

Abstract
Cancer cells usually show adaptations to their metabolism that facilitate their growth, invasiveness, and metastasis. Therefore, reprogramming the energy metabolism is one of the current key foci of cancer research and treatment. Although aerobic glycolysis-the Warburg effect-has been thought to be the dominant energy metabolism in cancer, recent data indicate a different possibility, specifically that oxidative phosphorylation (OXPHOS) is the more likely form of energy metabolism in some cancer cells. Due to the heterogeneity of epithelial ovarian cancer, there are different metabolic preferences among cell types, study types (in vivo/in vitro), and invasiveness. Current knowledge acknowledges glycolysis to be the main energy provider in ovarian cancer growth, invasion, migration, and viability, so specific agents targeting the glycolysis or OXPHOS pathways have been used in previous studies to attenuate tumor progression and increase chemosensitization. However, chemoresistant cell lines exert various metabolic preferences. This review comprehensively summarizes the information from existing reports which could together provide an in-depth understanding and insights for the development of a novel targeted therapy which can be used as an adjunctive treatment to standard chemotherapy to decelerate tumor progression and decrease the epithelial ovarian cancer mortality rate.
Keywords: Chemoresistance, chemosensitivity, epithelial ovarian cancer, glycolysis, oxidative phosphorylation
Introduction
Epithelial ovarian cancer (EOC) is one of the most common causes of cancer death in women for two main reasons: its most frequent presentation occurs at an advanced-stage and its high recurrence rate [1]. Despite the use of chemotherapy and targeted therapy, the ovarian cancer mortality rate remains as high as 70% [2]. The factors that affect the disease prognosis and survival of EOC are its invasiveness, metastatic properties, and treatment response [3,4]. The extent of its invasiveness and metastatic properties reflect the staging of EOC [2]. As regards the treatment response, several factors can contribute to chemoresistance after a period of treatment for EOC, including an increased elimination of the active form of chemotherapy and the development of drug-resistant genes [5-7]. In the past decade, a new theory called “deregulation of cellular energetics” has been posited that focuses on the metabolic support of the growth, proliferation, invasion, and metastasis of cancer cells, and this has become one of the biological targets of cancer treatment development [8]. The Warburg effect, defined as the preference of cancer cells to use glycolysis even in the presence of oxygen, became one of those targets [9]. Prior studies demonstrated that the Warburg effect might be associated with the resistance of most cancer cells to treatment [10-12]. However, current evidence suggests that not every tumor exhibits the Warburg effect [13,14]. EOC, a heterogeneous form of cancer, may use another pathway, such as oxidative phosphorylation (OXPHOS) [14,15].
Apart from the current regimens of chemotherapy and targeted therapy, such as antiangiogenesis and poly-ADP ribose polymerase (PARP) inhibitors, a treatment that modulates cancer metabolism has been proposed to improve the treatment response of EOC [16-20]. In this review, we comprehensively summarize studies on the metabolic changes in EOC compared to normal ovarian cells from in vitro, in vivo and clinical studies. Consistencies and controversies from the reports on the metabolic characters that are associated with the invasiveness and chemoresistant properties of EOC, and the effects of metabolic interventions on EOC progression and treatment response are also summarized and discussed. This comprehensive review will enhance the fundamental overarching understanding pertinent to the metabolism of EOC and highlight mechanistic insights for the development of novel drug regimens to target this. Advances in drug therapies may assist in reducing the tumor invasiveness, the tumor metastasis, and the mortality rate of EOC via the metabolic pathways.
Search strategy and selection criteria
The PubMed database was searched using the keywords “ovarian cancers”, “glycolysis”, and “oxidative” from its inception to September 2020. The search was limited to original articles published in English.
A general consideration of ovarian cancer metabolism regarding glycolysis and the oxidative phosphorylation pathway
The pathways and regulation of glycolysis and OXPHOS are depicted in Figure 1. Under normal physiological conditions, glycolysis consists of a multistep pathway of glucose breakdown, followed by the conversion of phosphoenolpyruvate (PEP) to pyruvate via the enzyme pyruvate kinase M1 (PKM1). Pyruvate is then moved to the mitochondria and enters the tricarboxylic acid (TCA) cycle via mitochondrial pyruvate carrier 1 (MPC1). This is followed by a respiratory chain consisting of five complexes, resulting in the release of thirty-six ATP molecules per single glucose molecule [21]. In the mitochondria, OXPHOS occurs resulting in the production of ATP, the energy for this production [21]. Under hypoxic conditions, PEP is converted to pyruvate by pyruvate kinase M2 (PKM2), pyruvate is subsequently changed into lactate by lactate dehydrogenase (LDH) and moves out of the cells via monocarboxylate transporter 4 (MCT4) (Figure 1) [22].
Figure 1.
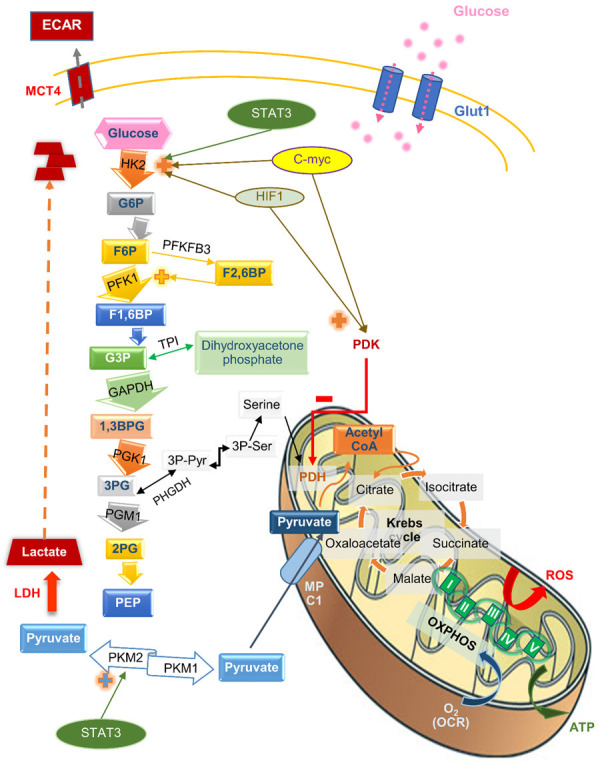
The pathways and regulation of glycolysis, the TCA cycle, and oxidative phosphorylation. The figure shows glucose metabolism from its uptake into the cell, the metabolic pathways, including the glycolytic pathway in the cell cytoplasms, the tricarboxylic acid cycle, and the mitochondrial oxidative phosphorylation, and its regulatory enzymes and metabolites. Abbreviations: 1, 3BPG, 1, 3-bisphosphoglycerate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; 3PPyr, 3 phosphopyruvate; 3PSer, 3 phosphoserine; ECAR, extracellular acidification rate; F1, 6BP, fructose-1,6-bisphosphate; F6P, fructose-6-phosphate; G3P, glyceraldehyde-3-phosphate; G6P, glucose-6-phosphate; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HIF1, hypoxia-induced transcription factor 1; HK2, hexokinase 2; LDH, lactate dehydrogenase; MCT4, monocarboxylate transporter 4; MPC1, mitochondrial pyruvate carrier 1; OCR, oxygen consumption rate; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PEP, phosphoenolpyruvate; PFK1, phosphofructokinase 1; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; PGM1, phosphoglucomutase 1; PHGDH, phosphoglycerate dehydrogenase; PK, pyruvate kinase; ROS, reactive oxygen species; TPI, triosephosphate isomerase.
Even under normoxic conditions, some cancer cells utilize glycolysis without any of the glucose residues entering the TCA cycle [22]. Since glycolysis allows the diversion of glycolytic intermediates into various biosynthetic pathways, glycolytic enzymes also support cell growth [8,23]. For instance, hexokinase 2 (HK2) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) enzymes can regulate mTOR and apoptosis, while the phosphoglycerate mutase 1 (PGAM1) enzyme can induce the formation of fibrillar actin (F-actin) and also cell migration [23]. In some cancers, F-actin is a crucial structure of cancer cell membrane protrusions (lamellipodia), which facilitate cell migration and the formation of dynamic protrusions (invadopodia), contributing to extracellular matrix penetration [24].
Although many studies have reported a metabolic shift to the aerobic glycolysis of cancer cells in the past decades, there is evidence to show that not all cancer types are dependent on glycolysis [17,18]. This includes EOC, which is one of the most heterogeneous cancers. It is now known that different histological types of EOC exhibit different kinds of metabolic change according to the specific conditions of this cancer, including chemoresistant vs. chemosensitive, and more aggressive vs. less aggressive.
Metabolic changes in the epithelial ovarian cancer cell lines
Previous in vitro studies investigated metabolic changes in the EOC cells and compared them to normal ovarian epithelial cells (Table 1). In addition, the metabolic alterations of EOC cells have been compared within a variety of histologic types and also the invasiveness levels in the cells. There is growing evidence to show that metabolic changes in EOC cells mainly involve OXPHOS rather than the glycolytic pathway (Table 1).
Table 1.
Metabolic changes in epithelial ovarian cancer: in vitro studies
| Study models | Major findings | Interpretation | References | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Glycolytic-related findings | OXPHOS-related findings | |||||||||
|
|
|
|||||||||
| Glycolytic enzymes | Basal ECAR | Glycolytic capacity | Others | OXPHOS enzymes | Basal OCR | Respiratory capacity | Others | |||
| -Human ovarian HGSC cells (OVCAR3 (S), human OCCC cells (TOV-21G (C)) | ↑↑ PFKP | (S): ↑↑ PDHB, IDH2, IDH3A, OGDHL, ND2, UQCRH | -HGSC and clear cells had an increase in mitochondrial complex I and complex III activity compared to normal cells. | [26] | ||||||
| ↓↓ CYB | ||||||||||
| -Normal ovarian epithelial cells (control) | ↑↑ PKM | (C): ↑↑ IDH2, OGDHL, ND2, CYB | ||||||||
| ↓↓ IDH3A | ||||||||||
| -Human HGSC cells (OVCAR3) | ↔ | ↔ | ↑↑ | ↑↑ | -Human OCCC had an increase in both glycolysis and OXPHOS metabolism compared to other carcinoma and normal cells, while HGSC had higher OXPHOS only when compared to other carcinoma and normal cells. | [25] | ||||
| -Normal ovarian epithelial cells (control) | ||||||||||
| -Human HGSC cells (OVCAR3) | ↔ | ↔ | ↑↑ | ↑↑ | ||||||
| -Human ovarian carcinoma cells OVCA420 (control) | ||||||||||
| -Human OCCC cells (ES-2, TOV-21G) | ↑↑ (ES2) | ↑↑ | ↑↑ | ↑ (ES2) | ||||||
| -Normal ovarian epithelial cells (control) | ↑ (TOV-21G) | ↑↑ (TOV-21G) | ||||||||
| -Human OCCC cells (ES-2, TOV-21G) | ↑ (ES2) | ↔ (ES2) | ↑↑ | ↑↑ | ||||||
| -Human non-OCCC cells, OVCA420 (control) | ↔ (TOV-21G) | ↑↑ (TOV-21G) | ||||||||
| -Human ovarian E/OCCC cells (SKOV3) | ↓↓ PFKP | ↑ PDHB | -Endometrioid or OCCC may have shown glycolysis impairment. | [26] | ||||||
| -Normal ovarian epithelial cells (control) | ↑↑ PKM | ↔ IDH2 | ||||||||
| ↑↑ IDH3A, IDH3B | ||||||||||
| ↔ OGDHL | ||||||||||
| ↑↑ ND2, ND5, CYB | ||||||||||
| ↓↓ UQCRH | ||||||||||
| -Human ovarian carcinoma cells (OVCA420, OVCA429, OVCA433, DOV-13) | ↓ (DOV13) | ↑↑* | ↑↑* | ↓↓ (OVCA420, OVCA433) | -Most human nonspecific adenocarcinoma cells had higher OXPHOS than normal cells. | [25] | ||||
| -Normal ovarian epithelial cells (control) | ||||||||||
| -More invasive human E/OCCC (SKOV3, SKOV3ip1) | ↔ lactate | ↑ | ↑ | ↑ pyruvate uptake | -More invasive human E/OCCC cells had higher OXPHOS compared to less invasive HGSCs. | [28] | ||||
| -Less invasive human HGSC (OVCAR3) (control) | ↑ ATP | |||||||||
| Mouse ovarian surface epithelial (MOSE) cells | ↑ | ↑↑ glucose uptake | ↑ PDK1 | ↓ | ↓ ATP | -Mouse aggressive ovarian cells had higher glycolysis and lower OXPHOS compared to mouse benign ovarian epithelial cells. | [27] | |||
| -MOSE-L = late-aggressive phenotype | ↑↑ lactate | ↑ CS | ||||||||
| -MOSE-E = early benign phenotype (control) | ↔ PDH | |||||||||
The superscript *indicates all cell lines except OVCA420. The arrow ↑ or ↓ denotes significant increase or decrease in the metabolic-related finding with a P value <0.05. The arrow ↑↑ or ↓↓ denotes significant increase or decrease in the metabolic-related finding with a P value <0.01. Abbreviations: CS; citrate synthase, CYB; cytochrome b reductase 1, E; endometrioid adenocarcinoma, ECAR; extracellular acidification rate, HGSC; high-grade serous carcinoma, IDH2; isocitrate dehydrogenase [NADP], IDH3A; isocitrate dehydrogenase [NAD] subunit alpha, IDH3B; isocitrate dehydrogenase [NAD] subunit beta, ND; NADH-ubiquinone oxidoreductase, OCCC; ovarian clear cell carcinoma, OCR; oxygen consumption rate, OGDHL; 2-oxoglutarate dehydrogenase-like, OXPHOS; oxidative phosphorylation, PDH; pyruvate dehydrogenase, PDK1; pyruvate dehydrogenase kinase 1, PFKP; phosphofructokinase platelet, PKM; pyruvate kinase muscle isozyme, UQCRH; ubiquinol-cytochrome c reductase hinge protein.
