

Abstract
Clinical applications of precision oncology require accurate tests that can distinguish true cancer-specific mutations from errors introduced at each step of next-generation sequencing (NGS). To date, no bulk sequencing study has addressed the effects of cross-site reproducibility, nor the biological, technical and computational factors that influence variant identification. Here we report a systematic interrogation of somatic mutations in paired tumor–normal cell lines to identify factors affecting detection reproducibility and accuracy at six different centers. Using whole-genome sequencing (WGS) and whole-exome sequencing (WES), we evaluated the reproducibility of different sample types with varying input amount and tumor purity, and multiple library construction protocols, followed by processing with nine bioinformatics pipelines. We found that read coverage and callers affected both WGS and WES reproducibility, but WES performance was influenced by insert fragment size, genomic copy content and the global imbalance score (GIV; G > T/C > A). Finally, taking into account library preparation protocol, tumor content, read coverage and bioinformatics processes concomitantly, we recommend actionable practices to improve the reproducibility and accuracy of NGS experiments for cancer mutation detection.
As costs continue to decrease, increasing numbers of researchers and clinicians are using NGS to profile clinical samples1. Currently, a panoply of bulk sample processing protocols, library preparation methods, sequencing technologies and bioinformatics pipelines are used to detect mutations relevant to cancer. Furthermore, samples can arrive at the testing laboratory in different states (that is, as formalin-fixed, paraffin-embedded (FFPE) samples rather than fresh samples), the amount of input DNA can be variable and tumor purity is rarely consistent across clinical samples, presenting substantial challenges in regard to sequencing assays, instrumentation and analytical tools. All of these technical challenges probably underlie the estimated irreproducibility rates of 51–89% in preclinical research1–3.
Previous studies have successfully addressed individual components of somatic variant calling in isolation—for example, bioinformatics pipelines4,5 or sample factors6 have been studied individually. Many reports have compared bioinformatics pipelines/callers based on the accuracy and consistency of mutation detection7–9, while others have compared other components such as assay development10, library preparation11 and biosample resources12,13. The majority of these studies used either in silico approaches with simulated ground truth or real tumor samples10,14–16 for benchmarking17. The datasets or samples from previous studies either do not accurately represent real human tumor biopsies (in silico) or are not sustainable (real tumor) for multiple benchmark studies. Although benchmarking with tumor tissue sounds more realistic for cancer mutation detection, the existence of spatial heterogenicity of tumor tissue18,19 (that is, different slices of tissue may have different mutation profiles) and limited sample quantity cannot support the comprehensive studies required for reliable assessment of detection reproducibility.
Here we develop systematic methods to evaluate performance using representative cell-line reference samples and datasets. We profile previously characterized20–22 and commercially available cell lines (breast cancer versus matched normal cell lines). We outline an experimental design where we sought to test the influence of each variable within a typical NGS-based, tumor-profiling workflow. Our design included different biospecimen types (fresh versus FFPE), input amounts and library preparation methods, and different Illumina sequencing instruments, sequencing centers and bioinformatics pipelines, which allowed us to investigate how these experimental and analytical elements interact to affect mutation detection.
Results
Study design.
To pinpoint factors affecting somatic variant calling, a matched pair of breast cancer cell lines (HCC1395 and HCC1395BL) was selected for profiling23–26. Although practical constraints precluded an exhaustive examination of all possible combinations of all possible variables, those chosen for study here reflect our real-world assessment of commonly encountered factors exerting substantial effects on the final result (Fig. 1a and Extended Data Fig. 1).
Fig. 1 |. Study design and read quality.
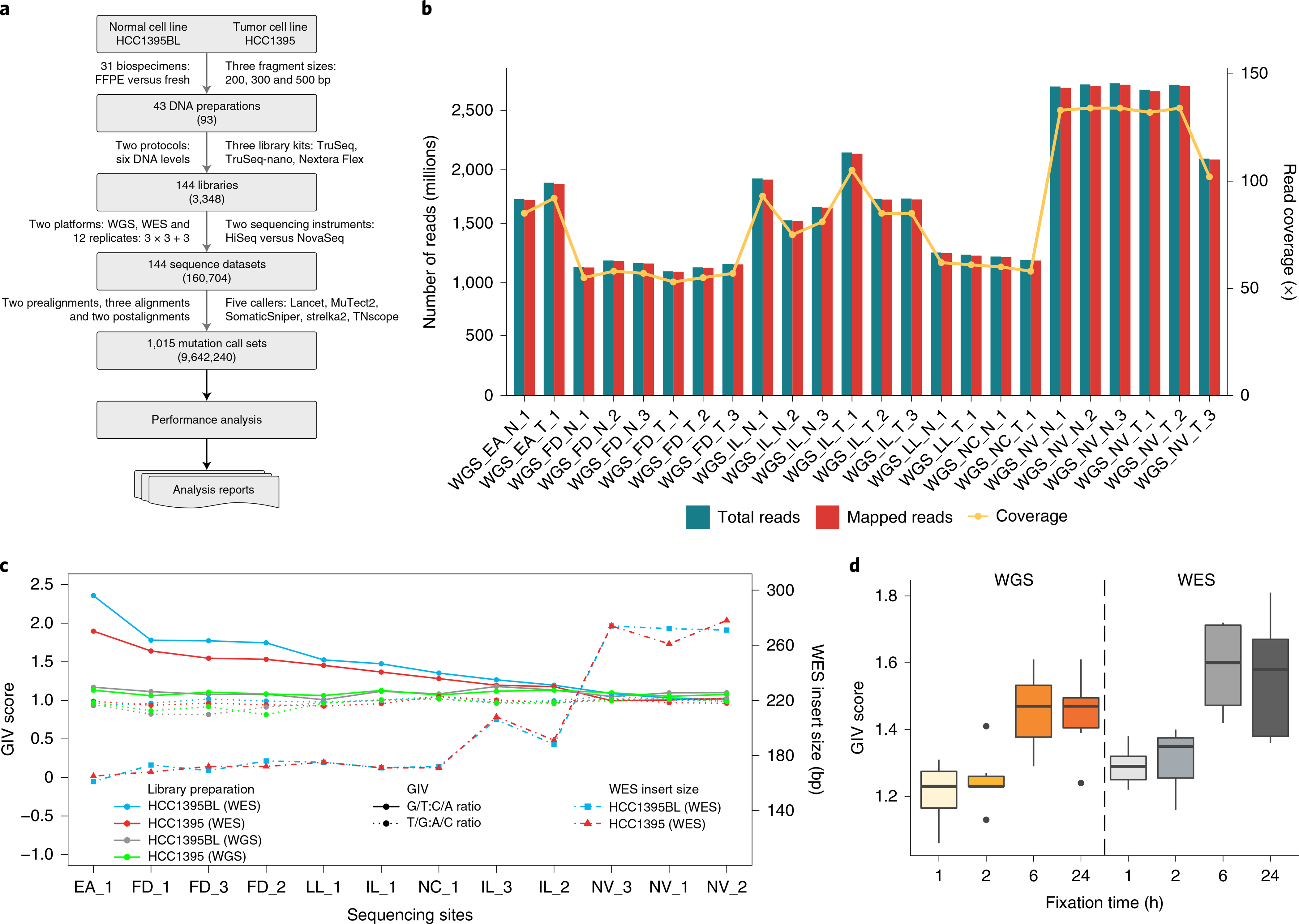
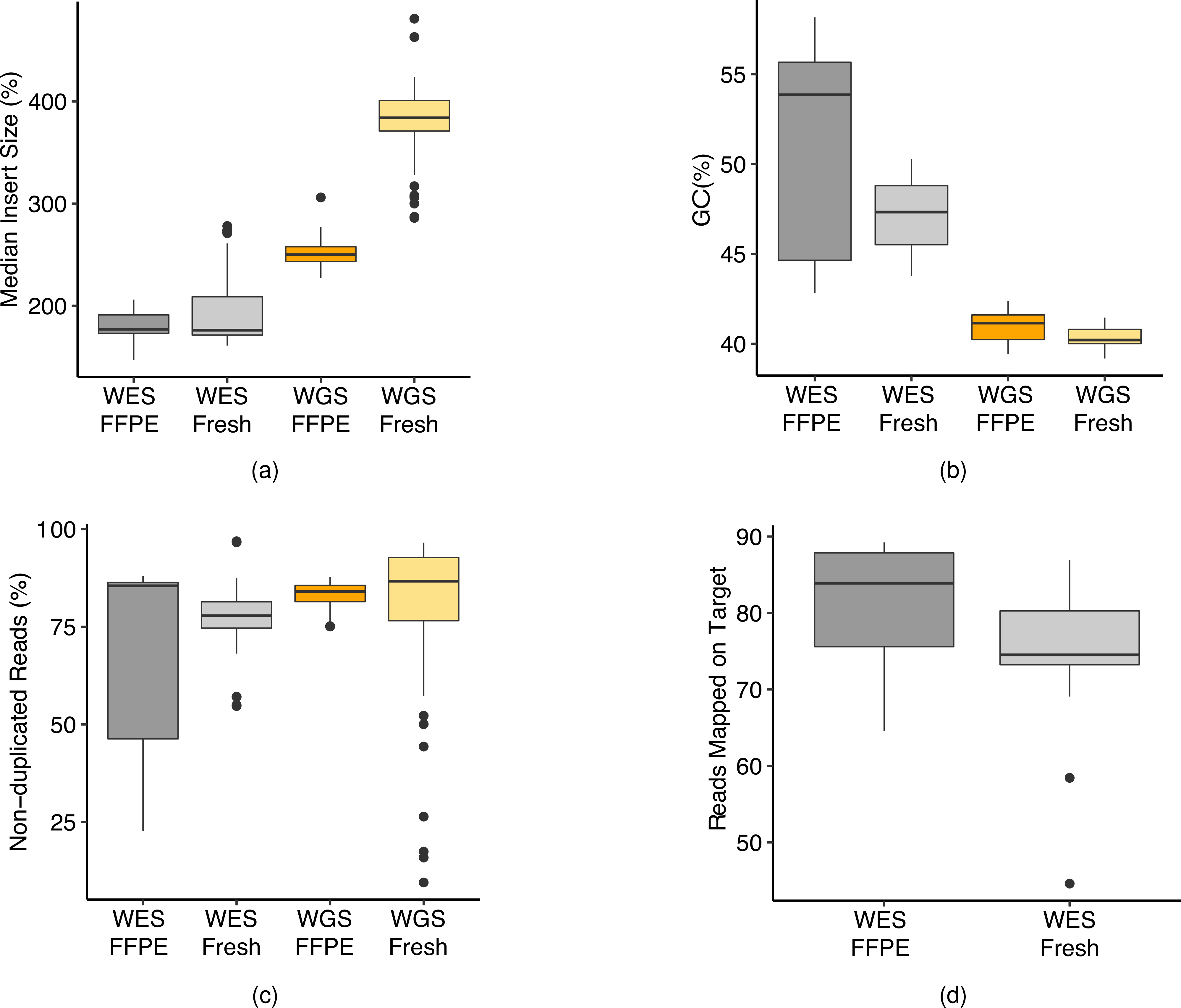
a, Study design used to capture nonanalytical and analytical factors affecting cancer mutation detection. DNA was extracted from either fresh cells or FFPE-processed cells and fragmented at three intended sizes. Libraries with various levels of DNA input (either from random shotgun or exome capture) were generated with three different library preparation kits and run on WGS and WES in parallel following recommended protocols (Methods). Twelve replicates were performed at six sequencing centers: three centers (FD, IL and NV) prepared WGS and WES libraries in triplicate; three centers (EA, LL and NC) prepared a single WGS and WES library (3×3 + 3); and 144 libraries were sequenced on either a HiSeq or NovaSeq instrument. Two prealignments (BFC and Trimmomatic), three alignments (BWA, Bowtie and NovoAlign) and two postalignments (GATK and no-GATK) were evaluated. A total of 1,015 mutation call sets were generated. Numbers in parentheses represent possible combinations at that level. Further details on the experiment design are given in Extended Data Fig. 1. b, Read yields (blue), mapping statistics (red) and genome coverage (yellow line) from 12 repeated WGS runs. c, GIV of G > T/C > A and T > /A > C mutation pairs in WES and WGS runs. Six centers used a range of time spans (80–300 s) for DNA shearing. As a result, average insert DNA fragment size ranged from 161 to 274 bp. d, Distribution of GIV score for FFPE DNA with four different fixation times (1, 2, 6 and 24 h) analyzed with WES or WGS: FFX and FFPE on WES platform and FFG and FFPE on WGS platform. Box-and-whisker plots shows the first and third quartiles as well as median values. The upper and lower whiskers extend from the hinge to the largest or smallest value no further than 1.5 × interquartile range from the hinge. For detailed statistics regarding minima, maxima, center, bounds of box and whiskers, and percentiles related to this figure, please refer to Supplementary Table 4.
