

Abstract
The gut microbiota can affect human metabolism, immunity, and other biologic pathways through the complex gut-kidney axis (GKA), and in turn participate in the occurrence and development of kidney disease. In this study, 39 patients with stage 4-5 chronic kidney disease (CKD) and 40 healthy individuals were recruited and 16S rDNA sequencing was performed to analyze the V3-V4 conserved regions of their microbiota. A total of 795 operational taxonomic units (OTUs) shared between groups or specific to each group were obtained, among which 255 OTUs with significant differences between the two groups were identified (P<0.05). Adonis differential analysis showed that the diversity of gut microbiota was highly correlated with CKD stages 4-5. Additionally, 61 genera with differences in the two groups were identified (P<0.05) and 111 species with significant differences in the phyla, classes, orders, families, and genera between the two groups were identified (P<0.05). The differential bacterial genera with the greatest contribution were, in descending order: c_Bacteroidia, o_Bacteroidales, p_Bacteroidetes, c_Clostridia, o_Clostridiales, etc. Those with the greatest contribution in stages 4-5 CKD were, in descending order: p_Proteobacteria, f_Enterobacteriaceae, o_Enterobacteriales, c_Gammaproteobacteria, c_Bacilli, etc. The results suggest that the diversity of the microbiota may affect the occurrence, development, and outcome of the terminal stages of CKD.
Keywords: Chronic kidney disease stages 4-5, 16S rDNA sequencing, gut microbiota
Introduction
Lederberg et al. [1] first proposed the concept of the human microbiome, which is a generic term for the symbiotic, commensal, and pathogenic microbes in the human body. The broad category of human genes includes a combination of the human genome and the microbiome; therefore, the metabolic function of humans has characteristics of both humans as well as microbes. As a large number of gut microbiota encode 150 times as many genesas the total genes in human cells, the gut microbiota is called “the second genome of human body” [2] and has drawn widespread attention. In many cases, microbes are presumed to be the cause of variation and can affect the host phenotype [3]. The gut microbiota, as an “endogenous organ” of the human body, play an important role in health and disease development [4]. The gut microbiota can affect metabolism, immunity, and other biologic pathways through the complex gut-kidney axis (GKA) [5], a mechanism in a new biological concept, and participate in the occurrence and development of kidney disease. Only when the human genome and microbiome are unified can the stability of the human ecosystem be guaranteed.
Recent studies have shown that the gut microbiota, including its gene expression products, play an indispensable role in multiple physiologic functions, such as regulating host bowel movement and immune system response and development, maintaining the balance of the host gut microecosystem, and affecting the digestion and absorption of nutrients. Atherosclerosis, hypertension, type 2 diabetes, inflammatory bowel disease, obesity, cardiovascular disease, chronic kidney disease (CKD), and other diseases have been confirmed to be related to structural disorders of the microbiota [6]. The increasing prevalence of CKD, its high treatment cost, as well as its numerous and severe complications [7] make it necessary to identify the endogenous and exogenous risk factors of CKD to better delay its progression. In this study fecal samples from patients with CKD and healthy individuals (control) were collected; high-throughput 16S rDNA sequencing analysis of the V3-V4 regions of the gut microbiota was conducted; and the species richness, composition, systematic evolution, and other information regarding the microbiota between the groups were analyzed and compared. The main structural differences in the gut microbiota between the CKD and control groups were further explored to determine the gut microbiota primarily responsible for the occurrence of CKD.
Materials and methods
Inclusion and exclusion criteria
Inclusion criteria: (1) patients met the diagnostic criteria for CKD in Kidney Disease: Improving Global Outcomes (KDIGO), 2012 [8] and (2) patients were diagnosed with stage 4-5 CKD (eGFR <29 ml/min/1.73 m2) and had not received dialysis therapy. CKD is defined as a kidney injury (renal dysfunction or structural abnormality) with various causes, with or without a decrease in glomerular filtration rate (GFR <60 ml/min/1.73 m2), with clinical manifestations such as abnormal kidney pathology or kidney injury, abnormal blood and urine composition, or abnormal findings in imaging examinations for at least 3 months.
Exclusion criteria: (1) patient also had constipation, diarrhea, or other intestinal diseases; (2) patient had received agents containing cathartic ingredients, such as antibiotics, urinalysis, and laxative drugs, within 4 weeks prior to sampling; (3) patient had a family history of kidney disease or other infectious diseases such as viral hepatitis or tuberculosis; (4) patient had smoking and drinking habits or could not communicate normally and work with others; (5) patient had tumors, acute and chronic infections, or active complications of autoimmune diseases; (6) the patient had a history of blood transfusion within the last 3 months, or had taken adrenocorticosteroids and immunosuppressants within 2 weeks prior to sampling.
The inclusion criteria for the control group: (1) participants with eGFR >90 ml/min/1.73 m2; (2) no abnormality by routine urine and kidney imaging examinations; (3) no history of chronic diseases that would cause damage to the kidneys, such as hypertension or diabetes; (4) no history of kidney transplantation; (5) no active inflammation; and (6) had not received any antimicrobial agents or immunosuppressants within 3 months, and no obvious infections such as viral, bacterial, or fungal infections. Informed consent was obtained from all subjects, and the study was approved by the medical ethics committee of Fujian Provincial Hospital (K2018-01-025).
General information
A total of 39 patients with stage 4-5 CKD who were admitted to the Fujian Provincial Hospital from April 2018 to September 2019 and who met the screening criteria were enrolled; the group included 19 men and 20 women, with an average age of 56.52±15.72 years old. Forty healthy individuals from the physical examination center of Fujian Provincial Hospital were recruited as the control group; this group included 18 males and 22 females, with a mean age of 56.35±10.96 years old. The age and gender were comparable between the two groups (all P>0.10). Routine blood and biochemical index data were gathered, and blood trimethylamine n-oxide (TMAO, MEIMIAN, mm-2294h1) levels were detected by ELISA (Table 1).
Table 1.
