

Abstract
Background
Tumor mutational burden (TMB) is emerging as a promising biomarker in immune checkpoint inhibitor (ICI) therapy. Despite whole-exome sequencing (WES) being the gold standard for quantifying TMB, TMB is determined by selected targeted panels in most cases, and WES-derived TMB data are lacking due to the greater cost and complexity. Determining TMB thresholds is another issue that needs attention.
Methods
A total of 309 patients who had received ICI therapy, representing five cancers (listed in “Results”), were recruited. Among them, 269 patients were evaluable for survival analysis. Tumor and matched blood samples from the patients were analyzed using WES and somatic mutations were determined. TMB is defined as the total number of somatic nonsynonymous mutations in the tumor exome in our study. The patients were divided into different TMB subgroups according to a common fixed number (10 mutations/Mb) or the top tertile within each tumor type.
Results
The distribution of WES-derived median TMBs was highly variable across different tumor types, ranging from 2.71 (cholangiocarcinoma) to 2.97 (nervous system tumor), 3.69 (gastric cancer), 4.31 (hepatocellular carcinoma), and 4.64 [colorectal cancer (CRC)] mutations/Mb. In CRC, the survival benefit of TMB-high patients was significant using both the top tertile and the 10 mutations/Mb threshold. In hepatocellular carcinoma, the 10 mutations/Mb threshold showed an advantage over the top tertile threshold. Among patients with nervous system tumors, cholangiocarcinoma, and gastric cancer, no obvious survival differences were observed between the TMB-high and TMB-low groups with either TMB stratification approach.
Conclusions
The TMB threshold criterion may vary for different cancers. Our data suggest that TMB is unable to predict ICI benefit across all cancer types in Chinese patients. However, it may be an effective biomarker for predicting the clinical benefit of ICI therapy for patients with CRC.
Keywords: Tumor mutational burden (TMB), whole-exome sequencing (WES), immunotherapy
Introduction
Immune checkpoint inhibitor (ICI) therapy has demonstrated a significant clinical benefit in patients with advanced-stage cancers (1-3). However, the majority of patients who receive ICIs fail to show a therapeutic response. For instance, the overall response rate of non-small-cell lung cancer (NSCLC) to ICIs is only approximately 20% (4). Therefore, effective biomarkers that can better predict the clinical efficacy of ICI therapy are urgently needed.
One of the promising biomarkers under investigation is tumor mutational burden (TMB). Previous studies have shown that the TMB is correlated with neoantigen load (5). Therefore, it is rational to use TMB to predict immunotherapeutic success, and this idea is supported by evidence from clinical studies. For instance, in NSCLC and melanoma, patients with a high TMB are more likely to respond to ICIs than those with a low TMB (6-8). On the other hand, the prognostic effects of TMB in solid tumors varies in patients who do not receive ICI therapy and may depend on tumor type (9). Moreover, the threshold of TMB is different for each tumor type (10). The clinical application potential of high TMB for identifying patients who could benefit from immunotherapy makes it critical to set the threshold appropriately. However, how to set the threshold is an unsettled issue.
Two approaches have been popular in studies reported to date. The first uses an empirical, fixed number (with 10 mut/Mb being the most frequently used number, despite its use lacking a strong theoretic rationale) (11,12), and the second uses a percentage line in a cohort or cancer type (13). Importantly, before the threshold can be set, the TMB has to be measured accurately. Next-generation sequencing (NGS)-based technologies including whole-exome sequencing (WES) and gene panels can be used to quantify TMB. WES can detect all mutations in the whole exome regions, whereas gene panels focus only on selected cancer-related genetic sites and are frequently used in diagnosis. Gene panels represent a lower cost approach; however, TMB determination by panels can sometimes be misleading.
