinical setting. Patients at risk of cardiovascular disease, including myocardial infarction and stroke, can benefit from antiplatelet therapies that reduce the risk of occlusion of a blood vessel by a thrombus, leading to ischemia. Antiplatelet drugs interfere with mechanisms of platelet activation or aggregation and include aspirin, P2Y12 receptor blockers (such as clopidogrel and ticagrelor), and αIIbβ3 antagonists.7 Antiplatelet drugs have gained visibility in the context of the COVID-19 pandemic,8 because overall, 17% of COVID-19 patients suffer from venous thromboembolisms.9 Although antiplatelet drugs are widely used in the clinics, there is a subtle balancing act between the desirable antiplatelet effects of therapy in patients and a higher risk of bleeding.10
In a normal physiology setting, the premature activation of platelets is inhibited by endogenous substances secreted by endothelial cells, namely, prostacyclin (PGI2) and nitric oxide (NO). PGI2 and NO inhibit platelet function through activation of adenylyl cyclase and guanylyl cyclase, respectively, and the corresponding cyclic nucleotide-dependent protein kinase A (PKA) and G (PKG) signaling.11 Vasodilator-stimulated phosphoprotein (VASP) is a key player in platelet signaling pathways through its role as an actin cytoskeleton regulator,12 and phosphorylation of VASP (by PKA and PKG) is a gold standard of platelet quiescence.13
Protein post-translational modifications are key players in signal transduction thanks to their ability to change protein activity, localization, and interactions. Arginine methylation (ArgMe) of proteins consists of the transfer of a methyl group (CH3) from S-adenosyl-l-methionine onto the side chain guanidino nitrogen of arginine, in an enzymatic reaction catalyzed by protein arginine methyltransferases (PRMTs).14 The addition of a CH3 group can affect hydrogen bonding interactions of the recipient arginine and produces bulkier and more hydrophobic methylarginine residues.15 There are three types of PRMTs, each responsible for a different ArgMe end-product: Type I PRMTs lead to asymmetric dimethylarginine (ADMA); Type II PRMTs produce symmetric dimethylarginine (SDMA); and Type III PRMTs form monomethyl arginine (MMA) only. MMA is produced by all three PRMT types and is often seen as a stable, but intermediate, product in Types I and II PRMT reactions. Type I PRMTs include PRMT1, −2, −3, −4, −6, and −8. PRMT5 and −9 are type II PRMTs. PRMT7 is the only type III PRMT.16 It is accepted that most of the ArgMe activity in mammalian cells can be attributed to PRMT1.17,18
PRMTs are key epigenetic regulators or “writers” of epigenetic methylation marks and PRMT inhibitors have been developed over the last two decades to pharmacologically modulate PRMT activity and regulate gene expression.14 Recently, PRMT inhibitors have entered clinical trials in the oncology setting (identifiers: NCT03666988, NCT02783300, NCT03614728, NCT03573310, NCT03854227, and NCT04089449).14 Most PRMT inhibitors in clinical trials are administered orally and therefore systemically.19,20 This raises the obvious question of possible off-target, side effects of ArgMe inhibition in any cell type. There is little documentation available on this issue, but preclinical experiments with rats and dogs revealed moderate changes to hematological profiles, including on the number of platelets, when the animals where treated with the Type I PRMT inhibitor GSK3368715 for up to 28 days.19 It is therefore timely to investigate any side effects of PRMT inhibitors on platelet function. Furthermore, the extent and role of ArgMe has not been explored in platelets. The aims of the present paper are (1) to reveal the relevance of ArgMe in platelets by describing the platelet “arginine methylome” and (2) to investigate the effect of ArgMe inhibition on platelet function by incubating platelets with PRMT inhibitors.
Results
The Platelet Arginine Methylome Reveals Key Roles in Platelet Function
To gain an understanding of the scope of protein ArgMe in platelets, we systematically searched available proteomics data sets of platelets for ArgMe. We searched the ProteomeXchange repository for the term “platelet” and identified 13 ProteomeXchange data sets (PXD) suitable for further analysis (Figure 1A and Suppl. Table S1). Using our recently published proteomics workflow,21 we identified proteins modified by ArgMe in each of the 13 projects. We then defined the platelet arginine methylome as those ArgMe sites that were common to three or more projects and we counted 96 sites in 64 proteins that fulfilled this criterium (Suppl. Table S2), including in each of the two components of the αIIbβ3 receptor.
Figure 1.

Revealing the platelet arginine methylome. (A) Decision pipeline toward a systematic analysis of ProteomeXchange data sets of the platelet proteome. (B) STRING analysis of the platelet arginine methylome showing highly interconnected protein–protein interaction networks. Included in this panel are the proteins that were interconnected (54 out of 64); for a full list, see Suppl. Table S2. ITGA2B and ITGB3 in the middle of the panel are the genes coding for the αIIb and β3 components of the fibrinogen receptor, respectively.
Analysis of GO terms enriched in this subset of 64 proteins against the platelet proteome background (8527 proteins identified in the 13 PXD projects analyzed) revealed enrichment in GO terms associated with platelet activation (Suppl. Table S3). GO terms of platelet aggregation, adhesion, degranulation, exocytosis, and secretion, to name a few, were strongly and specifically enriched in the platelet arginine methylome compared with the whole platelet proteome, indicating relevant roles of ArgMe in platelet function. STRING analysis of the arginine methylome mapped out highly interconnected protein networks (Figure 1B). VASP was identified in only 2 PXD projects and was therefore not included in our arginine methylome. Given the central role of VASP in platelet signaling, we nevertheless immunoprecipitated VASP from human platelets and found that, first, VASP was recognized by anti-ArgMe antibodies (Suppl. Figure S1) and, second, mass spectrometry analysis identified a novel ArgMe site at VASP R10 (Suppl. Figure S1 and Suppl. Table S4).
ArgMe Can Be Inhibited in Platelets
To provide direct evidence that platelet proteins can be modified by ArgMe, we first tested for expression of the enzyme responsible for S-adenosylmethionine (SAM) synthesis (SAM synthase, MAT2A) and of PRMT1 in platelets. We observed bands at the expected molecular weights (MW), (Figure 2A, left). We also searched for protein ArgMe using Western blot, and we routinely observed several protein bands recognized by antibodies specific for mono- and di-ArgMe in platelet lysates (Figure 2A, right), although the identity of these proteins remains unknown.
Figure 2.

