

Abstract
Background: Myocardial infarction (MI) is the principal cause of mortality globally. Fraxetin (Fra) has anti-oxidative and anti-inflammatory properties. Nevertheless, the functional action of Fra in the progression of MI has never been elucidated. Method: The in vivo model of MI was set up by ligating left anterior descending artery. The gene expression was tested by qRT-PCR and WB. The 2,3,5-triphenyltetrazolium chloride staining was applied to assess MI size. The cell viability was tested by MTT assay. Commercial kits were utilized to detect the activity of serum LDH and the levels of Fe2+, malondialdehyde (MDA), and glutathione (GSH). Results: Fra treatment could reduce the infraction size and restrain ferroptosis in rats with MI. Moreover, Fra reduced the activity of serum LDH, the accumulation of iron and the MDA level, and increased GSH and glutathione peroxidase 4 (GPX4) in rats with MI. Furthermore, Fra protected H9C2 myocardial cells against OGD/R-induced ferroptosis by up-regulating HO-1. Moreover, Fra activated phosphorylation of AKT and Nrf2 nuclear accumulation in MI in vivo and in vitro models. Notably, silencing Nrf2 enhanced the ferroptosis in H9C2 cells induced by OGD/R, while LY, an inhibitor of AKT phosphorylation, diminished the inhibition of Fra. Conclusion: Fra attenuated MI-induced ferroptosis via AKT/Nrf2/HO-1 signaling, providing a potential therapeutic agent for MI.
Keywords: Myocardial infarction, fraxetin, ferroptosis, AKT, nuclear factor erythroid 2-related factor 2, heme oxygenase-1
Introduction
MI is the most pervasive cardiac disease clinically, which severely threatens public health in terms of high mortality throughout the world [1]. MI triggered by coronary artery occlusion (ischemia and hypoxia) leads to the loss of cardiomyocytes, resulting in heart failure [2]. Restoring coronary artery circulation in time is currently the most effective treatment for MI, which may further aggravate the myocardial damage and increase the size of MI [3]. To date, the number of patients with MI is increasing every year. Therefore, novel and effective therapeutic strategies for MI are urgently needed.
The loss of cardiomyocytes occurs during MI through various necrocytosis processes, like autophagy, and ferroptosis [4]. Ferroptosis is a recently discovered type of necrocytosis [5]. Differently from the other types of necrocytosis, ferroptosis is characterized by iron-dependent lipid peroxide [6]. Several studies have revealed that ferroptosis is a key mechanism for the development of MI [7]. During myocardial injury, the free iron is excessively accumulated in the mitochondria, which leads to lipid peroxidation and ferroptotic cell death [8,9]. It has been reported that ferrostatin-1 (the ferroptosis inhibitor; Fer-1), mitochondria-targeted antioxidant MitoTEMPO, ZPP IX (a HO-1 antagonist) and iron chelation could protect against doxorubicin-induced cardiomyopathy by repressing ferroptosis [10]. Similarly, the inhibition of ferroptosis by Fer-1 could protect against diabetes mellitus MIRI in vivo and in vitro by reducing endoplasmic reticulum stress [11]. A recent study has revealed that HUCB-derived MSC exosome can attenuate myocardial injury in mice with acute MI by repressing ferroptosis [12]. Thus, targeting ferroptosis is considered as a therapeutic strategy for MI.
Fraxetin (7,8-dihydroxy-6-methoxy coumarin; Fra) is the active constituent of Chinese herbs--cortex fraxini, which has exerted protective effect against various human diseases [13]. Fra has been reported to restrain the multiplication and promote the apoptosis of colorectal carcinoma cells through the Janus kinase 2/signal transducer and activator of transcription 3 signaling [14]. Besides, Fra treatment could promote the metabolism of ethanol and inhibit lipid peroxidation, as well as reduce the levels of TNF-α and IL-1β, thereby alleviating ethanol-induced hepatic fibrosis [15]. However, there are few studies on the pharmacological action of Fra in the progress of MI. In this research, we elucidated the function of Fra in the progress of MI in vivo and in vitro. We first reported how Fra protected against MI-induced ferroptosis by inhibiting AKT/Nrf2/HO-1 signaling. Our findings have revealed that Fra promises to be a potential regent for MI.
Materials and methods
Animal experiments
Male Wistar rats (200-250 g) were obtained from Slac Laboratory Animal Center (Shanghai, China) and kept in cages. They were free to eat chow and drink tap water from the laboratory. The use and handling of the animals were ratified by the Institutional Animal Ethics Committee of the First Affiliated Hospital of Zhejiang Chinese Medical University (156945364).
The rats were randomized into 5 groups: Sham (n=6), Fra (25 mg/kg, n=6), MI (n=6), Fra (5 mg/kg) + MI group (n=6) and Fra (25 mg/kg) + MI groups (n=6). Fra was provided by Baoman Biotechnology Co., Ltd. (Shanghai, China, D0019). The rats in Fra (25 mg/kg), Fra (5 mg/kg) + MI, and Fra (25 mg/kg) + MI groups were intraperitoneally injected with 25, 5 or 25 mg/kg Fra daily respectively for 14 continuous days, while the rats in the other groups received an intraperitoneal injection of the equal amount of physiological saline. The MI was induced at 30 min after the last injection.
Establishment of MI
The rats were anesthetized with 1% pentobarbital and then lied on its back. Thereafter, the left precordial area of the rats were shaved and disinfected, followed by trachea intubation for artificial ventilation. After the left thoracotomy, the heart was fully exposed and the left coronary artery (LAD) was ligated with a 6-0 prolene suture at 2-3 mm from its origin between the pulmonary artery conus and the left atrial appendage. After 30 min, the suture was gently removed to allow reperfusion for 2 h. The marked elevation of ST-segment of the electrocardiogram indicated that the MI model was successfully established.
The body weight of rats was recorded at 0 and 17 d after the study initiation. At the 3 d after LAD ligation, the LVEDP, maximum rate of left ventricular pressure rise and fall (± dp/dtmax) and LVSP were evaluated. Subsequently, the rats were euthanized, and the organ index was evaluated.
2,3,5-TTC staining
After the rats were euthanized, the heart was excised, rinsed and cut into 3-mm slices. The sections were incubated for 30 min with 0.5% TTC solution, which can stain the infarct areas white and non-infarct areas red. The slices were fixed, pictured and analyzed using Image J (NIH, Rockville, MD, USA). The infarct area was presented as a percentage of the total left ventricular zone (%).