Comparison of clinical biochemical data between the stage 4-5 CKD group and the control group
| Item | Stage 4-5 CKD group (n = 39) | Control group (n = 40) | Statistic (T value or Z value or χ2 value) | P value |
|---|---|---|---|---|
| Sex (male/female) | 19 (51.3%)/20 (48.7%) | 18 (45.0%)/22 (55.0%) | 0.110* | 0.074 |
| Age (years) | 56.52±15.72 | 56.35±10.96 | -0.054# | 0.957 |
| BMI (kg/m2) | 21.21 (13.46~6.66) | 23.50 (21.51~25.63) | 1.504 | 0.022 |
| WBC (×109/L) | 6.63±2.14 | 6.18±1.77 | -1.009# | 0.316 |
| NE (%) | 73.40 (63.30~76.90) | 56.35 (49.23~61.83) | 2.857 | 0.000 |
| Hb (g/L) | 78.00 (68.00~96.00) | 139.00 (132.00~148.00) | 4.105 | 0.000 |
| ALB (g/L) | 31.05±5.85 | 44.73±3.44 | 12.695# | 0.000 |
| PLT (×109/L) | 203.00 (154.00~248.00) | 228.50 (203.50~271.50) | 1.609 | 0.011 |
| ALT (U/L) | 12.00 (6.00~15.00) | 18.00 (11.00~23.00) | 1.652 | 0.009 |
| AST (U/L) | 14.00 (11.00~19.00) | 18.0000 (15.00~20.75) | 1.729 | 0.005 |
| TG (mmol/l) | 1.05 (0.81~2.14) | 1.51 (1.17~1.85) | 1.849 | 0.002 |
| CHOL (mmol/l) | 3.76 (3.26~4.82) | 5.08 (4.35~5.53) | 2.282 | 0.000 |
| LDL-C (mmol/L) | 2.55 (1.96~3.23) | 3.69 (3.06~4.13) | 2.307 | 0.000 |
| HDL-C (mmol/L) | 1.05±0.34 | 1.23±0.30 | 2.473# | 0.016 |
| FPG (mmol/l) | 4.78 (3.94~5.60) | 5.00 (4.75~5.49) | 1.256 | 0.085 |
| URIC (umol/l) | 425.00 (315.00~538.00) | 350.50 (292.00~404.50) | 1.615 | 0.011 |
| eGFR (ml/min/1.73 m2) | 5.35 (4.37~7.37) | 94.10 (80.21~104.40) | 4.444 | 0.000 |
| TMAO (mg/l) | 4.29 (2.31~8.87) | 2.48 (3.08~5.53) | 1.401 | 0.039 |
= χ2 value;
= T value;
BMI (kg/m2) = body mass index; WBC = white blood cell; NE = neutrophil; Hb = hemoglobin; ALB = albumin; PLT = platelet count; ALT = alanine aminotransferase; AST = aspartate aminotransferase; TG = triglyceride; CHOL = cholesterol; LDL-C = low-density lipoprotein cholesterol; HDL-C = high-density lipoprotein cholesterol; FPG = fasting plasma glucose; eGFR = epidermal growth factor receptor; TMAO = trimethylamine N-oxide. The CKD-EPI equation was calculated using the formula eGFR (ml/min/1.73 m2) (Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF 3rd, Feldman HI, Kusek JW, Eggers P, Van Lente F, Greene T and Coresh J; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration). A new equation to estimate glomerular filtration rate. Ann Intern Med 2009; 150: 604-612).
Fecal sample collection and DNA extraction
Patients were prohibited from drinking and taking drugs one day before the examination and were asked to fast for eight hours before the examination. The samples were prepared according to the manual of procedures for the human microbiome project (Version 12.0, Accession: phd003190.2). The samples were then stored at -80°C immediately after packing. DNA extraction was performed in strict accordance with the specifications of the MoBio PowerSoil DNA Isolation Kit. The universal primers 27F/1492R for the 16S rRNA gene (27F: 5’-AGAGTTTGATCMTGGCTCAG-3’; 1492R: 5’-TACGGYTACCTTGTTACGACTT-3’) were used for the PCR amplification of the extracted DNA samples, and the quality control of PCR products was performed using a Thermo NanoDrop 2000 and agarose gel electrophoresis. The 27F/1492R primers were synthesized by Lifetech/Thermo.
16S rRNA gene sequencing analysis of its V3-V4 regions
After PCR amplification, purification, and library quality screening, the 16S rDNA samples were quantified using Qubit and the samples were mixed for normalization in corresponding proportions according to the data volume requirement of each sample. The specific variable regions V3-V4 were amplified using specific primers for the 16S rDNA to obtain an amplified fragment of approximately 425 bp. MiSeq PE300 was used for sequencing after adaptor linkage. Paired reads obtained by double-ended sequencing were spliced into a sequence using PANDAseq (V2.9) software using overlapping relationships, and long reads within the hypervariable region were obtained. The spliced reads were then processed using an internal writing program, as follows: (1) reads with an average quality value <20 were removed; (2) reads with over 3 bases containing N were removed; and (3) the length of reads ranged from 250 to 500 nt. Clean reads were obtained, and the length, distribution, and quantity were analyzed [9]. After splicing, further 16S rDNA analysis was performed using the following primer sequences: U341, CCTACGGGRSGCAGCAG; U806, GGACTACVVGGGTATCTAATC [10,11]. Sequencing was performed by Shanghai Ruiyi Biotechnology Co., Ltd.
Operational taxonomic unit (OTU) clustering analysis
To facilitate diversity analysis of the downstream species, OTUs were introduced for reads clustering. Clean reads with the same sequences were classified as a type of tag. The richness (number of reads) of each tag was counted, and the tags were ranked according to their richness. Singletons (sequences with only one corresponding reads) that might have been caused by sequencing errors were removed and were not used in the OTU clustering analysis. Usearch software (V7.0.1090) was used for sequence clustering, setting a similarity cutoff of 0.97. After chimera filtering, the final OTUs for the species classification were obtained. All clean reads were then aligned to the OTU sequences, and the reads organized into OTUs were extracted to obtain the final mapped reads [12]. The Ribosomal Database Project (RDP, http://rdp.cme.msu.edu) was used to align the representative sequences and 16S rDNA of the known species. An OTU richness table was obtained based on the sequence number of each OTU after classification according to the species and annotations [13,14].