Our experimental design covered many real-world scenarios that occur in research or clinical laboratories, including FFPE, heterogeneity of tumor biopsy, limited specimen DNA, ever-changing NGS machine models and analytical tools. We generated 1,015 call sets to evaluate the cross-center and cross-platform reproducibility of mutation detection, as well as the performance of mutation calling under various conditions
Survey of read quality.
Whole-genome sequencing was performed at six sequencing centers, generating a total of 42 sequencing results from the standard TruSeq PCR-free libraries prepared from 1,000 ng of input DNA. The cross-platform comparison included three different platforms: HiSeq 4000, HiSeq X10 and NovaSeq S6000. All sequencing centers and platforms produced high-quality data, as demonstrated by base call Phred quality scores >Q30, and >99.8% of reads mapped to the reference genome (GRCh38). Variation was observed in the quantity of reads per sample generated, with one center consistently delivering much higher cover ages (100×) (Novartis (NV)) than others (50×): (Illumina (IL), Fudan University (FD), European Infrastructure for Translational Medicine (EA), National Cancer Institute (NC) and Loma Linda University (LL)). This was driven by both sequencing platform yield differences and run-pooling schemes and target coverage (Fig. 1b). Among the WGS libraries prepared using fresh cells, insert size distribution and G/C content were uniform (40–43% G/C). Moreover, all WGS libraries had very low adapter contamination (<0.5%). Less than 10% of reads mapped redundantly for most libraries, indicating the high complexity of the WGS libraries. Similar mapping statistics were observed in NovaSeq WGS runs (Supplementary Table 1).
Similarly, WES performed across six sequencing centers (EA, FD, IL, LL, NC and NV) using three different HiSeq models (HiSeq 1500, HiSeq 2500 and HiSeq 4000) generated sequencing results where 99% of reads mapped successfully (Supplementary Table 2). The largest variations in sequencing yield and coverage on target were seen between sequencing centers, and even sometimes between different replicates of the same cell line at the same sequencing center. These variations were due largely to uneven library pooling and sequencing yield differences between platforms. This confounding factor was readily identified by statistical analysis and could be removed with downsampling to produce equivalent coverage for each run. WES libraries demonstrated much higher adapter contamination among all six centers, compared with WGS data. The WES libraries had higher G/C content (44–54%) compared with WGS libraries (40–43%). Those libraries with more serious adapter contamination also had much higher G/C content than other WES libraries. Generally, regions with more reads on the target region had higher G/C content (Extended Data Fig. 2a).
In terms of library preparation kits, the average percentage of mapped reads ranged from 96 to 99.9 across TruSeq PCR-free, TruSeq-Nano and Nextera Flex libraries prepared with 250, 100, 10 or 1 ng of DNA input. However, the percentage of nonredundant reads was very low (<20%) for TruSeq-Nano with 1 ng input, presumably due to PCR amplification (Extended Data Fig. 2b). Nevertheless, the percentage of nonredundant reads for 1 ng with Nextera Flex (also a PCR-based protocol) was reasonably good (~70, comparable to the performance of the TruSeq-Nano 100-ng protocol). In addition, overall G/C content was not affected by DNA input amount or library preparation kit. Thus, the Nextera Flex Library preparation may be superior to the TruSeq-Nano for lower-input DNA amounts. The FFPE libraries were prepared from cells fixed with formaldehyde at four time intervals. These samples yielded results with a high percentage of mapped reads and nonredundant read frequencies that were comparable to the results generated from WGS libraries prepared with fresh cells (Supplementary Table 1). More detail on data quality can be found in our companion paper23.
Evaluation of DNA quality.
The GIV is a commonly used indicator of DNA damage27 and was thus utilized to monitor DNA quality in NGS runs. We found high GIV scores for the G > T/C > A mutation pair in WES of cell lines HCC1395 and HCC1395BL. The GIV score for G > T/C > A was inversely correlated with insert fragment size (Supplementary Table 3), which was also found to be associated with DNA shearing time: longer shearing time produced shorter DNA fragments. Insert fragment size and G/C content were also inversely correlated, suggesting increased off-target (non-exome region) reads when larger DNA fragments were sequenced. We observed high G > T/C > A scores (>1.5) when insert fragment size was between 160 and 180 base pairs (bp); when insert fragment size was >200 bp we observed little or no imbalance (Fig. 1c). In contrast, we did not observe this imbalance in WGS runs (Extended Data Fig. 2c), for which insert size was normally >300 bp (Supplementary Table 1 and Extended Data Fig. 3a). We did not observe such imbalance in other mutation pairs, such as T > G/A > C (Fig. 1c). Previous reports demonstrate that excessive acoustic shearing results in 8-oxoguanine damage28. Therefore, the high ratio of G > T/C > A observed in some WES runs was probably an artifact of oxidative DNA damage during the fragmentation process.
Formaldehyde also causes the deamination of guanine. Thus, the GIV score of G > T/C > A is a good indicator of FFPE-induced DNA damage29. Consistent with this, we observed ‘dose’-dependent GIV imbalance in FFPE sample WGS runs (Fig. 1d). Taken together, these results indicate that WES is more sensitive to site-to-site library preparation variation than WGS. We propose that WGS, rather than WES, is mosre suitable for FFPE samples.
Reproducibility of cancer mutation detection.
To assess the reproducibility of cancer mutation detection with WES and WGS, we performed a total of 12 repeats of WES and WGS at six sequencing centers (Fig. 1a and Extended Data Fig. 1). We used three mutation callers (MuTect2 (ref. 30), Strelka2 (ref. 31) and SomaticSniper32) on alignments from three aligners (Bowtie2 (ref. 33), Burrows–Wheeler aligner (BWA)34 and NovoAlign) to generate a total of 108 variant call format (VCF) files from WES and WGS analyses separately.
In this study, as shown in Fig. 2a, both BWA and NovoAlign demonstrated a substantial pool of calls that were agreed upon under every repeated WGS or WES run called by three callers (MuTect2, Strelka2 and SomaticSniper). We did not observe substantial differences among results from the three aligners on WES. However, calling results from WGS with Bowtie2 as the aligner tended to have fewer consistent single-nucleotide variant (SNV) calls than BWA and Novalign, suggesting that mutation calling from Bowtie2 alignment was conservative (Fig. 2a).
Fig. 2 |. Mutation calling reproducibility.

a, In mutation-calling reproducibility, SNV overlaps across 108 VCF results from 12 repeated WGS and WES runs analyzed with three aligners (BWA, Bowtie2 and NovoAlign) and three callers (MuTect2, Strelka2 and SomaticSniper). b, SNV overlaps across 36 VCF results from 12 repeated WGS and WES runs as analyzed by MuTect2, Strelka2 and SomaticSniper from only BWA alignments. c, SNV overlaps in each of the 12 repeated WES and WGS runs, analyzed by Strelka2 from BWA alignments. The y axis shows that the probability of SNVs that were missed is equal to, or less than, the percentage of the 12 call sets depicted on the x axis. d, Effect summary of the model for WES or WGS. Effect tests were performed to evaluate the importance of each independent variable in a fixed-effect linear model fitted for WES or WGS. F-statistics and corresponding P values were calculated for variables. Both P values and LogWorth (−log10 (P values)) are plotted. The lower-order effects are identified with a caret. The sample size used to derive statistics was 36 for both WES and WGS.
We then fixed alignment to the widely used BWA and compared the results from our three callers, observing the differences in WES versus WGS run performance. SomaticSniper yielded more consistent SNV calls in WES than in WGS, but MuTect2 and Strelka2 showed greater divergence. In contrast, both MuTect2 and Strelka2 were more consistent when used for WGS rather than WES (Fig. 2b). Further examination of Strelka2 results from BWA alignments of 12 repeated WES and WGS runs confirmed this observation (Fig. 2c). Here we also introduced the O-score, a metric to measure the reproducibility of repeated analyses (Supplementary Methods). O-scores for Strelka2 and MuTect2 for WES runs were not only notably lower than those for WGS runs, but also more variable. Unexpectedly, even though its overall O-score was much lower than for Strelka2 and MuTect2 in WGS runs, SomaticSniper showed better consistency in calling results in WES than in WGS (Extended Data Fig. 4a). We also compared WGS runs with HiSeq versus WGS runs with NovaSeq using BWA for alignment and Strelka2 for calling. Both platforms were remarkably similar in terms of reproducibility, indicating that results from the HiSeq and NovaSeq platforms are comparable (Extended Data Fig. 4b). Taken together, Strelka2 had the best reproducibility in WGS repeated runs but the worst in WES repeated runs, whereas MuTect2 had the best reproducibility in WES repeated runs.
Factors influencing the reproducibility of cancer mutation detection.
After establishing the O-score to measure the reproducibility of NGS platform mutation detection, we also used it to determine which variables contribute most to variation between different repeated WGS or WES analyses separately. We included not only callers but machine model, read coverage, nonduplicated reads, G/C content, insert size and two GIV scores (G > T/C > A and T > G/A > C). The combination of these parameters represented most of the variation in WGS and WES runs, as a very high proportion of variance in the O-score could be predicted by these variables. For WGS, the combination of these eight individual variables accounted for >99% of O-score variance (R2 > 0.99) (Extended Data Fig. 5a). On the other hand, individual variables and five interaction terms (callers × coverage, callers × percentage GC, callers × machine model, callers × GIV(G > T/C > A) and callers × nonduplicated reads) were significant for O-score variance in WES runs (Extended Data Fig. 5b).