Measurement of LDH activity
The blood of rats was obtained before the rats were euthanized. After centrifugation, the serum was obtained and assayed for LDH activity via an LDH detection kit (Solarbio, Beijing, China, BC0685).
Following OGD/R, H9C2 cells were collected and incubated for 1 h with LDH release solution. After that, the supernatant of H9C2 cells were reacted for 30 min with LDH detection solution under dark conditions, and then examined for the absorbance at 490 nm.
Cell culture
Rat cardiomyocytes H9C2 of the American Type Culture Collection (ATCC; Manassas, VA, USA, iCell-r012) were cultivated in DMEM medium (Solarbio, Beijing, China, 31600) comprising 100 μM Fra and 10% FBS for 24 h, and then incubated for 8 h in serum-, glucose- and sodium pyruvate-free DMEM medium in an anaerobic chamber with 95% N2/5% CO2. After this, the cells grew for 1 h in the medium containing 10% FBS under 5% CO2/95% air at 37°C.
Cell transfection
si-ctrl, si-HO-1 and si-Nrf2 plasmids were obtained from GenePharma (Shanghai, China). The sequence of HO-1 was amplified and fused into the pcDNA-3.1 vector to generate pcDNA-HO-1 constructs. Cells were transfected by LipofectamineTM 2000 (Invitrogen, Carlsbad, CA, the States, 11668019) based on manufacturer’s recommendations.
MTT assay
H9C2 cells were seeded into 96-well plates. After incubation under indicated conditions, the cells were reacted for 4 h with 10 μl MTT solution (M1020) from Solarbio, followed by 10-minute incubation with Formazan solution on a shaker. The absorbance was determined at 490 nm via a universal microplate spectrophotometer.
Fe2+, MDA and GSH measurement
The levels of Fe2+, MDA and GSH in tissue homogenates and cell lysates were assessed via iron assay (BioAssay Systems, Hayward, CA, the States, DIFE-250), lipid peroxidation MDA assay (Beyotime, Shanghai, China, S0131M) and GSH assay kits (Solarbio, Beijing, China, G8180), respectively.
qRT-PCR
The total RNA was obtained via Trizol (Solarbio) digestion in light of the manufacturer’s recommendations. The purity and concentration of the extracted RNA were assessed using Nanodrop2000 (Thermo Fisher Scientific, Waltham, MA, United States). The RNA was transformed into cDNA via SuperScript III Reverse Transcriptase (Life Technologies, Gaithersburg, MD, United States), following the product manual. qRT-PCR assay was conducted to test the glutathione peroxidase 4 (GPX4) and HO-1 using Power SYBR Green Master Mix (Life technologies), with GAPDH as an internal control. 2-ΔΔCt method was applied to test the GPX4 and HO-1. The primers used in this research: GPX4 forward: 5’-GCT GTG CGC GCT CCA T-3’, and reverse: 5’-CCA TGT GCC CGT CGA TGT-3’; HO-1 forward: 5’-GGC TGT GAA CTC TGT CTC-3’, and reverse: 5’-GGC ATC TCC TTC CAT TCC-3’; GAPDH forward: 5’-GAT GGT GAA GGT CGG TGT GAA-3’, and reverse: 5’-TTG AAC TTG CCG TGG GTA GAG-3’.
WB
TP from myocardial tissues and H9C2 cells was isolated via RIPA assay in keeping with the manufacturer’s specifications. The isolated proteins were quantitated via BCA Protein Assay Kit (Solarbio, Beijing, China, PC0020). The proteins were segregated by 12% SDS-PAGE, and then transferred onto polyvinylidene fluoride film. The films were sealed by 5% fat-free milk, and probed with the primary antibodies against GPX4 (ab205720, 1:5000), HO-1 (ab223349, 1:1000), Nrf2 (ab62352, 1:1000), pAKT (ab40795, 1:1000), AKT (ab38449, 1:1000), Lamin B (ab32535, 1:500) and GAPDH (ab8245, 1:5000) at 4°C for one night, followed by 1-hour immunoblot with the secondary antibodies at indoor temperature. All the antibodies were acquired from Abcam (Cambridge, MA, United States). The bands were developed by using an enhanced chemiluminescence system, and analyzed via Image J.
Statistical analysis
SPSS 20.0 (SPSS Inc., Chicago, IL, the States) was used for data analyses. The data were shown as the mean ± standard deviation in represent of three independent tests. One-way ANOVA was used for comparison among multiple groups, represented by F. LSD test was used for pairwise comparison between groups. The statistical pictures were drawn using GraphPad Prism 8.0. P<0.05 was statistically significant.
Results
Influence of Fra administration on body weight and organ index of rats
Firstly, we evaluated the impact of Fra on rat body weight and organ index and found that there was no difference in body weight among groups (Supplementary Table 1). Similarly, the rats in different groups showed no significant difference in organ indices (heart index, liver index, kidney index, lung index and spleen index) (Supplementary Table 2).
Effect of Fra on hemodynamics in MI rat hearts
Compared with the sham group, MI rats demonstrated myocardial dysfunction, as evidenced by increased LVEDP, reduced maximum rate of left ventricular pressure rise, fall (± dp/dtmax) and left ventricular systolic pressure (LVSP) (Table 1). Fra administration, especially at a high concentration (25 mg/kg), markedly improved myocardial dysfunction in rats with MI. Therefore, the concentration of 25 mg/kg was selected for the following experiments.
Table 1.
Hemodynamics for each group at the day 17 (three days after MI)
| Group | LVSP (mmHg) | LVEDP (mmHg) | +dp/dtmax (mmHg/ms) | -dp/dtmax (mmHg/ms) |
|---|---|---|---|---|
| Sham | 131.55±8.59 | 2.34±0.31 | 5.61±0.51 | -5.25±0.45 |
| Fra | 132.98±10.54 | 2.28±0.35 | 5.46±0.62 | -5.41±0.52 |
| MI | 90.36±8.25* | 5.62±0.55* | 3.65±0.35* | -3.32±0.35* |
| Fra (5 mg/kg) + MI | 110.56±8.69# | 4.15±0.51# | 4.51±0.42# | -4.30±0.41# |
| Fra (25 mg/kg) + MI | 122.69±13.44# | 3.48±0.45# | 5.05±0.65# | -4.93±0.52# |
| F | 18.390 | 25.890 | 13.970 | 21.090 |
| P | <0.001 | <0.001 | <0.001 | <0.001 |
Note: Values are presented as mean ± SD. MI, myocardial infarction; Fra, fraxetin; LVSP, left ventricular systolic pressure; LVEDP, left ventricular end-diastolic pressure; ±dp/dtmax, maximum rate of left ventricular pressure rise and fall.