Platelet proteins are modified by ArgMe and incubation of platelets with PRMT inhibitors reduces ArgMe levels. (A) Detection of MAT2A (expected MW: 43.6 kDa), PRMT1 (expected MW of the main isoform: 42.6 kDa), and ArgMe in platelet lysates using Western blot. (B) Incubation of platelets for 4 h, but not 2 h, with 1 mM AMI-1 led to ArgMe inhibition. (C) Time-course of ArgMe inhibition by 10 μM furamidine, showing reduced ArgMe after 2–3 h of incubation. (D) Dose–response of ArgMe signals with increasing concentrations of furamidine, after a 4-h incubation period. (E) Dose–response of VASP phosphorylation at Ser-157 with increasing concentrations of AMI-1, after a 4-h incubation period. Representative blot (n = 3) and quantification of pS157-VASP relative to actin are shown. Stars indicate statistical significance (* p < 0.05).
We then incubated platelets with Type I PRMT inhibitors in vitro, and we observed reduced protein ArgMe profiles after 3–4 h incubation with μM concentrations of furamidine and AMI-1 (Figure 2B,C and Suppl. Figure S2). For this reason, subsequent functional experiments (see below) were done following incubation of platelets with Type I PRMT inhibitors for 3–4 h. We observed a dose–response relationship in the inhibition of ArgMe, shown by reduced intensity of ArgMe bands with increasing concentrations of furamidine (Figure 2D). Antibodies #8015 and #8711 were raised against different epitopes, and it was therefore not surprising that they seemed to recognize different proteins, which was also shown in the original publication.22 Incubation of platelets with PRMT inhibitors led to increased VASP phosphorylation at S157, a marker of platelet quiescence, although at lower levels than those observed after incubation with established platelet inhibitors such as PGI2 (Figure 2E).
Inhibition of ArgMe Impairs Platelet Aggregation
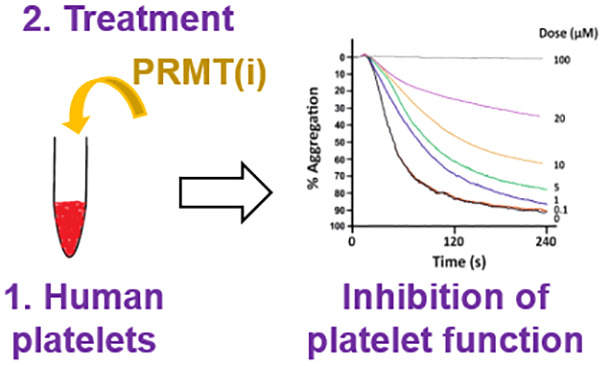
Based on the GO term enrichment analysis of our platelet arginine methylome and on our biochemical data, we hypothesized that ArgMe plays a role in platelet aggregation. To test this hypothesis, we performed platelet aggregometry experiments in the presence of increasing amounts of PRMT1 inhibitors. We chose GSK3368715 because it is currently in clinical trials (identifier: NCT03666988).19 We also used furamidine, AMI-1 and MS023 as Type I PRMT inhibitors that have been developed in preclinical studies and used widely.23,24
We found dose-dependent inhibition of platelet aggregation after incubation of platelets with Type I PRMT inhibitors in vitro for 4 h. We observed impaired platelet aggregation at low μM furamidine doses in response to thrombin (Figure 3A) or collagen (Suppl. Figure S3). We calculated IC50 values for the inhibition of platelet aggregation after thrombin stimulation in the low to mid μM range for furamidine, GSK3368715, MS023 (Figure 3B,C), and AMI-1 (Suppl. Figure S4). Because the strongest effects were observed with furamidine, subsequent experiments were performed using this Type I PRMT inhibitor.
Figure 3.

Furamidine causes a dose-dependent inhibition of platelet aggregation. (A) Effect of furamidine on platelet aggregation stimulated by thrombin (0.1 U/ml). Platelet suspensions were incubated for 4 h with the indicated concentrations of furamidine prior to stimulation. Traces are average of at least 6 independent experiments. (B,C) Visualization of and calculated IC50 values for furamidine, MS023 and GSK3368715. (D) The total inhibition of platelet aggregation by 80 μM furamidine (F) can be partly recovered by removing the inhibitor. Negative (-ve) control is platelets incubated with DMSO for 4 h at 37 °C. Positive (+ve) control is platelets incubated with furamidine (80 μM) for 4 h. Note the recovery of ca. 33.5% aggregation in platelets incubated first with 80 μM furamidine for 2 h and then with DMSO for 2 h, compared with platelets incubated first with DMSO and then with furamidine. Individual data for four different donors are shown. Stars indicate statistical significance (** p < 0.005).
Importantly, the inhibition of platelet aggregation by furamidine was at least partly reversible. Indeed, incubation of platelets with a maximum inhibitory dose of furamidine (80 μM) for 2 h, followed by 2 h incubation in the absence of furamidine, recovered an average of 33.5 ± 7.9% platelet aggregation across four donors (Figure 3D, see also Suppl. Figure S5 for representative aggregation curves). The reversibility of furamidine effects strongly suggested that platelets remained active and functional during incubation. To further support this view, we first analyzed lactate dehydrogenase (LDH) release from platelets incubated with furamidine. LDH analysis showed that platelet integrity was maintained at all concentrations assayed (Suppl. Figure S6). Second, we visualized mitochondria in platelets, and we found no changes in mitochondrial membrane potential in platelets treated with 80 μM furamidine (Suppl. Figure S7).
Furamidine Treatment Leads to Altered Platelet Spreading Phenotype
To assess for any effect of furamidine on platelet morphology, we assayed spreading on fibrinogen or collagen coated surfaces. We observed only very small and donor-dependent differences in total platelet numbers or cell surface area in samples treated with furamidine, with a trend toward slightly increased adhesion to fibrinogen and collagen with increased doses of the inhibitor (Figure 4A–D). Consistent with this, treated platelets spread mainly through stress fibers and lamellipodia (78%) with a minority of actin nodules (16%), compared to a combined phenotype (33% stress fibers and 61% actin nodules) in untreated platelets (Figure 4E).
Figure 4.

Mild changes in spreading after incubation of platelets with furamidine. (A–D) Number of platelets and platelet surface area on spreading onto fibrinogen (A,B) or collagen (C,D) in the absence (control) or in the presence of increasing concentrations of furamidine for 4 h. Data are shown as the mean ± SD (error bars), with the data points from each donor (n = 3 donors for fibrinogen and n = 4 donors for collagen, five slides analyzed from each donor) linked to make donor-specific comparisons. Donors are color coded. Statistical comparisons are shown between samples from the same donor. Stars indicating statistical relevance (* p < 0.05; ** p < 0.005) are colored according to the donor where any difference between treated and control samples was found to be significant (ANOVA with post hoc Tukey tests). (E) Representative images of platelets spreading on fibrinogen after treatment with a range of furamidine concentrations (4 h); all images are from the same donor. Inset: visualization of spreading phenotype: actin nodules (dark gray), stress fibers (light gray), and mixed phenotype or nonclassifiable (white).
