

Abstract
A Western-style, high-fat diet promotes cardiovascular disease, in part because it is rich in choline, which is converted to trimethylamine (TMA) by the gut microbiota. However, whether diet-induced changes in intestinal physiology can alter the metabolic capacity of the microbiota remains unknown. Using a mouse model of diet-induced obesity, we show that chronic exposure to a high-fat diet escalates Escherichia coli choline catabolism by altering intestinal epithelial physiology. A high-fat diet impaired the bioenergetics of mitochondria in the colonic epithelium to increase the luminal bioavailability of oxygen and nitrate, thereby intensifying respiration-dependent choline catabolism of E. coli. In turn, E. coli choline catabolism increased levels of circulating trimethlamine N-oxide, which is a potentially harmful metabolite generated by gut microbiota.
A Western-style, high-fat diet is often associated with cardiovascular disease, and one explanation for this is that members of the gut microbiota catabolize dietary choline into trimethylamine (TMA) (1), which is absorbed in the intestine and oxidized in the liver to trimethylamine N-oxide (TMAO), a metabolite that promotes atherosclerosis (2). A critical but unexplored aspect of the TMAO pathway is how the interaction between diet-impaired host physiology and microbial communities affects TMA production. Gene clusters responsible for TMA production are commonly found in obligately anaerobic Clostridia (phylum Firmicutes) and facultatively anaerobic Enterobacteriaceae (phylum Proteobacteria) (1, 3), but only the latter taxon features a substantial increase in abundance in the feces of individuals on a high-fat diet (4–7). In addition to altering the microbiota composition, a high-fat diet also changes host physiology because saturated fatty acids impair mitochondrial bioenergetics by inducing hydrogen peroxide production in the mitochondria (8, 9). Notably, in the colon, high mitochondrial oxygen consumption is vital for maintaining epithelial hypoxia (10), which preserves anaerobiosis to drive dominance of obligately anaerobic bacteria (11–13) while suppressing growth of facultative anaerobic Enterobacteriaceae (14). Therefore, we wanted to determine whether diet-impaired mitochondrial bioenergetics would escalate microbial choline catabolism by increasing the abundance of Enterobacteriaceae, which was modeled with E. coli.
To address this question, we first investigated whether diet-impaired mitochondrial bioenergetics were responsible for the increased Enterobacteriaceae abundance observed in individuals on a high-fat diet (4–7). We used mice from The Jackson Laboratory (C57BL/6J) that do not carry endogenous Enterobacteriaceae (15), which provided experimental control over this taxon. Inoculation of mice reared on a low- or high-fat diet (10% or 60% fat, respectively) with a single dose of E. coli Nissle 1917 (family Enterobacteriaceae), a commensal human isolate marketed as a probiotic (16), resulted in significantly higher fecal E. coli carriage in the latter group (Fig. 1A). This mirrors an increase in abundance of Enterobacteriaceae in the human fecal microbiota, driven by a high-fat diet (6, 7).
Fig. 1. A high-fat diet changes epithelial physiology to increase the luminal availability of host-derived respiratory electron acceptors.
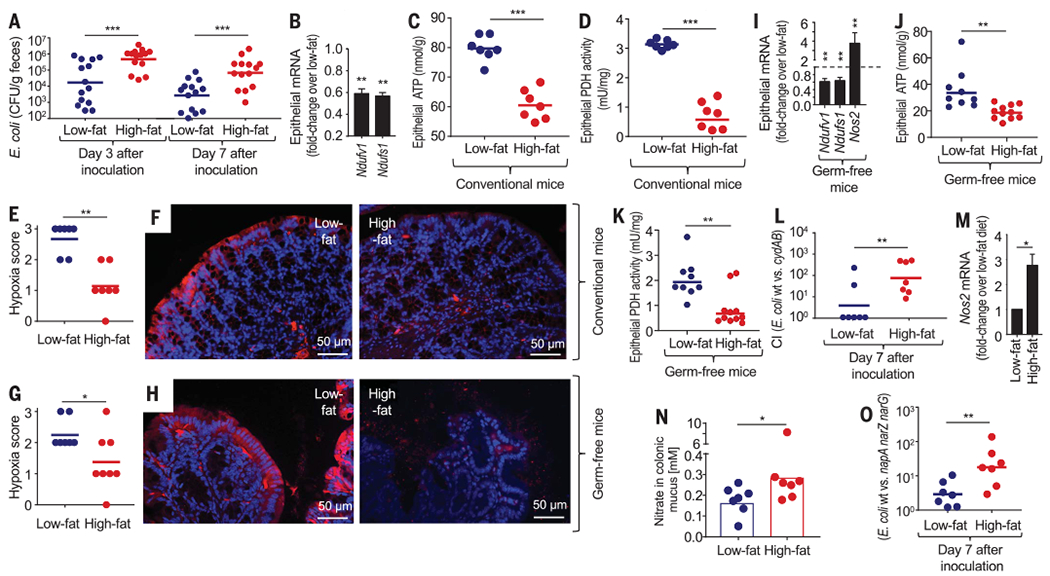