P<0.05 vs. sham group;
P<0.05 vs. MI group.
Fra decreased infarct size and ferroptosis in MI rats
As displayed in Figure 1A, large infarct size was discovered in rats with MI, which was strikingly improved following Fra administration. Similarly, the activity of serum LDH was increased in MI rats compared with that in sham group. In comparison to the MI group, Fra treatment reduced the activity of serum LDH in rats with MI (Figure 1B). Consistently, the levels of Fe2+ and MDA were enhanced, while the GSH was reduced in the cardiac tissues of rats with MI. Notably, these changes caused by MI were obviously blocked by Fra treatment (Figure 1D-F). Besides, it was found that the mRNA and protein levels of GPX4 were decreased in their cardiac tissues, which were reversed after Fra treatment (Figure 1G, 1H).
Figure 1.
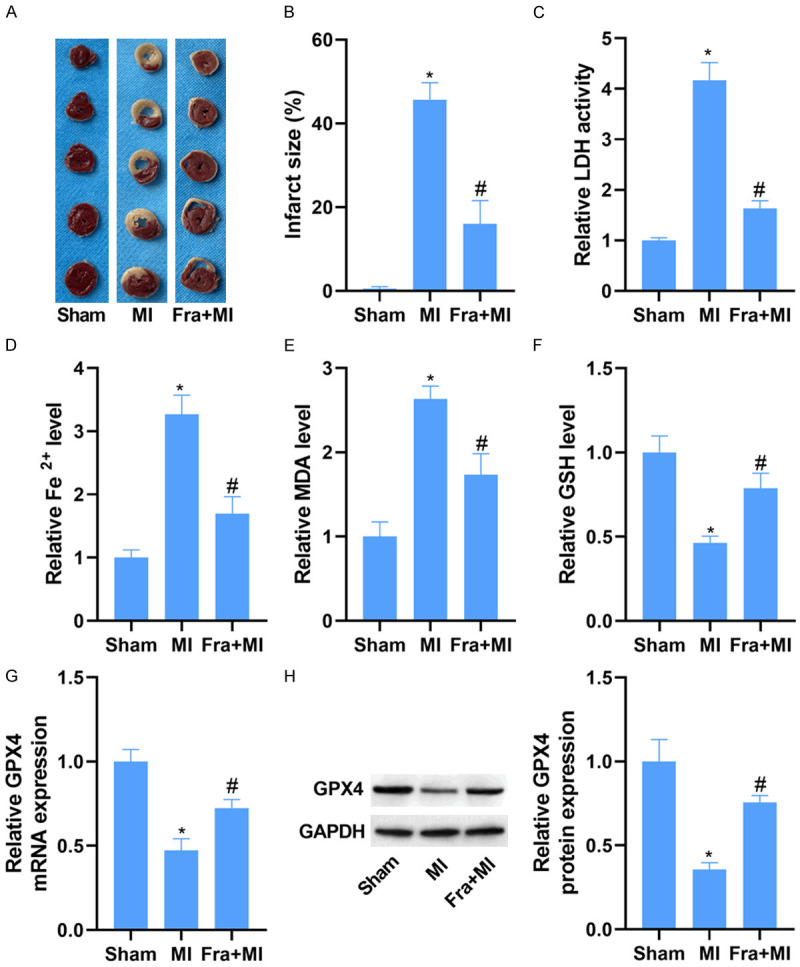
Fra decreases infarct size and ferroptosis due to MI. Male Wistar rats were intraperitoneally injected with 25 mg/kg Fra or normal saline before LAD ligation. A. 2,3,5-triphenyltetrazolium chloride staining of the heart tissues of different groups. B. Quantitative analysis of infarct size. C. LDH detection kit was applied to evaluate the activity of serum LDH in different groups. D-F. The levels of Fe2+, MDA and GSH in the myocardial tissues of different groups were evaluated using commercial kits. G and H. qRT-PCR and WB analysis of GPX4 mRNA and protein expression in the myocardial tissues of different groups. *P<0.05 vs. sham group; #P<0.05 vs. MI group.
Fra protected H9C2 cardiomyocytes against OGD/R-induced ferroptosis in vitro
Given the cardioprotection of Fra in vivo, we further explored the effect of Fra on MI in vitro. We treated H9C2 cells with increasing doses (0, 10, 50, 100 and 200 μM) of Fra to determine the toxic effect of Fra on cells. As determined by MTT assay, Fra appeared to be non-toxic to H9C2 cells at concentrations ranging from 10 to 100 μM, while it reduced the viability of H9C2 cells at a concentration of 200 μM (Supplementary Figure 1A). Meanwhile, Fra had no effect on LDH activity at concentrations ranging from 10 to 100 μM, but increased the activity of LDH at a concentration of 200 μM, which further revealed that Fra up to 100 μM exerted no toxicity (Supplementary Figure 1B). Thus, the dose of 100 μM was selected for the following studies.
To confirm the cardioprotection of Fra in vitro, H9C2 cells were cultured for 24 h in DMEM medium containing 100 μM Fra and subjected to OGD/R. As expected, OGD/R reduced H9C2 cell viability in the MTT test, and this reduction was conversed following Fra treatment (Figure 2A). The activity of LDH was enhanced in H9C2 cells stimulated by OGD/R, which was blocked by Fra treatment (Figure 2B). The ferroptosis indexes were also determined, including Fe2+, MDA, and GSH levels, and mRNA and protein expressions of GPX4. As displayed in Figure 2C-E, the Fe2+ and MDA levels were enhanced, but the GSH was decreased in H9C2 cells stimulated by OGD/R. Nevertheless, Fra management counteracted OGD/R-induced elevation of Fe2+ and MDA and reduction of GSH. Moreover, the mRNA and protein levels of GPX4 were conspicuously declined in H9C2 cells stimulated by OGD/R, which were overturned after Fra management (Figure 2F, 2G). Besides, it was found that the ferroptosis inhibitor, Fer-1, could diminish OGD/R-induced reduction of cell viability and increase of LDH activity (Supplementary Figure 2).
Figure 2.