Various levels of glycolytic activity have been observed among human EOC cell lines when compared to the activity of normal human ovarian cells. Specifically, ES-2, OVCA429, OVCA433, and TOV-21G reveal higher glycolytic enzyme-related gene expression levels, higher basal extracellular acidification rate ECAR, and higher glycolytic capacity than do normal ovarian cells [25,26]. OVCAR3 shows a higher level of PFK and PKM [26], but non-significantly different basal ECAR and glycolytic capacity compared to normal ovarian cells [25]. Conversely, the SKOV3 and DOV-13 cell lines show lower phosphofructokinase (PFK) levels and basal ECAR than normal ovarian cells respectively [25,26]. In mouse EOC cells, the glycolytic activity is lower than it is in normal mouse ovarian cells, as indicated by the lower levels of glucose uptake, basal ECAR, and lactate [27].
When the OXPHOS activity was compared between human EOC cells and normal human ovarian cells, the results showed that cells from high-grade serous adenocarcinoma (HGSC), clear cell carcinoma (CCC), and other ovarian adenocarcinomas displayed higher OXPHOS activity than the OXPHOS activity in normal cells, as indicated by the higher levels of OXPHOS enzymes, the basal oxygen consumption rate (OCR), and the respiratory capacity [25,26]. In contrast, a previous study on mouse ovarian cells revealed that the OXPHOS activity was lower in mouse EOC cells when compared with the OXPHOS activity of benign mouse ovarian epithelial cells, as indicated by the higher basal OCR and ATP levels [27].
When considering the invasiveness of human EOC cells, the lactate level (the end product of anaerobic glycolysis) is similar between the more invasive EOC cell line, SKOV3, and the less invasive EOC cell line, OVCAR3 [28]. However, the OXPHOS activity was found to be higher in the SKOV3 cell line when compared to the OXPHOS activity of the OVCAR3 cell line, as indicated by the higher basal OCR and respiratory capacity levels [28].
In summary, the results from in vitro studies reveal that OXPHOS activity is more dominant in human EOC cells than in normal human ovarian cells and similarly, it is more dominant in more invasive human EOC cells than in less invasive human EOC cells. However, the glycolytic activity of EOC cell lines varies in comparison to normal ovarian cells. The different levels of OXPHOS activity between human EOC cells and mouse EOC cells suggests that the metabolic changes in EOC are species-specific, which implies that they may be mediated by differences in genetic regulation. Further studies are required to identify the exact mechanisms that result in the contradictory findings between human and mouse EOC cells.
Metabolic changes in epithelial ovarian cancer cells: clinical evidence
Clinical evidence from previous studies investigating metabolic changes in ovarian cancer tissues and serum samples from patients is listed in Table 2. All the studies showed that the EOC tissues and serum from EOC patients showed more glycolytic activity than did the normal ovarian tissues and serum from healthy women, as indicated by the higher GLUT expression levels, glycolytic enzymes, and hypoxia-induced transcription factor 1α (HIF1α), a glycolysis-regulating factor [26,29-32]. In terms of OXPHOS, a previous study demonstrated an increase in the enzymes involved in the TCA cycle and OXPHOS, including pyruvate dehydrogenase (PDH), citrate synthase (CS), and isocitrate dehydrogenase (IDH) in human EOC tissues when compared to normal human ovarian tissues [26].
Table 2.
Metabolic changes in epithelial ovarian cancer: clinical studies
| Tumor tissues or serum samples from human | Major findings | Interpretation | References | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Glycolytic-related findings | OXPHOS-related findings | ||||||
|
|
|
||||||
| Glut expression | Glycolytic enzymes | Others | OXPHOS enzymes | Others | |||
| -150 EOC tissues (stage I-IV) | ↑ PKM2 | ↑↑ iNOS, eNOS | -Human EOC tissues showed higher glycolytic shift than normal ovarian tissues. | [29] | |||
| -10 normal ovarian tissues (control) | |||||||
| -38 EOC tissues | ↑ | ↑ HK2 | ↑ HIF1α, STAT3 | -Tissues and serum of EOC patients showed higher levels of glycolysis compared to tissues and serum of healthy people. | [31] | ||
| -22 normal ovarian tissues (control) | ↑ LDHA | ↓ SIRT3 | |||||
| -44 serum samples from EOC patients | ↑ | ↑ HK2 | ↑ HIF1α, STAT3 | ||||
| -35 serum samples from healthy people (control) | ↑ LDHA | ↓ SIRT3 | |||||
| -78 EOC tissues | ↑↑ | ↑↑ HK2 | ↑↑ FOXM1 | -Human EOC tissues had higher glycolysis compared to normal ovarian tissues. | [30] | ||
| -35 normal ovarian tissues (control) | |||||||
| -7 EOC tissues | ↑ PFK | ↑ PDHB, CS, IDH2, OGDHL | ↑ mitochondrial DEPs | -Human EOC tissues had an increase in OXPHOS compared to normal ovarian tissues. | [26] | ||
| -11 normal ovarian tissues (control) | ↑ PKM | ↑ UQCRH, CYB | |||||
| -282 HGSC TMAs | ↑↑ | ↑ HK2 | -HGSC tumor tissues had higher glucose metabolism compared to other histological types. | [35] | |||
| -98 non-HGSC TMAs (control) | ↔ PKM2 | ||||||
| ↓ LDHA | |||||||
| -139 OCCC microarray dataset | ↑↑ HK1 | -OCCC had higher glycolysis phenotype compared to other non-OCCC adenocarcinoma. | [33] | ||||
| -24 non-OCCC microarray dataset (control) | ↑↑ LDHA | ||||||
| -8 OCCC and 67 high-grade EOC tissues | ↑ | ↑ PKM2 | -High-grade EOCs and OCCC had higher glycolysis than normal and low-grade EOC tissues. | [34] | |||
| -8 normal ovarian TMAs and 29 low-grade EOC TMAs (control) | ↑ LDHA | ||||||
| -32 HGSC tissues | ↑ BEC index | -HGSC tumor tissues was more dependent on OXPHOS than LGSC tissues. | [36] | ||||
| -15 LGSC tissues (control) | |||||||
| -11 EOC tissues (stage I-III) | ↑ G6P* | ↔ citrate | ↔ pyruvate | -Human primary EOC and metastatic omental tissues had higher glycolysis than normal ovarian tissues. | [32] | ||
| -7 MOC tissues in the omentum (stage III-IV) | ↑↑ lactate | ||||||
| -12 normal ovarian tissues (control) | |||||||
| -1 tissue from recurrent stage IV PPC | ↑ PDK1 | -In recurrent stage IV PPC sample had lower OXPHOS shifting enzyme than stage I primary EOC tissue. | [60] | ||||
| -1 tissue from stage I EOC (control) | ↓ PDH | ||||||
The superscript *indicates only in metastatic ovarian cancer tissues. The arrow ↑ or ↓ denotes a significant increase or decrease in the metabolic-related finding with a P value <0.05. The arrow ↑↑ or ↓↓ denotes a significant increase or decrease in the metabolic-related finding with a P value <0.01. Abbreviations: BEC index; BioEnergetic Cellular index, CS; citrate synthase, CYB; cytochrome b reductase, DEPs; differentially expressed proteins, EOC; epithelial ovarian cancer, FOXM1; forkhead box protein M1, G6P; glucose-6-phosphate, Glut; glucose transporter, HGSC; high-grade serous adenocarcinoma, HIF; hypoxia-induced transcription factor, HK; hexokinase, IDH2; isocitrate dehydrogenase 2, LDHA; lactate dehydrogenase A, LGSC; low-grade serous adenocarcinoma, MOC; metastatic ovarian cancer, NOS; nitric oxide synthase, OCCC; ovarian clear cell carcinoma, OGDHL; 2-oxoglutarate dehydrogenase-like, OXPHOS; oxidative phosphorylation, PDH; pyruvate dehydrogenase, PDK; pyruvate dehydrogenase kinase, PFK; phosphofructokinase, PKM; pyruvate kinase muscle isozyme, PPC; primary peritoneal carcinoma, SIRT3; sirtuin 3, STAT3; signal transducer and activator of transcription 3, TMA; tissue microarray, UQCRH; ubiquinol-cytochrome c reductase hinge protein.
When the various histologic types of EOC cells were compared the results showed that HGSC and CCC show more glycolytic activity than other variants, as indicated by their higher GLUT expression levels and glycolytic enzyme levels [33-35]. Also, a gene set enrichment analysis (GSEA) revealed that the hepatocyte nuclear factor 1β (HNF-1β) gene, which is strongly associated with anaerobic glucose catabolism, was increased in the CCC group when compared with that of the non-CCC group [33]. As regards OXPHOS, HGSC was more dependent on OXPHOS than were low-grade serous carcinoma tissues (LGSC), as indicated by a higher BioEnergetic Cellular index and a non-dimensional ratio, measures used to express the mitochondrial activity [36].
In summary, clinical evidence indicates an increase in glycolytic activity in EOC tissues when compared to the glycolytic activity in normal ovarian tissues. Additionally, the more invasive histologic types of EOC, such as HGSC and CCC, display more glycolytic activity than the less invasive EOC types do. These findings suggest that the different histologic types of EOC may each have a unique metabolic sequence from specific regulators. However, there are only a few prior studies comparing the OXPHOS activity between EOC tissue and normal ovarian tissue and between the more invasive EOC and the less invasive EOC. Therefore, further studies with larger sample sizes and more outcome measurements are required to determine the overall changes in the cancer metabolic pathways.