A retrospective study comparing TMB -derived from a 284-gene panel and from WES found that the panel resulted in a three-fold overestimation of TMB compared to WES (14). Another study found a discrepancy in panel-derived TMB data depending on whether a subset of high-TMB samples were included in the dataset or not, and this discrepancy affected the correlation between the panel-derived and WES-derived TMB (15). A previous study established a quantitative correlation model between TMB based on whole exome sequencing and TMB based on targeted panel sequencing, but the samples were human tumor cell lines, rather than actual tumor samples, so the relationship between TMB derived from WES and survival of ICI patients was also lacking (16). Although it is agreed that WES is technologically superior to gene panels and has been regarded as the gold standard in the measurement of TMB, data on WES-derived TMB are lacking, even in several major tumors, due to limitations imposed by the cost and availability of this technique in cancer diagnosis and research. The above issues must be addressed before TMB can serve as an effective biomarker for predicting response across diverse cancer types.
In the present study, we used WES to examine the TMBs of 309 patients across 5 different cancers, with at least 30 eligible cases for each cancer. We aimed to establish the scale of WES-derived TMB in these cancers in Chinese patients and evaluate the value of WES-derived TMB in immunotherapy by examining its association with patient survival.
We present the following article in accordance with the REMARK reporting checklist (available at https://dx.doi.org/10.21037/atm-21-4227).
Methods
Patients
Patients from the First Affiliated Hospital of Sun Yat-sen University who received ICIs as either a monotherapy or combination therapy and had their genomic changes profiled using WES were eligible for inclusion in this study. Immunotherapy agents included nivolumab, atezolizumab, pembrolizumab, ipilimumab, and toripalimab. Tumor type was taken into consideration, as we aimed to enroll at least 30 eligible cases for each cancer. According to this requirement, we screened out five different types of cancer: cholangiocarcinoma, nervous system tumor, gastric cancer, liver cancer and colorectal cancer. The clinical data of the patients were extracted from hospital records or obtained through telephone follow-up. In total, 309 patients were profiled by WES across five tumor types, 269 of whom were evaluable for survival analysis. All procedures performed in this study involving human participants were in accordance with the Declaration of Helsinki (as revised in 2013). This study was approved by the institutional review board of the First Affiliated Hospital of Sun Yat-sen University. All participants signed an informed consent form.
WES analysis
The tumor samples used for WES analysis were formalin-fixed paraffin-embedded (FFPE). Patient-matched peripheral blood samples were stored in Streck blood collection tubes and used as a normal control for germline filtering. DNA was extracted from the FFPE samples using the MagMAX FFPE DNA/RNA Ultra kit (cat# A31881, ThermoFisher, Waltham, MA, USA) and from the peripheral whole blood samples using the Maxwell RSC blood DNA kit (cat# AS1400, Promega, Madison, WI, USA). The recovered DNA was sheared to approximately ~200 bp using a Covaris E220 Focused-ultrasonicator and then hybridized to whole exome probes from the Agilent SureSelect XT Human All Exon V7 kit (cat# 5991-9039, Agilent, Santa Clara, CA, USA). After enrichment, the captured DNA was amplified, end-repaired, and attached to sequencing adapters and barcodes using the SureSelect XT HS and Low Input Library Preparation Kit for ILM (Pre-PCR) (cat# G9704, Agilent, Santa Clara, CA, USA). The libraries were normalized, pooled, and loaded on the Illumina NovaSeq-6000 Sequencing System to generate 150×150-bp paired-end reads. Raw image analysis and base-calling were carried out using Illumina onboard RTA3 program with default parameters.
Variant calling pipeline
After adapter trimming and low-quality read filtering, reads were aligned to the NCBI human genome reference assembly hg19 using the Burrows-Wheeler aligner algorithm and further processed using the Genome Analysis Toolkit (GATK, version 3.5). Regions needing realignment were identified using the GATK Realigner Target Creator. Single-nucleotide variants (SNVs) and indels were determined with MuTect/ANNOVAR/dbNSFP31 and VarscanIndel software, respectively. Somatic mutations in tumors were identified through comparison with peripheral blood from the same patient during mutation calling. The variant-calling thresholds were set at allelic depth (AD) ≥3 and variant allele frequency (VAF) ≥5. The somatic mutations were then filtered and annotated using the Variant Effect Predictor (VEP) package (hg19 version).