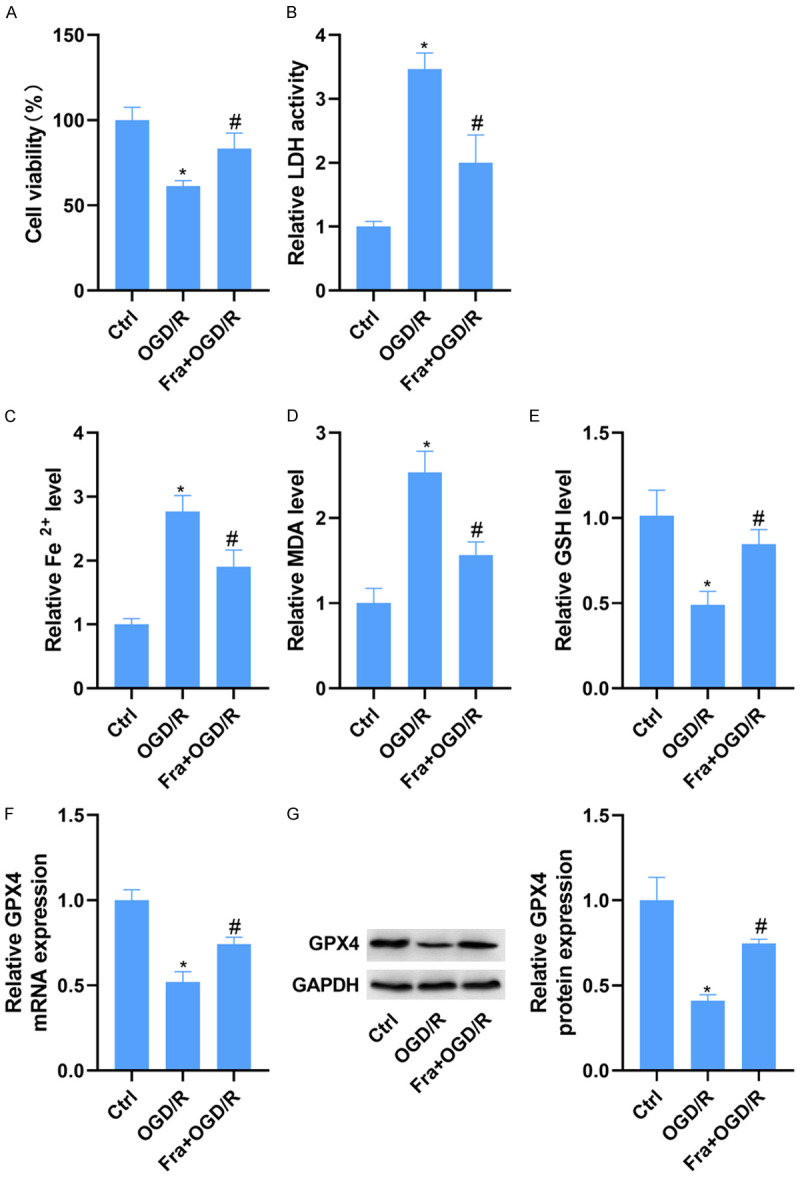
Fra protects H9C2 cardiomyocytes against OGD/R-induced ferroptosis in vitro. H9C2 cells were cultured in DMEM medium containing 100 μM Fra for 24 h and then subjected to OGD/R. A. The viability of H9C2 cells was determined using MTT assay. B. LDH detection kit was applied to evaluate the activity of LDH in H9C2 cells. C-E. The levels of Fe2+, MDA and GSH in H9C2 cells were evaluated using commercial kits. F and G. qRT-PCR and WB analysis of GPX4 mRNA and protein expression in H9C2 cells. *P<0.05 vs. ctrl group; #P<0.05 vs. OGD/R group.
Fra inhibited OGD/R-induced H9C2 cardiomyocyte ferroptosis by up-regulating HO-1
To further determine the mechanism by which Fra protected against OGD/R-induced ferroptosis in vitro, the expression of HO-1 was evaluated using qRT-PCR and WB. As a result, the mRNA and protein levels of HO-1 were increased in H9C2 cells under OGD/R, and the increased levels of HO-1 induced by OGD/R were reinforced under Fra treatment (Figure 3A, 3B). To affirm whether Fra inhibits OGD/R-induced H9C2 cardiomyocyte ferroptosis by up-regulating HO-1, we altered HO-1 in H9C2 cells by si-HO-1 and pcDNA-HO-1 transfection. As expected, the mRNA and protein levels of HO-1 were conspicuously reduced in those transfected with si-HO-1 compared with those transfected with si-ctrl (Figure 3C, 3D). Accordingly, the levels were strikingly elevated in H9C2 cells transfected with pcDNA-HO-1 compared with those transfected with the pcDNA-ctrl (Figure 3E, 3F).
Figure 3.
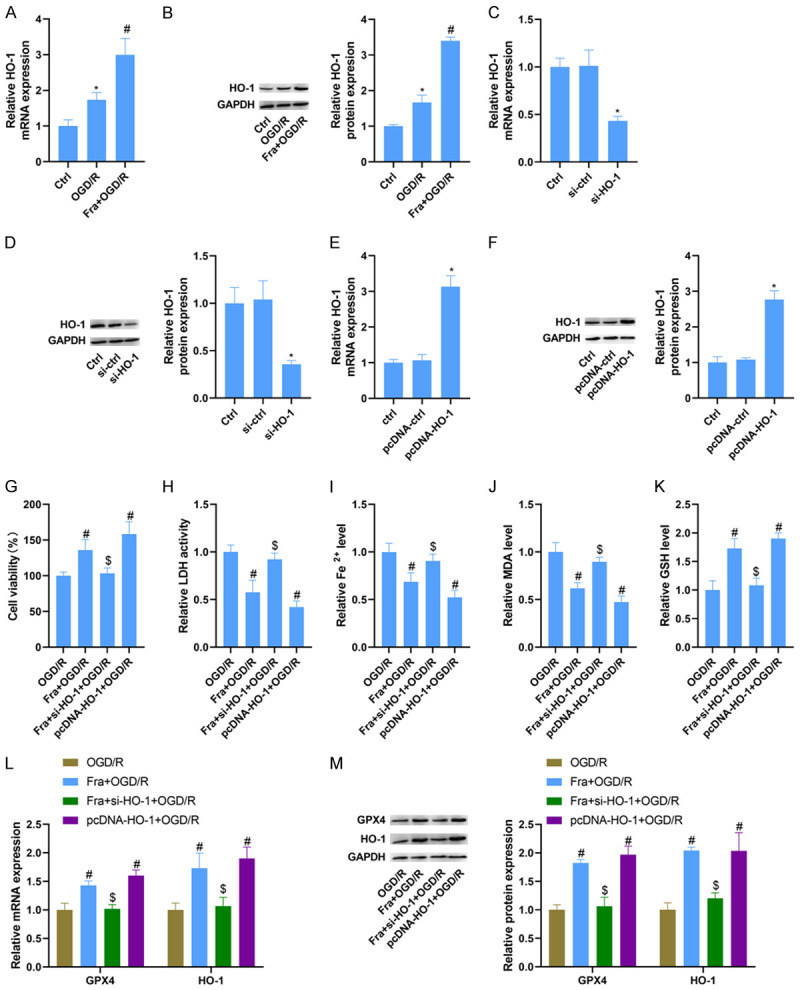
Fra inhibits OGD/R-induced H9C2 cardiomyocyte ferroptosis by upregulating HO-1. H9C2 cells were cultured in DMEM medium containing 100 μM Fra for 24 h and then subjected to OGD/R. A and B. qRT-PCR and WB analysis of HO-1 mRNA and protein expression in H9C2 cells treated with OGD/R alone or in combination with Fra. C and D. qRT-PCR and WB analysis of HO-1 mRNA and protein expression in H9C2 cells transfected with si-ctrl or si-HO-1. E and F. qRT-PCR and WB analysis of HO-1 mRNA and protein expression in H9C2 cells transfected with pcDNA-HO-1 or pcDNA-ctrl. G. MTT assay was performed to determine the cell viability. H. LDH detection kit was applied to evaluate the activity of LDH. I-K. The levels of Fe2+, MDA and GSH were detected using commercial kits. L and M. qRT-PCR and WB analysis of GPX4 and HO-1 mRNA and protein expression in H9C2 cells. *P<0.05 vs. si-ctrl group or pcDNA-ctrl group; #P<0.05 vs. OGD/R group; $P<0.05 vs. Fra + OGD/R group.