The effects of glycolytic interventions on epithelial ovarian cancer
The impact of glycolytic intervention on the different aspects of EOC, growth, invasion, migration, and apoptosis, has been investigated in in vitro and in vivo studies. Glycolytic intervention can be classified in three main ways: 1) the knockdown of the glycolytic enzyme genes at the rate-limiting steps, specifically hexokinase (HK2) and pyruvate kinase (PKM2); 2) miRNA-induced changes in the glycolytic activity of the EOC cells, and 3) substance-induced changes in the glycolytic activity of the EOC cells. The relevant studies are comprehensively summarized in Table 3 (in vitro reports) and Table 4 (in vivo reports).
Table 3.
The eEffects of glycolytic interventions on epithelial ovarian cancer cell metabolism and invasive properties: in vitro studies
| Human ovarian carcinoma cells | Intervention (dose, duration) | Major findings | Interpretation | References | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Glycolytic-related findings | Oncological outcomes | |||||||||||
|
|
|
|||||||||||
| Glucose uptake | Glycolytic enzymes | Lactate | Others | Growth | Invasion | Migration | Apoptosis | Others | ||||
| A2780CP, ES-2, OVCAR3, SKOV3 | -HK2 knockdown (shHK2/siHK2, 48 h) | ↓ HK2 | ↓ | ↓ | ↓ | ↓ | ↓ stemness markers | -Inhibition of glycolytic enzyme, HK2, caused a decrease in cell proliferation, invasion, migration, and stemness properties. | [38] | |||
| -HK2 overexpression (pCMV6-DDK-HK2 transfection, 72 h) | ↑ HK2 | ↑ | ↓ VEGF | |||||||||
| OVCAR3, SKOV3 | -PKM2 knockdown (siRNA-PKM2, 24-72 h) | ↓ PKM2 | ↓ | ↓ | ↑ | ↓ viability | -PKM2 knockdown inhibited ovarian cancer cell viability, invasion, and migration. | [37] | ||||
| A2780, SKOV3 | -MiR603 overexpression (mimic miR603 siRNA transfection, 48 h) | ↓ | ↓ HK2 | ↓ | ↓ | ↓ | ↓ | -miR603 upregulation from ginsenoside inhibited glycolysis, cell proliferation, invasion, and migration. | [18] | |||
| ↔ PKM2 | ||||||||||||
| -Ginsenoside (80 µg/mL (SKOV3)/40 µg/mL (A2780), 48 h) + miR603 inhibitor (inhibitor miR603 siRNA transfection, 48 h) | ↑ | ↑ HK2 | ↑ | ↑ | ↑ | |||||||
| -Ginsenoside (control) | ||||||||||||
| A2780, SKOV3 | -Ginsenoside (80 µg/mL (SKOV3)/40 µg/mL (A2780), 48 h) OR mimic miR532-3p (siRNA transfection 80 nM, 24 h) | ↓ | ↓ HK2 | ↓ | ↓ | -miR532-3p upregulation from ginsenoside inhibited glycolysis and cell growth. | [40] | |||||
| ↓ PKM2 | ||||||||||||
| -MiR532-3p inhibitor (siRNA transfection 100 nM, 24 h) followed by ginsenoside | ↑ | ↑ HK2 | ↑ | ↑ | ||||||||
| ↑ PKM2 | ||||||||||||
| -Ginsenoside (control) | ||||||||||||
| A2780, Hey | -Cryptotanshinone (5/10 µmol/L, 48 h) | ↓ | ↓ HK2 | ↓ | ↓ Glut1 | ↓ viability | -Cryptotanshinone inhibited glycolysis and cell viability. | [31] | ||||
| ↓ LDHA | ||||||||||||
| A2780, SKOV3 | -Resveratrol (25-800 µM, 72 h) | ↓ | ↓ | ↓ | ↓ | ↑ | ↓ viability | -Resveratrol inhibited glycolysis, cell growth, migration, and invasion. | [42] | |||
| A2780, Hey | -Bufalin (5/10 nM, 48 h) | ↓ | ↓ HK2 | ↓ | ↓ Glut4 | ↓ | ↓ viability | -Bufalin inhibited glycolysis, cell growth and proliferation. | [61] | |||
| ↓ LDHB | ||||||||||||
| SKOV3 | -MiR144 overexpression (mimic miR144 siRNA transfection, 36/48 h) | ↓ | ↓ | ↓ Glut1 | ↓ | ↓ viability | -MiR-144 inhibited glycolysis and suppressed cell growth and viability. | [39] | ||||
| OVCAR3, SKOV3 | -NOS inhibitor (0.75-10 mM, 24 h) | ↓ | ↓ | ↑ | ↓ viability | -A low dose of NO promoted glycolysis & cell viability. | [29] | |||||
| -NO donor (0.05-100 µM, 24 h) | ↑ | ↑ | ↓ | ↑ viability | ||||||||
| -NO donor (500-1000 µM, 24 h) | ↓ | ↓ | ↓ viability | |||||||||
| CAOV3, OVCA432 | -Lysophosphatidic acid (10 µM, 12 h) +5-3H glucose | ↑ HK2 | ↑ | ↑ glycolytic flux | -Lysophosphatidic acid up-regulated glycolysis, which may affect cancer cell growth. | [62] | ||||||
| -control | ||||||||||||
| -Lysophosphatidic acid +2-DG (1.25 mM (CAOV3)/3 mM (OVCA432)) | ↓ glycolytic flux | ↓ | ||||||||||
| -Lysophosphatidic acid alone (control) | ||||||||||||
| Ovca429, Ovca433 | -MiR203 overexpression (mimic miR203 transfection) | ↑ | ↑ | ↑ | ↑ | -MiR-203 promoted glycolysis, cell growth, and migration. | [41] | |||||
| -Anti-miR-203 | ↓ | ↓ | ↓ | ↓ | ||||||||
| OVCAR3, SKOV3, SKOV3ip1 | -High concentrated pyruvate (10 mM, 12 h) | ↑ | ↑ | ↑ pyruvate uptake | ↑ | -The high pyruvate concentration resulted in higher metabolism, and contributed to the motility in SKOV3 cells. | [28] | |||||
| -Original pyruvate concentration (1 mM) (control) | ||||||||||||
The arrow ↑ or ↓ denotes a significant increase or decrease in the glycolytic-related finding and oncological activities with a P value <0.05. Abbreviations: HK2; hexokinase 2, LDH; lactate dehydrogenase, miR; microRNA, NO; nitric oxide, NOS; nitric oxide synthase, PKM2; pyruvate kinase M2, VEGF; vascular endothelial growth factor.
Table 4.
The effects of glycolytic interventions on epithelial ovarian cancer cell metabolism and its invasive properties: in vivo studies
| Study models | Intervention (dose, duration) | Major findings | Interpretation | References | |||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Glycolytic-related findings | Oncological outcome-related findings | ||||||||
|
|
|
||||||||
| Glucose uptake | Glycolytic enzymes | Others | Growth | Volume/weight | Others | ||||
| Mice sc injected with ES-2 cells | -SC or ip inj. shHK2 ES-2 (2 × 106 cells, once) | ↓ HK2 | ↓ | ↓ | -HK2 knockdown mice had significantly lower tumor weight and tumor growth rate than that of control mice. | [38] | |||
| Mice sc injected with SKOV3 cells | -Intratumoral inj. agomiR603 (5 nM q 3 days × 7 cycles) | ↓ | ↓ HK2 | ↓ | -miR603 inhibited glycolysis and tumor growth. | [18] | |||
| -Intratumoral inj. agomiR-NC (control) | |||||||||
| Mice sc injected with Hey cells | -Cryptotanshinone ip inj. (10 mg/kg, once) | ↓ | ↓ HK2, LDHA | ↓ Glut1 | ↓ | ↓ | -Cryptotanshinone inhibited glycolysis and tumor growth. | [31] | |
| ↓ STAT3 | |||||||||
| ↓ HIF1α | |||||||||
| Mice sc injected with SKOV3 cells | -CHIP overexpression | ↓ PKM2 | ↓ | ↓ | -CHIP overexpression inhibited glycolytic shifting enzyme, PKM2, and tumor growth. | [44] | |||
| -CHIP suppression (shCHIP transfection) | ↑ PKM2 | ↑ | ↑ | ||||||
| Mice sc injected with SKOV3 cells | -PKM2 inhibitor, shikonin ip inj. (10 mg/kg, 5 days/week × 3 weeks) | ↓ PKM2 | ↓ | -Glycolytic shifting enzyme inhibitor, shikonin, inhibited tumor growth. | [47] | ||||
| Mice sc injected with SKOV3 cells | -Mimic miR144 (miR144 mimics-containing lentivirus transfection, 16 days) | ↓ Glut1 | ↓ | ↓ | -MiR-144 reduced glucose uptake and suppressed tumor growth. | [39] | |||
| Mice sc injected with A2780 cells | -Bufalin ip inj. (10 mg/kg, AD) | ↓ | ↓ HK2 | ↓ Glut4 | ↓ | ↓ | ↓ ITGB2 | -Bufalin decreased glycolysis and could inhibit cell growth, tumor volume and weight. | [61] |
| ↓ LDHB | ↓ FAK | ||||||||
| Mice sc injected with ID8 cells | -Lysophosphatidic acid sc inj. (0.4 µmol, AD × 20 days) | ↑ HIF1α | ↑ | -Lysophosphatidic acid-induced HIF1α expression and tumor growth in mice. | [63] | ||||
| Mice sc injected with A2780 and SKOV3 cells | -FOXM1 silencing (shFOXM1 transfection) | ↓ | ↓ HK2 | ↓ Glut1 | ↓ | -Decreased glycolysis by silencing FOXM1 gene could inhibit tumor volume and weight. | [30] | ||
| Mice intrabursally injected with M909 cells | -JQ1 ip inj. (50 mg/kg/day, 4 weeks) | ↓ | ↓ LDHA | ↓ lactate | ↓ | ↓ | ↑ apoptosis | -JQ1 inhibited glycolysis, tumor growth, and increased apoptosis. | [43] |
| ↓ c-myc | |||||||||
The arrow ↑ or ↓ denotes a significant increase or decrease in the glycolytic-related finding and oncological activities with a P value <0.05. Abbreviations: AD; alternate day, CHIP; carboxyl terminus of Hsc70-interacting protein, FAK; focal adhesion kinase, FOXM1; forkhead box protein M1, HIF; hypoxia-induced transcription factor, HK2; hexokinase 2, inj.; injection, ip; intraperitoneal, ITGB2; integrin beta 2, LDH; lactate dehydrogenase, miR; microRNA, PKM2; pyruvate kinase M2, sc; subcutaneous, STAT3; signal transducer and activator of transcription 3.
Genetic modification by knocked down glycolytic enzyme genes at the rate-limiting steps, HK2 and PKM2, to inhibit the glycolysis pathway has been studied. The in vitro and in vivo studies showed that glycolytic inhibition disrupted EOC growth as indicated by a decrease in the cell/colony counts, tumor sizes, and tumor weights [37,38]. Glycolytic inhibition can increase the apoptosis, decrease the invasion and migration, and reduce the VEGF protein expression in EOC cells [37,38].