Complexity analysis of samples and differential analysis between groups
Alpha diversity (such as Observed species index, Chao1 index, PD whole tree index, Shannon index, Simpson index and Goods coverage index) and beta diversity (such as Unifrac, MRPP analysis, PCoA analysis and Adonis analysis) were analyzed using QIIME (V1.9.1) [15,16], and the iterative algorithm was used for variance calculation based on the weighted species classification richness information and unweighted species classification richness information, respectively. A distance box plot between groups was drawn. The Wilcoxon test function in the R language (V3.5.1) stats package was used for differential analysis between two groups, and the Kruskal-Wallis test function in the R language was used for differential analysis between more than two groups. Linear discriminant analysis (LDA) effect size (LEfSe) analysis was performed using LEfSe Tools (V1.0). Principal coordinates analysis (PCoA) was performed using the dudi.pco function of the ade4 package in R (V3.5.1). An Adonis analysis was performed using the Adonis function of the vegan package in R (V3.5.1). The corrplot package of R language (V3.5.1) was used to draw a Spearman correlation heatmap among the different species. The 16S rDNA functions were predicted using the PICRUSt (V1.0.0) software.
Results
OTU analysis
The richness of the OTUs could preliminarily indicate the species richness of the samples. The rank-sum test indicated that the number of final OTUs in the stage 4-5 CKD group was lower than that in the control group, demonstrating that the samples in the stage 4-5 CKD group had fewer unique species than those in the control group (Tables 2 and S1).
Table 2.
Differential analysis of clean reads, mapped reads, and final OTUs (median, M) between the stage 4-5 CKD group and the control group
| Item | Stage 4-5 CKD group | Control group | Z value | Significance |
|---|---|---|---|---|
| Clean Reads | 36260.0 | 35204.0 | 1.396 | 0.041 |
| Mapped Reads | 32124.0 | 27809.5 | 2.760 | <0.001 |
| Final OTUs | 120.0 | 187.5 | 2.398 | <0.001 |
Species richness analysis
According to the results of the species annotation, histograms showing the species profile of each sample were plotted with respect to the phyla, classes, orders, families, and genera. The relative richness histograms of the species could be used to intuitively deduce the species with a relatively higher richness and the corresponding proportion in each sample at different classification levels, as shown in Figure 1.
Figure 1.

Profiling histograms of the species at the level of the phylum (A), class (B), order (C), family (D), and genus (E) in the different groups. The horizontal axis is the grouping (A is the stage 4-5 CKD group, B is the healthy control group) and the vertical axis is the proportion of relative richness. The colors correspond to the identity of the different species and the lengths of the color blocks indicate the relative richness proportions of these species.
Alpha diversity analysis
Alpha diversity reflects the species diversity of a single sample, including the observed species index, Chao1 index, and the PD whole tree index [15]. The observed species and Chao1 indices reflected the species richness, wherein more species were not detected if the species diversity was higher in each sample. The Shannon and Simpson indices reflect the species diversity in the community. The PD whole tree index reflects the difference in the preservation of the evolutionary history of the species found in the samples. A larger PD whole tree index indicates a greater difference in the preservation of evolutionary history. The Goods coverage index is an intuitive representation of the sample sequencing depth. Values closer to one mean that the sequencing data covered most of the species in the samples. The Chao1, Observe, PD whole tree, Shannon, and Simpson indices of the fecal colonies in the stage 4-5 CKD group were lower than those of the control group (all P<0.05), indicating that the control group had higher species diversity (Tables 3 and S2).
Table 3.
Alpha diversity analysis of samples (median, M) between stage 4-5 CKD group and the control group
| Item | Stage 4-5 CKD group | Control group | Z value | Significance |
|---|---|---|---|---|
| Chao1 | 132.8000 | 209.5417 | 2.060 | <0.001 |
| Observe | 99.0000 | 173.0000 | 2.182 | <0.001 |
| PD whole tree | 9.2921 | 12.5580 | 2.173 | <0.001 |
| Shannon | 3.5848 | 4.3331 | 1.966 | 0.001 |
| Simpson | 0.8254 | 0.8934 | 1.612 | 0.011 |
| Goods coverage | 0.9986 | 0.9979 | 1.857 | 0.002 |
Beta diversity analysis
Beta diversity analysis is an analysis of species diversity between pairs of samples [17]. PCA analysis and Adonis analysis are two methods that can analyze the overall differences among different groups. The Adonis analyses (n≥5 per group) performed in this study indicated significant differences in microbiota structure between groups (P<0.05), as shown in Table 4 and Figure 2. Through Adonis analyses, the total variance can be decomposed into a semi-metric (such as Unifrac) or a metric distance matrix (such as Euclidean). The dilution of sample differences caused by different grouping or environmental factors was analyzed using a linear model, and a significance analysis was conducted using a permutation test. Therefore, it could be inferred that the diversity of intestinal microbes was highly correlated with the incidence of stages 4-5 CKD.
Table 4.
Differential analysis of the species multi-response permutation procedure (MRPP) between the groups
| Group | Statistic | Observe Delta | Expect Delta | Significance |
|---|---|---|---|---|
| Weighted unifrac | 0.076952 | 0.370465 | 0.401349 | 0.001 |
| Unweighted unifrac | 0.044941 | 0.544606 | 0.570233 | 0.001 |
Note: Observe Delta: Observe Delta value corresponding to the Unifrac distance index; Expect Delta: Expect Delta value corresponding to the Unifrac distance index. An A>0 indicates that the difference between groups was more significant than that within groups, while an A<0 indicated the opposite result. A smaller observed delta value indicates that the difference between the groups was smaller, and a smaller expected delta means that the difference within groups was smaller.
Figure 2.

Adonis differential analysis shows the distribution of samples between the groups (n≥5 per group). The horizontal and vertical coordinates represent the first and second principal coordinates, respectively, and the percentage represents the contribution of the corresponding principal coordinates to the sample differences. The points in the figures represent each sample. (A) is the stage 4-5 CKD group and (B) is the control group. The value distributions of the different groups on the first and second principal coordinates are exhibited in the horizontal and vertical box plots, respectively. (A) The Unweighted UniFrac and (B) Weighted UniFrac.