Deletion of HO-1 diminished Fra-induced elevation of cell viability in H9C2 cells stimulated by OGD/R, while overexpression of HO-1 caused an increase in cell viability (Figure 3G). In line with this, up-regulation of HO-1 reduced the LDH activity of H9C2 cells stimulated by OGD/R, while knockdown of HO-1 abolished Fra-induced inhibition of LDH activity (Figure 3H). But beyond that, inhibiting HO-1 blocked the reduction of Fe2+ and MDA levels and the increase of GSH in H9C2 cells induced by Fra after OGD/R, but its upregulation could reduce the level of Fe2+ and MDA, and increase the level of GSH (Figure 3I-K). As tested by qRT-PCR and WB, the increased GPX4 and HO-1 induced by Fra was counteracted by HO-1 knockdown in H9C2 cells after OGD/R. Also, upregulation of HO-1 increased the mRNA and protein levels of GPX4 and HO-1 in H9C2 cells after OGD/R (Figure 3L, 3M).
Fra activated phosphorylation of AKT and Nrf2 nuclear accumulation
To explore whether AKT/Nrf2/HO-1 signal is involved in Fra’s cardioprotective effect against MI, the nuclear Nrf2, HO-1 and pAKT/AKT were detected using WB. It was found that MI enhanced the first two and repressed AKT phosphorylation, compared with the sham group. Notably, pretreatment of Fra led to the increased nuclear Nrf2 and HO-1 and the phosphorylation of AKT in MI rats (Figure 4A). Consistently, the nuclear Nrf2 and HO-1 were increased, but the pAKT level was reduced in H9C2 cells after OGD/R, and silencing Nrf2 could block the elevation of nuclear Nrf2 and HO-1 induced by OGD/R. Fra administration promoted the nuclear Nrf2 and HO-1 and AKT phosphorylation in H9C2 cells following OGD/R, which was block by 10 µM of LY (LY294002, an inhibitor of AKT phosphorylation) pre-treatment for 30 min (Figure 4B). Besides, it was found that silencing Nrf2 reduced but upregulation of Nrf2 by pcDNA-Nrf2 increased the mRNA and protein levels of HO-1, as determined by qRT-PCR and WB (Supplementary Figure 3).
Figure 4.
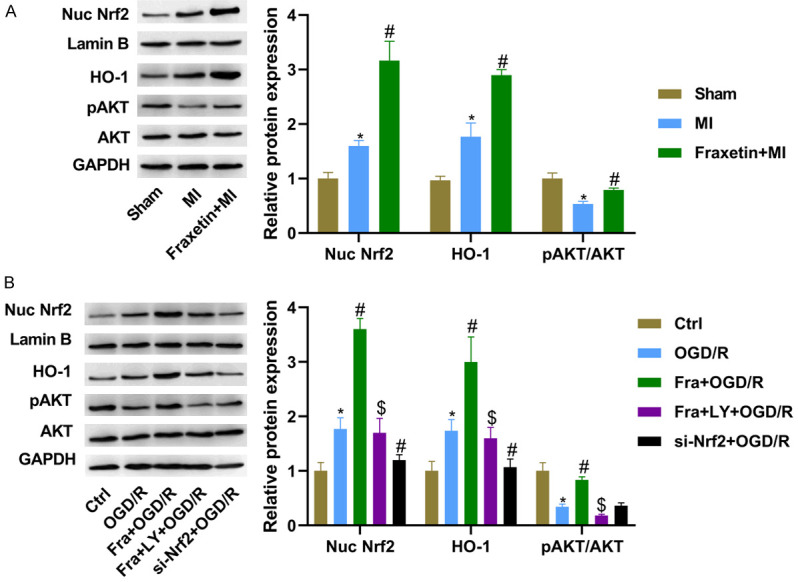
Fra activates phosphorylation of AKT and Nrf2 nuclear accumulation. A. WB analysis of nuclear Nrf2, HO-1, pAKT and AKT protein expression in the heart tissues of rats in different groups. B. WB analysis of nuclear Nrf2, HO-1, pAKT and AKT protein expression in H9C2 cells. *P<0.05 vs. sham group or ctrl group; #P<0.05 vs. MI or OGD/R group; $P<0.05 vs. Fra + OGD/R group.