Several miRNAs have demonstrated effects on the glycolytic activity of the EOC cells. The overexpression of miR-603, miR-532-3p, and miR-144 by miRNA transfection or treatment with a miRNA upregulator, ginsenoside 20(S)-Rg3 resulted in glycolytic disruption and impaired EOC cell growth, invasion, and migration [18,39,40]. Likewise, studies in mice revealed that either the intratumoral injection of miR-603 or the subcutaneous injection of miR-144 transfected-cancer cells led to reduced glucose uptake in ovarian tumors, limiting ovarian tumor growth, and tumor weight [18,39]. However, it has been shown that miR-203 promotes glycolysis, cancer cell growth, and migration in the ovca433 and ovca429 cell lines [41]. These findings suggest that miRNAs can modulate glycolysis differently depending on their type, causing either the upregulation or downregulation of glycolysis.
There are many substances extracted from herbs, animals, and fruits that can inhibit glycolysis. For example, crypsotanshinone, a Chinese herbal compound found in Danshen (Salvia miltiorrhiza) and bufalin-a substance isolated from the venom of a Chinese toad, have been shown to have a role in glycolytic inhibition and reducing EOC cell growth and viability, ovarian tumor growth, and ovarian tumor weight [29,31]. Resveratrol, a natural ingredient found in the skin of peanuts, grapes, blueberries, mulberries, and raspberries, may decrease glucose uptake, and lactate levels, and reduce invasion, migration, and increase the apoptosis of the EOC cells [42]. Other substances such as nitric oxide synthase (NOS) inhibitors, nitric oxide, carboxyl terminus of Hsc70-interacting protein (CHIP), and JQ1, a small molecule which selectively inhibits proteins and downregulates c-Myc, have also been found to exhibit glycolytic inhibitory effects [24,29,43,44]. In support of these findings, previous in vitro and in vivo studies reveal that those substances increased the apoptosis of EOC cells and impaired ovarian tumor growth, respectively [29,43].
In summary, both in vitro and in vivo studies demonstrated that glycolysis has a crucial role in EOC. In fact, when glycolysis is inhibited in vitro, EOC cell growth, invasion, migration, and viability are restricted. Likewise, when glycolysis is disrupted in vivo, tumor growth or tumor volume decreases significantly. These findings suggest that the modulation of the glycolysis pathway could be beneficial in therapeutic interventions in EOC.
The comparative effects of glycolytic and OXPHOS interventions on epithelial ovarian cancer
When glycolytic and OXPHOS interventions were compared, all the in vitro studies showed that EOC cell growth, invasion and migration depend mainly on glycolysis rather than OXPHOS (Table 5). In fact, various glycolytic inhibition methods, including glycolytic enzyme-related gene suppression, glycolytic enzyme inhibition, and glycolytic-related protein silencing resulted in a significant reduction in EOC cell growth, invasion, and migration [34,44-48].
Table 5.
The comparative effects of glycolytic and OXPHOS interventions on epithelial ovarian cancer cell metabolism and its invasive properties: in vitro studies
| Human ovarian carcinoma cells | Intervention (dose, duration) | Major findings | Interpretation | References | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||
| Glycolytic-related findings | OXPHOS-related findings | Oncological outcomes | |||||||||||||
|
|
|
|
|||||||||||||
| Glucose uptake | Glycolytic enzymes | Lactate | Others | OXPHOS enzymes | OCR | ROS | Other | Growth | Invasion | Migration | Others | ||||
| A2780, SKOV3 | -PTTG suppression (PTTG-shRNA, 25 mmol/L, 48 h) | ↓ | ↓ HK | ↓ | ↓ Glut1 | ↑ | ↑ | ↑ ATP | ↓ | -PTTG suppression caused a decrease in glycolysis and an increase in OXPHOS, and resulted in decreased cell proliferation. | [34] | ||||
| ↓ PFK | ↓ c-myc | ||||||||||||||
| ↓ PKM2 | |||||||||||||||
| ↓ LDHA | |||||||||||||||
| -PTTG-shRNA +2-DG (100 mM, 12-72 h) | ↓ decreased ECAR | ↑ | ↑ decreased viability | ||||||||||||
| -Control-shRNA +2-DG | |||||||||||||||
| -PTTG-shRNA + oligomycin (2 µg/mL, 12-72 h) | ↔ ECAR | ↓↓ | ↓ viability | ||||||||||||
| -Control-shRNA + oligomycin | |||||||||||||||
| Hey, SKOV3 | -JQ1 (500-1000 nM, 24 h) | ↓ | ↓ LDHA | ↓ | ↓ c-myc | ↓ | ↓ ATP | ↓ | ↑ apoptosis | -JQ1 inhibited c-myc, glycolysis, and cell proliferation. | [43] | ||||
| ↔ necrosis | |||||||||||||||
| CP90, OV90 | -MICU1 silencing (siRNA-MICU1 transfection, 48-96 h) | ↓ | ↓ p-PDH | ↑ | ↑ | ↑ complex III | ↓ | ↓ | ↓ | ↓ motility | -Silencing MICU1 decreased glycolysis, cell growth, and invasiveness but increased OXPHOS shifting enzymes and OXPHOS metabolism. | [45] | |||
| ↑ PDH | |||||||||||||||
| OVCAR3, SKOV3 | -MPC1 inhibitor (20 µM, 1 week) | ↑ | ↑ ECAR | ↓ mt-pyruvate | ↔ | ↑ | ↑ stemness markers | -MPC1 inhibitor resulted in increased glycolysis, stemness markers, migration, and motility. | [48] | ||||||
| ↓ ATP | ↑ motility | ||||||||||||||
| ↑ viability | |||||||||||||||
| A549, SKOV3 | -FAM210B silencing (siRNA transfection, 48 h) | ↓ | ↓ | ↓ ECAR | ↓ PDK4 | ↑ | ↔ | ↑ | ↑ viability | -Loss of mitochondrial protein FAM210B caused an increased OXPHOS and promoted cell aggressiveness. | [64] | ||||
| ↓ p-PDH | ↑ EMT | ||||||||||||||
| OVCAR3, SKOV3 | -NOS inhibitor (10 mM, 48 h) | ↓ | ↑ HK2 | ↓ | ↓ | ↑ | ↓ pyruvate uptake | ↓ | -NO could induce glycolysis, impaired OXPHOS, and cell growth. | [46] | |||||
| -NO donor (100 µM, 24 h) | ↑ | ↑ | ↓ | ↓ complex II, III, IV | |||||||||||
| CP70, SKOV3 | -PKM2 inhibitor, shikonin (3 µM, 5 h) | ↓ | ↓ PKM2 | ↓ | ↓ ECAR | ↔ | ↓ | ↓ | -Inhibition of PKM2 caused a decrease in glycolysis, cell proliferation, and migration. | [47] | |||||
| -Control (7 h) | |||||||||||||||
| SKOV3 | -Ad-CHIP (CHIP 5 µM, 4 h) | ↔ HK | ↓ pyruvate | ↑ | ↓ | -CHIP decreased glycolysis and inhibited cell proliferation. | [44] | ||||||||
| ↓ PKM2 | ↓ ECAR | ||||||||||||||
| ↓ glycolytic capacity | |||||||||||||||
The arrow ↑ or ↓ denotes a significant increase or decrease in the metabolic-related finding or the cells’ oncologic activities, i.e. cell growth, invasion, migration and viability, with a P value <0.05. The arrow ↑↑ or ↓↓ denotes a significant increase or decrease in the metabolic-related finding or the cells’ oncologic activities with a P value <0.01. Abbreviations: Ad-; adenovirus infected, CHIP; carboxyl terminus of Hsc70-interacting protein, ECAR; extracellular acidification rate, EMT; epithelial-mesenchymal transition, HK; hexokinase, LDH; lactate dehydrogenase, MICU1; mitochondrial calcium uptake 1, miR; microRNA, MPC; mitochondrial pyruvate carrier, mt-; mitochondrial, NO; nitric oxide, NOS; nitric oxide synthase, OXPHOS; oxidative phosphorylation, OCR; oxygen consumption rate, p-PDH; phosphorylated-pyruvate dehydrogenase, PDH; pyruvate dehydrogenase, PDK; pyruvate dehydrogenase kinase, PFK; phosphofructokinase, PKM2; pyruvate kinase M2, PTTG; pituitary tumor-transforming gene, ROS; reactive oxygen species.
Two in vivo studies support the results of the in vitro studies mentioned above, specifically that tumor growth and volume increase occurred mainly as a result of glycolysis (Table 6) [45,49]. Another study suggested that apart from glycolysis, fatty acid oxidation might also have a role in EOC growth [50]. In that study, mice with HGSC of the ovary were fed with either a low-fat diet (10% of calories derived from fat) or a high-fat diet (60% of calories derived from fat), followed by metformin treatment. The results showed that the tumor weight in the high-fat diet-fed mice was significantly higher than it was in the low-fat diet-fed mice. Metabolomic profiling was also carried out in mice ovarian tumors. Metabolomics revealed that the n-3 and n-6 fatty acid levels were decreased, while several acylcarnitine and dicarboxylic acid levels were increased in the ovarian tumors of the high-fat diet-fed mice. This suggests that the greater fatty acid oxidation occurring in high-fat diet-fed mice than in low-fat diet-fed mice has an impact on tumor growth. Additionally, the ovarian tumors from the high-fat diet-fed mice exhibited an impairment in the succinate dehydrogenase (complex II) activity, consistent with a parallel decrease in lysolipids, fumarate, and malate. Interestingly, the metformin treatment had a more significant effect on the reduction of tumor growth in the high-fat diet-fed mice than it did in the low-fat diet-fed mice [50]. This might be due to the inhibition of mitochondrial complex I by metformin that could potentiate the response in complex II-induced tumor impairment in obese mice [50].
Table 6.
The comparative effects of glycolytic and OXPHOS interventions on epithelial ovarian cancer cell metabolism and invasive properties: in vivo studies
| Study in mouse models | Intervention (dose, duration) | Major findings | Interpretation | References | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| Glycolytic-related findings | OXPHOS-related findings | Oncological outcome-related findings | |||||||||
|
|
|
|
|||||||||
| Glycolytic enzymes | Others | OXPHOS enzymes | Metabolites | Others | Growth | Volume/weight | Others | ||||
| Ovaries intrabursally implanted with OV90 | -MICU1 silencing (shMICU1 transfection, 48-72 h) | ↓ LDH | ↓ lactate | ↓ p-PDH | ↓ | ↓ | ↑ apoptosis | -Silencing MICU1 caused decreasing glycolysis, tumor growth, and mouse survival. | [45] | ||
| ↑ PDH | ↑ median survival | ||||||||||
| Serous EOC with somatic deletion of Brca1 and p53 inactivation | -High-fat diet (obese) | ↓ F6P, pyruvate | ↑ succinate | ↑ | -Obese mice had less glycolytic activity but had more tumor growth than lean mice. | [50] | |||||
| -Low-fat diet (control) | ↓ fumarate, malate | ||||||||||
| -Metformin (200 mg/kg, oral gavage × 4 weeks) in obese mice | ↑ G6P, F6P, FBP, 2,3-DPG, PEP, lactate | ↑ α-ketoglutarate, fumarate, malate | ↓ | ↓ | ↑ caspase-3 | ||||||
| ↓ VEGF | -Metformin induced glycolysis, decreased angiogenesis, cell proliferation, and invasive properties. | ||||||||||
| -Metformin (200 mg/kg, oral gavage × 4 weeks) in lean mice (control) | ↓ succinate | ↓ MMP 9 | |||||||||
| ID8 cells intraperitoneal injection | -SP-/- (SPARC gene) | ↑ HK2 | ↑ G6P, F6P, pyruvate, lactate | ↑ SUCLG1, SUCLG2, ME2, IDH1 | ↑ succinate | ↑ ROS | ↑ | -Absence of SPARC genes increased glycolysis and OXPHOS activity and supported tumor growth. | [49] | ||
| -SP+/+ (control) | ↑ TPI1 | ↓ fumarate, citrate | |||||||||
| ↑ PGK1 | ↑ complex I, III, IV, V | ||||||||||
| ↑ PHGDH | |||||||||||
The arrow ↑ or ↓ denotes a significant increase or decrease in the metabolic-related finding or tumor oncologic activities, with a P value <0.05. Abbreviations: 2,3-DPG; 2,3-diphosphoglycerate, EOC; epithelial ovarian cancer, F6P; fructose-6-phosphate, FBP; fructose 1,6-bisphosphate, G6P; glucose-6-phosphate, HK2; hexokinase 2, IDH; isocitrate dehydrogenase, ip; intraperitoneal, LDH; lactate dehydrogenase, ME; malic enzyme, MICU1; mitochondrial calcium uptake 1, MMP; matrix metalloproteinase, OXPHOS; oxidative phosphorylation, p-PDH; phosphorylated-pyruvate dehydrogenase, PDH; pyruvate dehydrogenase, PEP; phosphoenolpyruvate, PGK1; phosphoglycerate kinase 1, PHGDH; phosphoglycerate dehydrogenase, ROS; reactive oxygen species, SPARC; secreted protein acidic and rich in cysteine, SUCLG; succinyl-CoA ligase, TPI1; triosephosphate isomerase, VEGF; vascular endothelial growth factor.