Mice were reared and maintained on a low- or high-fat diet. (A) Mice were inoculated with E. coli strain Nissle 1917, and colony-forming units (CFU) of E. coli Nissle 1917 in feces were determined at the indicated time points. (B to K and M) Preparations of colonic epithelial cells were used to isolate RNA and prepare cell lysates. (B) Fold change in mice on the high-fat diet compared with low-fat diet controls in epithelial transcripts was determined by quantitative real-time polymerase chain reaction (PCR) for genes encoding nicotinamide adenine dinucleotide and hydrogen (NADH):ubiquinone oxidoreductase core subunit V1 (Ndufv1) and NADH:ubiquinone oxidoreductase core subunit S1 (Ndufs1) (n = 6 biological replicates). (C) Cytosolic concentrations of ATP. (D) Cytosolic concentrations of pyruvate dehydrogenase (PDH) activity. (E to H) Mice were injected with pimonidazole 1 hour before euthanasia. Binding of pimonidazole was detected with hypoxyprobe-1 primary antibody and a Cy-3 conjugated goat anti-mouse secondary antibody (red fluorescence) in the sections of proximal colon that were counterstained with DAPI (4′,6-diamidino-2-phenylindole) nuclear stain (blue fluorescence). (E) Pimonidazole staining was quantified by scoring blinded sections of proximal colon from conventional mice. (F) Representative images of colonic sections from conventional mice are shown. (G) Pimonidazole staining was quantified by scoring blinded sections of proximal colon from germ-free mice. (H) Representative images of colonic sections from germ-free mice are shown. (I) Epithelial transcripts of the indicated genes determined by quantitative real-time PCR in samples from germ-free mice (n = 6 biological replicates). (J) Cytosolic concentrations of ATP in germ-free mice. (K) Cytosolic concentrations of PDH activity in germ-free mice. (L) Mice were inoculated with a 1:1 mixture of E. coli strain Nissle 1917 (wt); an isogenic cydAB mutant and CFU were determined at the indicated time point to calculate the competitive index (CI). (M) Fold change in epithelial Nos2 transcripts was determined by quantitative real-time PCR. Bars represent geometric means ± geometric error (n = 6). (N) Nitrate concentrations were determined in colonic mucus. (O) Mice were inoculated with a 1:1 mixture of E. coli strain Nissle 1917 (wt) and an isogenic napA narG narZ mutant and CFU were determined at the indicated time point to calculate the CI. (A, C to E, G, I, K, and L) Each dot represents data from one animal (biological replicate). *P < 0.05; ** P < 0.01; ***P < 0.001 using an unpaired two-tailed Student’s t test [(A) to (D) and (I) to (O)] or a one-tailed Mann-Whitney test [(E) and (G)].
Increased abundance of Enterobacteriaceae in fecal microbiota has been linked to mucosal inflammation (17) and shifts in epithelial metabolism (14); as a result, we investigated diet-induced mucosal responses in mice. Weight gain induced by a high-fat diet (fig. S1A) was associated with low-grade mucosal inflammation characterized by a reduction in colon length (fig. S1B), epithelial metaplasia (fig. S1C), an increased number of mitotic figures in the colonic epithelium (fig. S1, D and E), and reduced numbers of goblet cells (fig. S1, F and G), which was consistent with previous observations (18). A high-fat diet triggered similar responses in germ-free (Swiss Webster) mice (fig. S1, H to K), suggesting that the generation of low-grade mucosal inflammation was microbiota independent. In line with previous reports on saturated fatty acid–mediated impairment of mitochondrial bioenergetics (8, 9), a high-fat diet was associated with reduced mitochondrial activity in the epithelium, as indicated by diminished expression of mitochondrial markers in mRNA isolated from colonic epithelial cells (Fig. 1B), as well as reduced levels of adenosine triphosphate (ATP) (Fig. 1C) and pyruvate dehydrogenase in the colonic epithelium (Fig. 1D). A high-fat diet is rich in saturated fatty acids, which have been implicated in diet-impaired mitochondrial bioenergetics (8, 9). Consistent with the role of saturated fatty acids in reducing mitochondrial bioenergetics in the epithelium, palmitate treatment diminished expression of mitochondrial genes (fig. S2A) and reduced mitochondrial ATP production (fig. S2, B and C) in a human colonic epithelial (Caco-2) cancer cell line in vitro.