Fra inhibited OGD/R-induced H9C2 cardiomyocyte ferroptosis via AKT/Nrf2/HO-1 pathway
We further confirmed whether Fra inhibited OGD/R-induced H9C2 cardiomyocyte ferroptosis via AKT/Nrf2/HO-1 pathway. As a result, LY administration mitigated Fra-induced elevation of H9C2 cell viability stimulated by OGD/R. Also, we concluded that silencing Nrf2 could inhibit the vitality of H9C2 cells following OGD/R (Figure 5A). Meanwhile, Fra-induced inhibition of LDH activity in H9C2 cells after OGD/R was blocked by LY administration, and deletion of Nrf2 enhanced the activity of LDH in those following OGD/R (Figure 5B). In those cells stimulated with OGD/R, Fra-induced reduction of Fe2+ and MDA and elevation of GSH were abrogated after LY management. Knockdown of Nrf2 increased the levels of Fe2+ and MDA, but reduced the levels of GSH in H9C2 cells after OGD/R (Figure 5C-E). As tested by qRT-PCR and WB assay, Fra management enhanced the mRNA and protein levels of GPX4 in H9C2 cells stimulated by OGD/R, which was abolished by LY treatment. Besides, silencing Nrf2 lessened the mRNA and protein levels of GPX4 in H9C2 cells stimulated by OGD/R (Figure 5F, 5G).
Figure 5.
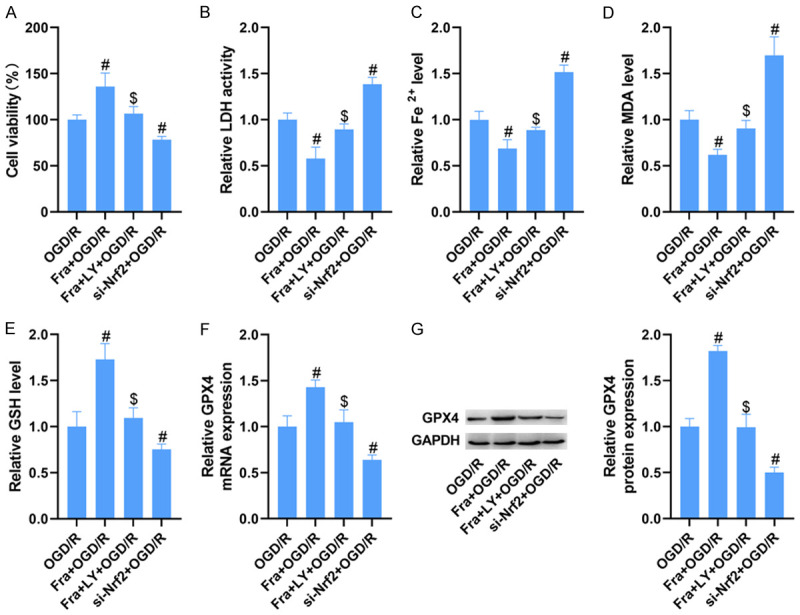
Fra inhibits OGD/R-induced H9C2 cardiomyocyte ferroptosis via AKT/Nrf2/HO-1 pathway. (A) MTT assay was performed to determine the cell viability. (B) LDH detection kits were applied to evaluate the activity of LDH. (C-E) The levels of Fe2+, MDA and GSH were detected using commercial kits. (F) qRT-PCR and (G) WB analysis of GPX4 mRNA and protein expression in H9C2 cells. #P<0.05 vs. OGD/R group; $P<0.05 vs. Fra + OGD/R group.
Discussion
MI brings an immense burden to patients and society, but there is no effective treatment for MI. Thus, seeking a novel and effective agent for MI therapy has never been more imperative. Fra has increasingly drawn the attention of researchers in the last years in terms of its pharmacological properties [16,17]. Although the significance of Fra has been well-established in the past years, surprisingly little attention has been devoted to investigating the role of Fra in the progression of MI. Here, we found that Fra administration strikingly improved the myocardial dysfunction, and reduced the infarct size in rats with MI. Moreover, Fra treatment reduced the LDH activity and increased the viability of H9C2 cells following OGD/R, indicating Fra’s cardioprotective effect on the progression of MI.
Ferroptosis has been recognized as a major contributor to the pathogenesis of MI [18]. By directly or indirectly repressing the expression of GPX4, MI causes the accumulation of ROS in mitochondria and then triggers ferroptosis, ultimately resulting in the loss of cardiac cells [19]. Similarly, repression of GPX4 by si-GPX4 or RSL3 leads to the accumulation of lipid peroxide, resulting in cell death by ferroptosis in H9C2 cells [20]. However, whether Fra exerts its cardioprotective effect via ferroptosis is still largely unknown. In our work, it was found that Fra administration could restrain MI-induced ferroptosis in vivo and in vitro, as evidenced by the reduced iron accumulation and MDA level, and the increased levels of GSH and GPX4. More significantly, the inhibitor of ferroptosis, Fer-1 could reverse OGD/R-induced inhibition of cell viability and LDH activity elevation, supporting the fact that Fra protected against MI by inhibiting ferroptosis.
Although there is a well-established link between Fra and MI-induced ferroptosis, relatively few have been mechanistically characterized. HO-1 is a representative cytoprotective enzyme that cleaves heme into porphyrin and iron, which serves as a major player in the progress of ferroptosis [21]. Inhibition of HO-1 could block magnesium isoglycyrrhizinate-mediated improvement of liver fibrosis by inhibiting hepatic stellate cell ferroptosis [22]. However, the opposite roles of HO-1 in ferroptosis were also demonstrated: HO-1 protects renal proximal tubule cells from erastin-induced ferroptosis [23]. This discrepancy might be due to the cracking of heme induced by HO-1, which not only produces the free radical scavengers dehydrobilirubin and cholerythrin, but also generates free iron that mediates ferroptosis through Fenton reaction. According to this study, overexpression of HO-1 protected H9C2 cells against OGD/R-induced ferroptosis, revealing that HO-1 acts as a negative regulator of ferroptosis. Interestingly, several studies have revealed that Fra exerts therapeutic effects in some diseases via upregulation of HO-1. In the research of Kundu et al., Fra can induce the expression of HO-1 by activating Akt/Nrf2 or AMP-activated protein kinase α/Nrf2 pathway in HaCaT cells [24]. In the research of Thuong et al., fraxetin has a direct protective effect against LDL oxidation at lower concentration, while Fra can activate and induce antioxidant enzymes through Nrf2/ARE at higher concentration [25]. Similarly, Fra also upregulated the expression of HO-1 in cells treated with OGD/R, while silencing HO-1 reduced the inhibitory effect of Fra on OGD/R induced hypertrophy, indicating that Fra could protect H9C2 cells from hypertrophy by enhancing HO-1. Similarly, Fra also upregulated the expression of HO-1 in cells treated with OGD/R, while silencing HO-1 reduced the inhibitory effect of Fra on OGD/R induced hypertrophy. The mechanism may be that Fra can protect H9C2 cells from hypertrophy by enhancing HO-1.