Because all 12 WGS runs were done with TruSeq PCR-free libraries and with the same amount of DNA input, the percentage of nonduplicated reads did not affect reproducibility. Although individual variables may influence WES run reproducibility, their effect levels are dependent on caller selection (Fig. 2d). Taken together, only a few factors (read coverage and callers) affected WGS reproducibility whereas several factors, including caller, read coverage, insert fragment size, GC content, GIV score (G > T/C > A) and their interactions, influenced WES run reproducibility. It is noteworthy that an older machine model (HiSeq 1500) was used in only one WES sequencing center, and this also yielded low insert fragment size and high GIV scores (G > T/C > A) in sequencing reads. Thus, the impact of machine model on WES run reproducibility could be a confounding factor. In contrast, the most influential factor in the performance of WGS was the caller, followed by read coverage.
Effect of nonanalytical and analytical factors on mutation calling.
To thoroughly investigate the influence of nonanalytical and analytical factors on cancer mutation calling, we set out to define the reference call set of somatic mutation in cell line HCC1395 for our benchmarking study23.
With three different library preparation protocols and varying DNA input amounts across multiple library preparations, we analyzed outcomes using combinations of the three callers and three aligners. MuTect2 was reliable, except for calling of the 1-ng TruSeq-Nano libraries (Fig. 3a), while the reliability of Strelka2 and SomaticSniper decreased by >50% for the 1-ng TruSeq-Nano libraries. We conclude that Nextera Flex library preparation might be a better option for a low-input DNA quantity.
Fig. 3 |. Nonanalytical factors affecting mutation calling.

a, Caller performance on three library preparation protocols with different DNA Inputs: 1, 10, 100, 250 and 1,000 ng. WGS sequencing on TruSeq and TruSeq-Nano libraries was performed at LL, while WGS sequencing on Nextera libraries was performed at IL. All sequencing experiments were performed with HiSeq 4000 and analyzed using three aligners (BWA, Bowtie2 and NovoAlign) and three callers (MuTect2, Strelka2 and SomaticSniper). b, Performance of MuTect2, Strelka2 and SomaticSniper on WGS with fresh DNA or FFPE DNA (24 h) from a BWA alignment.
Formalin-fixed, paraffin-embedded processing can have an effect on variant calling results35. In our study, samples of fresh cells and those processed with FFPE were called with Strelka2, SomaticSniper and MuTect2. Precision and recall for both MuTect2 and Strelka2 were greatly reduced when samples were subjected to FFPE processing (Fig. 3b). On the other hand, SomaticSniper demonstrated only a small decrease in both metrics but otherwise underperformed substantially compared with the other two callers.
Sometimes FFPE or samples of low tumor purity are all that are available for study. Bioinformatics can potentially reduce sample-related biases. To evaluate bioinformatics pipelines, reads were preprocessed using Trimmomatic36 or Bloom Filter Correction (BFC)37 to assess whether read trimming and error correction could affect WES variant call recall or precision (Methods). The application of error correction with BFC to FFPE samples increased precision but did not improve the recall rate (Extended Data Fig. 6a). However, precision was improved for fresh DNA samples subjected to BFC processing, but with a lower recall rate than results from Trimmomatic processing. Taken together, these results indicate that BFC is appropriate in cases of severe DNA damage but may not be worthwhile if there is only mild damage from a process such as sonication.
Formalin-fixed, paraffin embedding is also known to cause G > T/C > A artifacts35. Trimmomatic and BFC were investigated for their ability to detect these errors. Since the DNA damage causing the G > T/C > A mutation may not be confined to only low-quality base calls at the end of reads, Trimmomatic is not designed to remove this type of artifact. Trimmomatic processed data were more skewed toward C > A artifacts than were BFC-processed data, which showed changes more broadly across nucleotide transitions (Fig. 4a). BFC reduced C > A artifacts but introduced a few artifacts of other types, such as T > C mutations, indicating that caution should be exercised when using bioinformatics tools to correct FFPE artifacts.
Fig. 4 |. Bioinformatics for enhanced calling.
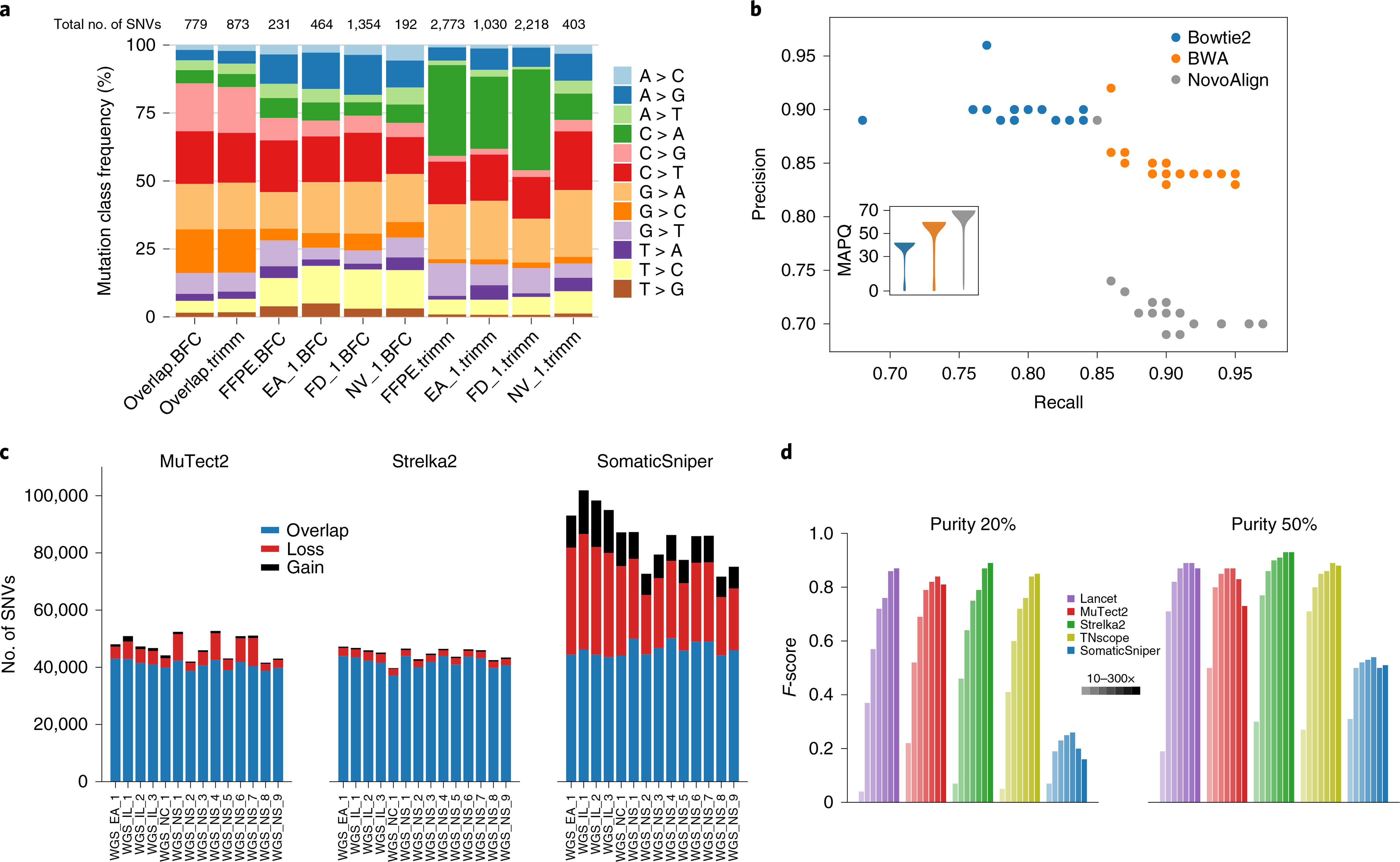
a, Distribution of mutation types called with Strelka2 on BWA alignments of four WES runs preprocessed by Trimmomatic or BFC. WES run on FFPE DNA (FFPE) or fresh DNA (EA_1, FD_1 and NV_1). Numbers of SNVs called from each process are shown at the top. Mutations shared across BFC datasets (overlap.BFC) and Trimmomatic datasets (overlap.trimm) are shown on the left. C > A and T > C artifacts were observed in the Trimmomatic and BFC datasets, respectively; both artifacts were minimized with repeats. b, Performance of mutation calling by Strelka2 on three alignments (Bowtie2, BWA and NovoAlign). Insert is a violin plot of mapping quality (MAPQ) scores from three alignments for an example WGS run. In total, 81 billion, 118 billion and 140 billion data points were used in violin plots for Bowtie2, BWA and NovoAlign, respectively. c, Effect of postalignment processing (indel realignment + BQSR) on mutation calling by MuTect2, Strelka2 and SomaticSniper). d, Effect of tumor purity (20 versus 50%) on five callers (Lancet, MuTect2, Strelka2, TNscope and SomaticSniper) with read coverage of 10×, 30×, 50×, 80×, 100×, 200× and 300×.
Calling accuracy was dependent on the choice of caller and aligner, as well as their interaction. Strelka2 results with BWA-aligned reads were balanced whereas those with Bowtie2-aligned reads seemed conservative. In contrast, Strelka2 results with NovoAlign-aligned reads appeared aggressive (Fig. 4b). When we examined mapping quality scores for the three alignments, those for BWA were usually between 50 and 60, for Bowtie between 40 and 50 and for NovoAlign between 60 and 70. Strelka2 was trained and modeled on the BWA alignment and thus works best in the bioinformatics context where it was developed31. Taken together, these results indicate that there may be a joint effect between aligner and caller, depending on how callers were developed and on which aligner’s dataset they were trained.
Next, the effect of the Genome Analysis Toolkit (GATK) local indel realignment, together with base quality score recalibration (BQSR), was queried. MuTect2 and Strelka2 identified a very similar number of SNVs regardless of whether the process was employed (Fig. 4c). Conversely, MuTect2 was modestly impacted and SomaticSniper was highly sensitive to postalignment processing, with some SNVs gained but far more lost. Relatedly, the precision and recall rates changed dramatically when this process was applied to SomaticSniper calling but not for calling by MuTect2 or Strelka2 (Extended Data Fig. 6b,c). Clearly our study confirms, with real-world experimental data, the importance of a full understanding of how the various components of mutation analysis by NGS methods work and interact with each other.
Tumor purity and coverage also play a role in caller performance. As expected, higher read coverage yielded more SNV calls (Fig. 4d). When tumor purity was high (>50%), 50× performed very similarly to 100× coverage across all callers tested; when tumor purity was low (<50%), calling was much more sensitive to sequencing depth. To test the performance of callers on samples of low tumor purity, we pooled reads from WGS triplicate runs on samples that were sequenced at 100× coverage to generate coverage of either 200× or 300× on each cell line. In addition to the three main callers used in this study, we also included two other tools, TNscope38 and Lancet39, to compare their capabilities in the detection of mutations from a mix with tumor DNA as low as 5%. With high tumor purity (>50%) we again observed that the accuracy of all callers, with the exception of SomaticSniper, declined slightly with higher read coverage, indicating that our truth set lacked <5% variant allele frequency (VAF) mutations. However, when tumor purity was 20% or lower, the benefit of higher coverage for mutation detection was apparent. For a sample with 20% tumor, Lancet, Strelka2 and TNscope performed similarly well, with 300× coverage (Fig. 4d). On the other hand, SomaticSniper performed poorly at any tumor purity level and increased read coverage did not rescue performance. These results indicate that tumor purity is much more influential than coverage in the ranges tested here.