To investigate whether mitochondrial bioenergetics impaired by a high-fat diet were linked to increased epithelial oxygenation in the colon, we visualized epithelial hypoxia with the exogenous hypoxic marker pimonidazole, which is reduced under hypoxic conditions to hydroxylamine intermediates that irreversibly bind to nucleophilic groups in proteins or DNA (19, 20). Pimonidazole staining revealed that for mice on a low-fat diet, the colonic epithelial surface was hypoxic, but hypoxia was eliminated in mice receiving a high-fat diet (Fig. 1, E and F). Consistent with the idea that saturated fatty acids act directly on the epithelium (fig. S2), prolonged high-fat intake also eliminated epithelial hypoxia (Fig. 1, G and H), diminished expression of mitochondrial markers in mRNA isolated from colonic epithelial cells (Fig. 1I), reduced ATP levels (Fig. 1J), and decreased levels of pyruvate dehydrogenase in the colonic epithelium (Fig. 1K) in germ-free mice. Taken together, this suggests that the mechanism triggering changes in epithelial oxygenation is microbiota independent.
Mice from The Jackson Laboratory remain Enterobacteriaceae-free because the vendor screens against the presence of this taxon using pathogen-free procedures (15). Because the niche of Enterobacteriaceae remains vacant in mice from The Jackson Laboratory, microbiota assembly can be completed by the designed engraftment of E. coli strains engineered to probe the contribution of predestined metabolic pathways to bacterial growth. We used this approach for precision editing of microbiota to determine the bioavailability of oxygen, a factor linked to growth of Enterobacteriaceae (14, 21–23). This was accomplished by comparing the fitness of the aerobic respiration-proficient E. coli strain Nissle 1917 with a genetically identical (isogenic) strain lacking cytochrome bd-II oxidase (cydAB mutant), an enzyme required for aerobic respiration under microaerophilic conditions (14). The use of these indicator strains to measure the bioavailability of oxygen has been validated in mouse models of antibiotic treatment and chemically induced colitis (14, 23). Increased recovery of an aerobic respiration–proficient wild type (E. coli Nissle 1917) over the aerobic respiration–deficient mutant (cydAB mutant) supported the idea that a high-fat diet increased the bioavailability of oxygen in the intestinal lumen (Fig. 1L and fig. S1L).
Reduced mitochondrial activity in colonic epithelial cells is associated with increased Nos2 expression (14), which was observed in mRNA isolated from the colonic epithelial cells of mice on a high-fat diet (Fig. 1, I and M). The Nos2 gene encodes inducible nitric oxide synthase (iNOS), an enzyme that generates nitric oxide, which is converted into nitrate in the intestinal mucous layer (17). Consistent with this chain of events, a high-fat diet increased the nitrate concentration in the colonic mucus layer of both conventional (Fig. 1N) and germ-free (fig. S1M) mice. To investigate whether a high-fat diet-induced increase in luminal nitrate availability provided a nitrate respiration–mediated fitness advantage for facultative anaerobic Enterobacteriaceae, we compared growth of wild type E. coli Nissle 1917 and an isogenic mutant lacking nitrate reductase activity encoded by the napFDAGHBC, narGHJI, and narZYWV operons (napA narG narZ mutant) (fig. S3A). Inoculation of conventional mice with a 1:1 mixture of both strains resulted in increased recovery of the wild type (E. coli Nissle 1917) over a nitrate respiration-deficient mutant (napA narG narZ mutant), suggesting that nitrate respiration provided a growth advantage for E. coli in mice on a high-fat diet (Fig. 1O and fig. S1N). Collectively, these data indicated that the elevated abundance of E. coli induced by a high-fat diet was driven by an increased bioavailability of host-derived respiratory electron acceptors, such as oxygen and nitrate, which fueled E. coli proliferation.