TMB calculation
The total number of mutations is defined as the number of somatic nonsynonymous mutations in the tumor exome, including SNVs and indels in the coding regions and splicing sites. The TMB is reported as the number of mutations per megabase (which equals the total number of mutations divided by 35 Mb).
Evaluation of microsatellite instability (MSI)
The MSI score (the number of unstable microsatellite sites/total valid sites) was calculated according to a previously reported method (17). An MSI score of <1% was defined as microsatellite stable (MSS), a score of 1–3.5% was defined as MSI-low (MSI-L), and a score of ≥3.5% was defined as MSI-high (MSI-H) (17).
Statistical analysis
Statistical analyses were performed using R (v3.6.1). Student’s t-test and the Chi-square test were used to assess continuous and categorical variables, respectively. Overall survival (OS) was measured from the date of ICI therapy starting to the date of death or the last follow-up. Patients with no evaluable survival data were excluded from the survival analysis. The Kaplan-Meier method was adopted to construct survival curves and P values were obtained by the log-rank test. P<0.05 was considered significant.
Results
Cohort description
This was a retrospective study of 309 patients who received ICI therapy between 2015 and 2020 and underwent genomic profiling by WES. The median length of follow-up was 12 months. The basic demographic and disease information of the patients is shown in Table 1. The cohort contained 217 (70.22%) males and 92 (29.78%) females, who ranged in age from 7 to 87 years, with a median age of 51 years. The study focused on the following five tumor types: cholangiocarcinoma (69, 22.33%), nervous system tumor (32, 10.36%), gastric cancer (19, 6.15%), hepatocellular carcinoma (111, 35.92%), and colorectal cancer (CRC; 78, 25.24%).
Table 1. Patient characteristics.
| Patient characteristics | Number |
|---|---|
| Sex, n (%) | |
| Male | 217 (70.22) |
| Female | 92 (29.78) |
| Age, median [range] | 51 [7–87] |
| Tumor type, n (%) | |
| Cholangiocarcinoma | 69 (22.33) |
| Nervous system tumor | 32 (10.36) |
| Gastric cancer | 19 (6.15) |
| Hepatocellular carcinoma | 111 (35.92) |
| Colorectal cancer | 78 (25.24) |
TMB distribution across the different cancer types
The WES-derived median TMBs were highly variable between the different tumors, ranging from 2.71 (cholangiocarcinoma) to 2.97 (nervous system tumor), 3.69 (gastric cancer), 4.31 (hepatocellular carcinoma), and 4.64 (CRC) (Figure 1). No significant difference in TMB was observed between the patients when grouped by age (P=0.81, ≤60 vs. >60) or sex (P=0.94, males vs. females). The median WES-derived TMB for the different cancer types was almost the same as the median panel-derived TMB in the literature (18,19).
Figure 1.
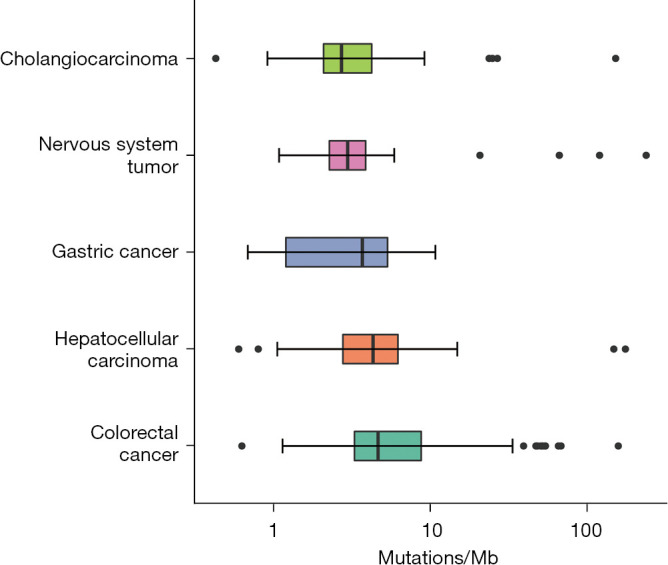
TMB distribution in different cancer types. TMB, tumor mutational burden.