Performance of WGS and WES across multiple sequencing centers.
Because this study leveraged six sequencing centers performing 12 WES and WGS experiments simultaneously, we were able to assess both inter- and intracenter reproducibility of the two sequencing platforms. Using our established call set as a reference, we defined resulting SNV calls from any of the NGS runs with the pair of cell lines into three categories: (1) repeatable (SNVs in the reference call set defined in the categories high confidence (‘HighConf’) and medium confidence (‘MedConf’)); (2) gray zone (SNVs defined in the categories low confidence (‘LowConf’) and ‘Unclassified’); and (3) nonrepeatable (SNVs not found in the reference call set) (Extended Data Fig. 7). Cross-center and cross-platform variations were very small for repeatable SNVs, indicating that all individual NGS runs, regardless of sequencing center or NGS platform, detected most ‘true’ mutations consistently. This ‘consistency’ dropped dramatically for SNVs in the gray zone, and further down to nearly zero if SNVs were nonrepeatable (Table 1).
Table 1 |.
Intra- and intercenter SNV calling reproducibility based on WGS and WES
| Concordance of SNVsa | MuTect2 | Strelka2 | SomaticSniper | ||||
|---|---|---|---|---|---|---|---|
| WESb | WGSb | WESb | WGSb | WESb | WGSb | ||
| Overall SNVs | Overall | 0.54 | 0.72 | 0.36 | 0.74 | 0.45 | 0.34 |
| Intracenter | 0.57 | 0.73 | 0.44 | 0.76 | 0.52 | 0.36 | |
| Intercenter | 0.54 | 0.72 | 0.35 | 0.74 | 0.44 | 0.34 | |
| Repeatable | Overall | 0.83 | 0.92 | 0.9 | 0.91 | 0.87 | 0.90 |
| Intracenter | 0.87 | 0.93 | 0.94 | 0.93 | 0.89 | 0.91 | |
| Intercenter | 0.82 | 0.91 | 0.89 | 0.91 | 0.86 | 0.90 | |
| Gray zone | Overall | 0.29 | 0.28 | 0.33 | 0.21 | 0.30 | 0.22 |
| Intracenter | 0.43 | 0.32 | 0.44 | 0.25 | 0.45 | 0.26 | |
| Intercenter | 0.26 | 0.28 | 0.31 | 0.21 | 0.27 | 0.21 | |
| Nonrepeatable | Overall | 0.01 | 0.02 | 0.03 | 0.07 | 0.13 | 0.08 |
| Intracenter | 0.02 | 0.03 | 0.06 | 0.10 | 0.21 | 0.10 | |
| Intercenter | 0.01 | 0.02 | 0.02 | 0.07 | 0.12 | 0.08 | |
Overall SNVs, all SNVs in a pair of NGS runs; repeatable SNVs, SNVs defined in categories HighConf and MedConf; gray zone SNVs, SNVs defined in categories LowConf and Unclassified; nonrepeatable SNVs, SNVs not defined in the four categories above.
Average Jaccard scores of NGS run pairs.
Taken together, these results indicate that there were two major sources for discordant SNV calls between any two different library preparations: (1) stochastic effects of sequence coverage on SNVs with low VAF, which are sensitive to read coverage and mainly represented by SNVs in the gray zone group; and (2) artifacts due to library preparation, mainly represented by SNVs in the nonrepeatable group. The benefit of high read coverage not only empowers the detection of mutations with low VAF, but also increases result reproducibility (for both WES and WGS), probably due to reduction of stochastic effects.
Using multivariate analysis, we were able to further dissect the source of variation driving the reproducibility of mutation detection by WES and WGS. Consistent with our results from O-score analysis (Fig. 2d), callers, read coverage and platforms were the major factors influencing the reproducibility of mutation detection. However, the subset of SNVs/indels (repeatable, nonrepeatable and gray zone) was the dominant source of inconsistent mutation calls (Extended Data Fig. 8a). In addition, we observed that the difference between inter-/intracenter variations was subject to callers (Extended Data Fig. 8b) and SNV/indels subsets (Extended Data Fig. 8c). Overall, intercenter variations for WES were larger than those for WGS whereas the difference in intracenter variation between WES and WGS was insignificant (Extended Data Fig. 8d).
Moreover, results from the Jaccard index score analysis confirmed our conclusions: for WES, Strelka2 was less reproducible than MuTect2 (Fig. 2b and Extended Data Fig. 4a). As shown in Fig. 4a, Strelka2 was very sensitive in detecting C > A mutation artifacts from excessive sonication. Thus, it was not surprising to see that many nonrepeatable SNVs detected by Strelka2 in WES were C > A mutations. To a lesser extent, MuTect2 also detected C > A mutation artifacts; in contrast, SomaticSniper did not detect this artifact (Supplementary Fig. 1).
Precision and recall rates from WES and WGS were also compared directly across all three callers and all 12 replicates. Mutations shared by two replicates generally had higher precision. Moreover, almost all mutations called by both MuTect2 and SomaticSniper were true (Fig. 5a). Although leveraging additional callers increased precision, this was at the cost of recall (Fig. 5b). Taken together, according to the precision metric, WGS clearly outperformed WES across replicates, callers and sequencing centers. These results demonstrate the importance of using sufficient library replicates during study design, rather than trying to compensate by using multiple callers.
Fig. 5 |. Biological repeats versus analytical repeats.

Precision (a) and recall (b) of overlapping SNVs/indels that were supported by biological repeats (library repeats) or analytical repeats (two different callers). Each row or column represents calling results from a WES or WGS run called by one of the three callers from a BWA alignment. All 12 repeats of WES and WGS from six sequencing centers (FD, IL, NV, EA, LL and NC) were included.
Finally we compared precision, recall and F-scores (as defined in Supplementary Methods) across a range of variant allele frequencies with three callers in both WES and WGS. Interestingly, Strelka2 exhibited the best performance on WGS samples but the worst on WES samples (Extended Data Fig. 9), which is consistent with results from the reproducibility study (Fig. 2b). On the other hand, even though SomaticSniper did not perform well overall it was not affected by C > A artifacts. However, for both WES and WGS runs we did observe that the limit of VAF calling by SomaticSniper was ~12%, and many false positives were misidentified by SomaticSniper at a higher VAF (Extended Data Fig. 10a,b). Therefore, the ‘better’ performance of SomaticSniper compared with that of Strelka2 on WES was driven by the insensitivity of the former to artifacts from the DNA fragmentation process (Fig. 1c and Extended Data Fig. 10a).
Discussion
We observed that each component of the sequencing and analysis process can affect the final outcome. The overall concordance and correlation of results from WES and WGS were good. Although WES had a better coverage/cost ratio than WGS, sequencing coverage of WES target regions was not even (Extended Data Fig. 10a,c). In addition, WES showed more batch effects/artifacts due to laboratory processing and thus had larger variation between runs, laboratories and probably among researchers preparing the libraries. As a result, WES was less reproducible than WGS. WGS had more uniform coverage and was less sensitive to different runs and laboratories. Our experimental design also allowed us to estimate inter-/intracenter variation for both WES and WGS platforms. Although WES had much larger intercenter variation than WGS, intracenter variation for both platforms was quite comparable (Extended Data Fig. 8d). Biological (library) repeats removed some artifacts due to random events (nonrepeatable calls) and thus offered much better calling precision than did a single test. Analytical repeats (two bioinformatics pipelines) also increased calling precision, but at the cost of increased false negatives (Fig. 5). We found that biological replicates are more important than bioinformatics replicates in cases where high specificity and sensitivity are needed.
Detection of cancer mutations is an integrated process. No individual component can be singled out in isolation as being more important than any other, and specific components affect and interact with each other. Every component and every combination of components can lead to false discovery (false positives or false negatives). Individual components of a cancer mutation calling pipeline should never be considered ‘plug and play’. Although the initial steps of NGS test validation may be performed in three separate stages (platform, test specific and informatics)40, our study demonstrates the complex interdependency of these stages on overall NGS test performance. Thus, final validation and verification studies should be performed using the entire sample-to-result pipeline. Detailed recommendations for cancer mutation detection experiments and analysis are provided in Table 2.
Table 2 |.
Recommendations for cancer mutation detection using NGS
| Process and pipeline | Recommendations |
|---|---|
| DNA input and library construction | Fragment size |
| • WGS, 300–600 bp | |
| • WES, 250–350 bp | |
| Fragment method | |
| • Size >250 bp, sonication | |
| • Size <200 bp, enzyme | |
| DNA input | |
| • TruSeq PCR-free, 200 to ∼1,000 ng | |
| • TruSeq-Nano, 10 to ∼200 ng | |
| • Nextera Flex, 1 to ∼100 ng | |
| NGS platform choice | WGS |
| • More reproducible | |
| • Limit of detection (LOD) >5% of VAF | |
| • Read coverage <100× (cost constraints) | |
| • Best choice if tumor content is high (>50%) | |
| WES | |
| • Less reproducible | |
| • More cost effective | |
| • High read coverage (>100×) | |
| • LOD could be as low as 1 to 2% of VAF | |
| Read coverage and quality assurance | Read coverage |
| • Tumor content >50% | |
| ○ 50× (WGS) | |
| ○ 100× (WES) | |
| • Tumor content 20 to 50% | |
| ○ >100× (WGS) | |
| ○ >200× (WES) | |
| QC metrics | |
| • Read redundancy <30% | |
| • Mappable reads >95% | |
| • GC content | |
| ○ 40 to 43% (WGS) | |
| ○ 45 to 48% (WES) | |
| • On-target 65 to ∼85% | |
| • GIV score <1.5 | |
| Read alignment | Alignment quality score range |
| • Bowtie2, 40 to 50 | |
| • BWA-MEM, 50 to 60 | |
| • NovoAlign, 60 to 70 | |
| No significant differences between BWA-MEM, Bowtie2 and NovoAlign | |
| Postalignment | GATK postalignment processing may not be needed for some mutation callers, such as Strelka2 |
| Mutation calling | • Strelka2 |
| ○ Reliable for WGS | |
| ○ Sensitive to artifacts with WES | |
| • MuTect2 | |
| ○ Reliable for both WES and WGS | |
| ○ Not suitable for WGS with high coverage (200×) | |
| Pipeline construction and change control | • Detection of cancer mutations is an inherently integrated process |
| • Every component and every combination of components is equally important | |
| • No plug and play | |
| • Final validation studies and revalidation should be performed using the entire sample-to-result pipeline |
We cannot conclude that results from our study are generalizable to cancer types other than breast cancer. However, the high complexity of chromosome loss/gains41, large number of somatic mutations23 and highly heterogeneous cell populations25 present in cell line HCC1395 resemble the abnormalities commonly seen in a hyperdiploid cancer genome.
In summary, this study provides datasets comparing DNA from fresh cells, FFPE DNA and tumor/normal DNA mixtures and the performance of various bioinformatics tools. Because these samples were prepared from a pair of well-characterized, renewable tumor/normal cell lines from the same donor, our results can serve as a reference for the NGS research community when performing benchmarking studies for the development of new NGS products, assays and informatics tools42. In Table 2 we provide recommendations regarding DNA fragmentation for WES runs, selection of NGS platforms and bioinformatics tools based on the nature of available biosamples and study objectives.