The cut gene cluster is present in various Enterobacteriaceae (1, 3) and encodes a microcompartment thought to protect the bacterial cell from acetaldehyde, a toxic intermediate generated by choline TMA-lyase (Fig. 2A). Orthologous microcompartments are used for the breakdown of ethanolamine and 1,2-propanediol by some Enterobacteriaceae, but catabolism of these substrates in the mouse intestine requires the presence of respiratory electron acceptors such as tetrathionate, nitrate, or oxygen (24, 25). Therefore, we wanted to investigate whether a high-fat diet-induced increase in the availability of respiratory electron acceptors would escalate the production of TMA in E. coli microcompartments. To this end, we used E. coli strain MS 200-1, which encodes the cut gene cluster involved in converting choline into TMA, acetate, and ethanol (26) (Fig. 2A). E. coli strain MS 200-1 was stably engrafted in mice from The Jackson Laboratory (C57BL6/J), resulting in fecal shedding at levels similar to those observed for a murine E. coli isolate (Mt1b1) or fecal shedding of endogenous Enterobacteriaceae from Charles River (C57BL6/NCrl) mice (fig. S4A). A high-fat diet increased fecal carriage of E. coli strain MS 200-1 (fig. S4B) by promoting growth with oxygen (fig. S4C) and nitrate (fig. S4D and fig. S3B) as electron acceptors. The cutC gene, encoding choline TMA-lyase, provided no growth benefit (Fig. 2B) or only a modest growth benefit (Fig. 2C) when E. coli strain MS 200-1 was cultured under conditions that mimicked the gut environment [i.e., growth in minimal medium supplemented with choline under microaerophilic (1% oxygen) or under anaerobic conditions, respectively]. Further, the presence of nitrate markedly enhanced cutC-dependent growth on choline as a carbon source (Fig. 2, B and C), which was associated with elevated expression of the cut genes (Fig. 2, D and E). Nitrate respiration-dependent growth of the cutC mutant on choline could be restored by introducing the cloned cutC gene on a plasmid (Fig. 2F and fig. S4E). The induction of cut gene expression by nitrate was further enhanced when E. coli was cultured under microaerobic conditions, compared with being cultured anaerobically (Fig. 2G and fig. S4, F and G). However, a nitrate respiration–deficient mutant (E. coli MS 200-1 napA narG narZ mutant) was unable to grow in minimal medium supplemented with choline and nitrate (Fig. 2H and fig. S4H), suggesting that growth on choline was dependent on nitrate respiration. These observations predicted that a high-fat diet–induced increase in the luminal concentration of host-derived nitrate would escalate choline catabolism by E. coli strain MS 200-1 in the digestive tract.
Fig. 2. E. coli choline catabolism requires nitrate respiration in vitro.
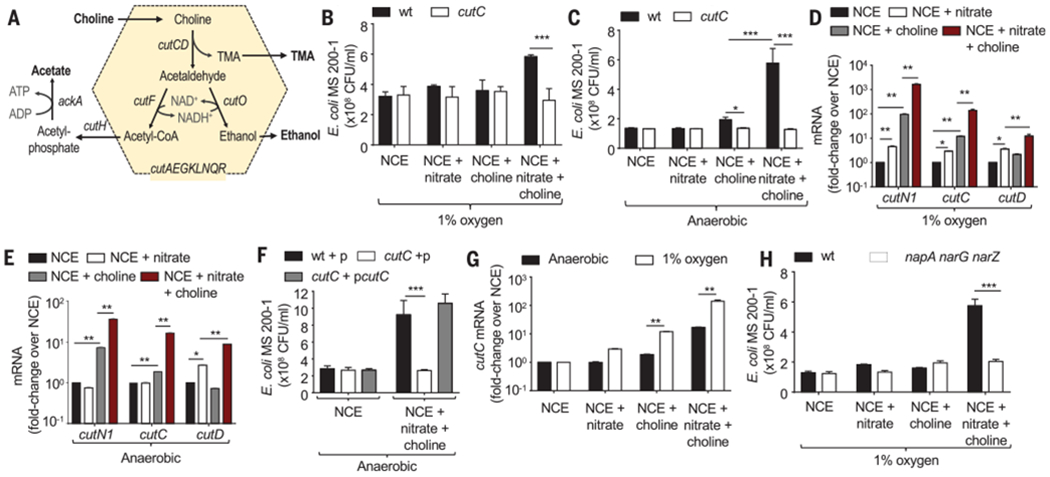
(A) Schematic of choline catabolism encoded by the cut gene cluster of E. coli MS 200-1. (B and C) In vitro growth of E. coli MS 200-1 (wt) and an isogenic cutC mutant in no-carbon essential (NCE) medium supplemented with the indicated nutrients in a hypoxia chamber with 1% oxygen (B) or in an anaerobic chamber (C). (D and C) Expression of the indicated genes was determined by quantitative real-time PCR in RNA isolated from E. coli MS 200-1, which was grown in the indicated media in a hypoxia chamber with 1% oxygen (D) or in an anaerobic chamber (E). (F) In vitro growth in an anaerobic chamber of E. coli MS 200-1 carrying a cloning vector (wt+p), a cutC mutant carrying a cloning vector (cutC+p), and a cutC mutant complemented with the cloned cutC gene (cutC+pcutC) in NCE medium supplemented with the indicated nutrients. (G) Expression of cutC was determined by quantitative real-time PCR in RNA isolated from E. coli MS 200-1 grown under the indicated conditions. (H) In vitro growth of E. coli MS 200-1 (wt) and an isogenic napA narG narZ mutant in NCE medium supplemented with the indicated nutrients, in a hypoxia chamber with 1% oxygen. (B to H) Bars represent geometric means ± geometric error. n = 4 biological replicates (average of triplicate technical replicate per biological replicate). *, P < 0.05; **, P < 0.01; ***, P < 0.001 using an unpaired two-tailed Student’s t test [(B), (C), (G), and (H)] or a one-way analysis of variance (ANOVA) followed by Tukey’s HSD test [(D) to (F)].