Metabolic characteristics that determine the chemoresistance of epithelial ovarian cancer
The metabolic activities of the chemoresistant and chemosensitive EOC cell lines have been compared, but the results remain inconclusive (Table 7). Five studies compared the metabolic activities between the chemoresistant and chemosensitive EOC cell lines, the results differing between the cell lines [19,36,51-53]. Chemoresistant serous C200 and PEO4 showed higher glycolytic activity and higher OXPHOS activity, with a higher proportion of OXPHOS activity than glycolysis, compared to chemosensitive serous A2780 and PEO1 [51]. Platinum-resistant HGSC PEA2 had lower overall glucose metabolism, as indicated by its lower levels of glycolytic enzymes and glycolytic capacity, lower ECAR, but higher OXPHOS than platinum-sensitive HGSC PEA1 [36]. A cisplatin-resistant ovarian adenocarcinoma, SKOV3/DDP, demonstrated higher glucose uptake and higher levels of glycolytic enzymes but lower ECAR and lactate levels than a cisplatin-sensitive cell line, SKOV3 [52]. In addition, SKOV3/DDP displayed higher OXPHOS than SKOV3, as indicated by the greater basal OCR and ROS [52]. These findings suggest that the resistant cell line SKOV3/DDP showed increased glycolysis, resulting in increased OXPHOS via the increased entering of substrates into the TCA cycle. In contrast, the chemoresistant ovarian adenocarcinoma variants C13, HeyA8MDR, and A2780/DDP had a lower OXPHOS levels, as indicated by the lower basal OCR and ROS, but higher glycolysis, as indicated by the higher glycolytic enzyme levels and the higher glucose uptake and lactate levels than in chemosensitive ovarian adenocarcinoma-OV2008, HeyA8, and A2780 [19,53].
Table 7.
Characteristics of epithelial ovarian cancer cell metabolism in relation to chemosensitive and chemoresistant cell lines
| Study Model | Major findings | Interpretation | References | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| Glycolytic-related findings | OXPHOS-related findings | |||||||||
|
|
|
|||||||||
| Glycolytic enzymes | Lactate | Basal ECAR | Others | OXPHOS enzymes | Basal OCR | ROS | Others | |||
| -Chemoresistant human serous ovarian carcinoma (C200, PEO4) | ↔ LDHa | ↑ | ↑ GLUT1 | ↑↑ CoxVb | ↑ | ↑ | ↑ OCR/ECAR | -Chemoresistant cells had higher metabolic states than chemosensitive cells. | [51] | |
| -Chemosensitive human ovarian carcinoma (A2780, PEO1) (control) | ↑ glycolytic capacity | ↑ ATP | ||||||||
| ↑ respiratory reserve | ||||||||||
| ↑↑ PGC-1α | ||||||||||
| -Human platinum-resistant HGSC (PEA2) | ↓ PFK | ↓ | ↓ glycolytic capacity | ↔ | ↑ | ↑ maximal OCR | -Platinum resistant cells had higher OXPHOS but had lower glycolysis than platinum-sensitive cells. | [36] | ||
| -Human platinum-sensitive HGSC (PEA1) (control) | ↓ GAPDH | ↑ BEC index | ||||||||
| -Cisplatin resistant human ovarian carcinoma (SKOV3/DDP) | ↑ PFK | ↓ | ↓ | ↑ Glut1 | ↔ PDK1 | ↑ | ↑ | ↑ ATP | -Cisplatin resistant cells had higher OXPHOS and glucose metabolism than cisplatin-sensitive cells. | [52] |
| -Cisplatin sensitive human ovarian carcinoma (SKOV3) (control) | ↑ LDHA | ↑ glucose uptake | ||||||||
| -Chemoresistant human ovarian carcinoma (C13, HeyA8MDR) | ↑ PFK | ↑ | ↑ glucose uptake | ↓ | ↑ ATP | -Chemoresistant cells had higher glycolysis compared to sensitive cells. | [19] | |||
| -Chemosensitive human ovarian carcinoma (HeyA8, OV2008) (control) | ↑ LDH | ↔ F2,6BP | ||||||||
| -Cisplatin resistant human ovarian carcinoma (A2780/DDP) | ↑ HK2 | ↑ | ↑ glucose uptake | ↓ | ↓ | ↓ citrate | -Cisplatin resistant cells had higher glycolysis than cisplatin sensitive cells. | [53] | ||
| -Cisplatin sensitive human ovarian carcinoma (A2780) (control) | ↑ LDH | |||||||||
The arrow ↑ or ↓ denotes a significant increase or decrease in the metabolic-related finding with a P value <0.05. The arrow ↑↑ or ↓↓ denotes a significant increase or decrease in the metabolic-related finding with a P value <0.01. Abbreviations: BEC index; BioEnergetic Cellular index, CoxVb; cytochrome c oxidase subunit Vb, ECAR; extracellular acidification rate, F2,6BP; fructose-2,6-bisphosphate, GAPDH; glyceraldehyde 3-phosphate dehydrogenase, HGSC; high-grade serous adenocarcinoma, HK2; Hexokinase 2, LDH; lactate dehydrogenase, OCR; oxygen consumption rate, OXPHOS; oxidative phosphorylation, PDK; pyruvate dehydrogenase kinase, PFK; Phosphofructokinase, PGC-1α; peroxisome proliferator-activated receptor gamma coactivator 1-alpha, ROS; Reactive oxygen species.
In summary, the studies on chemoresistant cell lines indicated that they could be classified into three groups according to their dominant metabolic activity: first, cells that show a prevalence of glycolysis, for example C13, HeyA8MDR, and A2780/DDP; second, cells that have a prevalence of OXPHOS, including HGSC PEA2 and SKOV3/DDP; finally, cells that are dominant in both glycolysis and OXPHOS, serous C200 and PEO4. These findings indicate the potential targets for therapeutic strategies via metabolic regulation depending on the type of resistance exhibited by the cells.
Effects of metabolic interventions on epithelial ovarian cancer chemoresistant cell lines
The effects of metabolic interventions, including glycolytic or OXPHOS inhibition, on the alterations of the chemoresistant or chemosensitive properties of the EOC cell lines are summarized in Table 8. Compared with the chemosensitive cell lines (A2780, PEA1, and SKOV), the chemoresistant cell lines (C200, HGSC, PEA2, and SKOV/DDP) were more tolerant of glycolytic stress [36,51,52]. In other words, these resistant cells could shift the glycolysis toward OXPHOS and maintain viability when they were cultured in glucose-free media or treated with a glycolytic inhibitor: 2-deoxyglucose (2-DG).
Table 8.
The Effects of the metabolic interventions on the epithelial ovarian cancer chemoresistant cell lines and the metabolic interventions that alter the chemosensitization properties: reports from in vitro studies
| Human Ovarian Cell Types | Intervention (Dose/Duration) | Major findings | Interpretation | References | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| Glycolytic-related findings | OXPHOS-related findings | Oncological outcomes | ||||||||||||
|
|
|
|
||||||||||||
| Glycolytic enzymes | Lactate | Basal ECAR | Others | OXPHOS enzymes | Basal OCR | ROS | Others | Proliferation | Apoptosis | Viability | ||||
| -Chemoresistant ovarian carcinoma (C200) | -Glucose-free media (48 h) | ↑↑ | ↑↑ glycolytic capacity | ↑↑ | ↑↑ max. respiration | ↑↑ | -Chemoresistant cells had higher metabolic state than chemosensitive cells, and could shift toward OXPHOS or glycolysis under stress conditions. | [51] | ||||||
| ↑↑ ATP | ||||||||||||||
| -Chemosensitive ovarian carcinoma (A2780) (control) | -Glucose-free media + pyruvate (1 mM, 48 h) | ↑↑ | ↑↑ glycolytic capacity | ↑↑ | ↑↑ max. respiration | ↑↑ | ||||||||
| ↑↑ ATP | ||||||||||||||
| -Glucose supplement (10 mM, 48 h) | ↑↑ | ↔ glycolytic capacity | ↑↑ | ↑↑ max. respiration | ||||||||||
| ↑ ATP | ||||||||||||||
| -2-DG (100-200 mM, 48 h) | ↑↑ | |||||||||||||
| -Oligomycin (0.1-2 µM, 48 h) | ↔ | |||||||||||||
| -Chemosensitive ovarian carcinoma (A2780) | -2-DG (6.25, 25, 100 mM, 48 h) | ↓↓ | ↑↑ glycolytic capacity | ↔ | ↑↑ max. respiration | ↓ | ↔ | -Chemosensitive cells might use glycolysis as the main pathway and could increase the metabolic state when using non-toxic concentration of cisplatin. | ||||||
| -Oligomycin (0.31-1.25 µM, 48 h) | ↑↑ | ↓↓ | ||||||||||||
| -Cisplatin (1 µM, 24-48 h) | ↑↑ | ↑↑ | ||||||||||||
| -Chemoresistant HGSC (PEO4) | -Cisplatin (10 mM, 48 h) + oligomycin (0.1 mM, 48 h) | ↓↓ | -Resistant cells had the ability to shift metabolism type, and decreased viability when exposed to a combined OXPHOS inhibitor with cisplatin. | |||||||||||
| -Cisplatin (control) | ||||||||||||||
| -Cisplatin (10 mM, 48 h) + 2-DG (100 mM, 48 h) | ↓↓ | |||||||||||||
| -Cisplatin (control) | ||||||||||||||
| -Chemoresistant serous ovarian carcinoma (C200) | -2-DG (6.25, 25, 100 mM, 48 h) | ↓↓ | ↑↑ | ↓ | ||||||||||
| -Oligomycin (0.31-1.25 µM, 48 h) | ↑↑ | ↓↓ | ||||||||||||
| -Cisplatin (10 mM, 48 h) + oligomycin (0.1 mM, 48 h) | ↓↓ | |||||||||||||
| -Cisplatin (control) | ||||||||||||||
| -Cisplatin (10 mM, 48 h) + 2-DG (100 mM, 48 h) | ↔ | |||||||||||||
| -Cisplatin (control) | ||||||||||||||