Methods
Cell lines and DNA extraction. Cell line HCC1395, breast carcinoma, human (Homo sapiens) cells (expanded from the American Type Culture Collection (ATCC) no. CRL-2324) were cultured in ATCC-formulated RPMI-1640 medium (ATCC, no. 30–2001) supplemented with fetal bovine serum (FBS; ATCC, no. 30–2020) to a final concentration of 10%. Cells were maintained at 37 °C with 5% carbon dioxide (CO2) and were subcultured every 2–3 days, as per ATCC recommended procedures, using 0.25% (w/v) trypsin/0.53 mM EDTA solution (ATCC, no. 30–2101), until appropriate densities were reached. HCC1395BL, B lymphoblast, EBV-transformed, human (H. sapiens) cells (expanded from ATCC, no. CRL-2325) were cultured in ATCC-formulated Iscove’s modified Dulbecco’s medium (ATCC, no. 30–2005) supplemented with FBS (ATCC, no. 30–2020) to a final concentration of 20%. Cells were maintained at 37 °C with 5% CO2 and were subcultured every 2–3 days, as per ATCC recommended procedures, using centrifugation with subsequent resuspension in fresh medium until appropriate densities were reached. Final cell suspensions were spun down and resuspended in PBS for nucleic acid extraction.
All cellular genomic material was extracted using a modified phenol-chloroform-iso-amyl alcohol extraction approach. Essentially, cell pellets were resuspended in Tris-EDTA (TE), subjected to lysis in a solution of 2% TritonX-100/0.1% SDS/0.1 M NaCl/10 mM Tris/1 mM EDTA and extracted with a mixture of glass beads and phenol-chloroform-iso-amyl alcohol. Following multiple rounds of extraction, the aqueous layer was further treated with chloroform-indoleacetic acid and finally underwent RNAse treatment and DNA precipitation using sodium acetate (3 M, pH 5.2) and ice-cold ethanol. The final DNA preparation was resuspended in TE and stored at −80 °C until use.
FFPE processing and DNA extraction.
Cell lines cultured in T75 flasks (Corning, no. 10–126-28) were harvested according to the supplier’s product specifications (https://www.atcc.org/). For each cell line, harvested materials were combined into a single 15-m, conical tube (Falcon, no. 14–959-53 A) and resuspended to a total volume of 1 ml with neutral buffered 10% formalin (StatLab, no. 28600). In separate vials, HistoGel specimen processing gel matrix (ThermoFisher, no. HG-4000–012) had been heated to 60 °C for 2 h to liquefy and then allowed to cool and equilibrate to 45 °C in a vendor-supplied thermal block (ThermoFisher, no. HGSK-2050–1). For each cell line, eight replicate, rectangular-shaped cell-block molds were set up (Fisherbrand, no. EDU00552). In each mold, 500 μl of 45 °C HistoGel was added and, to this, 100 μl of neutral buffered formalin-suspended cell line mixture was added. These were immediately stirred gently to ensure homogeneity of cells within the cooling HistoGel matrix, and then allowed to sit and solidify on the bench top for at least 5 min. Next, for each mold, a microspatula was used to carefully dislodge the formed HistoGel embedded cell mixtures, which were then carefully placed in nylon mesh bags (Thermo Scientific, no. 6774010) to prevent disaggregation during subsequent tissue processing. These formed HistoGel cell mixtures in nylon bags were placed in individual tissue-processing cassettes (Thermo Scientific, no. 1000957) and then submerged in a plastic pail filled with neutral buffered 10% formalin, to simulate pretissue processing time-in-formalin delay before batch tissue-processing steps.
The sequence described above was performed at 1-, 2-, 6- and 24-h time points before batch tissue processing. All cassettes were then placed in a tissue processor for a ‘routine’ tissue-processing run at the University of Toledo Medical Center Department of Pathology (Sakura Tissue Tek VIP 5 Tissue Processor; see Supplementary Table for routine run conditions). The processed formalin-fixed, paraffin-infiltrated cell blocks were then embedded in paraffin (Sakura Tissue Tek TEC 5 Tissue Embedding Station) to create FFPE cell blocks.
Each FFPE cell block was serially sectioned at 5-μm thickness with a microtome, and these ribbons of shaved material were placed in individual 15-ml conical tubes. A QIAamp DNA FFPE Tissue Kit (Qiagen) was used to extract FFPE DNA from each cell block, following a slightly modified protocol. The first xylene step for deparaffinization was removed due to the low yield and purity of DNA commonly experienced with clinical aspirate specimens or dyshesive specimens derived from cell culture specimens43. Instead, buffer ATL and proteinase K were directly added to the tubes with FFPE slices and incubated according to the supplier’s specifications. After digestion and lysis, specimens were cooled to room temperature and continuously inverted to let the paraffin solidify along the inner surface of the tubes. After cooling, the tubes were spun for 5 min at 1,200g until aqueous and paraffin layers become visible, when the aqueous layer was carefully transferred to a new 15-ml conical tube. The specimens were then incubated at 90° C for 1 h then again cooled to room temperature and briefly centrifuged to remove liquid from the cap. The remainder of the Qiagen QIAamp DNA FFPE Tissue Kit protocol, starting with the addition of Buffer AL and 100% ethanol with vortexing, was followed according to the supplier’s specifications. DNA was eluted from the QIAamp MinElute column using 100 ml of low-concentration EDTA TE buffer (0.1 mM EDTA, Tris-HCl buffer, 10 mM, pH 8.5). Quality control for specimens was performed using the following supplier’s instruments/kits: Thermo Scientific NanoDrop Spectrophotometer, Thermo Scientific Qubit fluorometer, absolute and relative quantitative PCR measures of DNA quality, Agilent HighSensitivity D5000 Tapestation and an Agilent Highsensitivity DNA Bioanalyzer chip. A representative selection of the prepared cell blocks had a portion of their microtome sections taken for microscopic evaluation with hematoxylin and eosin (H&E) staining, as well as immunohistochemistry. The routine H&E-stained glass slides were used for estimation of cellularity, evenness of dispersion of cells in the cell block and cytologic quality (viability and lack of degeneration in cellular membranes).
Immunohistochemistry for Pankeratin (Ventana, no. 760–2135) and CONFIRM-anti-CD45 (Ventana, no. 760–2505) was performed using a Benchmark Ultra Ventana Automated IHC slide-staining system. These two IHC stains were used to ensure that no cross-mixing of cellular materials occurred between the two cell lines during culture, harvesting and processing for FFPE (~10,000 cells assessed for each IHC staining/cell-line category).
DNA fragmentation and library preparation.
A TruSeq DNA PCR-Free LT Kit (Illumina, no. FC-121–3001) was used to prepare samples for WGS. DNA libraries for WES were first prepared with the Ovation Ultralow System V2 (NuGEN, no. 0347-A01), following the manufacturer’s instructions. Next, exonic regions of each library (750 ng) were captured using the SureSelectXT Reagent kit (Agilent Technologies, no. G9611A), the SureSelectXT Human All Exon V6 + UTR Capture Library (Agilent Technologies, no. 5190–8881) and the Ovation Target Capture Module (NuGEN, no. 0332–16), following the manufacturers’ instructions.
WGS libraries were prepared at six sites with the TruSeq DNA PCR-Free LT Kit (Illumina, no. FC-121–3001) according to the manufacturer;s protocol. Unless specified otherwise, 1 μg of DNA was used for the TruSeq PCR-free libraries. All sites used the same fragmentation conditions for WGS by utilizing Covaris with a targeted size of 350 bp. All replicated WGS and WES libraries were prepared on different days. The input amount of WGS runs with fresh DNA was 1 μg unless otherwise specified. Detailed parameters of DNA fragmentation for 24 WES libraries are presented in Supplementary Table 10.
The concentration of the TruSeq DNA PCR-Free libraries for WGS was measured by quantitative PCR with the KAPA Library Quantification Complete Kit (Universal) (Roche, no. KK4824). The concentration of all other libraries was measured by fluorometry either on a Qubit 1.0 fluorometer or a GloMax Luminometer with the Quant-iT dsDNA HS Assay kit (Thermo Fisher Scientific, no. Q32854). The quality of all libraries was assessed by capillary electrophoresis on either a Bioanalyzer 2100 or a TapeStation instrument (Agilent), in combination with either a High Sensitivity DNA Kit (Agilent, no. 5067–4626), a DNA 1000 Kit (Agilent, no. 5067–1504) or a TapeStation 4200 instrument (Agilent) with the D1000 assay (Agilent, nos. 5067–5582 and 5067–5583).
For the library preparation study, HCC1395 and HCC1395BL were diluted to 250, 100, 10 and 1 ng in Resuspension Buffer (Illumina). For the 250-ng samples, libraries were generated using the Truseq DNA PCR-free protocol as described above. For the remaining samples, libraries were generated using the Truseq DNA Nano (Illumina) protocol according to the manufacturer’s instructions. DNA was sheared as described above, and the following PCR cycles were performed: eight cycles for 100-ng input, ten cycles for 10-ng input and 12 cycles for 1-ng input. Nextera Flex (Illumina) libraries were also prepared from 1-, 10- and 100-ng inputs according to the manufacturer’s instructions, and amplified with 12, eight and five cycles of PCR, respectively.
For the tumor purity study, 1 μg of tumor/normal dilutions was made in the following ratios using Resuspension Buffer (Illumina): 1:0, 3:1, 1:1, 1:4, 1:9, 1:19 and 0:1; each ratio was diluted in triplicate. DNA was sheared using the Covaris S220 to target a 350-bp fragment size (peak power 140 w, duty factor 10%, 200 cycles/bursts, 55 s, temperature 4 °C). NGS library preparation was performed using the Truseq DNA PCR-free protocol (Illumina) following the manufacturer’s recommendations.
For the FFPE study, SureSelect (Agilent) WES libraries were prepared according to the manufacturer’s instructions for 200 ng of DNA input, including reduction of shearing time to 4 min. In addition, adapter-ligated libraries were split in half before amplification; one half was amplified for ten cycles and the other for 11 cycles, to ensure adequate yields for probe hybridization. Both halves were combined after PCR for the subsequent purification step. For WGS, NEBNext Ultra II (NEB) libraries were prepared according to the manufacturer’s instructions. However, input adjustments were made according to the delta-Cq (dCq) obtained for each sample using the TruSeq FFPE DNA Library Prep QC Kit (Illumina), to account for differences in sample amplifiability. A total of 33 ng of amplifiable DNA was used as input for each sample.
DNA sequencing.
Whole-genome libraries were sequenced on a HiSeq 4000 instrument (Illumina) at 2 × 150 bases read length with HiSeq 3000/4000 SBS chemistry (Illumina, no. FC-410–1003), and on a NovaSeq instrument (Illumina) at 2 × 150 bases read length using the S2 configuration (Illumina, no. PN 20012860). Whole-exome libraries were sequenced on a HiSeq 2500 instrument (Illumina) at 2 × 125 bases read length using HiSeq Rapid SBS v2 chemistry (Illumina, nos. FC-402–4021 and FC-402–4022). In all cases, sequencing was performed following the manufacturer’s instructions.
FASTQ sequence files for WGS and WES were generated from the Illumina sequencer images using either an Illumina RTA 1.18.66.3 (HiSeq 2500) or 2.7.7 (HiSeq 4000) and bcl2fastq 2.17.1.14 software.