To test this idea in vivo, mouse diets were supplemented with 1% choline, because the high-fat diet used in previous experiments (Fig. 1) contained only trace amounts of this nutrient (<0.1%). Mice were reared on a choline-supplemented low-fat (10% fat) diet or on a choline-supplemented high-fat (60% fat) diet and then inoculated with E. coli strain MS 200-1. Rearing animals on a choline-supplemented high-fat diet was associated with increased weight gain (fig. S5A), reduced colon length (fig. S5B), low-grade mucosal inflammation (fig. S5C), an increased number of mitotic figures in the colonic epithelium (fig. S5D), loss of epithelial hypoxia (fig. S5, D and E), and elevated fecal shedding of E. coli (Fig. 3A). The choline-supplemented high-fat diet–induced proliferation of E. coli was respiration dependent, as it was no longer observed when the ability to respire nitrate or oxygen under microaerobic conditions was genetically ablated (Fig. 3, A to C). The presence of 1% choline in the diet provided a cutC-dependent growth advantage for E. coli in mice on a high-fat diet but not in animals on a low-fat diet (Fig. 3D), suggesting that changes in the gut environment induced by a high-fat diet were necessary for E. coli to catabolize choline. To determine whether a high-fat diet–induced increase in the luminal nitrate concentration was required for the cutC-mediated growth advantage, mice were treated with aminoguanidine hydrochloride (AG), an inhibitor of the host enzyme iNOS. The choline-supplemented high-fat diet increased the nitrate concentration in the mucus layer, which was abrogated in mice receiving the AG treatment (Fig. 3E). The AG treatment eliminated the growth advantage conferred upon E. coli by cutC-mediated choline utilization (Fig. 3F), suggesting that the high-fat diet enhanced choline catabolism by elevating the availability of host-derived nitrate.
Fig. 3. Host-derived electron acceptors license E. coli choline catabolism in vivo.
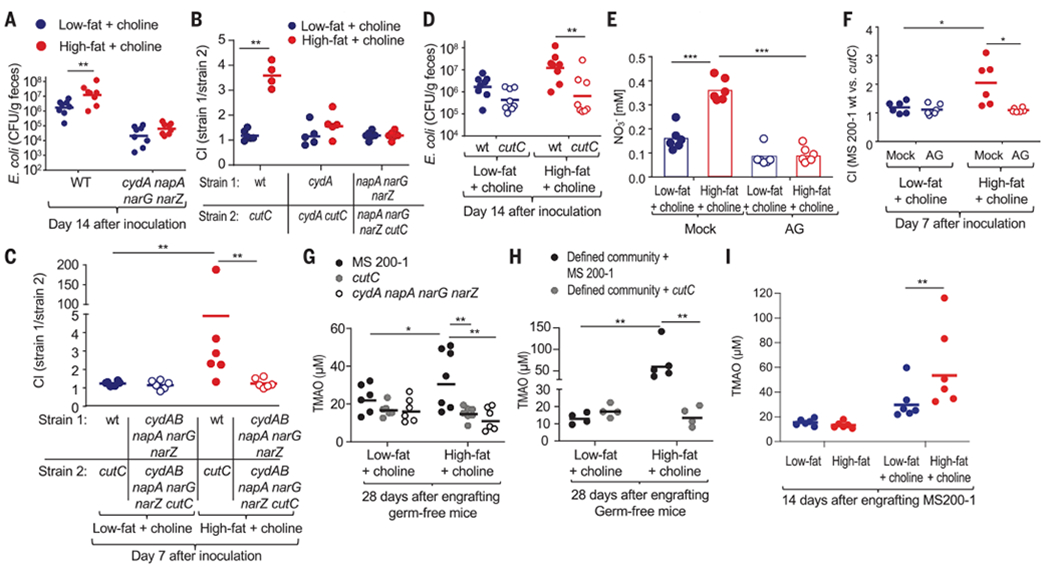
Mice were reared and maintained on a low-fat diet supplemented with 1% choline or a high-fat diet supplemented with 1% choline unless indicated otherwise. (A) Mice (C57BL/6J) were inoculated with E. coli strain MS 200-1 (wt) or an isogenic cydA napA narG narZ mutant, and CFU of E. coli in the feces were determined. (B and C) Mice (C57BL/6J) were inoculated with the indicated E. coli MS 200-1 strain mixtures, and the CI in the feces was determined 14 (B) or 7 (C) days later. (D) Mice (C57BL/6J) were inoculated with E. coli strain MS 200-1 (wt) or an isogenic cutC mutant, and CFU of E. coli in the feces were determined. (E and F) Mice (C57BL/6J) were mock-treated or received drinking water supplemented with the iNOS-inhibitor aminoguanidine (AG). (E) Nitrate concentrations were determined in colonic mucus. (F) Mice were inoculated with a 1:1 mixture of E. coli strain MS 200-1 (wt); an isogenic cutC mutant and CFU of each E. coli strain in the feces were determined to calculate the CI. (G) Germ-free (Swiss Webster) mice were mono-associated with the indicated E. coli strains, and TMAO levels in the plasma were determined 28 days later. (H) Germ-free (Swiss Webster) mice were engrafted with a defined microbial community containing either E. coli strain MS 200-1 or an isogenic cutC mutant. TMAO levels in the plasma were determined 28 days later. (I) Conventional (C57BL/6J) mice were engrafted with E. coli strain MS 200-1 and maintained on the indicated diet. TMAO levels in the plasma were determined 14 days later. (A to I) Each dot represents data from one animal (biological replicate). *P < 0.05; **P < 0.01; ***P < 0.001 using an unpaired two-tailed Student’s t test [(A) to (F), (H), and (I)] or a one-way ANOVA followed by Tukey’s HSD test (G).