| -Platinum resistant HGSC (PEA2) | -PEA2 and PEA1 in low-glucose medium (glucose 1 g/L, 72 h) | ↑ | -Platinum-resistant cells were more tolerate to glucose deprivation. | [36] | ||||||||||
| -Platinum sensitive HGSC (PEA1) (control) | ||||||||||||||
| -Platinum sensitive HGSC (PEA1) | -TRAP1 silencing (siRNA transfection, 72 h) | ↔ PFK | ↔ | ↔ glycolytic capacity | ↑ | ↑ | ↑ BEC index | -Increasing OXPHOS by TRAP1 silencing or OXPHOS inducer could induce platinum resistant in platinum sensitive HGSC. | ||||||
| -Cisplatin (20 µM, 24 h) | ↑↑ | ↓ | ||||||||||||
| -Cisplatin + OXPHOS inducer (FCCP 1.2 µM, 24 h) | ↓ | ↑↑ | ||||||||||||
| -Cisplatin (control) | ||||||||||||||
| -Cisplatin + TRAP1 silencing | ↓ | ↑ | ||||||||||||
| -Cisplatin (control) | ||||||||||||||
| -Cisplatin + TRAP1 silencing + metformin | ↓ | |||||||||||||
| -Cisplatin + TRAP1 silencing (control) | ||||||||||||||
| -Platinum resistant HGSC (PEA2) | -TRAP1 silencing (siRNA transfection, 72 h) | ↔ BEC index | -Using complex I inhibitor, metformin, could induce chemosensitivity in platinum resistant HGSC. | |||||||||||
| -Cisplatin (40 µM, 24 h) | ↑ | ↓ | ||||||||||||
| -Metformin (10 mM, 24 h) | ↔ | ↓ | ||||||||||||
| -Cisplatin + metformin | ↑ | ↓↓ | ||||||||||||
| -Cisplatin resistant carcinoma (SKOV3/DDP) | -2-DG (10 mM, 24 h) | ↑ | -Cisplatin resistant cells had higher OXPHOS, were less sensitive to glucose deprivation, but use of glycolysis or anti-apoptotic inhibitors could increase cell apoptosis. | [52] | ||||||||||
| -Cisplatin sensitive carcinoma (SKOV3) (control) | -OXPHOS inducer (FCCP 2.5 µM, 24 h) | ↑ | ↑ respiratory reserve | |||||||||||
| -Cisplatin sensitive ovarian carcinoma (SKOV3) | -Glucose-free media (24 h) | ↓ | ||||||||||||
| -Bcl-2 inhibitor (ABT737 10 µM, 24 h) | ↓ HIF-1α | ↔ | ↔ ATP | ↔ | ||||||||||
| -Cisplatin (6 µg/mL, 24 h) | ↓↓ | ↓↓ ATP | ↑↑ | |||||||||||
| -Bcl-2 inhibitor + cisplatin | ↓ | ↓↓ ATP | ↑↑ | |||||||||||
| -Cisplatin resistant ovarian carcinoma (SKOV3/DDP) | -Glucose deprivation (glucose-free medium) | ↓ | ||||||||||||
| -Glucose deprivation + 2-DG (10 mM, 24 h) | ↓ | |||||||||||||
| -Glucose deprivation (control) | ||||||||||||||
| -Bcl-2 inhibitor (ABT737 10 µM, 24 h) | ↓ HIF-1α | ↓ | ↓↓ ATP | ↑↑ | ||||||||||
| -Cisplatin (6 µg/mL, 24 h) | ↔ | ↓↓ ATP | ↑↑ | |||||||||||
| -Bcl-2 inhibitor + cisplatin | ↓↓ | ↓↓ ATP | ↑↑ | |||||||||||
| -Bcl-2 inhibitor + 2-DG | ↓ HK2 | ↓ Glut1 | ↓ PDHB | ↑ | ↑ | |||||||||
| ↓ HIF-1α | ↓ IDH1 | |||||||||||||
| -Chemosensitive ovarian carcinoma (HeyA8, OV2008) | -PFKFB3 inhibitor (PFK158, 10-15 µM, 30 min) | ↓ | ↓ glucose uptake | -Inhibition of PFKFB3 could resensitize chemoresistant cells. | [19] | |||||||||
| -PFKFB3 inhibitor (PFK158, 10 µM, 24 h) | ↔ | |||||||||||||
| -CBP (77 µM, 24 h) or PTX (0.2 µM, 24 h) | ↔ | |||||||||||||
| -PFKFB3 inhibitor + CBP or PTX | ↑↑ | |||||||||||||
| -Chemoresistant ovarian carcinoma (C13, HeyA8MDR) | -PFKFB3 inhibitor (PFK158, 10-15 µM, 30 min) | ↓ | ↓ glucose uptake | |||||||||||
| -PFKFB3 inhibitor (PFK158, 10 µM, 24 h) | ↔ | |||||||||||||
| -CBP (453 µM, 24 h) or PTX (3.59 µM, 24 h) | ↔ | |||||||||||||
| -PFKFB3 inhibitor + CBP or PTX | ↑↑ | |||||||||||||
| -Ovarian carcinoma (SKOV3, OVCAR3) | -MPC1 inhibitor (20 µM, 1 week | ↑ | ↑ glucose uptake | ↓ mt-pyruvate | ↑ | -MPC1 knockout promoted glycolysis and induced chemoresistance. | [48] | |||||||
| ↓ ATP | ||||||||||||||
| -MPC1 inhibitor + docetaxel (10 nM, 2 weeks) | ↑ | |||||||||||||
| -Ovarian carcinoma (SKOV3, COC1) | -BM-MSC-CM (miR-1180 abundant 24 h) | ↑ HK2 | ↑ | ↑ PDK1 | ↑ ATP | ↑ | ↑ | -MiR-1180 induced glycolysis and chemoresistance. | [17] | |||||
| -Standard medium (control) | ↑ PKM2 | |||||||||||||
| ↑ LDHA | ||||||||||||||
| -Glycolytic inducer (oligomycin 2 mg/mL, 24 h) | ↑ | |||||||||||||
| -BM-MSC-CM + cisplatin (0.5 mg/L, 24 h) | ↑ | ↑ | ||||||||||||
| -Cisplatin (control) | ||||||||||||||
| -Cisplatin (0.5 mg/L, 24 h) + oligomycin (2 mg/mL, 24 h) | ↑ | ↑ | ||||||||||||
| -Cisplatin (control) | ||||||||||||||
| -Anti-miR-1180 in BM-MSC-CM + cisplatin (0.5 mg/L, 40 min for ECAR, 5 days for proliferation) | ↓ | ↓ ATP | ↓ | ↓ | ||||||||||
| -control miRNA in BM-MSC-CM + cisplatin (control) | ||||||||||||||
| -Ovarian carcinoma (433, A2780, SKOV3, OVCAR3) | -Pim1 overexpression (72 h) | ↓ PGK1, PGAM1, ENO1, PKM, LDHA | ↑ | ↑ | ↓ GLUT1 | ↓ | ↑ | ↑ | -Inhibition of Pim1 reduced glycolysis and induced chemosensitization to cisplatin. | [65] | ||||
| -Pim1 silencing (shPim1/siPim1 transfection, 72 h) | ↓ | ↓ | ↔ HIF-1α | ↑ | ↓ | ↓ | ||||||||
| -Pim1 inhibitor (4 nM) + cisplatin (1 µM), 48 h | ↓ | |||||||||||||
| -Cisplatin alone (control) | ||||||||||||||
| -Ovarian carcinoma (CP90, OV90) | -MICU1 silencing (siRNA transfection, 48-96 h) | ↓ | ↓ p-PDH | ↑ | ↑ | ↑ complex III | ↓ | ↑ | ↓ | -Silencing MICU1 induced OXPHOS and enhanced cytotoxic effects of cisplatin in ovarian cancer cells. | [45] | |||
| -MICU1 silencing + cisplatin (10-20 µM)/topotecan (5-10 µM)/PTX (10-25 µM)/doxorubicin (1-2 µM), 48 h | ↔ | ↑ PDH | ↑ | ↓ | ||||||||||
| -Cisplatin/topotecan/paclitaxel/doxorubicin (control) | ↑ PDH | |||||||||||||
| -Normal ovarian epithelial cells (OSE) | -MICU overexpression (1 µg) | ↑ | ↑ p-PDH | ↓ | ↑ | -MICU overexpression induced glycolysis and chemoresistance in normal ovarian cells. | [45] | |||||||
| -MICU overexpression (1 µg) + cisplatin (2.5 µM), 48 h | ↓ PDH | |||||||||||||
| -Cisplatin (control) | ||||||||||||||
| -OCCC (RMG2, JHOC5) | -Knockdown HNF1ß (sh-HNF1ß transfection) | ↓ HK1 | ↓ | ↑ 3-PG | ↑ | ↑ citrate, 2-oxoglutarate | -Silencing HNF-1ß caused OCCC cells impaired adaptation to hypoxic conditions, increased resistance to glucose deprivation, and enhanced cytotoxicity of cisplatin in hypoxic conditions. | [33] | ||||||
| ↓ LDHA | ↑ PEP | ↔ ATP | ||||||||||||
| ↑ pyruvate | ↓ fumarate, malate | |||||||||||||
| ↓ MCT4 | ||||||||||||||
| -Hypoxic condition (2% O2, 24/48 h) | ↑ | |||||||||||||
| -Knockdown HNF-1ß + Hypoxic condition | ↓ | |||||||||||||
| -Control + hypoxic condition | ||||||||||||||
| -Knockdown HNF-1ß + glucose-free medium (24/48 h) | ↑ | |||||||||||||
| -Cisplatin (20-80 µM + sh-HNF1ß in hypoxic condition | ↓↓ | |||||||||||||
| -Cisplatin treated in control cells in hypoxic condition | ||||||||||||||
| -Cisplatin sensitive serous carcinoma (A2780, PEO1) | -Cisplatin (50 µM, 24 h) | ↓ HIF-1α | ↑ | ↑ | ↑ | -Cisplatin downregulated HIF-1α and induced apoptosis only in cisplatin sensitive cells. | [54] | |||||||
| -NAC (5 mM, 24 h) + cisplatin (20 µM, 72 h) | ↓ | |||||||||||||
| -Cisplatin (control) | -HIF-1α knockdown could resensitize cisplatin-resistant cells. | |||||||||||||
| -Cisplatin resistant serous carcinoma (A2780/CP, PEO4) | -Cisplatin (50 µM, 24 h) | ↓ LDHA | ↔ HIF-1α | ↔ | ↔ | ↓ | ||||||||
| -HIF-1α silencing (siHIF1 transfection, 72 h) + cisplatin (20 µM, 24 h) | ↑ | ↑ | ||||||||||||
| -Cisplatin (control) | ||||||||||||||
The arrow ↑ or ↓ denotes a significant increase or decrease in the metabolic-related finding or oncological activities with a P value <0.05. The arrow ↑↑ or ↓↓ denotes a significant increase or decrease in the metabolic-related finding or oncological activities with a P value <0.01. Abbreviations: 3-PG; 3-phosphoglycerate, BEC index; BioEnergetic Cellular index, BM-MSC-CM; Condition media of bone marrow-derived mesenchymal stem cells, CBP; carboplatin, ECAR; extracellular acidification rate, ENO; enolase, FCCP; carbonilcyanide p-triflouromethoxyphenylhydrazone, HGSC; high grade serous adenocarcinoma, HIF-1α; hypoxia-induced transcription factor, HK2; Hexokinase 2, HNF-1ß; Hepatocyte nuclear factor 1ß, IDH; isocitrate dehydrogenase, LDH; lactate dehydrogenase, MCT4; Monocarboxylate transporter 4, MICU1; Mitochondrial calcium uptake 1, miR; microRNA, MPC; mitochondrial pyruvate carrier, mt-; mitochondrial, NAC; N-acetyl cysteine, OCCC; Ovarian clear cell carcinoma, OCR; oxygen consumption rate, OXPHOS; oxidative phosphorylation, p-PDH; Phosphorylated-pyruvate dehydrogenase, PDH; pyruvate dehydrogenase, PDK; pyruvate dehydrogenase kinase, PEP; phosphoenolpyruvate, PFK; Phosphofructokinase, PFKFB3; 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, PGAM; phosphoglycerate mutase, PGK; phosphoglycerate kinase, PKM2; Pyruvate kinase M2, PTX; paclitaxel, ROS; Reactive oxygen species, TRAP-1; Tumor necrosis factor receptor-associated protein 1.