Effects of TMB on OS after anti-PD-L1 therapy
We examined the effects of different approaches to setting the TMB threshold on patient survival prediction. First, the top tertile TMB value in each of the five cancers was used as the divider. Within each cancer type, patients whose TMB values were in the top tertile were defined as the TMB-high group and those in the other two tertiles were defined as the TMB-low group. The OS trends in the TMB-high and TMB-low groups and the separation (the degree of separation of the survival curves) between the two groups varied between the different cancer types. For instance, in cholangiocarcinoma, the TMB-high group showed a survival disadvantage compared to the TMB-low group, although this difference did not reach the level of significance [P=0.13, hazard ratio (HR) =1.75, 95% confidence intervals (CI): 0.76–4.04] (Figure 2A). No obvious survival difference was observed between the TMB-high and TMB-low groups in nervous system tumor (P=0.29, HR =1.82, 95% CI: 0.56–5.93), gastric cancer (P=0.62, HR =0.61, 95% CI: 0.14–2.61), or hepatocellular carcinoma (P=0.90, HR =0.95, 95% CI: 0.40–1.99) (Figure 2B-2D). In CRC, the separation between the TMB-high and TMB-low groups was extremely significant and the TMB-high group showed a survival benefit after receiving ICI treatment [P=0.01, HR =0.31, 95% CI: 0.15–0.68] (Figure 2E).
Figure 2.

Kaplan-Meier curves of patients with five cancer types after ICI treatment, stratified by top tertile TMB value. (A) Cholangiocarcinoma, (B) nervous system tumor, (C) gastric cancer, (D) hepatocellular carcinoma, (E) colorectal cancer. Patients with TMB values in the top tertile were defined as the TMB-high group and the patients in the other two tertiles were defined as the TMB-low group. P value was obtained from two-sided log-rank test. TMB, tumor mutational burden; ICI, immune checkpoint inhibitor.
We next used a fixed number (TMB above or below 10 mutations/Mb) to stratify patients into TMB-high and TMB-low groups. In order to compare the prognosis of patients, patients with no overall survival time were excluded. Overall, the patient numbers in the TMB-high and TMB-low groups were imbalanced when this threshold was used (Table 2). Under this condition, patients with CRC still showed a significant difference between the TMB-high and TMB-low groups (P=0.002, HR =0.14, 95% CI: 0.06–0.31) (Figure 3A). Patients with hepatocellular carcinoma also showed a significant difference between two groups, with the TMB-high group demonstrating an apparent survival disadvantage (P=0.03, HR =2.97, 95% CI: 0.57–15.46) (Figure 3B). Only 4 of 62 (6.45%) patients with cholangiocarcinoma (7 cholangiocarcinoma patients had no overall survival), 4 of 27 (14.81%) patients with nervous system tumor (5 patients with nervous system tumor had no overall survival), and 1 of 16 (6.25%) patient with gastric cancer (3 patients with gastric cancer had no overall survival) had TMB >10 mutations/Mb (Table 2). The imbalance in patient numbers prevented a meaningful statistical comparison between the TMB-high and TMB-low groups, suggesting that the use of a fixed threshold number (TMB >10 mutation/Mb) may not be a good stratification approach for these types of cancers.