To investigate whether E. coli choline catabolism would alter circulating TMAO levels, germ-free mice were mono-associated with the E. coli MS 200-1 wild type, an isogenic cutC mutant, or an isogenic cydA napA narG narZ mutant (fig. S5G). TMAO plasma levels were significantly increased in mice mono-associated with the E. coli MS 200-1 wild type compared with the other groups, which supported our hypothesis that a high-fat diet escalates E. coli choline catabolism in vivo by supporting bacterial respiration (Fig. 3G). Next, we engrafted germ-free mice with a defined microbial community composed of cutC-negative bacteria (Bacteroides thetaiotaomicron, Bacteroides caecimuris, Limosilactobacillus reuteri, Clostridium innocuum, Clostridioides mangenotti, Clostridium cochlearium, and Clostridium sporogenes) (fig. S6) and either the E. coli MS 200-1 wild type or a cutC mutant (fig. S5H). TMAO plasma levels were significantly increased in mice engrafted with the defined microbial community containing a cutC-positive bacterium (E. coli MS 200-1 wild type) compared with the other groups (Fig. 3H), thus further corroborating the idea that a high-fat diet escalates E. coli choline catabolism in vivo. Since TMA producers are naturally present in conventional mouse microbiota (27), we wanted to determine whether engrafting mice with a single TMA-producing E. coli strain would measurably increase TMAO levels in the plasma during a high-fat diet. Compared with low-fat diet controls, inoculation of mice on the choline-supplemented high-fat diet with E. coli MS 200-1 resulted in an increased abundance of the cutC gene in the microbiota (1) (fig. S5I), an increased abundance of E. coli (fig. S5J), and a higher TMAO plasma level (Fig. 3I and fig. S5K). This increase in TMAO plasma levels was dependent on E. coli respiration, because it was no longer observed when mice were engrafted with an isogenic cydA mutant or an isogenic napA narG narZ mutant (fig. S5K). Collectively, these data suggested that an increased availability of host-derived electron acceptors promoted E. coli choline catabolism, so that this species was able to singlehandedly elevate levels of TMAO in the plasma of mice on the high-fat diet.
Finally, we wanted to determine whether treatment with drugs that reduce the availability of host-derived respiratory electron acceptors would lower circulating TMAO levels in mice engrafted with E. coli MS 200-1. Inhibition of host iNOS activity by AG treatment blunted growth of E. coli MS 200-1 in mice on the choline-supplemented high-fat diet (Fig. 4A) and reduced circulating TMAO levels (Fig. 4B). Next, we targeted peroxisome proliferator–activated receptor gamma (PPAR-γ), a nuclear receptor that maintains epithelial hypoxia and suppresses Nos2 expression in the colonic epithelium (14). To this end, mice were treated with 5-aminosalicylic acid (5-ASA), a PPAR-γ agonist (28) that activates mitochondrial bioenergetics specifically in the intestinal epithelium (29). In mice on a choline-supplemented high-fat diet, 5-ASA treatment restored epithelial hypoxia (Fig. 4, C and D), and diminished Nos2 expression in mRNA isolated from colonic epithelial cells (Fig. 4E). Furthermore, 5-ASA treatment of mice on a choline-supplemented high-fat diet abrogated the fitness advantage conferred by cutC-mediated choline catabolism in E. coli MS 200-1 (Fig. 4, F and G). The 5-ASA treatment blunted the increase in circulating TMAO in mice on the choline-supplemented high-fat diet (Fig. 4H).