The induction of chemosensitization with glycolytic inhibition using a glucose-free medium, and a PFK inhibitor in a chemoresistant cell line, 2-DG, was investigated. Treatment with a combination of a glycolytic inhibitor and conventional chemotherapy resulted in a significant decrease in cell viability when compared to chemotherapy alone in some resistant cell lines such as PEO4 (serous carcinoma), adenocarcinoma C13, and HeyA8MDR [19,51,54]. An in vivo study using a combined glycolytic inhibitor (PFK158) and chemotherapy (carboplatin/paclitaxel) package also demonstrated consistent results in HeyA8MDR tumor-bearing mice, the PFK158 inducing chemosensitivity [19]. Nevertheless, that combination did not significantly alter the cell viability in serous carcinoma C200 [51]. Previous clinical studies observed that OCCC also has a low sensitivity to chemotherapy [55,56]. Its chemoresistant property is possibly due to its higher HNF-1β, leading to more aerobic glycolysis, leading to a reduction in ROS and resulting in the chemoresistant property of the EOC cells [57,58]. After the knockdown of HNF-1β in OCCC (RMG2 and JHOC5), a glycolytic shift was inhibited, resulting in a more cisplatin-sensitive property of these EOC cells [33].
The induction of chemosensitization in the chemoresistant cell lines using a combination of chemotherapy and an OXPHOS inhibitor was also investigated. An OXPHOS inhibitor-oligomycin-can induce chemosensitization in the chemoresistant EOC cell lines: serous carcinoma, C200, and PEO4 [51]. Metformin is considered an OXPHOS inhibitor because it can inhibit mitochondrial complex I (NADH dehydrogenase) and subsequently decrease mitochondrial ATP production [16,59]. The combined treatment of metformin and cisplatin can decrease cell viability and increase apoptosis in the chemoresistant cell line [36]. Another study used a combination of cisplatin and a Bcl-2 inhibitor, which mimics the OXPHOS inhibitor. The results showed that the Bcl-2 inhibitor enhances the cisplatin-sensitive property of SKOV3/DDP, as indicated by the increased apoptosis of the pertinent cells [52].
In summary, the diminution of the dominant metabolic pathway used by chemoresistant EOC cells can enhance chemotherapy cytotoxicity, i.e. the increased chemosensitive property of the cells. This is indicated by the increased cell apoptosis, as well as the decreased cell proliferation and viability.
Conclusion
EOC preferentially uses aerobic glycolysis and mitochondrial OXPHOS to provide sufficient energy to the cancer cells and to promote cell proliferation, invasion, and metastasis. The primary dominant metabolic pathway used by EOCs can differ depending on the histological cell type, the tumor aggressiveness, and the tumor microenvironment. To overcome any chemoresistant properties, a potential strategic intervention is to attenuate the preferred primary metabolism of the EOC cells. Future studies are needed to determine the clinical usefulness of these strategies in EOC patients.
Future directions and clinical applications
Although there is a wealth of evidence from previous studies regarding the metabolic changes in EOC cells and the impact of the dominant metabolic pathway of EOC cells on their invasiveness and chemoresistance, most studies have had to interpret the results from either few metabolites or few metabolic enzymes in a single aspect. To increase the knowledge surrounding this complex area further, comprehensive studies investigating the whole network of the potential metabolic pathways, glycolysis, the TCA cycle, OXPHOS, lipid oxidation, and amino acid metabolism are required. Also, several prior studies by necessity were conducted solely in in vitro scenarios, so only a few chemotherapy regimens were investigated. Therefore, future investigations into the efficacy of a range of chemotherapy regimens are needed to improve our fundamental understanding regarding their effects on the metabolic changes in cancer cells. In addition, a better understanding of the metabolic reprogramming of EOC cells could lead to the development of novel targeted therapies which may be used as alternatives to the standard chemotherapy options to slow down tumor progression and decrease the EOC mortality rate.
Acknowledgements
This work was supported by Thailand Science Research and Innovation grant no. MRG6280014 (C.T.), by a Senior Research Scholar grant from the National Research Council of Thailand (S.C.), by a NSTDA Research Chair grant from the National Science and Technology Development Agency Thailand (N.C.), and by a Chiang Mai University Center of Excellence Award (N.C.).
Disclosure of conflict of interest
None.
References
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Chi DS, Berchuck A, Dizon DS, Yashar CM. Epithelial ovarian cancer. Principles and Practice of Gynecologic Oncology. 7th. Philadelphia: Lippincott Williams & Wilkins; 2017. pp. 611–675. [Google Scholar]
- 3.Heintz AP, Odicino F, Maisonneuve P, Quinn MA, Benedet JL, Creasman WT, Ngan HY, Pecorelli S, Beller U. Carcinoma of the ovary. FIGO 26th annual report on the results of treatment in gynecological cancer. Int J Gynaecol Obstet. 2006;95(Suppl 1):S161–192. doi: 10.1016/S0020-7292(06)60033-7. [DOI] [PubMed] [Google Scholar]
- 4.Fung-Kee-Fung M, Oliver T, Elit L, Oza A, Hirte HW, Bryson P. Optimal chemotherapy treatment for women with recurrent ovarian cancer. Curr Oncol. 2007;14:195–208. doi: 10.3747/co.2007.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ali AY, Farrand L, Kim JY, Byun S, Suh JY, Lee HJ, Tsang BK. Molecular determinants of ovarian cancer chemoresistance: new insights into an old conundrum. Ann N Y Acad Sci. 2012;1271:58–67. doi: 10.1111/j.1749-6632.2012.06734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crawford S. Is it time for a new paradigm for systemic cancer treatment? Lessons from a century of cancer chemotherapy. Front Pharmacol. 2013;4:68. doi: 10.3389/fphar.2013.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zahreddine H, Borden KL. Mechanisms and insights into drug resistance in cancer. Front Pharmacol. 2013;4:28. doi: 10.3389/fphar.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 9.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 10.Bhattacharya B, Mohd Omar MF, Soong R. The Warburg effect and drug resistance. Br J Pharmacol. 2016;173:970–979. doi: 10.1111/bph.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71:6921–6925. doi: 10.1158/0008-5472.CAN-11-1457. [DOI] [PubMed] [Google Scholar]
- 12.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 13.Kozlovski I, Siegfried Z, Amar-Schwartz A, Karni R. The role of RNA alternative splicing in regulating cancer metabolism. Hum Genet. 2017;136:1113–1127. doi: 10.1007/s00439-017-1803-x. [DOI] [PubMed] [Google Scholar]
- 14.Suh DH, Kim HS, Kim B, Song YS. Metabolic orchestration between cancer cells and tumor microenvironment as a co-evolutionary source of chemoresistance in ovarian cancer: a therapeutic implication. Biochem Pharmacol. 2014;92:43–54. doi: 10.1016/j.bcp.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Nayak AP, Kapur A, Barroilhet L, Patankar MS. Oxidative phosphorylation: a target for novel therapeutic strategies against ovarian cancer. Cancers (Basel) 2018;10:337. doi: 10.3390/cancers10090337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gentric G, Kieffer Y, Mieulet V, Goundiam O, Bonneau C, Nemati F, Hurbain I, Raposo G, Popova T, Stern MH, Lallemand-Breitenbach V, Müller S, Cañeque T, Rodriguez R, Vincent-Salomon A, de Thé H, Rossignol R, Mechta-Grigoriou F. PML-regulated mitochondrial metabolism enhances chemosensitivity in human ovarian cancers. Cell Metab. 2019;29:156–173. e110. doi: 10.1016/j.cmet.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gu ZW, He YF, Wang WJ, Tian Q, Di W. MiR-1180 from bone marrow-derived mesenchymal stem cells induces glycolysis and chemoresistance in ovarian cancer cells by upregulating the Wnt signaling pathway. J Zhejiang Univ Sci B. 2019;20:219–237. doi: 10.1631/jzus.B1800190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu J, Wang L, Chen W, Wang Y, Zhen S, Chen H, Cheng J, Zhou Y, Li X, Zhao L. miR-603 targeted hexokinase-2 to inhibit the malignancy of ovarian cancer cells. Arch Biochem Biophys. 2019;661:1–9. doi: 10.1016/j.abb.2018.10.014. [DOI] [PubMed] [Google Scholar]
- 19.Mondal S, Roy D, Sarkar Bhattacharya S, Jin L, Jung D, Zhang S, Kalogera E, Staub J, Wang Y, Xuyang W, Khurana A, Chien J, Telang S, Chesney J, Tapolsky G, Petras D, Shridhar V. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int J Cancer. 2019;144:178–189. doi: 10.1002/ijc.31868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagao A, Kobayashi M, Koyasu S, Chow CCT, Harada H. HIF-1-Dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int J Mol Sci. 2019;20:238. doi: 10.3390/ijms20020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apte S, Sarangarajan R. Cellular respiration and carcinogenesis. Humana Press; 2008. [Google Scholar]
- 22.Weinberg RA. Cancer cells exhibit an altered energy metabolism. In: Zayatz E, editor. The biology of Cancer. New York, USA: Garland Science, Taylor & Francis Group; 2014. pp. 53–55. [Google Scholar]
- 23.Snaebjornsson MT, Schulze A. Non-canonical functions of enzymes facilitate cross-talk between cell metabolic and regulatory pathways. Exp Mol Med. 2018;50:1–16. doi: 10.1038/s12276-018-0065-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Izdebska M, Zielińska W, Grzanka D, Gagat M. The role of actin dynamics and actin-binding proteins expression in epithelial-to-mesenchymal transition and its association with cancer progression and evaluation of possible therapeutic targets. Biomed Res Int. 2018;2018:4578373. doi: 10.1155/2018/4578373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dier U, Shin DH, Hemachandra LP, Uusitalo LM, Hempel N. Bioenergetic analysis of ovarian cancer cell lines: profiling of histological subtypes and identification of a mitochondria-defective cell line. PLoS One. 2014;9:e98479. doi: 10.1371/journal.pone.0098479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li N, Zhan X, Zhan X. The lncRNA SNHG3 regulates energy metabolism of ovarian cancer by an analysis of mitochondrial proteomes. Gynecol Oncol. 2018;150:343–354. doi: 10.1016/j.ygyno.2018.06.013. [DOI] [PubMed] [Google Scholar]
- 27.Anderson AS, Roberts PC, Frisard MI, McMillan RP, Brown TJ, Lawless MH, Hulver MW, Schmelz EM. Metabolic changes during ovarian cancer progression as targets for sphingosine treatment. Exp Cell Res. 2013;319:1431–1442. doi: 10.1016/j.yexcr.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caneba CA, Bellance N, Yang L, Pabst L, Nagrath D. Pyruvate uptake is increased in highly invasive ovarian cancer cells under anoikis conditions for anaplerosis, mitochondrial function, and migration. Am J Physiol Endocrinol Metab. 2012;303:E1036–1052. doi: 10.1152/ajpendo.00151.2012. [DOI] [PubMed] [Google Scholar]