Expression and Activation of Key Platelet Receptors Changes upon Treatment with Furamidine
To identify any changes in expression or activation of key platelet proteins upon treatment with PRMT inhibitors that could explain the observations above, we performed flow cytometry experiments using platelets incubated with low furamidine concentrations (10 μM). There were no changes in the expression of the collagen receptors integrin α-2 (CD49b) and glycoprotein VI (GPVI) (Figure 5A). Consistent with the latter, phosphor-tyrosine (pY) signaling was not significantly affected by furamidine (Suppl. Figure S8). However, we observed significant (although small) reductions in the expression of the α-chain of the vWF binding complex GPIb (CD42b) and the critical integrin αIIb (CD41) (Figure 5A). We tested the effect of furamidine on the activation of the αIIbβ3 receptor by a range of agonists and found that furamidine impaired αIIbβ3 activation by thrombin, collagen related peptide (CRP), ADP, and U46619, an agonist of the thromboxane receptor (Figure 5B).
Figure 5.

Furamidine treatment leads to changes in receptor expression and granule secretion. Analysis of the effect of 10 μM furamidine on the expression of (A) key platelet receptors, (B) activated αIIbβ3 receptor, (C) dense granule secretion, and (D) alpha granule secretion. Colors and joined lines denote pairs of control and furamidine-treated platelets and white and gray bars are control and furamidine (at 10 μM for 4 h) experiments, respectively. Data are shown as the mean ± SD (error bars), with the data points from each donor (n = 6 donors) color-coded and linked to make pairwise comparisons. Stars indicate statistical significance (* p < 0.05; ** p < 0.005, paired Student’s t test).
We also quantified secretion of dense and alpha granules following platelet activation, measured by CD63 and P-selectin, respectively. Both experiments showed a general decrease in antibody binding following furamidine incubation, and this decrease was statistically significant when platelets were stimulated with low and high doses of CRP (Figure 5C,D). Taken together, our results show that Type I PRMT inhibitors affect expression and activation of critical receptors and impair platelet function.
Discussion
Over the past few years, several groups have begun to identify the set of proteins modified by ArgMe, that is, the arginine methylome, in various cells and tissues;22,25−28 however, the extent and role of ArgMe in platelets has not yet been investigated. Our work indicates that many proteins are modified by ArgMe in platelets. From that standpoint, we decided to investigate the effect of ArgMe inhibition on platelet function. PRMT inhibitors have been hailed as novel, promising drugs for the treatment of several cancer types. This is based on remarkable success in the synthesis of specific inhibitors and on encouraging preclinical and clinical models of disease,14,29−31 which have led to clinical trials by major pharmaceutical companies. The research community has appreciated the need to monitor any side-effects of these emerging class of drugs,32 and our work raises the need to consider platelet inhibition as a possible off-target effect of the systemic administration of PRMT inhibitors in ongoing and subsequent clinical trials.
Incubation of platelets with PRMT inhibitors for 4 h led to reduced levels of global ArgMe as judged by western analysis. This suggests that there is PRMT activity in washed platelets and that PRMT inhibitors can permeate platelet membranes relatively rapidly. We tested several PRMT1 (or more generally Type I PRMT) inhibitors including AMI-1, MS023, GSK3368715, and furamidine. We expected that MS023 and GSK3368715 would inhibit ArgMe in platelets at the lowest concentrations, given their reported IC50 values against PRMT1 (30 and 3.1 nM, respectively).19,24 However, the largest effect on platelet aggregation was observed in the presence of low μM concentrations of furamidine, which has a reported PRMT1 IC50 value of 9.4 μM.23 We hypothesize that this is due to active transport of furamidine into platelets through the organic cation transporter 1,33 which is expressed in platelets.34 Platelets cannot be maintained in cell culture and rapidly lose viability in vitro, which limits the time that washed platelets can be incubated with PRMT inhibitors. An obvious question that needs to be addressed in the future is the effect of systemic administration of therapeutic doses of PRMT1 inhibitors on platelet function in animal models and clinical trials; especially given previous, limited reports of in vivo hematological effects of PRMT inhibitors in the μM range.19 One would expect that continued oral administration of PRMT inhibitors, for example, of GSK3368715, for the duration of weeks may lead to steady-state concentration inside short-lived platelets and consequently ArgMe inhibition. Full-body and tissue-specific PRMT knockouts (KO) have been generated and present a wide range of defects, from embryonic lethality (PRMT1 and −5 KO) to only minor phenotypes;35 however, to our knowledge platelet dysfunction has not been reported in these models. It may be possible that mice platelets are less affected by PRMT downregulation or inhibition and it would be interesting to generate platelet specific PRMT1 KOs in the future to shed light onto this standing matter.
While acknowledging the limitations of working with washed human platelets in vitro, we report moderate changes to platelet aggregation and mild changes to spreading and expression of key platelet receptors at low doses of furamidine. We observed impaired platelet aggregation in response to both thrombin and collagen. This is consistent with a decreased expression of αIIb at the cell membrane and reduced activation of the αIIbβ3 receptor after incubation of platelets with 10 μM furamidine, both at basal conditions and when platelets were stimulated with agonists (thrombin, CRP, ADP and a thromboxane analogue). Of note, both components of the αIIbβ3 receptor were found to be methylated in platelets, and it is tempting to speculate that inhibition of ArgMe may jeopardize αIIbβ3 docking at the cell membrane. It is known, for example, that ArgMe of certain membrane proteins, such as ion channels, stimulates protein trafficking to the cell membrane.36,37 Alternatively, ArgMe may be associated with receptor stability or heterodimerization. This can occur for instance through mediating protein–protein interactions as it has been suggested for the endothelial growth factor receptor (EGFR),38 or through cross-talk with neighboring modifications. For example, Y660 and K669 in β3 are modified by phosphorylation and ubiquitination, respectively,39,40 and lie in the vicinity of R659, which we have identified as methylated, and there are many examples of cross-talk between ArgMe and neighboring post-translational modifications that can affect protein interactions, localization, and stability.14 In any event and given the central role of the αIIbβ3 receptor in platelet function, further investigations of αIIbβ3 ArgMe are warranted. Some ArgMe sites, including those in both components of the αIIbβ3 receptor, were identified in extracellular domains. While less commonly reported than intracellular ArgMe sites, it is accepted that extracellular receptor domains, for example in EGFR, can be extensively methylated by PRMT1, presumably at the endoplasmic reticulum/Golgi compartments.38,41,42
We also observed a general trend toward a mild decrease of dense and alpha granule secretion after furamidine treatment, as judged by CD63 and P-selectin exposure. This is consistent with the inhibition of platelet aggregation,43 and also with the enrichment in the platelet arginine methylome of GO terms associated with platelet degranulation and secretion. We show very mild effects of furamidine on platelet adhesion. We observed a primed platelet phenotype upon treatment with furamidine, with a decrease in the number of actin nodules and an increase in the number of stress fibers. This is consistent with the enrichment in GO terms associated with the actomyosin cytoskeleton and stress fibers in the platelet arginine methylome. Proteins with these GO terms in the arginine methylome include myosins, tropomyosins, and actinins. We report minor changes in the number of platelets and platelet surface area, but only in a couple of donors and furamidine doses. To bring together the significant effects of furamidine on platelet aggregation and on receptor expression with the mild spreading phenotype, we reason that furamidine must affect signaling downstream of the receptors. There are potentially many pathways that could be affected (for example, cyclic nucleotide dependent signaling, CalDAG-GEFI, Src family tyrosine kinases, cytoskeletal substrates) and decreased receptor levels may not fully explain the observed phenotype. For instance, platelet spreading on fibrinogen could be maintained in the context of reduced αIIbβ3 activity if ArgMe inhibition enhances GPVI activation by immobilized fibrinogen.44 The combination of moderate effects of furamidine on platelet aggregation and a mild trend toward increasing platelet adhesion may be of interest toward repurposing PRMT inhibitors as potential, novel antiplatelet agents. However, this idea comes with its own caution note: it is conceivable that PRMT inhibitors also affect the morphology, signaling, and function of other cell entities, and these effects cannot currently be predicted, especially for long-term treatments.