Read processing and quality assessment.
The FASTQ files generated for WGS and WES from each sequencing center were transferred to central storage for read preprocessing and quality control. The Illumina bcl2fastq2 (v.2.17) was used to demultiplex and convert binary base calls and qualities to FASTQ format. FASTQC (v.0.11.2)44 was run on raw reads to assess base call quality, adapter content, G/C content, sequencing length and duplication level. In addition, FASTQ_screen (v.0.5.1) and miniKraken (v.0.10.0)45 were run to detect potential cross-contamination with other species. A multiQC (v.1.3) run report was generated for each sample set. The sequencing reads were trimmed of adapters and low-quality bases using Trimmomatic (v.0.30)36. The trimmed reads were mapped to the human reference genome GRCm38 (Read alignment) using BWA-mem (v.0.7.12)34 in paired-end mode. In addition, DNA Damage Estimator (v.3)27 was used to calculate GIV scores based on an imbalance between R1 and R2 variant frequency of the sequencing reads, to estimate the level of DNA damage introduced in the sample/library preparation processes. Postalignment quality control (QC) was performed based on BWA alignment BAM files; genome-mapped percentages and mapped reads duplication rates were calculated using BamTools (v.2.2.3) and Picard (v.1.84)46. Genome and exome target region coverage, as well as mapped reads insert sizes and G/C contents, were profiled using Qualimap (v.2.2)47 and custom scripts. Preprocessing QC reports were generated during each step of the process. MultiQC (v.1.3)48 was run to generate an aggregated report in html format. A standard QC metrics report was generated from a custom script.
To assess trimming and error correction effects on mutation call precision and recall, we chose a trimming software tool (Trimmomatic) and an error correction software package (BFC). For Trimmomatic we used MAXINFO: 50:0.97 to run against the same set as WES for benchmarking. BFC v.1.0–7-g69ab176 (ref. 37) was run with default parameters, apart from k-values, to provide corrected reads. Since BFC has an upper limit of 62 for k, FASTQ files with a larger optimal k value were processed with k = 62.
Read alignment.
For all alignments, we used the decoy version of the hg38 human reference genome (https://gdc.cancer.gov/about-data/data-harmonization-and-generation/gdc-reference-files; GRCh38.d1.dv1.fa) utilized by the Genomic Data Commons. For alignment comparisons we ran NovoAlign v.3.07.01 (Novocraft Technologies), Bowtie2 v.2.2.9 (ref. 49) and BWA-MEM v.0.7.17 (ref. 50). Bowtie2 was run using all default parameters while BWA-MEM was run with the –M flag for downstream Picard compatibility. Due to the prohibitively slow speed of NovoAlign, and to improve multithreading performance, we split each sample’s reads into 20 separate batches of equal size and then mapped each batch of 20 using 32 threads with NovoAlign and default parameters.
Read downsampling and pooling.
Sequencing reads were downsampled using SAMtools v.1.6 on the BioGenLink platform (BGL). A workflow was created in BGL called ‘Multi downsample BAM’, which runs the “SAMtools view” tool on all SAM or BAM files in a directory and includes an option to downsample reads by a given fraction corresponding to the “-s” parameter in SAMtools view. The workflow indexed the resulting BAM files using ‘SAMtools index’. The workflow was used to generate all downsampled BAM files and index files, and created a subset with defined read coverage.
BAM files from BWA34 alignment of three replicated runs of WGS with 100× coverage on HCC1395 and HCC1395BL were merged using SAMtools (v.1.8)51 for 200× or 300× coverage, respectively. Newly created BAM files were then indexed and regrouped using Picard Tools (v.2.17.11)46.
Assessment of reproducibility and O-score calculation.
We created and used ‘tornado’ plots to visualize the consistency of mutation calls derived from aligners, callers or repeated NGS runs. The height of the tornado represents the number of overlapping calls in the VCF files, in descending order. The top of each plot portrays SNVs called in every VCF file, while the bottom of each plot contains SNVs present in only one VCF file. The width of the tornado represents the number of accumulated SNVs in that overlapping category, which is scaled by the total number of SNVs in the corresponding subgroup. In addition, we established the following formula to measure reproducibility based on overlapping SNVs:
where n is the total number of VCF results in the pool set, i is the number of overlaps and Oi is the number of accumulated SNVs in the set with i number of overlappings.
Statistical analysis was performed to evaluate the sources of variance in WES and WGS O-scores (JMP Genomics 9.0). For WES, a primary fixed-effect linear regression was first used to screen for two-degree interaction terms significantly contributing to the outcome (F-test P < 0.05). All possible two-degree interactions, along with original variables, were included in this primary model, and five interaction terms (Callers × Mean Coverage Depth, Callers × Percentage GC, Machine model × Callers, Callers × GIV(G > T) and Callers × Percentage Nonduplicated Reads) were found significant (P < 0.05). A linear transform was applied to individual variables to rescale the data to range from −1 to +1. The final fixed-effect linear regression for WES included a total of 13 variables (eight original and five interactions). For WGS, we did not include any interactions because individual variables accounted for >99% of O-score variance. The coefficient of determination (R2) was calculated for both models. In addition, we calculated F-statistics and corresponding P values for variables included in the final model to measure their effects on O-score. Pairwise Pearson correlation coefficients between continuous variables were also calculated for both platforms.
Somatic SNV callers.
We used four somatic variant callers, MuTect2 (GATK 3.8–0)30, SomaticSniper (1.0.5.0)32, Lancet (1.0.7) and Strelka2 (2.8.4)31, which are readily available on the NIH Biowulf cluster, and ran each using the default parameters or parameters recommended in the user’s manual. Specifically, for MuTect2 we included flags for ‘-nct 1 -rf DuplicateRead -rf FailsVendorQualityCheck -rf NotPrimaryAlignment -rf BadMate -rf MappingQualityUnavailable -rf UnmappedRead -rf BadCigar’, to avoid the running exception for ‘Somehow the requested coordinate is not covered by the read’. For MuTect2, we used COSMIC v.82 as required inputs. For SomaticSniper we added a flag for ‘-Q 40 -G -L –F’, as suggested by its original author, to ensure quality scores and reduce probable false positives. For TNscope (201711.03) we used the version implemented in Seven Bridges’s CGC with the following command: ‘sentieon driver -i $tumor_bam -i $normal_bam -r $ref-algo TNscope-tumor_sample $tumor_sample_name-normal_sample $normal_sample_name -d $dbsnp $output_vcf’. For Lancet, we ran with 24 threads on the following parameters: ‘–num-threads 24–cov-thr 10–cov-ratio 0.005–max-indel-len 50 -e 0.005’. Strelka2 was run with 24 threads and the default configuration. The remainder of the software analyzed was run as a single thread on each computer node.
All mutation calling on WES data was performed with the specified genome region in a BED file for exome-capture target sequences. The high-confidence outputs or SNVs flagged as ‘PASS’ in the resulting VCF files were applied to our comparison analysis. Results from each caller used for comparison were all mutation candidates that users would otherwise consider as ‘real’ mutations detected by this caller.
The performance of SNV callers was compared using the following metrics:
GATK indel realignment and quality score recalibration.
The GATK (3.8–0)-IndelRealigner was used to perform indel adjustment with reference indels defined in the 1000 Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/GRCh38_reference_genome/other_mapping_resources/ALL.wgs.1000G_phase3.GRCh38.ncbi_remapper.20150424.shapeit2_indels.vcf.gz). The resulting BAM files were then recalibrated for quality with BaseRecalibrator and dbSNP build 146 as the SNP reference. Lastly, PrintReads was used to generate recalibrated BAM files.
Statistical methods.
Multivariate analyses with both two- and three-way interactions were conducted to explore the source of variation in Jaccard index (JMP Genomics 9.0). Five factors—caller (Strelka2, SomaticSniper and MuTect2), type (all-read versus downsample), SNV_subset (overall, In-truth, Not In-truth and Not defined), pair_group (inter-versus intracenter) and platform (WES versus WGS), as well as their interaction terms—were included in the model. A coefficient of determination (R2) of 0.98 was achieved in the model fitting. F-statistics and corresponding P values were calculated for all factors. We also performed Student’s t-test to evaluate Jaccard index changes for WES and WGS within and across pair groups.
Pearson correlation coefficients were calculated for the percentage of nonduplicated reads, effect mean coverage within target, percentage reads mapped on target, percentage GC, median insert size, GIV (G > T) and GIV (T > G).
Disclaimer.
This is a research study and is not intended to guide clinical applications. The views presented in this article do not necessarily reflect current or future opinion or policy of the US Food and Drug Administration. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services. Any mention of commercial products is for clarification and is not intended as endorsement.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All raw data (FASTQ files) are available on NCBI’s SRA database (SRP162370). The call set for somatic mutations in HCC1395, VCF files derived from individual WES and WGS runs, bam files for BWA-MEM alignments and source codes are available on NCBI’s ftp site (http://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/seqc/Somatic_Mutation_WG/).
Code availability
The code used to create figures and tables is deposited on GitHub under a BSD 2-Clause open-source license tagged at https://github.com/bioinform/somaticseq/tree/seqc2/utilities/Code_for_Figures/best_practices_manuscript. A snapshot can also be downloaded at https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/seqc/Somatic_Mutation_WG/tools/.
Extended Data
Extended Data Fig. 1 |. Study design to capture “wet lab” factors affecting sequencing quality.