Table 2. Numbers and percentages of TMB-high patients within each cancer type with the TMB cutoff set to 10 mutations/Mb.
| Tumor type (number of patients with overall survival)* | Numbers and percentages of TMB-high cases (n, %) |
|---|---|
| Cholangiocarcinoma (n=62) | 4 (6.45) |
| Nervous system tumor (n=27) | 4 (14.81) |
| Gastric cancer (n=16) | 1 (6.25) |
| Hepatocellular carcinoma (n=98) | 6 (6.12) |
| Colorectal cancer (n=66) | 17 (25.76) |
*, to compare patient outcomes with different TMB status, only patients with overall survival were included in this table. TMB, tumor mutational burden.
Figure 3.

Kaplan-Meier curves of patients with two cancer types after ICI treatment, stratified by 10 mutations/Mb TMB value. (A) Colorectal cancer, (B) hepatocellular carcinoma. Patients with >10 mutations/Mb TMB values were defined as the TMB-high group and the other patients were defined as the TMB-low group. P value was obtained from two-sided log-rank test. TMB, tumor mutational burden; ICI, immune checkpoint inhibitor.
Association between MSI status and TMB in CRC
In addition to TMB, MSI status has also been used as a predictive marker in ICI treatment of several cancers. Therefore, we next compared the performances of MSI status and TMB value as predictive markers of ICI therapy.
We first surveyed the proportions of MSI-high patients in the five cancer types (Table 3). When MSI-high was defined as an MSI score >3.5%, as described in a published work (17), no cases of hepatocellular carcinoma or gastric cancer were included in the MSI-high group; 4.2% of patients with cholangiocarcinoma and 6.2% of patients with nervous system tumor were in the MSI-high group. Of the five cancers, CRC had the highest percentage of MSI-high patients (19.2%).
Table 3. Numbers and percentages of MSI-high patients within each cancer type.
| Tumor type | Numbers and percentages of MSI-high cases (n, %) |
|---|---|
| Cholangiocarcinoma | 3 (4.35) |
| Nervous system tumor | 2 (6.25) |
| Gastric cancer | 0 |
| Hepatocellular carcinoma | 0 |
| Colorectal cancer | 15 (19.23) |
MSI, microsatellite instability.
A high MSI score is an effective immunotherapeutic indicator for patients with CRC (20,21). Among the patients with CRC in our cohort, those with MSI-high status had longer OS than those with MSI-low status (Figure 4A). However, only a limited number of patients (~19%) showed a potentially benefit when MSI-high was used as a marker. Next, we analyzed the correlation between MSI status and TMB status in patients with CRC. All MSI-high patients were in TMB-high group (using the top tertile threshold) (Figure 4B). The top tertile threshold was used for TMB stratification in this analysis, as it allowed more patients to be included and to potentially benefit from this marker.
Figure 4.

The correlation between TMB and MSI status in colorectal cancer. (A) Kaplan-Meier curves of patients with three MSI status. (B) Percentage of three MSI status in TMB-H and TMB-L. P value, Kruskal-Wallis test. TMB, tumor mutational burden; MSI, microsatellite instability; MSS, microsatellite stable; MSI-H, MSI-high; MSI-L, MSI-low.
Discussion
In this study, we analyzed the distribution of WES-derived TMBs and examined the association between TMB and OS in Chinese patients who received immunotherapy, across five major cancer types.
This article is a real-world study, so many patients did not evaluate TMB before ICI therapy. This also provides more real information for us to analyze the relationship between TMB and ICI drugs. Our data suggest that the TMB varies widely across the different tumor types (Figure 1). The median WES-derived TMB for the different cancer types was almost the same as the median panel-derived TMB in the literature, although some median TMB values showed a distinct difference between those derived from WES and from a gene panel (18,19). This observation indicates that relative differences in intrinsic DNA mutation potential among different cancers are reflected by the TMB detected by either WES or gene panel. However, it may also be attributable to the inherent defect of gene panels (i.e., small coverage of the genome) and heterogeneity between samples. More WES-derived TMB data should be collected to determine the role of TMB in clinical medication guidance.