Furamidine has been used as a specific PRMT1 inhibitor at low μM concentrations.23,45−48 Furamidine is also known to have other effects including as an antiprotozoal agent,49 DNA binding, and inhibitor of tyrosyl-DNA phosphodiesterase 1.50 These alternative activities of furamidine are unlikely to be relevant in the context of washed or circulating platelets, and furamidine treatment reduced the intensity of ArgMe bands, which supports the idea that the reported inhibition of platelet function is due to direct inhibition of PRMT activity. Furamidine is thought to bind to the substrate pocket on PRMT1 by mimicking the side-chain guanidino group of arginine with involvement of hydrogen bonding and electrostatic interactions23,51 and thus competes with the substrate and reversibly inhibits PRMT1. Consistent with this, the effects of maximal inhibitory doses of furamidine on platelet aggregation were at least partially reversed after a short wash-off.
Taken together, our results open new avenues for research on ArgMe in platelets. First, platelets, being anucleate cells, are a good model for research on ArgMe outside of the nucleus, and our data show that tens of proteins, including critical platelet receptors, are modified by ArgMe in human platelets. Second, there may be an opportunity to develop inhibitors of specific ArgMe sites of platelet proteins (e.g., αIIbβ3) with possible therapeutic applications. Third, we recommend that current and future clinical trials using PRMT inhibitors consider the investigation of platelet function and include the monitoring of possible adverse effects related to platelet inhibition.
Materials and Methods
Ethics Statement
This work was completed in accordance with the University of Hull and Hull York Medical School (HYMS) ethical guidelines. Work with human blood samples, including platelets, was approved by the HYMS ethics committee and was completed under the ‘project 1501: The study of platelet activation, signaling and metabolism’. All participants gave their informed consent prior to their inclusion in the study and the study conformed to the Declaration of Helsinki.
Bioinformatics Analysis of ProteomeXchange Data Sets
The bioinformatic analysis of PXD projects was completed following published protocols.21 Briefly, all proteomic raw data in ProteomeXchange as for May 2020 were systematically searched using the following inclusion criteria: (1) inclusion of platelet proteomic data and (2) the data being from human samples. Exclusion criteria were: (1) data sets where enrichment e.g. phosphoenrichment had been performed and (2) unclear labeling of files. Datasets that fulfilled the study criteria were downloaded and mined for protein ArgMe using MaxQuant (v1.6.14.0). ArgMe was set as a variable modification together with Met oxidation and N-terminal protein acetylation. Cys carbamidomethylation was set as a fixed modification. MaxQuant parameters were left as default in searches against the human proteome downloaded from Uniprot (April 2020). The platelet arginine methylome was defined as those ArgMe sites identified in three or more PXD projects, after manual curation of the data to remove likely contaminants (e.g., keratins, immunoglobulins). Enrichment in gene ontology (GO) terms in the platelet arginine methylome was assessed using GOrilla,52 using the platelet proteome as background set and the platelet arginine methylome as the target set. STRING analysis of protein networks was done online (https://string-db.org/) using high confidence interaction scores (0.7) and all available interaction sources.53
PRMT Inhibitors and Reagents
MS023 and furamidine were purchased from Tocris, AMI-1 was from Sigma-Aldrich, and GSK3368715 was purchased from Cambridge Bioscience. Stock solutions of all ArgMe inhibitors were at 50 mM concentration in DMSO. Thrombin, ADP, fibrinogen, and FITC-conjugated phalloidin were from Sigma-Aldrich. Collagen was purchased from Takeda. MitoTracker red CMXRos was from ThermoFisher Scientific. Other reagents were from Sigma-Aldrich unless indicated otherwise.
Platelet Isolation and Incubation with PRMT Inhibitors
Blood (20–80 mL) was collected from healthy donors through venepuncture into acid citrate dextrose (29.9 mM trisodium citrate, 113.8 mM glucose, 72.6 mM NaCl, 2.9 mM citric acid, pH 6.4) and centrifuged at 190g for 15 min at room temperature to obtain platelet rich plasma, as previously reported.54 Platelets were pelleted further by centrifugation at 800g for 12 min in the presence of 6 mM citric acid, washed (0.036 M citric acid, 0.01 M EDTA, 0.005 M glucose, 0.005 M KCl, 0.09 M NaCl, pH 6.5) and centrifuged as before. Pelleted platelets were resuspended in modified Tyrode’s buffer (150 mM NaCl, 5 mM HEPES, 0.55 mM NaH2PO4, 7 mM NaHCO3, 2.7 mM KCl, 0.5 mM MgCl2, 5.6 mM glucose, pH 7.4) and maintained at 37 °C for 30 min in preparation for experiments. Platelets were incubated with PRMT inhibitors or vehicle (DMSO) for 3–4 h at 37 °C unless indicated otherwise.
Platelet Aggregation
Platelet aggregation in response to agonists was recorded at a concentration of 3 × 108 platelets/ml under constant stirring conditions (1000 rpm) for 4 min at 37 °C using Born aggregometry.11,55 For the reversibility experiments, we incubated platelets with 80 μM furamidine (or vehicle) for 2 h and then on ice for 5 min. Platelets were then centrifuged for 50 s at 3600g. The supernatant was removed, and the platelets were resuspended in modified Tyrode’s buffer at room temperature. Only one wash was performed. Platelets were then incubated at 37 °C for the remaining 2 h with vehicle or 80 μM furamidine as appropriate. Aggregations were completed with 0.1 U/ml thrombin or 10 μg/mL collagen.