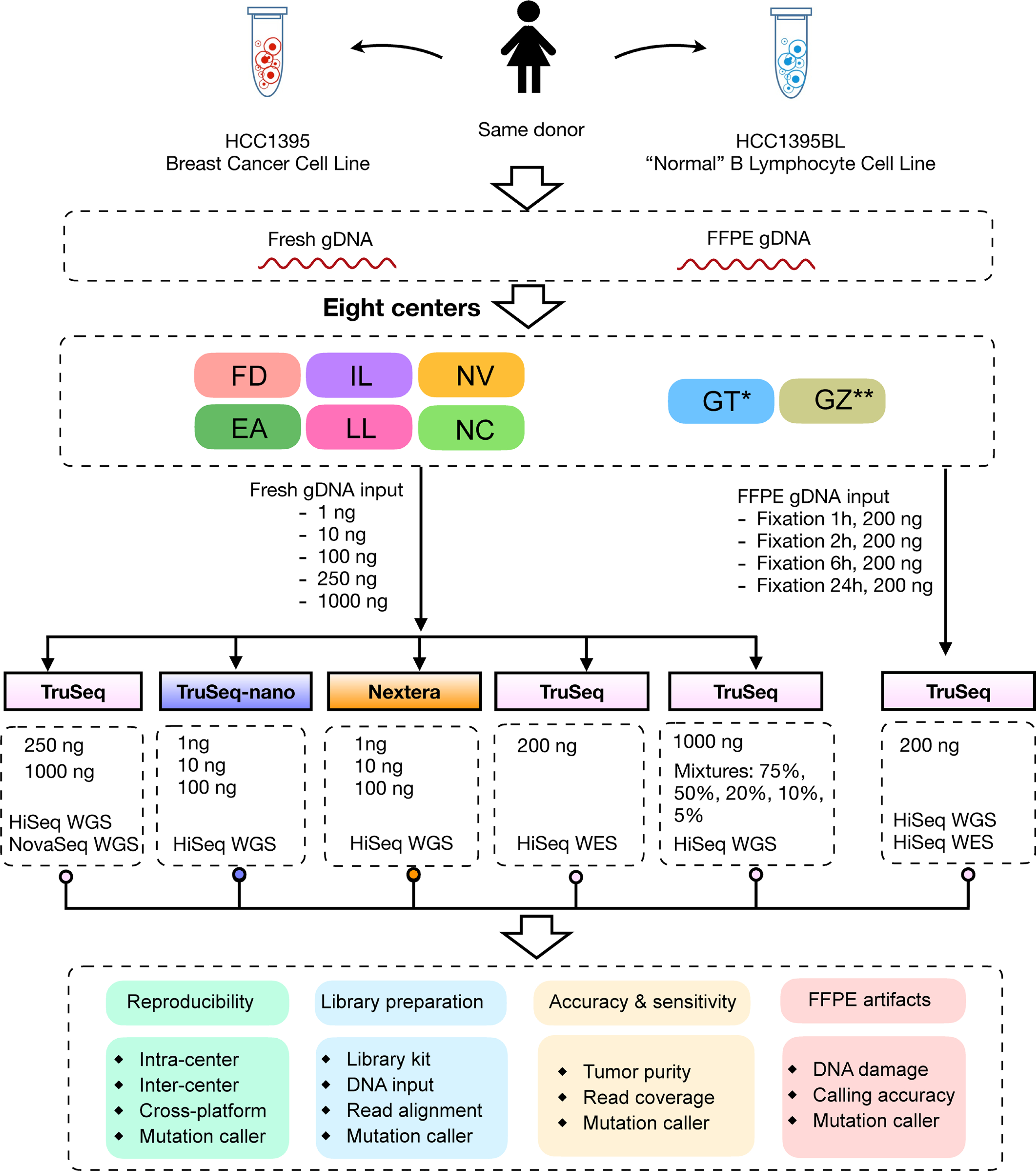
DNA was extracted from either fresh cells or FFPE processed cells (formalin fixation time of 1, 2, 6, or 24 hours). Both fresh DNA and FFPE DNA were profiled on WGS and WES platforms. For fresh DNA, six centers (Fudan University (FD), Illumina (IL), Novartis (NV), European Infrastructure for Translational Medicine (EA), National Cancer Institute (NC), and Loma Linda University (LL)) performed WGS and WES in parallel following manufacturer recommended protocols with limited deviation. Three of the six sequencing centers (FD, IL, and NV) generated library preparation in triplicate. For FFPE samples, each fixation time point had six blocks that were sequenced at two different centers (IL and GeneWiz (GZ)). Three library preparation protocols (TruSeq PCR-free, TruSeq-Nano, and Nextera Flex) were used with four different quantities of DNA input (1, 10, 100, and 250 ng) and sequenced by IL and LL. DNAs from HCC1395 and HCC1395BL were pooled at various ratios to create mixtures of 75%, 50%, 20%, 10%, and 5%. All libraries from these experiments were sequenced in triplicate on the HiSeq series by Genentech (GT). In addition, nine libraries using the TruSeq PCR-free preparation were run on a NovaSeq for WGS analysis by IL. Sample naming convention (example: WGS_FD_N_1): First field was used for sequencing study: Whole genome sequencing (WGS), Whole exome sequencing (WES), WGS on FFPE sample (FFG), WES on FFPE sample (FFX), WGS on library preparation protocol (LBP), WGS on tumor purity (SPP); Second field was used for sequencing centers, EA, FD, IL, LL, NC, NV, GT, and GZ or sequencing technologies, HiSeq (HS) and NovaSeq (NS); Third field was used for tumor (T) or normal (N); The last field was used for the number of repeats. *WGS performed only on Mixture (tumor purity) samples. ** WGS and WES performed only on FFPE samples.
Extended Data Fig. 2 |. Read mapping quality statistics.
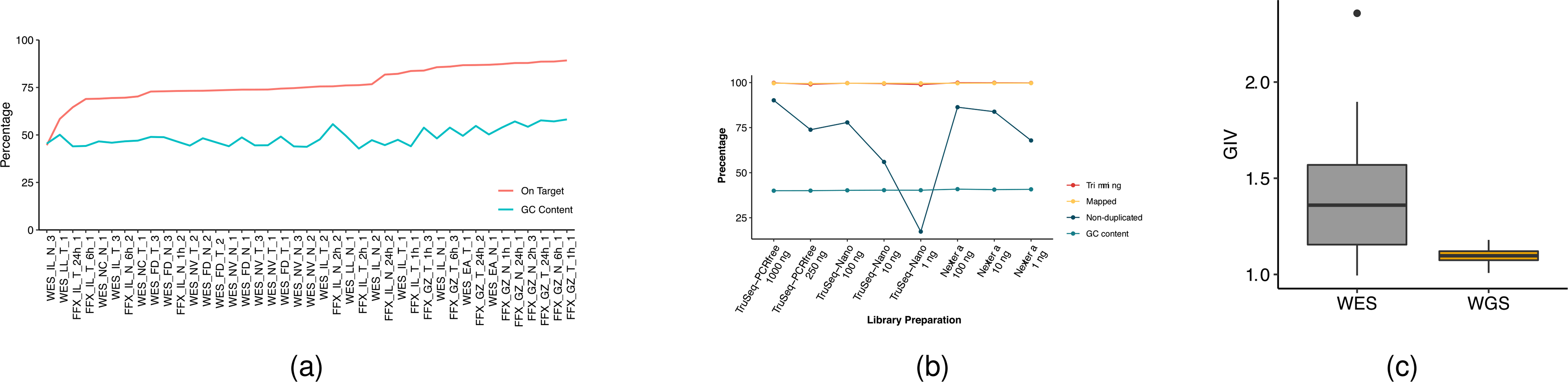
(a) Percentage of reads mapped to target regions (SureSelect V6 + UTR) and G/C content for WES runs on fresh or FFPE DNA. (b) Read quality from three WGS library preparation kits (TruSeq PCRfree, TruSeq-Nano, and Nextera Flex) on fresh or FFPE DNA. (c) Distribution of GIV scores in WGS and WES runs. For detailed statistics regarding the boxplot, please refer to Supplementary Table 5.
Extended Data Fig. 3 |. Overall read quality distribution for all WES and WGS runs.
(a) Median insert fragment size of WES and WGS run on fresh and FFPE DNA. (b) G/C read content for Wes and WGS runs. (c) Overall read redundancy for WES and WGS runs. Some outliers were observed in WGS on fresh DNA, which were from runs of TruSeq-Nano with 1 ng of DNA input. (d) Overall percentage of reads mapped to target regions for WES runs for fresh and FFPE DNA. For detailed statistics regarding the boxplot, please refer to Supplementary Table 6.
Extended Data Fig. 4 |. Mutation calling repeatability and O_Score distribution.
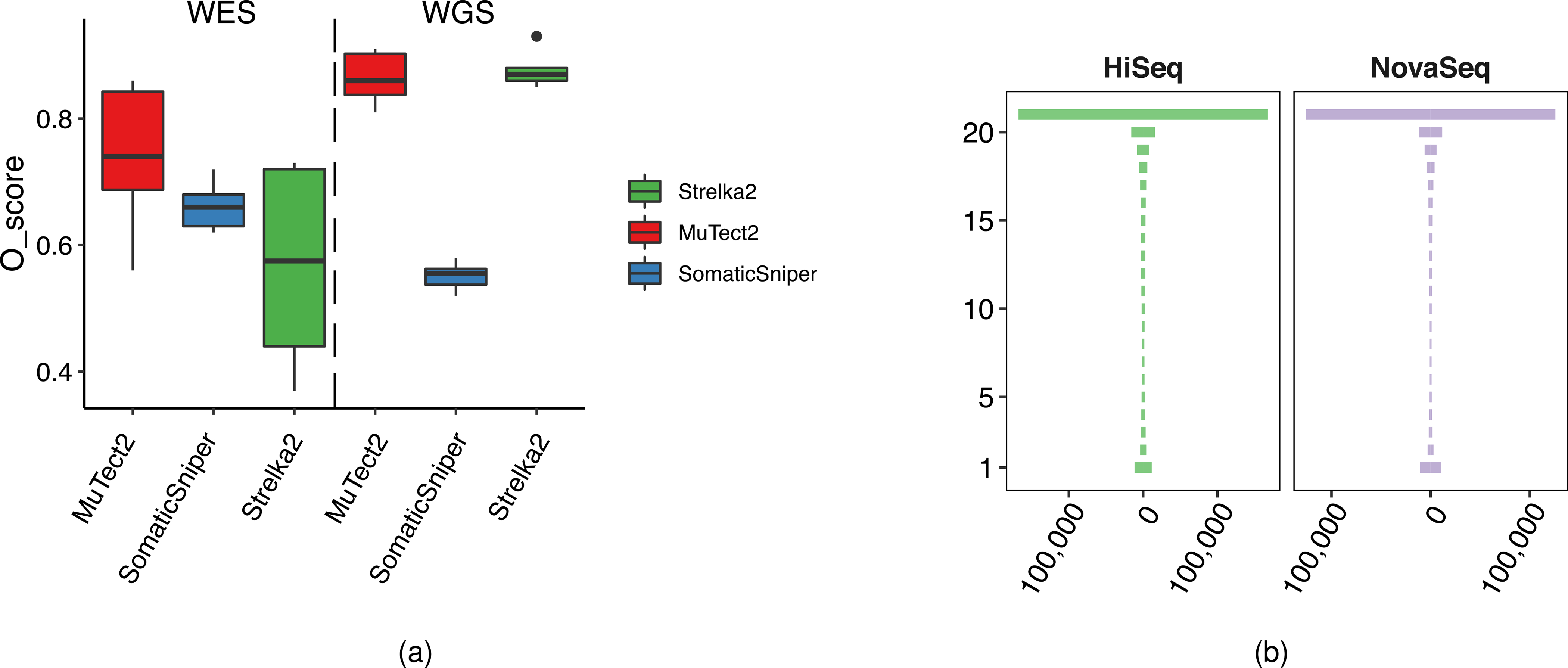
(a) Distribution of O_Score of three callers (MuTect2, Strelka2, and SomaticSniper) for twelve WGS and WES runs on BWA alignments. For detailed statistics regarding the boxplot, please refer to Supplementary Table 7. (b) “Tornado” plot of reproducibility between twelve WGS runs on the HiSeq series (2500, 4000, and X10) and nine WGS runs on the NovaSeq (S6000). SNVs/indels were called by Strelka2 on BWA alignments.
Extended Data Fig. 5 |. Source of variance in reproducibility measured by O_Score.

Actual by predicted plot of WGS (a) and WES (b). A total of 8 variables (WGS) or 13 variables (WES), including 2-degree interactions, were included in the fixed effect linear model. 36 samples were used to derive statistics for both WES and WGS. The central blue line is the mean. The shaded region represents the 95% confidence interval.
Extended Data Fig. 6 |. Effect of post alignment processing on precision and recall of WES and WGS run on FFPE DNA.