Using different threshold criteria (the top tertile or a fixed number of 10 mutations/Mb), we stratified patients and conducted subsequent survival analysis. In CRC, the survival benefit for TMB-high patients was clear using both the top tertile and 10 mutations/Mb threshold (Figures 2A,3A). Because more patients were in the top tertile than met the 10 mutations/Mb threshold, we recommend using this classification method in ICI therapy in CRC to maximize patient inclusion. However, in hepatocellular carcinoma, the top tertile threshold showed a disadvantage over the 10 mutations/Mb threshold. The 10 mutations/Mb threshold could separate patients to two groups with different survival benefit after ICI therapy, whereas the top tertile threshold failed to distinguish patient survival. When a threshold of 10 mutations/Mb was used, the survival benefit was opposite to that in patients with CRC. Among patients with hepatocellular carcinoma, those in the TMB-low group survived longer overall than those in the TMB-high group. In cholangiocarcinoma, nervous system tumor, and gastric cancer, there was no clear trend of survival benefit using either TMB stratification approach.
Considering the high variability in TMB among different tumors, it is rational to define TMB subgroups using a threshold criterion appropriate for individual tumors, rather than a fixed number across all types. In CRC, the survival benefit for TMB-high patients was clear, and the top tertile threshold should be used for maximum patient inclusion in ICI therapy. In hepatocellular cancer, using a fixed number of 10 mutations/Mb could distinguish patient survival. Meanwhile, in cholangiocarcinoma, nervous system tumor, and gastric cancer, a clear survival benefit could not be established in our patient cohort with either TMB threshold criterion. This suggests that TMB may not be an effective marker for patient selection in ICI therapy. However, the small number of patients in our study with these cancer subtypes prevented us from drawing a conclusion.
This is the first study in a Chinese population to describe the correlation between TMB status and OS after ICI therapy across multiple cancers. A strong correlation between TMB and ICI benefit in patients with NSCLC and melanoma has been confirmed by several clinical trials (6-8). However, this correlation has yet to be confirmed in other histological tumor types. Based on the results of KEYNOTE-177 (22), the United States Food and Drug Administration (FDA) approved ICIs for use in the treatment of MSI-high patients with advanced CRC. Following KEYNOTE-158, the FDA also approved the use of TMB as a marker for ICI treatment in solid tumors, which include CRC (23); however, the trial did not include any patients with CRC, which has prompted some controversy among clinicians. McGrail et al.’s meta-analysis using a cohort from The Cancer Genome Atlas reached a similar conclusion to ours, finding that a single TMB threshold had value in selected tumors but the conclusion could not be extended to all solid tumors blindly. Specifically, in CRC, McGrail et al. reported that TMB-high patients may benefit from ICI therapy but the benefit barely meet the minimal requirement of Log odd ratio <0 (24). The data from our single-cohort analysis suggest that TMB-high CRC patients have markedly longer OS than TMB-low patients after immunotherapy (P=0.01).
Conclusions
The TMB threshold criterion may differ for various cancers. Our data suggest that TMB cannot predict ICI benefit across all cancer types in Chinese patients. However, it may be an effective biomarker for predicting clinical benefit to ICI therapy in CRC.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All procedures performed in this study involving human participants were in accordance with the Declaration of Helsinki (as revised in 2013). This study was approved by the institutional review board of the First Affiliated Hospital of Sun Yat-sen University. Informed consent was obtained from all patients.
Reporting Checklist: The authors have completed the REMARK reporting checklist. Available at https://dx.doi.org/10.21037/atm-21-4227
Data Sharing Statement: Available at https://dx.doi.org/10.21037/atm-21-4227
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at https://dx.doi.org/10.21037/atm-21-4227). Dr. FX, Dr. JW, Dr. XZ, and Dr. JL report that they are from Genomicare Biotechnology (Shanghai) Co., Ltd., Shanghai, China. The other authors have no conflicts of interest to declare.