Platelet Spreading
For platelet spreading analysis, untreated or furamidine-treated platelets (2 × 107/ml) were spread on fibrinogen or collagen (both at 100 μg/mL) coated coverslips at 37 °C for the specified times. Platelets were fixed in 4% paraformaldehyde solution for 10 min and then permeabilized in 0.1% Triton X-100 for 5 min. Slides were stained with FITC-phalloidin for 1 h for F-actin visualization, washed, and imaged on a Zeiss Axio Imager fluorescence microscope with a 63× 1.4NA oil immersion objective.54,56 Platelet numbers, actin nodule/stress fiber ratios, and surface areas were measured using ImageJ (NIH).
Western Blot
Platelet lysates (30–60 μg in 1% SDS) were resolved by 10–12% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were probed with antibodies targeting mono-ArgMe (#8015 and #8711, Cell Signaling Technologies); di-ArgMe (#13522, Cell Signaling Technologies); VASP, α-MAT2A, and α-PRMT1 (Abcam); and phosphor-tyrosine (#05-321, Sigma), as appropriate, with GAPDH or β-actin (Thermo) as loading control. Blots were developed using enhanced chemiluminescence reagents (Luminata Forte, Millipore), and signals were visualized and captured on a ChemiDoc (Bio-Rad). Blots shown are representative of at least two independent experiments. Quantitative data drawn from Western blots are derived from at least n = 3 experiments. Densitometry analysis was done using ImageJ (NIH).
Mass Spectrometry Analysis of VASP
VASP was immunoprecipitated from platelets using specific antibodies (#ab109321, Abcam), and the immunoprecipitate was resolved through SDS-PAGE. The putative Coomassie-stained VASP band was digested with Lys-C protease after reduction with dithioerythritol and alkylation with iodoacetamide. The resulting peptides were analyzed by LC-MS/MS using an Orbitrap Fusion mass spectrometer with elution from a 50 cm PepMap column over a 35 min gradient. All MS/MS samples were analyzed using Mascot (version 2.6.1) and X! Tandem (version CYCLONE 2010.12.01.1). Searches was set up against the UniProt human FASTA database (June 2014, 20259 entries) using Lys-C as the protease. Mascot and X! Tandem were searched with a fragment ion mass tolerance of 0.50 Da and a parent ion tolerance of 3.0 ppm. Cys carbamidomethylation was specified in Mascot and X! Tandem as a fixed modification. Deamidation of Asn and Gln, methylation of Arg, oxidation of Met, and acetylation of protein N-termini were specified in Mascot as variable modifications. Glu → pyro-Glu, ammonia-loss, acetylation and Gln → pyro-Glu of the N-termini, deamidation of Asn and Gln, methylation of Arg and oxidation of Met were specified in X! Tandem as variable modifications. Peptide and protein identifications were filtered in Scaffold (version Scaffold_4.8.2) to require a global false discovery rate of <1% at both the protein and peptide level. Protein matches required a minimum of two unique peptide identifications. Peptide identifications were accepted if they could be established at greater than 90.0% probability to achieve an FDR less than 1.0% by the Scaffold Local FDR algorithm.
Flow Cytometry
For flow cytometry analysis of platelet receptors, platelets were incubated with 10 μM furamidine for a period of 4 h before they were fixed (20% paraformaldehyde, BD Phosflow) and labeled with antibodies. The antibodies were from BioLegend against the following targets: CD49b (#359309), GPVI (clone HIM3-4, #303302), CD42b (clone HIP1, #303903), CD41 (clone A2A9/6, #359805), active αIIbβ3 receptor (PAC1, #362803), CD63 (clone H5C6, 353005), and P-selectin (clone AK4, #304905). Analysis was completed using a LSR FortessaTM from BD Biosciences, and data were analyzed with FlowJo.
LDH Assays
Lactate dehydrogenase (LDH) assays were done following the instructions of the cytotoxicity detection KitPLUS (LDH) (Roche). Briefly, platelets were lysed in 1% Triton X-100 in 96-well plates (n = 3 independent experiments, 2 technical replicates); next, the LDH reaction mixture (diaphorase, NAD+ and sodium lactate) was added to each well, and the plate was incubated at 37 °C for 30 min in the dark. The reactions were stopped by 1 M HCl, and the plate was read at 490 nm with a correction of 680 nm using a spectrophotometer.
Statistical Analysis
Data were analyzed using paired student’s t tests (flow cytometry experiments), or one-way ANOVA (with post hoc Tukey for pairwise comparisons) as appropriate, with statistical significance defined as p < 0.05. Stars in figures define statistical significance.
Acknowledgments
A.J.M. acknowledges a University of Hull Ph.D. scholarship within the Biomarkers cluster. D.R.J.R. was a recipient of a British heart Foundation PhD scholarship FS/15/46/31606. J.S.K. was a recipient of a University of Hull Ph.D. scholarship. We acknowledge HEIF (HEFCE) funding to F.R. and P.B.A. and a Yorkshire Brain Tumour Charity Ph.D. studentship to A.B. and P.B.A. We thank former and current members of the Beltran-Alvarez and Rivero groups and of the Centre for Atherothrombosis and Metabolic Disease (Hull York Medical School), especially Simon Calaminus, Lloyd Atkinson, and Pooja Joshi. Mass spectrometry analysis of VASP was performed at the Centre of Excellence in Mass Spectrometry at the University of York (Adam Dowle, Tony Larson). We thank Kath Bulmer for outstanding technical assistance.
Glossary
Abbreviations:
- (ADP)
adenosine diphosphate
- (ArgMe)
arginine methylation
- (LDH)
lactate dehydrogenase
- (PGI2)
prostacyclin
- (PRMT)
protein arginine methyltransferase
- (vWF)
von Willebrand factor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00135.
VASP is modified by ArgMe in human platelets; incubation of platelets with 1 mM AMI-1 for 4 h leads to inhibition of protein ArgMe; furamidine causes the dose-dependent inhibition of platelet aggregation stimulated by collagen; AMI-1 inhibits platelet aggregation; representative example of raw aggregation curves in reversibility experiments; LDH assay demonstrates platelet integrity upon incubation with increasing concentrations of furamidine; furamidine does not impair mitochondrial membrane potential; furamidine does not lead to changes in phosphor-tyrosine (pY) signaling; identity of the 13 PXD projects included in our bioinformatics analysis; curated list of proteins modified by ArgMe in platelets, that is, the arginine methylome; GO enrichment analysis in the subset of proteins modified by ArgMe in three or more PXD projects; mass spectrometry identification of VASP and R10 methylation (PDF)
Author Contributions
F.R. and P.B.A. designed the research. N.K., B.G., F.R., and P.B.A. critically contributed funding, materials, and expertise including software for analysis of platelet spreading (NTK). A.J.M., D.R.J.R., J.S.K., A.B., F.R., and P.B.A. performed the research. A.J.M., D.R.J.R., A.B., B.G., F.R., and P.B.A. analyzed data. A.J.M., F.R., and P.B.A. wrote the paper.