Nrf2 is a crucial transcription factor of oxidative response, which maintains the balance between antioxidant and oxidant [26]. Under the normal condition, Kelch-like-ECH-related protein 1 (Keap 1) sequesters Nrf2 in the cytoplasm, resulting in its degradation [27]. Under the oxidative condition, the Keap 1-Cul3 ubiquitination system is disrupted, and the specific serine or threonine residues of Nrf2 is phosphorylated by several kinases, such as PI3K/AKT and AMP-activated protein kinase kinases, thereby leading to the liberation of Nrf2 from Keap-1 [28,29]. Then, Nrf2 translocates into the nucleus, initiating anti-oxidative gene transcription [30]. Of note, Nrf2 has been confirmed to modulate ferroptosis via activating multiple target gene expression that participate in the metabolism of GSH, iron and lipids [31]. In vitro and in vivo tests have revealed that activation of Nrf2 induces the transcription of quinone oxidoreductase-1, HO-1, and ferritin heavy chain-1, and then restrains ferroptosis induced by erastin and sorafenib in HCC cells via interacting with transcriptional coactivator small v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog proteins [32]. Moreover, Nrf2 relieves ALI and inhibits IIR-induced ferroptosis, through upregulating the expression of a subunit of the cystine/glutamate transporter x and HO-1, suggesting that Nrf2 acts as a central switch in regulating the expression of HO-1 [33]. Our research revealed that Nrf2 positively modulated the expression of HO-1, which was consistent with the previous study [33].
Remarkably, it is reported that the activation of AKT/Nrf2/HO-1 signal is tightly linked with the progression of cardiac disease [34]. Previously, glaucocalyxin A protects H9C2 cells from hypoxia/reoxygenation-stimulated myocardial injury by activating the AKT/Nrf2/HO-1 signal path [35]. Additionally, platycodin D treatment induces the activation of AKT/Nrf2/HO-1 signaling, and then restrains H9C2 cell oxidant stress and apoptosis after hypoxia/reoxygenation stimulation [36]. Nevertheless, there still remains considerable uncertainty as to whether Fra attenuates MI-induced ferroptosis via AKT/Nrf2/HO-1 signal. In the present research, MI repressed the phosphorylation of AKT and increased nuclear Nrf2 and HO-1 expression. However, pretreatment of Fra led to the increased nuclear Nrf2 and HO-1 and AKT phosphorylation in MI in vivo and in vitro models. This result is in good agreement with the previous research which has suggested that Fra induced HO-1 by activating the AKT/Nrf2 signaling pathway [24]. Notably, silencing Nrf2 could block OGD/R-induced elevation of nuclear Nrf2 and HO-1 levels. LY management reversed Fra-caused elevation of nuclear Nrf2 and HO-1, and phosphorylation of AKT in H9C2 cells following OGD/R, revealing the involvement of AKT/Nrf2/HO-1 signaling in Fra’s protective effect on MI. Additionally, we concluded that knockdown of Nrf2 promoted OGD/R-induced ferroptosis in H9C2 cells, fitting the established notion of Nrf2 as a central regulator of ferroptosis. More importantly, LY treatment abrogated Fra’s effect on OGD/R-induced ferroptosis, providing evidence that Fra inhibits OGD/R-induced H9C2 cardiomyocyte ferroptosis via AKT/Nrf2/HO-1 pathway. In this study, we only carried out basic experiments in vivo and in vitro, and the role of Fra in acute myocardial infarction needs to be further studied through clinical practice in the future.
Taken together, we first clarified the cardioprotective effect of Fra on MI progression. More significantly, our data highlighted a mechanism, involving AKT/Nrf2/HO-1 signaling, through which Fra could protect against MI-induced ferroptosis. Thus, Fra may become a potential therapeutic reagent for MI, and our study provided the theoretical basis for its clinical application.
Acknowledgements
This research was funded by Medical health science and technology project of Zhejiang Provincial Health Commission (2020KY427) and the Ningbo natural fund project (2018A610393).
Disclosure of conflict of interest
None.
Abbreviations
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- WB
western blot
- MTT
methylthiazolyldiphenyl-tetrazolium bromide
- LDH
lactate dehydrogenase
- OGD/R
oxygen-glucose deprivation and reoxygenation
- HO-1
heme oxygenase-1
- Nrf2
nuclear factor erythroid 2-related factor 2
- ZPP
zinc protoporphyrin
- MIRI
myocardial ischemia/reperfusion injury
- HUCB
human umbilical cord blood
- MSC
mesenchymal stem cell
- TNF-α
tumor necrosis factor-α
- IL-1β
interleukin-1β
- LVEDP
left ventricular end-diastolic pressure
- LVSP
left ventricular systolic pressure
- TTC
triphenyltetrazolium chloride
- TP
total protein
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- ROS
reactive oxygen species
- HCC
hepatocellular carcinoma
- ALI
acute lung injury
- IIR
intestinal ischemia reperfusion
Supporting Information
References
- 1.Anderson JL, Morrow DA. Acute myocardial infarction. N Engl J Med. 2017;376:2053–2064. doi: 10.1056/NEJMra1606915. [DOI] [PubMed] [Google Scholar]
- 2.Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth universal definition of myocardial infarction (2018) J Am Coll Cardiol. 2018;72:2231–2264. doi: 10.1016/j.jacc.2018.08.1038. [DOI] [PubMed] [Google Scholar]
- 3.Neri M, Riezzo I, Pascale N, Pomara C, Turillazzi E. Ischemia/reperfusion injury following acute myocardial infarction: a critical issue for clinicians and forensic pathologists. Mediators Inflamm. 2017;2017:7018393. doi: 10.1155/2017/7018393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, Li B. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34. doi: 10.1186/s13045-019-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshimura C, Nagasaka A, Kurose H, Nakaya M. Efferocytosis during myocardial infarction. J Biochem. 2020;168:1–6. doi: 10.1093/jb/mvaa051. [DOI] [PubMed] [Google Scholar]
- 8.Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861:1893–1900. doi: 10.1016/j.bbagen.2017.05.019. [DOI] [PubMed] [Google Scholar]