Fig. 4. Restoring normal epithelial physiology blunts an E. coli–induced increase in circulating TMAO levels.
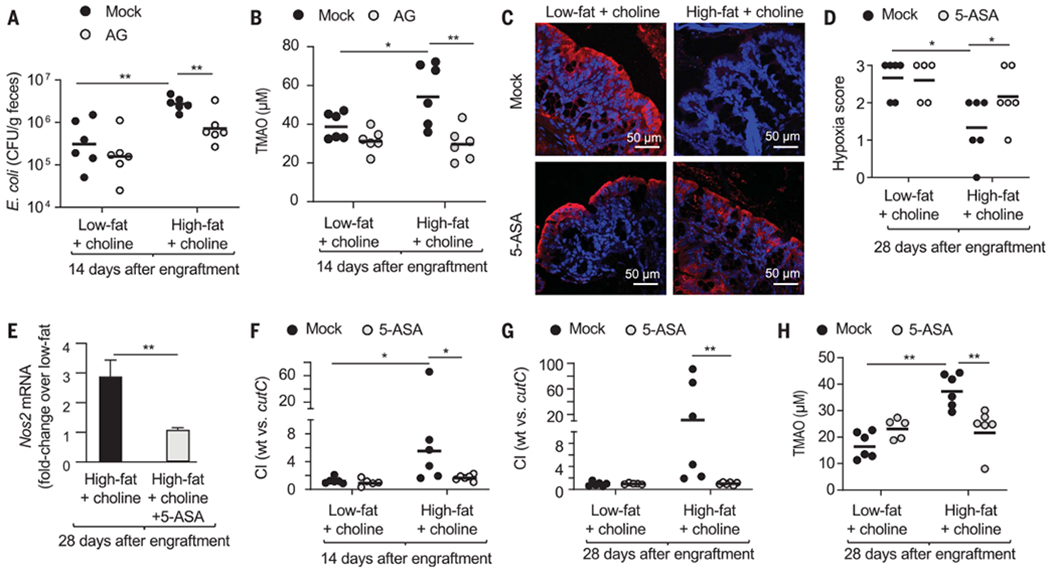
(A and B) Mice (C57BL/6J) maintained on a low- or high-fat diet supplemented with 1% choline were engrafted with E. coli MS 200-1 and received drinking water supplemented with AG or no supplementation (mock). (A) CFU of E. coli in the feces were determined. (B) TMAO levels in the plasma were determined. (C to H) Mice (C57BL/6J) maintained on a low- or high-fat diet supplemented with 1% choline were engrafted with a mixture of E. coli MS 200-1 wild type (wt) and cutC mutant, and received chow supplemented with 5-ASA or no 5-ASA supplementation (mock). (C and D) Mice were injected with pimonidazole one hour before euthanasia. Binding of pimonidazole was detected using hypoxyprobe-1 primary antibody and a Cy-3 conjugated goat anti-mouse secondary antibody (red fluorescence) in the sections of proximal colon that were counterstained with DAPI nuclear stain (blue fluorescence). (C) Representative images of colonic sections from conventional mice are shown. (D) Pimonidazole staining was quantified by scoring blinded sections of proximal colon from conventional mice. (E) Fold change in epithelial Nos2 transcripts was determined by quantitative real-time PCR. Bars represent geometric means ± geometric error. (F and G) The CI was determined 14 (F) and 28 (G) days after engraftment. (H) TMAO levels in the plasma were determined. (A, B, D, and F to H) Each dot represents data from one animal (biological replicate). *P < 0.05; **P < 0.01 using an unpaired two-tailed Student’s t test [(A), (B), and (E)to (H)] or a one-tailed Mann-Whitney test (D).
Taken together, our data suggest that high-fat diet–induced low-grade mucosal inflammation is associated with diet-impaired mitochondrial bioenergetics in the colonic epithelium, thereby eliminating epithelial hypoxia. The consequent increase in the availability of respiratory electron acceptors, such as oxygen and nitrate, drives the proliferation of E. coli and other Enterobacteriaceae, explaining some of the changes in the composition of microbiota observed in individuals on a high-fat diet (4–7). Our results also suggest that E. coli strains carrying the cut operon catabolize choline only when high-fat diet–mediated changes in host physiology increase the luminal concentration of host-derived nitrate and oxygen. The emerging picture suggests that epithelial hypoxia ensures the microbiota remains beneficial by limiting the availability of respiratory electron acceptors, which limits E. coli choline catabolism. However, a prolonged high-fat diet impairs this host control mechanism, thereby elevating Enterobacteriaceae abundance while simultaneously escalating E. coli choline catabolism to elevate circulating TMAO levels. Dose response meta-analysis implicates this metabolite in increasing the relative risk for all-cause mortality in patients by 7.6% per each 10-μmol/liter increment of TMAO (30).