Significant difference analysis
LEfSe analysis was used to estimate the effect of the richness of each component (species) on the differences and to determine the community or species that had significant impact on sample classification. LEfSe analysis emphasizes statistical significance and biologic correlations [18]. The clustering tree diagram showing significant species differences is displayed in Figure 3. The microbial groups that significantly contributed to the species differences were analyzed by linear regression analysis, and the LDA scores are shown in Figure 4. Compared with the stage 4-5 CKD group, the bacterial genera with the greatest contribution to the species differences in the control group were, in descending order: c_Bacteroidia, o_Bacteroidales, p_Bacteroidetes, c_Clostridia, o_Clostridiales, f_Ruminococcaceae, f_Prevotellaceae, g_Prevotella, g_Faecalibacterium, g_Roseburia, etc. Compared with the control group, the bacterial genera with the greatest contribution to the stage 4-5 CKD group were as follows, in descending order: p_Proteobacteria, f_Enterobacteriaceae, o_Enterobacteriales, c_Gammaproteobacteria, c_Bacilli, o_Lactobacillales, g_Escherichia_Shigella, g_Enterococcus, f_Enterococcaceae, and f_Lactobacillaceae.
Figure 3.

In the microbiota clustering tree diagrams, different colors represent different groups (A is the stage 4-5 CKD group, B is the control group). The colored nodes indicate the microbiota that play an important role in each group. The yellow nodes represent the microbiota that do not play an important role in each group. The names of the species are listed in the legend on the right.
Figure 4.
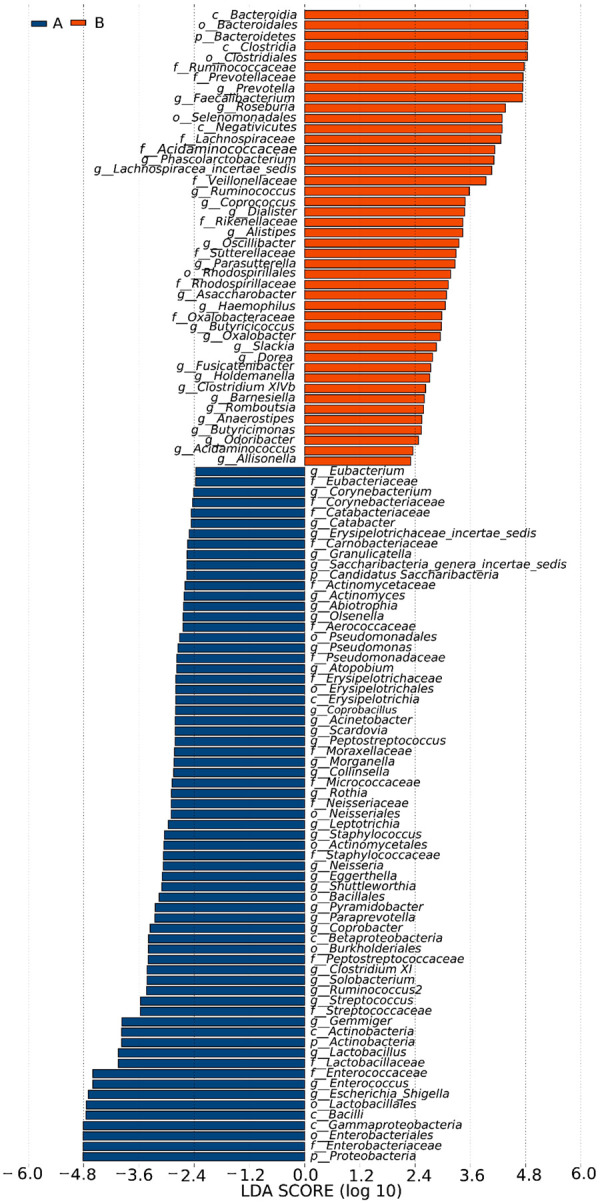
LEfSe analysis was used to statistically resolve the microbiota with significant effects in the different groups. The LDA threshold was set as 2. (A) The stage 4-5 CKD group and (B) the control group.
Differential analysis of the OTUs between groups
In the stage 4-5 CKD group and the control group, a total of 795 OTUs shared between groups or specific to each group were obtained by the rank-sum test, among which 255 OTUs with significant differences between the two groups were identified (P<0.05) (Table S3). Heatmaps, box plots, and PCA statistical graphs were constructed to show the differential OTUs, as shown in Figure 5.
Figure 5.

The heatmap, box plot, and PCA graphs of differential OTUs in the microbiota (A is the stage 4-5 CKD group and B is the control group). (A) Horizontal clustering indicates similar richness of the species in each sample. A closer distance and a shorter branch suggest that the composition of the two species is more similar between samples. Vertical clustering represents the expression similarity of all species in the different samples. A closer distance and a shorter branch length indicate that the composition and richness of species between samples are more similar. (B) The horizontal coordinate indicates the names of the OTUs and the vertical coordinate shows the log2 value of the relative richness. (C) The horizontal coordinate represents the first principal component, and % represents its contribution to the sample differences. The vertical coordinate represents the second principal component, and % represents its contribution to the sample differences. Every point in the graph represents a sample.
Differential analysis of the genera between groups
In this experiment, 61 genera with differences between the stage 4-5 CKD group and the control group were found (P<0.05), as shown in Table S4. Heatmaps, box plots, and PCA statistical graphs were plotted to show the differential genera (Figure 6). A total of 111 species with obvious differences between groups were found at the phylum, class, order, family, and genus levels (P<0.05), as shown in Table S5. Heatmaps, box plots, and PCA statistical graphs were constructed to show the species differences at various levels (Figure 7).
Figure 6.

The heatmap, box plot, and PCA graphs of the differential species at the genus level in the microbiota (A is the stage 4-5 CKD group and B is the control group). (A) Horizontal clustering indicates similar richness of the species in each sample. A closer distance and a shorter branch suggest that the composition of the two species was more similar between samples. Vertical clustering represents the expression similarity of all species in different samples. A closer distance and a shorter branch length indicate that the composition and richness of species between samples are more similar. (B) The horizontal coordinate indicates the names of the genera and the vertical coordinate shows the log2 value of the relative richness. (C) The horizontal coordinate represents the first principal component and % represents its contribution to the sample differences. The vertical coordinate represents the second principal component and % represents its contribution to the sample differences. Every point in the graph represents a sample.