The authors declare the following competing financial interest(s): FR and PBA are authors of patent application P031106WO: Arginine methylation inhibitors as novel antiplatelet agents, January 2018.
This paper was published ASAP on August 9, 2021, with a version of the Supporting Information that was missing four tables. The SI was corrected and reposted August 11, 2021.
Special Issue
Published as part of the ACS Pharmacology & Translational Science special issue “Epigenetics 2022”.
Supplementary Material
References
- Kaushansky K. (2005) The molecular mechanisms that control thrombopoiesis. J. Clin. Invest. 115 (12), 3339–3347. 10.1172/JCI26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Hoffmeister K. M.; Falet H. (2016) Glycans and the platelet life cycle. Platelets 27 (6), 505–511. 10.3109/09537104.2016.1171304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romaniuk M. A.; Croci D. O.; Lapponi M. J.; Tribulatti M. V.; Negrotto S.; Poirier F.; Campetella O.; Rabinovich G. A.; Schattner M. (2012) Binding of galectin-1 to αIIbβ3 integrin triggers “outside-in” signals, stimulates platelet activation, and controls primary hemostasis. FASEB J. 26 (7), 2788–98. 10.1096/fj.11-197541. [DOI] [PubMed] [Google Scholar]
- Ruggeri Z. M.; Mendolicchio G. L. (2007) Adhesion mechanisms in platelet function. Circ. Res. 100 (12), 1673–85. 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- van den Kerkhof D. L.; van der Meijden P. E. J.; Hackeng T. M.; Dijkgraaf I. (2021) Exogenous Integrin αIIbβ3 Inhibitors Revisited: Past, Present and Future Applications. Int. J. Mol. Sci. 22 (7), 3366. 10.3390/ijms22073366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice P.; Legrand C.; Fauvel-Lafeve F. (2004) Platelet adhesion and signaling induced by the octapeptide primary binding sequence (KOGEOGPK) from type III collagen. FASEB J. 18 (12), 1339–47. 10.1096/fj.03-1151com. [DOI] [PubMed] [Google Scholar]
- Eikelboom J. W.; Hirsh J.; Spencer F. A.; Baglin T. P.; Weitz J. I. (2012) Antiplatelet drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 141 (2 Suppl), e89S–e119S. 10.1378/chest.11-2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talasaz A. H.; Sadeghipour P.; Kakavand H.; Aghakouchakzadeh M.; Kordzadeh-Kermani E.; Van Tassell B. W.; Gheymati A.; Ariannejad H.; Hosseini S. H.; Jamalkhani S.; Sholzberg M.; Monreal M.; Jimenez D.; Piazza G.; Parikh S. A.; Kirtane A. J.; Eikelboom J. W.; Connors J. M.; Hunt B. J.; Konstantinides S. V.; Cushman M.; Weitz J. I.; Stone G. W.; Krumholz H. M.; Lip G. Y. H.; Goldhaber S. Z.; Bikdeli B. (2021) Recent Randomized Trials of Antithrombotic Therapy for Patients With COVID-19: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 77 (15), 1903–1921. 10.1016/j.jacc.2021.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez D.; García-Sanchez A.; Rali P.; Muriel A.; Bikdeli B.; Ruiz-Artacho P.; Le Mao R.; Rodríguez C.; Hunt B. J.; Monreal M. (2021) Incidence of VTE and Bleeding Among Hospitalized Patients With Coronavirus Disease 2019: A Systematic Review and Meta-analysis. Chest 159 (3), 1182–1196. 10.1016/j.chest.2020.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara D.; Tarafdar A.; Celikag M.; Patinha D.; Gulacsy C. E.; Hounslea E.; Warren Z.; Ferreira B.; Koeners M. P.; Caggiano L.; Pula G. (2020) NADPH oxidase 1 is a novel pharmacological target for the development of an antiplatelet drug without bleeding side effects. FASEB J. 34 (10), 13959–13977. 10.1096/fj.202001086RRR. [DOI] [PubMed] [Google Scholar]
- Aburima A.; Berger M.; Spurgeon B. E. J.; Webb B. A.; Wraith K. S.; Febbraio M.; Poole A. W.; Naseem K. M. (2021) Thrombospondin-1 promotes hemostasis through modulation of cAMP signaling in blood platelets. Blood 137 (5), 678–689. 10.1182/blood.2020005382. [DOI] [PubMed] [Google Scholar]
- Hubberstey A. V.; Mottillo E. P. (2002) Cyclase-associated proteins: CAPacity for linking signal transduction and actin polymerization. FASEB J. 16 (6), 487–99. 10.1096/fj.01-0659rev. [DOI] [PubMed] [Google Scholar]
- Romero S.; Le Clainche C.; Gautreau A. M. (2020) Actin polymerization downstream of integrins: signaling pathways and mechanotransduction. Biochem. J. 477 (1), 1–21. 10.1042/BCJ20170719. [DOI] [PubMed] [Google Scholar]
- Samuel S. F.; Barry A.; Greenman J.; Beltran-Alvarez P. (2021) Arginine methylation: the promise of a ’silver bullet’ for brain tumours?. Amino Acids 53 (4), 489–506. 10.1007/s00726-020-02937-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain K.; Clarke S. G. (2019) PRMT7 as a unique member of the protein arginine methyltransferase family: A review. Arch. Biochem. Biophys. 665, 36–45. 10.1016/j.abb.2019.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurita-Lopez C. I.; Sandberg T.; Kelly R.; Clarke S. G. (2012) Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming ω-NG-monomethylated arginine residues. J. Biol. Chem. 287 (11), 7859–70. 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.; Frankel A.; Cook R. J.; Kim S.; Paik W. K.; Williams K. R.; Clarke S.; Herschman H. R. (2000) PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 275 (11), 7723–30. 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- Wright T.; Wang Y.; Bedford M. T. (2021) The Role of the PRMT5-SND1 Axis in Hepatocellular Carcinoma. Epigenomes. 2021 Mar 5 (1), 2. 10.3390/epigenomes5010002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoriw A.; Rajapurkar S. R.; O’Brien S.; Gerhart S. V.; Mitchell L. H.; Adams N. D.; Rioux N.; Lingaraj T.; Ribich S. A.; Pappalardi M. B.; Shah N.; Laraio J.; Liu Y.; Butticello M.; Carpenter C. L.; Creasy C.; Korenchuk S.; McCabe M. T.; McHugh C. F.; Nagarajan R.; Wagner C.; Zappacosta F.; Annan R.; Concha N. O.; Thomas R. A.; Hart T. K.; Smith J. J.; Copeland R. A.; Moyer M. P.; Campbell J.; Stickland K.; Mills J.; Jacques-O’Hagan S.; Allain C.; Johnston D.; Raimondi A.; Porter Scott M.; Waters N.; Swinger K.; Boriack-Sjodin A.; Riera T.; Shapiro G.; Chesworth R.; Prinjha R. K.; Kruger R. G.; Barbash O.; Mohammad H. P. (2019) Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 36 (1), 100–114.e25. 