- 29.Li L, Zhu L, Hao B, Gao W, Wang Q, Li K, Wang M, Huang M, Liu Z, Yang Q, Li X, Zhong Z, Huang W, Xiao G, Xu Y, Yao K, Liu Q. NOS-derived nitric oxide promotes glycolysis by inducing pyruvate kinase M2 nuclear translocation in ovarian cancer. Oncotarget. 2017;8:33047–33063. doi: 10.18632/oncotarget.16523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Yun Y, Wu B, Wen L, Wen M, Yang H, Zhao L, Liu W, Huang S, Wen N, Li Y. FOXM1 promotes reprogramming of glucose metabolism in epithelial ovarian cancer cells via activation of GLUT1 and HK2 transcription. Oncotarget. 2016;7:47985–47997. doi: 10.18632/oncotarget.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y, Cao Y, Chen L, Liu F, Qi Z, Cheng X, Wang Z. Cryptotanshinone suppresses cell proliferation and glucose metabolism via STAT3/SIRT3 signaling pathway in ovarian cancer cells. Cancer Med. 2018;7:4610–4618. doi: 10.1002/cam4.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fong MY, McDunn J, Kakar SS. Identification of metabolites in the normal ovary and their transformation in primary and metastatic ovarian cancer. PLoS One. 2011;6:e19963. doi: 10.1371/journal.pone.0019963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amano Y, Mandai M, Yamaguchi K, Matsumura N, Kharma B, Baba T, Abiko K, Hamanishi J, Yoshioka Y, Konishi I. Metabolic alterations caused by HNF1beta expression in ovarian clear cell carcinoma contribute to cell survival. Oncotarget. 2015;6:26002–26017. doi: 10.18632/oncotarget.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Duan W, Li X, Liu J, Li D, Ye L, Qian L, Yang A, Xu Q, Liu H, Fu Q, Wu E, Ma Q, Shen X. PTTG regulates the metabolic switch of ovarian cancer cells via the c-myc pathway. Oncotarget. 2015;6:40959–40969. doi: 10.18632/oncotarget.5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xintaropoulou C, Ward C, Wise A, Queckborner S, Turnbull A, Michie CO, Williams ARW, Rye T, Gourley C, Langdon SP. Expression of glycolytic enzymes in ovarian cancers and evaluation of the glycolytic pathway as a strategy for ovarian cancer treatment. BMC Cancer. 2018;18:636. doi: 10.1186/s12885-018-4521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matassa DS, Amoroso MR, Lu H, Avolio R, Arzeni D, Procaccini C, Faicchia D, Maddalena F, Simeon V, Agliarulo I, Zanini E, Mazzoccoli C, Recchi C, Stronach E, Marone G, Gabra H, Matarese G, Landriscina M, Esposito F. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell Death Differ. 2016;23:1542–1554. doi: 10.1038/cdd.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miao Y, Lu M, Yan Q, Li S, Feng Y. Inhibition of proliferation, migration, and invasion by knockdown of Pyruvate Kinase-M2 (PKM2) in ovarian cancer SKOV3 and OVCAR3 cells. Oncol Res. 2016;24:463–475. doi: 10.3727/096504016X14685034103671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siu MKY, Jiang YX, Wang JJ, Leung THY, Han CY, Tsang BK, Cheung ANY, Ngan HYS, Chan KKL. Hexokinase 2 regulates ovarian cancer cell migration, invasion and stemness via FAK/ERK1/2/MMP9/NANOG/SOX9 signaling cascades. Cancers (Basel) 2019;11:813. doi: 10.3390/cancers11060813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan JY, Yang Y, Xie JY, Lu YL, Shi K, Huang YQ. MicroRNA-144 mediates metabolic shift in ovarian cancer cells by directly targeting Glut1. Tumour Biol. 2016;37:6855–6860. doi: 10.1007/s13277-015-4558-9. [DOI] [PubMed] [Google Scholar]
- 40.Zhou Y, Zheng X, Lu J, Chen W, Li X, Zhao L. Ginsenoside 20(S)-Rg3 inhibits the Warburg effect via modulating DNMT3A/MiR-532-3p/HK2 pathway in ovarian cancer cells. Cell Physiol Biochem. 2018;45:2548–2559. doi: 10.1159/000488273. [DOI] [PubMed] [Google Scholar]
- 41.Xiaohong Z, Lichun F, Na X, Kejian Z, Xiaolan X, Shaosheng W. MiR-203 promotes the growth and migration of ovarian cancer cells by enhancing glycolytic pathway. Tumour Biol. 2016;37:14989–14997. doi: 10.1007/s13277-016-5415-1. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Tong L, Luo Y, Li X, Chen G, Wang Y. Resveratrol inhibits the proliferation and induces the apoptosis in ovarian cancer cells via inhibiting glycolysis and targeting AMPK/mTOR signaling pathway. J Cell Biochem. 2018;119:6162–6172. doi: 10.1002/jcb.26822. [DOI] [PubMed] [Google Scholar]
- 43.Qiu H, Jackson AL, Kilgore JE, Zhong Y, Chan LL, Gehrig PA, Zhou C, Bae-Jump VL. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget. 2015;6:6915–6930. doi: 10.18632/oncotarget.3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shang Y, He J, Wang Y, Feng Q, Zhang Y, Guo J, Li J, Li S, Wang Y, Yan G, Ren F, Shi Y, Xu J, Zeps N, Zhai Y, He D, Chang Z. CHIP/Stub1 regulates the Warburg effect by promoting degradation of PKM2 in ovarian carcinoma. Oncogene. 2017;36:4191–4200. doi: 10.1038/onc.2017.31. [DOI] [PubMed] [Google Scholar]
- 45.Chakraborty PK, Mustafi SB, Xiong X, Dwivedi SKD, Nesin V, Saha S, Zhang M, Dhanasekaran D, Jayaraman M, Mannel R, Moore K, McMeekin S, Yang D, Zuna R, Ding K, Tsiokas L, Bhattacharya R, Mukherjee P. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat Commun. 2017;8:14634. doi: 10.1038/ncomms14634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Caneba CA, Yang L, Baddour J, Curtis R, Win J, Hartig S, Marini J, Nagrath D. Nitric oxide is a positive regulator of the Warburg effect in ovarian cancer cells. Cell Death Dis. 2014;5:e1302. doi: 10.1038/cddis.2014.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chao TK, Huang TS, Liao YP, Huang RL, Su PH, Shen HY, Lai HC, Wang YC. Pyruvate kinase M2 is a poor prognostic marker of and a therapeutic target in ovarian cancer. PLoS One. 2017;12:e0182166. doi: 10.1371/journal.pone.0182166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Han G, Li X, Kan Q, Fan Z, Li Y, Ji Y, Zhao J, Zhang M, Grigalavicius M, Berge V, Goscinski MA, Nesland JM, Suo Z. Mitochondrial pyruvate carrier function determines cell stemness and metabolic reprogramming in cancer cells. Oncotarget. 2017;8:46363–46380. doi: 10.18632/oncotarget.18199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naczki C, John B, Patel C, Lafferty A, Ghoneum A, Afify H, White M, Davis A, Jin G, Kridel S, Said N. SPARC inhibits metabolic plasticity in ovarian cancer. Cancers (Basel) 2018;10:385. doi: 10.3390/cancers10100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han J, Wysham WZ, Zhong Y, Guo H, Zhang L, Malloy KM, Dickens HK, Huh G, Lee D, Makowski L, Zhou C, Bae-Jump VL. Increased efficacy of metformin corresponds to differential metabolic effects in the ovarian tumors from obese versus lean mice. Oncotarget. 2017;8:110965–110982. doi: 10.18632/oncotarget.20754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dar S, Chhina J, Mert I, Chitale D, Buekers T, Kaur H, Giri S, Munkarah A, Rattan R. Bioenergetic adaptations in chemoresistant ovarian cancer cells. Sci Rep. 2017;7:8760. doi: 10.1038/s41598-017-09206-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Y, Gao W, Zhang Y, Wu S, Liu Y, Deng X, Xie L, Yang J, Yu H, Su J, Sun L. ABT737 reverses cisplatin resistance by targeting glucose metabolism of human ovarian cancer cells. Int J Oncol. 2018;53:1055–1068. doi: 10.3892/ijo.2018.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou L, Liu L, Chai W, Zhao T, Jin X, Guo X, Han L, Yuan C. Dichloroacetic acid upregulates apoptosis of ovarian cancer cells by regulating mitochondrial function. Onco Targets Ther. 2019;12:1729–1739. doi: 10.2147/OTT.S194329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ai Z, Lu Y, Qiu S, Fan Z. Overcoming cisplatin resistance of ovarian cancer cells by targeting HIF-1-regulated cancer metabolism. Cancer Lett. 2016;373:36–44. doi: 10.1016/j.canlet.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goff BA, Sainz de la Cuesta R, Muntz HG, Fleischhacker D, Ek M, Rice LW, Nikrui N, Tamimi HK, Cain JM, Greer BE, Fuller AF Jr. Clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy in stage III disease. Gynecol Oncol. 1996;60:412–417. doi: 10.1006/gyno.1996.0065. [DOI] [PubMed] [Google Scholar]
- 56.Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, Suzuki M, Sato I, Taguchi K. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer. 2000;88:2584–2589. [PubMed] [Google Scholar]
- 57.Mandai M, Amano Y, Yamaguchi K, Matsumura N, Baba T, Konishi I. Ovarian clear cell carcinoma meets metabolism; HNF-1beta confers survival benefits through the Warburg effect and ROS reduction. Oncotarget. 2015;6:30704–30714. doi: 10.18632/oncotarget.5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amano T, Chano T, Yoshino F, Kimura F, Murakami T. Current Position of the molecular therapeutic targets for ovarian clear cell carcinoma: a literature review. Healthcare (Basel) 2019;7:94. doi: 10.3390/healthcare7030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. doi: 10.7554/eLife.02242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lim HY, Ho QS, Low J, Choolani M, Wong KP. Respiratory competent mitochondria in human ovarian and peritoneal cancer. Mitochondrion. 2011;11:437–443. doi: 10.1016/j.mito.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 61.Li H, Hu S, Pang Y, Li M, Chen L, Liu F, Liu M, Wang Z, Cheng X. Bufalin inhibits glycolysis-induced cell growth and proliferation through the suppression of Integrin beta2/FAK signaling pathway in ovarian cancer. Am J Cancer Res. 2018;8:1288–1296. [PMC free article] [PubMed] [Google Scholar]
- 62.Mukherjee A, Ma Y, Yuan F, Gong Y, Fang Z, Mohamed EM, Berrios E, Shao H, Fang X. Lysophosphatidic acid up-regulates hexokinase II and glycolysis to promote proliferation of ovarian cancer cells. Neoplasia. 2015;17:723–734. doi: 10.1016/j.neo.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ray U, Roy Chowdhury S, Vasudevan M, Bankar K, Roychoudhury S, Roy SS. Gene regulatory networking reveals the molecular cue to lysophosphatidic acid-induced metabolic adaptations in ovarian cancer cells. Mol Oncol. 2017;11:491–516. doi: 10.1002/1878-0261.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun S, Liu J, Zhao M, Han Y, Chen P, Mo Q, Wang B, Chen G, Fang Y, Tian Y, Zhou J, Ma D, Gao Q, Wu P. Loss of the novel mitochondrial protein FAM210B promotes metastasis via PDK4-dependent metabolic reprogramming. Cell Death Dis. 2017;8:e2870. doi: 10.1038/cddis.2017.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu Y, Deng Y, Zhu J, Duan Y, Weng W, Wu X. Pim1 promotes cell proliferation and regulates glycolysis via interaction with MYC in ovarian cancer. Onco Targets Ther. 2018;11:6647–6656. doi: 10.2147/OTT.S180520. [DOI] [PMC free article] [PubMed] [Google Scholar]