Figure 7.

The heatmap, box plot, and PCA graphs of differential species at all levels in the microbiota (A is the stage 4-5 CKD group and B is the control group). (A) Horizontal clustering indicates similar richness of the species in each sample. A closer distance and a shorter branch suggest that the composition of the two species is more similar between samples. Vertical clustering represents the expression similarity of all species among the different samples. A closer distance and a shorter branch length indicate that the composition and richness of species between samples are more similar. (B) The horizontal coordinate shows the names of the species and the vertical coordinate shows the log2 value of the relative richness. (C) The horizontal coordinate represents the first principal component and % represents its contribution to the sample differences. The vertical coordinate represents the second principal component and % represents its contribution to the sample differences. Every point in the graph represents a sample.
Spearman correlation coefficient analysis of the dominant species
The top 30 significantly different species with the largest contribution to species richness were selected at all levels of flora through LEfSe analysis, and a Spearman’s heatmap was drawn using R software, which represented the interaction pattern between dominant species, such as the symbiosis and antagonistic relationships of each flora. The important patterns and relationships between the dominant species can be intuitively observed in Figure 8A.
Figure 8.

A. Shows the correlation coefficients of the top 30 species according to differential richness. The blue on the right represents a positive correlation and the red shows a negative correlation. A darker shade indicates a stronger correlation between species. The left prefix “k_”, “p_”, “c_”, “o_”, “f_” before the species shows that they are respectively annotated to the kingdoms, phyla, classes, orders, and families. B. The horizontal coordinate is the log value obtained through the LDA of the KEGG pathway that has a significant effect on the different groups, which represents the enrichment degree of the KEGG pathway in the different groups.
LEfSe analysis of the KEGG pathways
The LEfSe analysis method was used to estimate the effect of the metabolic pathways (KEGG pathway) of each component on the species differences and to identify the metabolic pathways that had significant differences in the sample classification (the default screening condition was set as LDA >2). The results suggested that the relevant metabolic and other functions changed along with alterations in the relevant flora structures (Figure 8B).
Discussion
The human gut microbiota form an intestinal microecosystem and their steady-state structure failure can cause clinical symptoms [19]. The homeostasis of the intestinal microecosystem is related to the richness of the flora present in the tissues. For example, in obese individuals, the ratio of intestinal Firmicutes/Bacteroides was increased, while the richness of polymorphic Bacteroidetes decreased significantly [20]. Additionally, dietary patterns can also lead to changes in the steady-state structure of the intestinal microecosystem, such as changes in the composition of the gut microbiota induced by intermittent fasting [21]. Drug abuse, such as antibiotic abuse, may also cause the translocation of gut microbiota, resulting in colony instability and inflammation [22]. An imbalance in the human gut microbiota caused by multiple risk factors can further affect kidney function [23].
The mechanism by which the gastrointestinal tract and kidney interact with each other through the microbiota remains unclear. The microbiota mainly play a bridging role in the interaction between the gut and the kidney through the GKA mechanism, which is a two-way information exchange system that involves structural changes in the gut microbiota, intestinal barrier, inflammatory response, immune response, material and energy absorption, and the presence of certain metabolites [23-25]. The gut microbiota are a core component of the GKA. A chronically unstable intestinal microecology is prone to cause an imbalance in the gut microbiota, leading to a significant decrease in the number of beneficial bacteria such as Bifidobacteria and Lactobacillus and an increase in the number of pathogenic bacteria such as enterobacteria and Enterococcus [26]. However, intestinal barrier dysfunction may permit intestinal bacteria and endotoxins such as lipopolysaccharides to continuously enter the blood circulation through the intestinal tract in trace amounts and form a subclinical level of endotoxemia, which activates the mononuclear-macrophage system, releases a large number of inflammatory cytokines, and induces a systemic inflammatory response [27]. The systemic inflammatory response is an independent risk factor for the deterioration brought about by CKD and is also a risk factor for cardiovascular disease (CVD) complications caused by CKD. Therefore, the occurrence and development of CKD can be delayed by improving the systemic inflammatory response [28]. Administration of probiotics can slow the decline in kidney function in patients with non-dialysis CKD [29,30].
The gut microbiota are also one of the sources of uremic toxins. Due to an increase in the number of pathogenic bacteria that can produce uremic toxins in CKD, a large number of enterogenous uremic toxins, such as para-cresol sulfate (PCS) and indoxyl sulfate (IS), accumulate in the blood, while levels of short-chain fatty acids (SCFAs), which are beneficial to the body and can regulate blood pressure under normal conditions to protect kidney cells, show a decreasing trend [31]. Anaerobic bacteria in the gut microbiota ferment tyrosine and tryptophan in food to produce para-cresol and indoles, which are then metabolized in the liver to produce PCS and IS [32]. The injection of PCS into uninephrectomized B-6 mice resulted in increased tubulointerstitial oxidative stress levels, inducing the expression of inflammatory factors, damaging renal tubular cells, aggravating renal fibrosis [33], inducing CpG island methylation of the Klotho gene, and reducing Klotho expression in renal tubular cells. In rats that underwent 5/6 nephrectomy, oral administration of IS significantly increased their oxidative stress levels, showing more obvious glomerulosclerosis, and leading to the progression of CKD [25]. The accumulation of both IS and PCS can lead to the occurrence of renal osteopathy [34]. IS and PCS are mainly secreted and excreted by organic anion transporters on the basal membrane side of renal tubules, and they are not easily cleared through dialysis. IS and PCS are closely related to CKD progression and the occurrence of fatal CVD [35]. IS and PCS may be involved in the pathogenesis of CVD in a variety of ways, such as through stimulation of inflammation and the oxidative stress response, stimulating the heart, damaging vascular endothelial cells, increasing platelet activity, and inducing the production of atherosclerotic lesions [36]. Intestinal adsorbents such as AST-120 can remove enterotoxins and delay the deterioration of kidney function in uremic rats [37].