(English Language Editor: J. Reynolds)
References
- 1.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 2015;373:1627-39. 10.1056/NEJMoa1507643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Travert C, Tomasini P, Jeanson A, et al. First-line pembrolizumab in programmed death ligand 1 positive non-small cell lung cancer. Transl Cancer Res 2019;8:2514-6. 10.21037/tcr.2019.05.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg JE, Hoffman-Censits J, Powles T, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 2016;387:1909-20. 10.1016/S0140-6736(16)00561-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443-54. 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chabanon RM, Pedrero M, Lefebvre C, et al. Mutational Landscape and Sensitivity to Immune Checkpoint Blockers. Clin Cancer Res 2016;22:4309-21. 10.1158/1078-0432.CCR-16-0903 [DOI] [PubMed] [Google Scholar]
- 6.Carbone DP, Reck M, Paz-Ares L, et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N Engl J Med 2017;376:2415-26. 10.1056/NEJMoa1613493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Allen EM, Miao D, Schilling B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207-11. 10.1126/science.aad0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Y, Li H, Huang Z, et al. Predictive values of genomic variation, tumor mutational burden, and PD-L1 expression in advanced lung squamous cell carcinoma treated with immunotherapy. Transl Lung Cancer Res 2020;9:2367-79. 10.21037/tlcr-20-1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao D, Xu H, Xu X, et al. High tumor mutation burden predicts better efficacy of immunotherapy: a pooled analysis of 103078 cancer patients. Oncoimmunology 2019;8:e1629258. 10.1080/2162402X.2019.1629258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu HX, Wang ZX, Zhao Q, et al. Tumor mutational and indel burden: a systematic pan-cancer evaluation as prognostic biomarkers. Ann Transl Med 2019;7:640. 10.21037/atm.2019.10.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ready N, Hellmann MD, Awad MM, et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J Clin Oncol 2019;37:992-1000. 10.1200/JCO.18.01042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N Engl J Med 2018;378:2093-104. 10.1056/NEJMoa1801946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51:202-6. 10.1038/s41588-018-0312-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen A, Garner C, Reddy SK, et al. Three-fold overestimation of tumor mutation burden using 248 gene panel versus whole exome. J Clin Oncol 2018;36:12117. 10.1200/JCO.2018.36.15_suppl.12117 [DOI] [Google Scholar]
- 15.Wu HX, Wang ZX, Zhao Q, et al. Designing gene panels for tumor mutational burden estimation: the need to shift from 'correlation' to 'accuracy'. J Immunother Cancer 2019;7:206. 10.1186/s40425-019-0681-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Wang R, Fang H, et al. Influence of low tumor content on tumor mutational burden estimation by whole-exome sequencing and targeted panel sequencing. Clin Transl Med 2021;11:e415. 10.1002/ctm2.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niu B, Ye K, Zhang Q, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014;30:1015-6. 10.1093/bioinformatics/btt755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. 10.1186/s13073-017-0424-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yarchoan M, Albacker LA, Hopkins AC, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019;4:e126908. 10.1172/jci.insight.126908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182-91. 10.1016/S1470-2045(17)30422-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overman MJ, Lonardi S, Wong KYM, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J Clin Oncol 2018;36:773-9. 10.1200/JCO.2017.76.9901 [DOI] [PubMed] [Google Scholar]
- 22.André T, Shiu KK, Kim TW, et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N Engl J Med 2020;383:2207-18. 10.1056/NEJMoa2017699 [DOI] [PubMed] [Google Scholar]
- 23.Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol 2020;21:1353-65. 10.1016/S1470-2045(20)30445-9 [DOI] [PubMed] [Google Scholar]
- 24.McGrail DJ, Pilié PG, Rashid NU, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol 2021;32:661-72. 10.1016/j.annonc.2021.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as