(a) precision and recall of mutation calls by Strelka2 on BWA alignments. A single library of FFPE DNA (FFX) and three libraries of fresh DNA (EA_1, FD_1, and NV_1) were run on a WES platform. Resulting reads were either processed by the BFC tool or by Trimmomatic. processed FASTQ files were then aligned by BWA and called by Strelka2. precision and recall were derived by matching calling results with the truth set. (b) precision and recall of mutation calls by three callers, Mutect2 (blue), Strelka2 (green), and SomaticSniper (red), on BWA alignments without or with GATK post alignment process (indel realignment & BQSR).
Extended Data Fig. 7 |. Jaccard index scores to measure reproducibility of SNVs called by three callers.

Box plot of Jaccard scores of inter-center, intra-center, and overall pair of SNV call sets from two WGS or WES runs. SNVs were divided into three groups; Repeatable: SNVs defined in the truth set of the reference call set; Gray zone: SNVs not defined as “truth” in the reference call set; Non-Repeatable: SNVs were not in the reference call set. For detailed statistics regarding the boxplot, please refer to Supplementary Table 8.
Extended Data Fig. 8 |. Sources of variation in Jaccard index.

(a) Summary of factor effects. Twenty-five factors, including five original factors, ten 2-way interactions, and ten 3-way interactions were evaluated in the model. Both P values (derived from F-test) and their LogWorth (−log10 (P value)) are included in the summary plot. The factors are ordered by their LogWorth values. (b) Least square means of caller*pair_group*platform interaction. The height of the markers represents the adjusted least square means, and the bars represent confidence intervals of the means. (c) Least square means SNV_subset*pair_group*platform interaction. The height of the markers represents the adjusted least square means, and the bars represent confidence intervals of the means. 3168 samples were used to derive these statistics. (d) Student’s t-test for platform*pair_group interaction with SNV calls from three callers, MuTect2, Strelka2, and SomaticSniper. The left two panels compare Jaccard indices between intra-center and inter-center for WGS and WES, respectively. The right two panels compare Jaccard indices between WGS and WES for inter-center and intra-center pairs, respectively. Prob > |t| is the two-tailed test P value, and Prob > t is the one-tailed test P value.
Extended Data Fig. 9 |. WGS vs. WES platform-specific mutations and allele frequency calling accuracy.

Cumulative VAF plot of precision (a), recall (b), and F-Score (c) for three callers (MuTect2, Strelka2, and SomaticSniper) on WES and WGS runs.
Extended Data Fig. 10 |. Mutation allele frequency and coverage depth in WES and WGS sample.

Scatter plot of allele frequency and coverage depth by three callers, MuTect2, Strelka2, and SomaticSniper in one example WES sample (a) or WGS sample (b). (c) Boxplot of read depth on called mutations in WES or WGS. For detailed statistics regarding the boxplot, please refer to Supplementary table 9.
Supplementary Material
Acknowledgements
We thank G. Sivakumar (Novartis) and S. Chacko (Center for Information Technology, National Institutes of Health (NIH)) for their assistance with data transfer, and J. Ye (Sentieon) for providing the Sentieon software package. We also thank D. Goldstein (Office of Technology and Science at the National Cancer Institute (NCI); L. Amundadottir (Division of Cancer Epidemiology and Genetics, NCI, NIH) for sponsorship and usage of the NIH Biowulf cluster; R. Phillip (Center for Devices and Radiological Health, US Food and Drug Administration) for advice on study design; and Seven Bridges for providing storage and computational support on the Cancer Genomic Cloud (CGC). The CGC has been funded in whole or in part with federal funds from the NCI, NIH (contract no. HHSN261201400008C and ID/IQ agreement no. 17×146 under contract no. HHSN261201500003I). Y. Zhao, J.L., T.-W.S., K.T., J.S., Y.K., A.R., B.T. and P.J. were supported by the Frederick National Laboratory for Cancer Research and through the NIH fund (NCI contract no. 75N910D00024). Research carried out in Charles Wang’s laboratory was partially supported by NIH grant no. S10OD019960 (to C.W), the Ardmore Institute of Health (grant no. 2150141 to C.W) and a Charles A. Sims gift. L.S. and Y. Zheng were supported by the National Key R&D Project of China (no. 2018YFE0201600), the National Natural Science Foundation of China (no. 31720103909) and Shanghai Municipal Science and Technology Major Project (no. 2017SHZDZX01). E.R. was supported by the European Union through the European Regional Development Fund (project no. 2014–2020.4.01.15–0012). J.N. and U.L. were supported by grants from the Swedish Research Council (no. 2017–00630/2019–01976). The work carried out at Palacky University was supported by grant no. LM2018132 from the Czech Ministry of Education, Youth and Sports. C.X. and S.T.S. were supported by the Intramural Research Program of the National Library of Medicine, NIH. This work also used the computational resources of the NIH Biowulf cluster (http://hpc.nih.gov). Original data were also backed up on servers provided by the Center for Biomedical Informatics and Information Technology, NCI. In addition, we thank the following individuals for their participation in working group discussions: M. Ashby, O. Aygun, X. Bian, P. Bushel, F. Campagne, T. Chen, H. Chuang, Y. Deng, D. Freed, P. Giresi, P. Gong, Y. Guo, C. Hatzis, S. Hester, J. Keats, E. Lee, Y. Li, S. Liang, T. McDianiel, J. Pandey, A. Pathak, T. Shi, J. Trent, M. Wang, X. Xu and C. Zhang. The following individuals from the University of Toledo Medical Center helped to troubleshoot or set up the FFPE protocol: A. Al-Agha, T. Cummins, C. Freeman, C. Nowak, A. Smigelski, J. Yeo and V. Kholodovych.
Footnotes
Competing interests
L.F. was an employees of Roche Sequencing Solutions Inc. L.K., K.L. and M.M. are employees of ATCC, which provided cell lines and derivative materials. E.J., O.D.A., T.T., A.M., A.N., A.G. and G.P.S. are employees of Illumina Inc. V.P. and M.S. are employees of Novartis Institutes for Biomedical Research. T.H., E.P and R. Kalamegham are employees of Genentech (a member of the Roche group). Z.L. is an employee of Sentieon Inc. R.K. is an employee of Immuneering Corp. C.E.M. is a cofounder of Onegevity Health. All other authors claim no competing interests.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-021-00994-5.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41587-021-00994-5.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-021-00994-5.
Reprints and permissions information is available at www.nature.com/reprints.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply 2021
References
- 1.Glasziou P, Meats E, Heneghan C & Shepperd S What is missing from descriptions of treatment in trials and reviews? Brit. Med. J 336, 1472–1474 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasilevsky NA et al. On the reproducibility of science: unique identification of research resources in the biomedical literature. PeerJ 1, e148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Begley CG & Ellis LM Drug development: raise standards for preclinical cancer research. Nature 483, 531–533 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Alioto TS et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun 6, 10001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Griffith M et al. Genome Modeling System: a knowledge management platform for genomics. PLoS Comput. Biol 11, e1004274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chalmers ZR et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu H, DiCarlo J, Satya RV, Peng Q & Wang Y Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics 15, 244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghoneim DH, Myers JR, Tuttle E & Paciorkowski AR Comparison of insertion/deletion calling algorithms on human next-generation sequencing data. BMC Res. Notes 7, 864 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Q et al. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 5, 91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simen BB et al. Validation of a next-generation-sequencing cancer panel for use in the clinical laboratory. Arch. Pathol. Lab. Med 139, 508–517 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Linderman MD et al. Analytical validation of whole exome and whole genome sequencing for clinical applications. BMC Med. Genomics 7, 20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zook JM et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol 32, 246–251 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Zook JM et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 3, 160025 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin M-T et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol 141, 856–866 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh RR et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J. Mol. Diagn 15, 607–622 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Griffith M et al. Optimizing cancer genome sequencing and analysis. Cell Syst. 1, 210–223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olson ND et al. precisionFDA Truth Challenge V2: calling variants from short- and long-reads in difficult-to-map regions. Preprint at bioRxiv 10.1101/2020.11.13.380741 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morrissy AS et al. Spatial heterogeneity in medulloblastoma. Nat. Genet 49, 780–788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Araf S et al. Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia 32, 1261–1265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stephens PJ et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462, 1005–1010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalyana-Sundaram S et al. Gene fusions associated with recurrent amplicons represent a class of passenger aberrations in breast cancer. Neoplasia 14, 702–708 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J et al. INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 26, 108–118 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang LT et al. Establishing reference data and call sets for benchmarking cancer mutation detection using whole-genome sequencing. Preprint at bioRxiv 10.1101/625624 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen X et al. A multi-center cross-platform single-cell RNA sequencing reference dataset. Sci. Data 8, 39 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen W et al. A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples. Nature Biotechnol. https://www.nature.com/articles/s41587-020-00748-9 (2020). [DOI] [PMC free article] [PubMed]
- 26.Zhao Y et al. Whole genome and exome sequencing reference datasets from a multi-center and cross-platform benchmark study. Preprint at bioRxiv 10.1101/2021.02.27.433136 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Liu P, Evans TC & Ettwiller LM DNA damage is a pervasive cause of sequencing errors, directly confounding variant identification. Science 355, 752–756 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Costello M et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Do H & Dobrovic A Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin. Chem 61, 64–71 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Cibulskis K et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saunders CT et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Larson DE et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ivanov M et al. Towards standardization of next-generation sequencing of FFPE samples for clinical oncology: intrinsic obstacles and possible solutions. J. Transl. Med 15, 22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H BFC: correcting Illumina sequencing errors. Bioinformatics 31, 2885–2887 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Freed D, Pan R & Aldana R TNscope: accurate detection of somatic mutations with haplotype-based variant candidate detection and machine learning filtering. Preprint at bioRxiv 10.1101/250647 (2018). [DOI] [Google Scholar]
- 39.Narzisi G et al. Lancet: genome-wide somatic variant calling using localized colored DeBruijn graphs. Commun. Biol 1, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gargis AS et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat. Biotechnol 30, 1033–1036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y-C et al. Comprehensive assessment of somatic copy number variation calling using next-generation sequencing data. Preprint at bioRxiv 10.1101/2021.02.18.431906 (2021). [DOI] [Google Scholar]
- 42.Sahraeian SME, Fang LT, Mohiyuddin M, Hong H & Xiao W Robust cancer mutation detection with deep learning models derived from tumor-normal sequencing data. Preprint at bioRxiv 10.1101/667261 (2019). [DOI] [Google Scholar]
- 43.Tian SK et al. Optimizing workflows and processing of cytologic samples for comprehensive analysis by next-generation sequencing: Memorial Sloan Kettering Cancer Center experience. Arch. Pathol. Lab. Med 140, 1200–1205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.FastQC (Babraham Bioinformatics, accessed 2 July 2021); https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- 45.Wood DE & Salzberg SL Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Picard (Broad Institute, accessed 2 July 2021); http://broadinstitute.github.io/picard/ [Google Scholar]
- 47.Okonechnikov K, Conesa A & García-Alcalde F Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ewels P MultiQ. C. Aggregate results from bioinformatics analysis across many samples into a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013). [Google Scholar]
- 51.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data (FASTQ files) are available on NCBI’s SRA database (SRP162370). The call set for somatic mutations in HCC1395, VCF files derived from individual WES and WGS runs, bam files for BWA-MEM alignments and source codes are available on NCBI’s ftp site (http://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/seqc/Somatic_Mutation_WG/).