Trimethylamine n-oxide (TMAO) is also a uremic toxin. Choline and carnitine, which are found in certain foods such as red meat and shellfish, are the main raw materials for the production of TMAO. Under the action of trimethylamine lyase produced by the gut microbiota, this kind of food is metabolized as trimethylamine, and TMAO is generated through the catalysis of liver flavin-containing monooxygenase 3 and is ultimately cleared through the kidney. Long-term administration of TMAO or choline in a mouse model has been shown to lead to renal fibrosis and elevated levels of tubular injury markers [38]. Although TMAO is a small molecule that can be effectively cleared by dialysis, its harmful effects cannot be ignored. Current studies show that the elevated levels of TMAO in the blood of patients with CKD is related to the formation of foam cells, atherosclerosis, and the occurrence of CVD [39] and that TMAO is also an independent predictor of coronary atherosclerosis [40]. CKD patients with high levels of TMAO in their blood have poor prognosis, a low probability of long-term survival, and a high risk for CVD complications [41].
Mounting evidence suggests that immune response is an important pathophysiological mechanism of the GKA [32,42]. Bifidobacteria in the gut microbiota can promote the expression of the tight junction proteins ZO1 and occludin in intestinal epithelial cells, maintaining the integrity of the intestinal mucosal barrier. In addition, segmented filamentous bacteria such as Bacteroides and Clostridium difficile can regulate the proliferation and differentiation of intestinal immune-regulatory T cells, auxiliary T lymphocytes, and intestinal mucosa-secreted IgA, resulting in a strong innate and adaptive immune response of the intestinal tract [43,44]. Kidney function is closely related to the immune system; for instance, the existence of different forms of inflammation and infection, the overstimulation of the immune system, and the dominant Thl immune response leads to an increased possibility of proliferative and crescentic glomerulonephritis [45]. An imbalance in the ratio of gut microbiota in mice fed a high-salt diet for a long term was accompanied by an impaired intestinal immune gene expression, kidney injury, as well as CVD [46]. Long-term dysbacteriosis aggravates microinflammation in patients with CKD as the microbiota and its metabolites can participate in the immune inflammatory response in CKD, causing the activation of the systemic immune system [47]. The levels of C-reactive protein in patients with CKD were decreased but their immune-related indices increased after supplementation with probiotics (such as Bifidobacterium and Bactrium) [48]. Patients with CKD often have weakened adaptive immunity, making them susceptible to infection, increasing the risk and incidence of complications such as CVD. However, fecal microbiota transplantation can improve this situation; for example, patients with CKD are prone to develop Clostridium difficile-associated diarrhea but fecal microbiota transplantation with beneficial bacteria can reduce the incidence of enteritis and diarrhea in these patients. It was suggested that the connection between dysbacteriosis and immune inflammation in the gut microbiota accelerates the progression of CKD [49].
The results of this study suggest that the richness of Bacteroidia, Bacteroidales, and Bacteroidetes in the intestinal tract of healthy individuals was higher than those in patients with CKD and that Bacteroidetes played a dominant role in the intestinal microbiota of healthy adults [50]. Bacteroidia, Bacteroidales, and Bacteroidetes in the natural orifices can produce butyric acid, propionic acid, acetic acid, and other SCFAs by fermentation, and SCFAs can inhibit inflammation and reduce kidney injury [51]. Clostridia are gram-positive bacteria that can limit the colonization of pathogenic Enterobacteriaceae in the intestinal tract of newborns [52] and also reduce the incidence of urinary tract infections. Roseburia is a gram-positive anaerobic bacterium that can prevent atherosclerosis and alleviate the symptoms of CKD and CVD [53]. The dominant genus in the control group was Prevotella while Bacteroides was the normal microbiota in the oral, gut, and female genital tracts [54]. Prevotella is associated with carbohydrate metabolism and low path ogenicity and its richness can also be associated with gastrointestinal symptoms.
In the CKD group, 67 differential genera were identified; therefore, we believe that the initiation of CKD was related to a disorder in the gut microbiota. The richness of Proteobacteria, Enterobacteriaceae, Gammaproteobacteria, and Bacilli in patients with CKD stage 4-5 was dramatically higher than that of the control individuals, suggesting that CKD had a characteristic microbiota structure. Proteobacteria, Enterobacteriaceae, and Enterobacteriales in patients with CKD may be bound to uremic toxins [26,55]. A study has reported that the increased richness of Gammaproteobacteria was associated with the occurrence of colitis [56]. Moreover, the classes Bacilli and Gammaproteobacteria and order Enterobacteriales have shown adverse effects on a mouse model of conventional adenoma of colon cancer at an early stage [57]. However, the increased proportion of facultative anaerobes such as Proteobacteria is a potential factor for intestinal microdysbiosis and a risk factor for kidney diseases [58].
In the present study, we found that changes in the microecology of gut microbiota can lead to the dysregulation of gut microbiota, changes in specific metabolites, and induction of systemic inflammatory responses, which in turn can cause immune disorders and atherosclerosis, thus accelerating the progression of CKD and the occurrence of CVD complications. With the progression of CKD, the imbalance of the gut microbiota is aggravated, and a vicious circle of positive feedback is created. For example: the abundance of Proteobacteria, Enterobacteriaceae, Gammaproteobacteria and Bacilli in patients with stage 4-5 CKD was significantly higher than that of the control group, suggesting that CKD had a characteristic microbiota structure. Proteobacteria, Enterobacteriaceae, and Enterobacteriales in CKD patients may be associated with the uremic toxins (e.g. para-cresol sulfate, indoxyl sulfate, trimethylamine n-oxide). When the intestinal flora is out of balance, the accumulation of enterogenous uremic toxins such as para-cresol sulfate, indoxyl sulfate, trimethylamine n-oxide lead to the change of intestinal microecological, bacterial translocation, metabolites change, causing systemic inflammatory response, and then leading to immune disorders, and atherosclerosis. This accelerates the progression of CKD and the occurrence of cardiovascular complications. The decline in renal function slows down the clearance of uremia toxins and accumulates in the blood, forming a vicious cycle, which in turn leads to the deterioration of CKD patients and a high incidence of cardiovascular complications. Of course, the mechanism may be more complex than this. We will further explore the mechanism of the responsible gut microbiota in the future. Our team will conduct animal experiments on bacteria associated with CKD4-5 stage in the future, such as studying the effect of fecal bacteria transplantation on mice with chronic kidney disease, exploring the therapeutic effect of fecal bacteria transplantation on chronic renal failure, and screening the inflammatory cytokines and related signal pathways involved in the treatment of chronic renal failure by fecal bacteria transplantation. This study will provide a theoretical basis for further study of the influence mechanism of fecal bacteria transplantation on chronic renal failure and the treatment of chronic renal failure by regulating gut microbiota.