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- Fong J. Y.; Pignata L.; Goy P. A.; Kawabata K. C.; Lee S. C.; Koh C. M.; Musiani D.; Massignani E.; Kotini A. G.; Penson A.; Wun C. M.; Shen Y.; Schwarz M.; Low D. H.; Rialdi A.; Ki M.; Wollmann H.; Mzoughi S.; Gay F.; Thompson C.; Hart T.; Barbash O.; Luciani G. M.; Szewczyk M. M.; Wouters B. J.; Delwel R.; Papapetrou E. P.; Barsyte-Lovejoy D.; Arrowsmith C. H.; Minden M. D.; Jin J.; Melnick A.; Bonaldi T.; Abdel-Wahab O.; Guccione E. (2019) Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 36 (2), 194–209.e9. 10.1016/j.ccell.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onwuli D. O.; Rigau-Roca L.; Cawthorne C.; Beltran-Alvarez P. (2017) Mapping arginine methylation in the human body and cardiac disease. Proteomics: Clin. Appl. 11 (1–2), 1600106. 10.1002/prca.201600106. [DOI] [PubMed] [Google Scholar]
- Guo A.; Gu H.; Zhou J.; Mulhern D.; Wang Y.; Lee K. A.; Yang V.; Aguiar M.; Kornhauser J.; Jia X.; Ren J.; Beausoleil S. A.; Silva J. C.; Vemulapalli V.; Bedford M. T.; Comb M. J. (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell Proteomics. 13 (1), 372–87. 10.1074/mcp.O113.027870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L.; Yan C.; Qian K.; Su H.; Kofsky-Wofford S.; Lee W.; Zhao X.; Ho M.; Ivanov I.; Zheng Y. G. (2014) Diamidine compounds for selective inhibition of protein arginine methyltransferase 1. J. Med. Chem. 57 (6), 2611–2622. 10.1021/jm401884z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eram M. S.; Shen Y.; Szewczyk M.; Wu H.; Senisterra G.; Li F.; Butler K. V.; Kaniskan H.Ü.; Speed B. A.; Dela Seña C.; Dong A.; Zeng H.; Schapira M.; Brown P. J.; Arrowsmith C. H.; Barsyte-Lovejoy D.; Liu J.; Vedadi M.; Jin J. (2016) A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 11 (3), 772–781. 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan V.; Guo A.; Trudgian D.; Thomas B.; Acuto O. (2015) Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat. Commun. 6, 6758. 10.1038/ncomms7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen S. C.; Sylvestersen K. B.; Mund A.; Lyon D.; Mullari M.; Madsen M. V.; Daniel J. A.; Jensen L. J.; Nielsen M. L. (2016) Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signaling 9 (443), rs9. 10.1126/scisignal.aaf7329. [DOI] [PubMed] [Google Scholar]
- Musiani D.; Bok J.; Massignani E.; Wu L.; Tabaglio T.; Ippolito M. R.; Cuomo A.; Ozbek U.; Zorgati H.; Ghoshdastider U.; Robinson R. C.; Guccione E.; Bonaldi T. (2019) Proteomics profiling of arginine methylation defines PRMT5 substrate specificity. Sci. Signaling 12 (575), eaat8388. 10.1126/scisignal.aat8388. [DOI] [PubMed] [Google Scholar]
- Lim Y.; Lee J. Y.; Ha S. J.; Yu S.; Shin J. K.; Kim H. C. (2020) Proteome-wide identification of arginine methylation in colorectal cancer tissues from patients. Proteome Sci. 18, 6. 10.1186/s12953-020-00162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrold J.; Davies C. C. (2019) PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret?. Trends Mol. Med. 25 (11), 993–1009. 10.1016/j.molmed.2019.05.007. [DOI] [PubMed] [Google Scholar]
- Bryant J. P.; Heiss J.; Banasavadi-Siddegowda Y. K. (2021) Arginine Methylation in Brain Tumors: Tumor Biology and Therapeutic Strategies. Cells 10 (1), 124. 10.3390/cells10010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.; Schapira M.; Arrowsmith C. H.; Barsyte-Lovejoy D. (2021) Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discovery 20 (7), 509–530. 10.1038/s41573-021-00159-8. [DOI] [PubMed] [Google Scholar]
- Hu H.; Qian K.; Ho M. C.; Zheng Y. G. (2016) Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert Opin. Invest. Drugs 25 (3), 335–58. 10.1517/13543784.2016.1144747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X.; Ju W.; Wu H.; Tidwell R. R.; Hall J. E.; Thakker D. R. (2009) Transport of dicationic drugs pentamidine and furamidine by human organic cation transporters. Drug Metab. Dispos. 37 (2), 424–30. 10.1124/dmd.108.024083. [DOI] [PubMed] [Google Scholar]
- Kim M. S.; Pinto S. M.; Getnet D.; Nirujogi R. S.; Manda S. S.; Chaerkady R.; Madugundu A. K.; Kelkar D. S.; Isserlin R.; Jain S.; Thomas J. K.; Muthusamy B.; Leal-Rojas P.; Kumar P.; Sahasrabuddhe N. A.; Balakrishnan L.; Advani J.; George B.; Renuse S.; Selvan L. D.; Patil A. H.; Nanjappa V.; Radhakrishnan A.; Prasad S.; Subbannayya T.; Raju R.; Kumar M.; Sreenivasamurthy S. K.; Marimuthu A.; Sathe G. J.; Chavan S.; Datta K. K.; Subbannayya Y.; Sahu A.; Yelamanchi S. D.; Jayaram S.; Rajagopalan P.; Sharma J.; Murthy K. R.; Syed N.; Goel R.; Khan A. A.; Ahmad S.; Dey G.; Mudgal K.; Chatterjee A.; Huang T. C.; Zhong J.; Wu X.; Shaw P. G.; Freed D.; Zahari M. S.; Mukherjee K. K.; Shankar S.; Mahadevan A.; Lam H.; Mitchell C. J.; Shankar S. K.; Satishchandra P.; Schroeder J. T.; Sirdeshmukh R.; Maitra A.; Leach S. D.; Drake C. G.; Halushka M. K.; Prasad T. S.; Hruban R. H.; Kerr C. L.; Bader G. D.; Iacobuzio-Donahue C. A.; Gowda H.; Pandey A. (2014) A draft map of the human proteome. Nature 509 (7502), 575–81. 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc R. S.; Richard S. (2017) Arginine Methylation: The Coming of Age. Mol. Cell 65 (1), 8–24. 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- Baek J. H.; Rubinstein M.; Scheuer T.; Trimmer J. S. (2014) Reciprocal changes in phosphorylation and methylation of mammalian brain sodium channels in response to seizures. J. Biol. Chem. 289 (22), 15363–73. 10.1074/jbc.M114.562785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran-Alvarez P.; Espejo A.; Schmauder R.; Beltran C.; Mrowka R.; Linke T.; Batlle M.; Pérez-Villa F.; Pérez G. J.; Scornik F. S.; Benndorf K.; Pagans S.; Zimmer T.; Brugada R. (2013) Protein arginine methyl transferases-3 and −5 increase cell surface expression of cardiac sodium channel. FEBS Lett. 587 (19), 3159–65. 10.1016/j.febslet.2013.07.043. [DOI] [PubMed] [Google Scholar]