- 9.Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, Cheng Q, Zhang P, Dai W, Chen J, Yang F, Yang HT, Linkermann A, Gu W, Min J, Wang F. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020;39:210–225. doi: 10.1089/dna.2019.5097. [DOI] [PubMed] [Google Scholar]
- 12.Song Y, Wang B, Zhu X, Hu J, Sun J, Xuan J, Ge Z. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol Toxicol. 2021;37:51–64. doi: 10.1007/s10565-020-09530-8. [DOI] [PubMed] [Google Scholar]
- 13.Xu H, Zhang J, Wang Q, Li Y, Zhang B. Fraxetin inhibits the proliferation of RL95-2 cells through regulation of metabolism. Int J Clin Exp Pathol. 2020;13:1500–1505. [PMC free article] [PubMed] [Google Scholar]
- 14.Ren S, Xing Y, Wang C, Jiang F, Liu G, Li Z, Jiang T, Zhu Y, Piao D. Fraxetin inhibits the growth of colon adenocarcinoma cells via the Janus kinase 2/signal transducer and activator of transcription 3 signalling pathway. Int J Biochem Cell Biol. 2020;125:105777. doi: 10.1016/j.biocel.2020.105777. [DOI] [PubMed] [Google Scholar]
- 15.Chen X, Ying X, Sun W, Zhu H, Jiang X, Chen B. The therapeutic effect of fraxetin on ethanol-induced hepatic fibrosis by enhancing ethanol metabolism, inhibiting oxidative stress and modulating inflammatory mediators in rats. Int Immunopharmacol. 2018;56:98–104. doi: 10.1016/j.intimp.2018.01.027. [DOI] [PubMed] [Google Scholar]
- 16.Liao JC, Wei ZX, Zhao C, Ma ZP, Cai DZ. Inhibition of osteoclastogenesis for periprosthetic osteolysis therapy through the suppression of p38 signaling by fraxetin. Int J Mol Med. 2018;42:1257–1264. doi: 10.3892/ijmm.2018.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu B, Wang R, Li S, Wang Y, Song F, Gu Y, Yuan Y. Antifibrotic effects of fraxetin on carbon tetrachloride-induced liver fibrosis by targeting NF-kappaB/IkappaBalpha, MAPKs and Bcl-2/Bax pathways. Pharmacol Rep. 2019;71:409–416. doi: 10.1016/j.pharep.2019.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Ravingerova T, Kindernay L, Bartekova M, Ferko M, Adameova A, Zohdi V, Bernatova I, Ferenczyova K, Lazou A. The molecular mechanisms of iron metabolism and its role in cardiac dysfunction and cardioprotection. Int J Mol Sci. 2020;21:21. doi: 10.3390/ijms21217889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol. 2017;403:143–170. doi: 10.1007/82_2016_508. [DOI] [PubMed] [Google Scholar]
- 20.Park TJ, Park JH, Lee GS, Lee JY, Shin JH, Kim MW, Kim YS, Kim JY, Oh KJ, Han BS, Kim WK, Ahn Y, Moon JH, Song J, Bae KH, Kim DH, Lee EW, Lee SC. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019;10:835. doi: 10.1038/s41419-019-2061-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sui M, Jiang X, Chen J, Yang H, Zhu Y. Magnesium isoglycyrrhizinate ameliorates liver fibrosis and hepatic stellate cell activation by regulating ferroptosis signaling pathway. Biomed Pharmacother. 2018;106:125–133. doi: 10.1016/j.biopha.2018.06.060. [DOI] [PubMed] [Google Scholar]
- 23.Adedoyin O, Boddu R, Traylor A, Lever JM, Bolisetty S, George JF, Agarwal A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol. 2018;314:F702–F714. doi: 10.1152/ajprenal.00044.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kundu J, Chae IG, Chun KS. Fraxetin induces heme oxygenase-1 expression by activation of Akt/Nrf2 or AMP-activated protein kinase alpha/Nrf2 pathway in HaCaT cells. J Cancer Prev. 2016;21:135–143. doi: 10.15430/JCP.2016.21.3.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thuong PT, Pokharel YR, Lee MY, Kim SK, Bae K, Su ND, Oh WK, Kang KW. Dual anti-oxidative effects of fraxetin isolated from fraxinus rhinchophylla. Biol Pharm Bull. 2009;32:1527–1532. doi: 10.1248/bpb.32.1527. [DOI] [PubMed] [Google Scholar]
- 26.Vomund S, Schafer A, Parnham MJ, Brune B, von Knethen A. Nrf2, the master regulator of anti-oxidative responses. Int J Mol Sci. 2017;18:12. doi: 10.3390/ijms18122772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bellezza I, Giambanco I, Minelli A, Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res. 2018;1865:721–733. doi: 10.1016/j.bbamcr.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 28.Fei B, Liu Z, Xie L, Lv L, Zhu W, Liu J, Dai Y, She W. Panax notoginseng saponins protect auditory cells against cisplatininduced ototoxicity by inducing the AKT/Nrf2 signalingmediated redox pathway. Mol Med Rep. 2020;22:3533–3540. doi: 10.3892/mmr.2020.11390. [DOI] [PubMed] [Google Scholar]
- 29.Ding X, Jian T, Li J, Lv H, Tong B, Li J, Meng X, Ren B, Chen J. Chicoric acid ameliorates nonalcoholic fatty liver disease via the AMPK/Nrf2/NFkappaB signaling pathway and restores gut microbiota in high-fat-diet-fed mice. Oxid Med Cell Longev. 2020;2020:9734560. doi: 10.1155/2020/9734560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen QM, Maltagliati AJ. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol Genomics. 2018;50:77–97. doi: 10.1152/physiolgenomics.00041.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi: 10.1016/j.redox.2019.101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu R, Jiang H. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging (Albany NY) 2020;12:12943–12959. doi: 10.18632/aging.103378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang HJ, Chen RC, Sun GB, Yang LP, Zhu YD, Xu XD, Sun XB. Protective effects of total flavonoids from clinopodium chinense (benth. ) O. Ktze on myocardial injury in vivo and in vitro via regulation of Akt/Nrf2/HO-1 pathway. Phytomedicine. 2018;40:88–97. doi: 10.1016/j.phymed.2018.01.004. [DOI] [PubMed] [Google Scholar]
- 35.Peng Z, Zhang R, Pan L, Pei H, Niu Z, Wang H, Lv J, Dang X. Glaucocalyxin a protects H9c2 cells against hypoxia/reoxygenation-induced injury through the activation of Akt/Nrf2/HO-1 pathway. Cell Transplant. 2020;29:963689720967672. doi: 10.1177/0963689720967672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y, Che J, Zhao H, Tang J, Shi G. Platycodin D inhibits oxidative stress and apoptosis in H9c2 cardiomyocytes following hypoxia/reoxygenation injury. Biochem Biophys Res Commun. 2018;503:3219–3224. doi: 10.1016/j.bbrc.2018.08.129. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.