We preliminarily found that gut microbiota diversity was highly correlated with CKD stages 4-5. Hence, in the future, fecal microbiota transplantation may become a promising treatment for patients with CKD to improve symptoms and delay progression.
Acknowledgements
This work was supported by a grant from the National Natural Science Foundation of China (No. 81874379), Fujian Province Medical Innovation Foundation (No. 2019-CXB-3, 2019-CXB-4), and the Fujian Province Joint Funds for the Innovation of Science and Technology (2018Y9011), China.
Disclosure of conflict of interest
None.
Table S1
Table S2
Table S3
Table S4
Table S5
References
- 1.Lederberg J. Infectious history. Science. 2000;288:287–293. doi: 10.1126/science.288.5464.287. [DOI] [PubMed] [Google Scholar]
- 2.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, Arumugam M, Kultima JR, Prifti E, Nielsen T, Juncker AS, Manichanh C, Chen B, Zhang W, Levenez F, Wang J, Xu X, Xiao L, Liang S, Zhang D, Zhang Z, Chen W, Zhao H, Al-Aama JY, Edris S, Yang H, Wang J, Hansen T, Nielsen HB, Brunak S, Kristiansen K, Guarner F, Pedersen O, Doré J, Ehrlich SD MetaHIT Consortium; Bork P, Wang J MetaHIT Consortium. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 3.Brinker P, Fontaine MC, Beukeboom LW, Falcao Salles J. Host, symbionts, and the microbiome: the missing tripartite interaction. Trends Microbiol. 2019;27:480–488. doi: 10.1016/j.tim.2019.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Byndloss MX, Baumler AJ. The germ-organ theory of non-communicable diseases. Nat Rev Microbiol. 2018;16:103–110. doi: 10.1038/nrmicro.2017.158. [DOI] [PubMed] [Google Scholar]
- 5.Ticinesi A, Milani C, Guerra A, Allegri F, Lauretani F, Nouvenne A, Mancabelli L, Lugli GA, Turroni F, Duranti S, Mangifesta M, Viappiani A, Ferrario C, Dodi R, Dall’Asta M, Del Rio D, Ventura M, Meschi T. Understanding the gut-kidney axis in nephrolithiasis: an analysis of the gut microbiota composition and functionality of stone formers. Gut. 2018;67:2097–2106. doi: 10.1136/gutjnl-2017-315734. [DOI] [PubMed] [Google Scholar]
- 6.Gentile CL, Weir TL. The gut microbiota at the intersection of diet and human health. Science. 2018;362:776–780. doi: 10.1126/science.aau5812. [DOI] [PubMed] [Google Scholar]
- 7.Buyadaa O, Magliano DJ, Salim A, Koye DN, Shaw JE. Risk of rapid kidney function decline, all-cause mortality, and major cardiovascular events in nonalbuminuric chronic kidney disease in type 2 diabetes. Diabetes Care. 2020;43:122–129. doi: 10.2337/dc19-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kidney Disease: Improving Global Outcomes (KDIGO) Hepatitis C Work Group. KDIGO 2018 clinical practice guideline for the prevention, diagnosis, evaluation, and treatment of hepatitis C in chronic kidney disease. Kidney Int Suppl (2011) 2018;8:91–165. doi: 10.1016/j.kisu.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. doi: 10.1186/1471-2105-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Qian PY. Conservative fragments in bacterial 16S rRNA genes and primer design for 16S ribosomal DNA amplicons in metagenomic studies. PLoS One. 2009;4:e7401. doi: 10.1371/journal.pone.0007401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zakrzewski M, Goesmann A, Jaenicke S, Junemann S, Eikmeyer F, Szczepanowski R, Al-Soud WA, Sorensen S, Puhler A, Schluter A. Profiling of the metabolically active community from a production-scale biogas plant by means of high-throughput metatranscriptome sequencing. J Biotechnol. 2012;158:248–258. doi: 10.1016/j.jbiotec.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 12.Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–998. doi: 10.1038/nmeth.2604. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, Brown CT, Porras-Alfaro A, Kuske CR, Tiedje JM. Ribosomal database project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42:D633–642. doi: 10.1093/nar/gkt1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kemp PF, Aller JY. Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol Ecol. 2004;47:161–177. doi: 10.1016/S0168-6496(03)00257-5. [DOI] [PubMed] [Google Scholar]
- 16.Yu Z, Yang J, Liu L, Zhang W, Amalfitano S. Bacterioplankton community shifts associated with epipelagic and mesopelagic waters in the Southern Ocean. Sci Rep. 2015;5:12897. doi: 10.1038/srep12897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Cao J, Li JR, Yang F, Li Z, Li LX. Comparative analysis of the gastrointestinal microbial communities of bar-headed goose (Anser indicus) in different breeding patterns by high-throughput sequencing. Microbiol Res. 2016;182:59–67. doi: 10.1016/j.micres.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Li L, Butcher J, Stintzi A, Figeys D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome. 2019;7:154. doi: 10.1186/s40168-019-0767-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan RC, Wang Z, Usyk M, Sotres-Alvarez D, Daviglus ML, Schneiderman N, Talavera GA, Gellman MD, Thyagarajan B, Moon JY, Vazquez-Baeza Y, McDonald D, Williams-Nguyen JS, Wu MC, North KE, Shaffer J, Sollecito CC, Qi Q, Isasi CR, Wang T, Knight R, Burk RD. Gut microbiome composition in the hispanic community health study/study of latinos is shaped by geographic relocation, environmental factors, and obesity. Genome Biol. 2019;20:219. doi: 10.1186/s13059-019-1831-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Cabo R, Mattson MP. Effects of intermittent fasting on health, aging, and disease. N Engl J Med. 2019;381:2541–2551. doi: 10.1056/NEJMra1905136. [DOI] [PubMed] [Google Scholar]