Supplementary Material
ACKNOWLEDGMENTS
We thank E. Balkus for providing E. coli strain MS 200-1 and A. Goodman for providing Bacteroides thetaiotaomicron strain VPI-5482.
Funding:
W.Y. was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) by the Ministry of Education 2020R1A6A3A03037326. Y.L. was supported by Vaadia-BARD Postdoctoral Fellowship FI-505-2014. E.E.O. was supported by Public Health Service Grant TR001861. C.S. was supported by the Dorothy Beryl and Theodore Roe Austin Pathology Research Fund and T32AI112541. N.J.F. was supported by T32DK007673-07. N.G.S. was supported by T32ES007028-46. E.G. and B.J.B. were supported by the U.S. Department of Agriculture (USDA) Project 2032-51530-025-00D. Work in A.J.B.’s laboratory was supported by USDA/NIFA award 2015-67015-22930; by Crohn’s and Colitis Foundation of America Senior Investigator Award 650976; and by Public Health Service Grants AI044170, AI096528, AI112445, AI112949, AI146432, and AI153069. Work in M.X.B.’s laboratory was supported by V Scholar grant V2020-013 from The V Foundation for Cancer Research, Vanderbilt Digestive Disease Pilot and Feasibility grant P30 058404, ACS Institutional Research Grant IRG-19-139-59, VICC GI SPORE grant P50CA236733, United States-Israel Binational Science Foundation grant 2019136, and Vanderbilt Institute for Clinical and Translational Research Grant VR53102 and VR54267.
Competing interests:
J.C.R. is a founder, scientific advisory board member, and stockholder of Sitryx Therapeutics as well as a member of the scientific advisory boards of Caribou Biosciences and Nirogy Therapeutics. J.C.R. has also consulted for Merck and Pfizer within the past 3 years and has received research support from Incyte Corp., Calithera Biosciences, and Tempest Therapeutics. All other authors declare no competing interests.
Data and materials availability:
All data are available in the main text or supplementary materials.
REFERENCES AND NOTES
- 1.Martínez-del Campo A et al. , mBio 6, e00042–e15 (2015).25873372 [Google Scholar]
- 2.Wang Z et al. , Nature 472, 57–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Y et al. , Proc. Natl. Acad. Sci. U.S.A 111, 4268–4273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Devkota S et al. , Nature 487, 104–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fei N, Zhao L, ISME J. 7, 880–884 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez-Medina M et al. , Gut 63, 116–124 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Anitha M et al. , Cell. Mol. Gastroenterol. Hepatol 2, 328–339 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kakimoto PA, Tamaki FK, Cardoso AR, Marana SR, Kowaltowski AJ, Redox Biol. 4, 375–380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardoso AR, Kakimoto PA, Kowaltowski AJ, PLOS ONE 8, e77088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kelly CJ et al. , Cell Host Microbe 17, 662–671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Litvak Y, Byndloss MX, Bäumler AJ, Colonocyte metabolism shapes the gut microbiota. Science 362, eaat9076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Litvak Y, Bäumler AJ, Immunity 51, 214–224 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Tiffany CR, Bäumler AJ, Am. J. Physiol. Gastrointest. Liver Physiol 317, G602–G608 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byndloss MX et al. , Science 357, 570–575 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Velazquez EM et al. , Nat. Microbiol 4, 1057–1064 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nissle A, DMW-Deutsche Medizinische Wochenschrift 51, 1809–1813 (1925). [Google Scholar]
- 17.Winter SE et al. , Science 339, 708–711 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gulhane M et al. , Sci. Rep 6, 28990 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terada N, Ohno N, Saitoh S, Ohno S, Histochem. Cell Biol 128, 253–261 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Kizaka-Kondoh S, Konse-Nagasawa H, Cancer Sci. 100, 1366–1373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rivera-Chávez F et al. , Cell Host Microbe 19, 443–454 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lopez CA et al. , Science 353, 1249–1253 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cevallos SA et al. , mBio 10, e02244–e19 (2019).31575772 [Google Scholar]
- 24.Thiennimitr P et al. , Proc. Natl. Acad. Sci. U.S.A 108, 17480–17485 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faber F et al. , PLOS Pathog. 13, e1006129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Craciun S, Balskus EP, Proc. Natl. Acad. Sci. U.S.A 109, 21307–21312 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z et al. , Cell 163, 1585–1595 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rousseaux C et al. , Carcinogenesis 34, 2580–2586 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cevallos SA et al. , mBio 12, e03227–e20 (2021).33468700 [Google Scholar]
- 30.Schiattarella GG et al. , Eur. Heart J 38, 2948–2956 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or supplementary materials.