- Liao H. W.; Hsu J. M.; Xia W.; Wang H. L.; Wang Y. N.; Chang W. C.; Arold S. T.; Chou C. K.; Tsou P. H.; Yamaguchi H.; Fang Y. F.; Lee H. J.; Lee H. H.; Tai S. K.; Yang M. H.; Morelli M. P.; Sen M.; Ladbury J. E.; Chen C. H.; Grandis J. R.; Kopetz S.; Hung M. C. (2015) PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J. Clin. Invest. 125 (12), 4529–43. 10.1172/JCI82826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck P. V.; Zhang B.; Murray B.; Kornhauser J. M.; Latham V.; Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations. Nucleic Acids Res. 43, D512–20. 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimov V.; Barrio-Hernandez I.; Hansen S. V. F.; Hallenborg P.; Pedersen A. K.; Bekker-Jensen D. B.; Puglia M.; Christensen S. D. K.; Vanselow J. T.; Nielsen M. M.; Kratchmarova I.; Kelstrup C. D.; Olsen J. V.; Blagoev B. (2018) UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 25 (7), 631–640. 10.1038/s41594-018-0084-y. [DOI] [PubMed] [Google Scholar]
- Wu C. C.; MacCoss M. J.; Mardones G.; Finnigan C.; Mogelsvang S.; Yates J. R. 3rd; Howell K. E. (2004) Organellar proteomics reveals Golgi arginine dimethylation. Mol. Biol. Cell 15 (6), 2907–19. 10.1091/mbc.e04-02-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein D. M.; Buck E. (2015) Old dog, new tricks: extracellular domain arginine methylation regulates EGFR function. J. Clin. Invest. 125 (12), 4320–2. 10.1172/JCI85001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golebiewska E. M.; Poole A. W. (2015) Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 29 (3), 153–62. 10.1016/j.blre.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangin P. H.; Onselaer M. B.; Receveur N.; Le Lay N.; Hardy A. T.; Wilson C.; Sanchez X.; Loyau S.; Dupuis A.; Babar A. K.; Miller J. L.; Philippou H.; Hughes C. E.; Herr A. B.; Ariëns R. A.; Mezzano D.; Jandrot-Perrus M.; Gachet C.; Watson S. P. (2018) Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica. 103 (5), 898–907. 10.3324/haematol.2017.182972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iderzorig T.; Kellen J.; Osude C.; Singh S.; Woodman J. A.; Garcia C.; Puri N. (2018) Comparison of EMT mediated tyrosine kinase inhibitor resistance in NSCLC. Biochem. Biophys. Res. Commun. 496 (2), 770–777. 10.1016/j.bbrc.2018.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.; Mi Y.; Song J.; Zhang P.; Liu Y. (2018) PRMT1-RBM15 axis regulates megakaryocytic differentiation of human umbilical cord blood CD34+ cells. Exp. Ther. Med. 15 (3), 2563–2568. 10.3892/etm.2018.5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel S. F.; Marsden A. J.; Deepak S.; Rivero F.; Greenman J.; Beltran-Alvarez P. (2018) Inhibiting Arginine Methylation as a Tool to Investigate Cross-Talk with Methylation and Acetylation Post-Translational Modifications in a Glioblastoma Cell Line. Proteomes 6 (4), 44. 10.3390/proteomes6040044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Li X.; Ge J.; Liu M.; Pang X.; Liu J.; Luo C.; Xu Y.; Zhao Q. (2021) The methyltransferase PRMT1 regulates γ-globin translation. J. Biol. Chem. 296, 100417. 10.1016/j.jbc.2021.100417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick D. A.; Bakunov S. A.; Bakunova S. M.; Wenzler T.; Brun R.; Tidwell R. R. (2014) Antiprotozoal activity of dicationic 3,5-diphenylisoxazoles, their prodrugs and aza-analogues. Bioorg. Med. Chem. 22 (1), 559–76. 10.1016/j.bmc.2013.10.050. [DOI] [PubMed] [Google Scholar]
- Antony S.; Marchand C.; Stephen A. G.; Thibaut L.; Agama K. K.; Fisher R. J.; Pommier Y. (2007) Novel high-throughput electrochemiluminescent assay for identification of human tyrosyl-DNA phosphodiesterase (Tdp1) inhibitors and characterization of furamidine (NSC 305831) as an inhibitor of Tdp1. Nucleic Acids Res. 35 (13), 4474–84. 10.1093/nar/gkm463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Qian K.; Yan C.; He M.; Jassim B. A.; Ivanov I.; Zheng Y. G. (2017) Discovery of Decamidine as a New and Potent PRMT1 Inhibitor. MedChemComm 8 (2), 440–444. 10.1039/C6MD00573J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E.; Navon R.; Steinfeld I.; Lipson D.; Yakhini Z. (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinf. 10, 48. 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D.; Franceschini A.; Wyder S.; Forslund K.; Heller D.; Huerta-Cepas J.; Simonovic M.; Roth A.; Santos A.; Tsafou K. P.; Kuhn M.; Bork P.; Jensen L. J.; von Mering C. (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–52. 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley D. R. J.; Khalil J. S.; Naseem K. M.; Rivero F. (2020) Biochemical and immunocytochemical characterization of coronins in platelets. Platelets 31 (7), 913–924. 10.1080/09537104.2019.1696457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley D. R. J.; Khalil J. S.; Pieters J.; Naseem K. M.; Rivero F. (2020) Coronin 1 Is Required for Integrin β2 Translocation in Platelets. Int. J. Mol. Sci. 21 (1), 356. 10.3390/ijms21010356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson L.; Yusuf M. Z.; Aburima A.; Ahmed Y.; Thomas S. G.; Naseem K. M.; Calaminus S. D. J. (2018) Reversal of stress fibre formation by Nitric Oxide mediated RhoA inhibition leads to reduction in the height of preformed thrombi. Sci. Rep. 8 (1), 3032. 10.1038/s41598-018-21167-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.