- 22.Fritsch J, Abreu MT. Exposing the Achilles heel of antibiotic therapy for pouchitis using microbial function and composition. Gastroenterology. 2020;158:470–472. doi: 10.1053/j.gastro.2019.11.303. [DOI] [PubMed] [Google Scholar]
- 23.Al Khodor S, Shatat IF. Gut microbiome and kidney disease: a bidirectional relationship. Pediatr Nephrol. 2017;32:921–931. doi: 10.1007/s00467-016-3392-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meijers BK, Evenepoel P. The gut-kidney axis: indoxyl sulfate, p-cresyl sulfate and CKD progression. Nephrol Dial Transplant. 2011;26:759–761. doi: 10.1093/ndt/gfq818. [DOI] [PubMed] [Google Scholar]
- 25.Jansen J, Jansen K, Neven E, Poesen R, Othman A, van Mil A, Sluijter J, Sastre Torano J, Zaal EA, Berkers CR, Esser D, Wichers HJ, van Ede K, van Duursen M, Burtey S, Verhaar MC, Meijers B, Masereeuw R. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. Proc Natl Acad Sci U S A. 2019;116:16105–16110. doi: 10.1073/pnas.1821809116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaziri ND, Zhao YY, Pahl MV. Altered intestinal microbial flora and impaired epithelial barrier structure and function in CKD: the nature, mechanisms, consequences and potential treatment. Nephrol Dial Transplant. 2016;31:737–746. doi: 10.1093/ndt/gfv095. [DOI] [PubMed] [Google Scholar]
- 27.Caruso R, Lo BC, Nunez G. Host-microbiota interactions in inflammatory bowel disease. Nat Rev Immunol. 2020;20:411–426. doi: 10.1038/s41577-019-0268-7. [DOI] [PubMed] [Google Scholar]
- 28.Esgalhado M, Kemp JA, Azevedo R, Paiva BR, Stockler-Pinto MB, Dolenga CJ, Borges NA, Nakao LS, Mafra D. Could resistant starch supplementation improve inflammatory and oxidative stress biomarkers and uremic toxins levels in hemodialysis patients? A pilot randomized controlled trial. Food Funct. 2018;9:6508–6516. doi: 10.1039/c8fo01876f. [DOI] [PubMed] [Google Scholar]
- 29.Wang IK, Wu YY, Yang YF, Ting IW, Lin CC, Yen TH, Chen JH, Wang CH, Huang CC, Lin HC. The effect of probiotics on serum levels of cytokine and endotoxin in peritoneal dialysis patients: a randomised, double-blind, placebo-controlled trial. Benef Microbes. 2015;6:423–430. doi: 10.3920/BM2014.0088. [DOI] [PubMed] [Google Scholar]
- 30.Tsai YL, Lin TL, Chang CJ, Wu TR, Lai WF, Lu CC, Lai HC. Probiotics, prebiotics and amelioration of diseases. J Biomed Sci. 2019;26:3. doi: 10.1186/s12929-018-0493-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Treuren W, Dodd D. Microbial contribution to the human metabolome: implications for health and disease. Annu Rev Pathol. 2020;15:345–369. doi: 10.1146/annurev-pathol-020117-043559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meijers B, Evenepoel P, Anders HJ. Intestinal microbiome and fitness in kidney disease. Nat Rev Nephrol. 2019;15:531–545. doi: 10.1038/s41581-019-0172-1. [DOI] [PubMed] [Google Scholar]
- 33.Summers S, Quimby JM, Phillips RK, Stockman J, Isaiah A, Lidbury JA, Steiner JM, Suchodolski J. Preliminary evaluation of fecal fatty acid concentrations in cats with chronic kidney disease and correlation with indoxyl sulfate and p-cresol sulfate. J Vet Intern Med. 2020;34:206–215. doi: 10.1111/jvim.15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto S, Fukagawa M. Uremic toxicity and bone in CKD. J Nephrol. 2017;30:623–627. doi: 10.1007/s40620-017-0406-x. [DOI] [PubMed] [Google Scholar]
- 35.Madan S, Mehra MR. Gut dysbiosis and heart failure: navigating the universe within. Eur J Heart Fail. 2020;22:629–637. doi: 10.1002/ejhf.1792. [DOI] [PubMed] [Google Scholar]
- 36.Yang K, Du C, Wang X, Li F, Xu Y, Wang S, Chen S, Chen F, Shen M, Chen M, Hu M, He T, Su Y, Wang J, Zhao J. Indoxyl sulfate induces platelet hyperactivity and contributes to chronic kidney disease-associated thrombosis in mice. Blood. 2017;129:2667–2679. doi: 10.1182/blood-2016-10-744060. [DOI] [PubMed] [Google Scholar]
- 37.Chen YC, Wu MY, Hu PJ, Chen TT, Shen WC, Chang WC, Wu MS. Effects and safety of an oral adsorbent on chronic kidney disease progression: a systematic review and meta-analysis. J Clin Med. 2019;8:1718. doi: 10.3390/jcm8101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li W, Tan L, Li X, Zhang X, Wu X, Chen H, Hu L, Wang X, Luo X, Wang F, Xu C, Chen Q, Jin R, Wang QK. Identification of a p.Trp403* nonsense variant in PHEX causing X-linked hypophosphatemia by inhibiting p38 MAPK signaling. Hum Mutat. 2019;40:879–885. doi: 10.1002/humu.23743. [DOI] [PubMed] [Google Scholar]
- 39.Chen S, Henderson A, Petriello MC, Romano KA, Gearing M, Miao J, Schell M, Sandoval-Espinola WJ, Tao J, Sha B, Graham M, Crooke R, Kleinridders A, Balskus EP, Rey FE, Morris AJ, Biddinger SB. Trimethylamine N-oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab. 2019;30:1141–1151.e1145. doi: 10.1016/j.cmet.2019.08.021. [DOI] [PubMed] [Google Scholar]
- 40.Fu BC, Hullar MAJ, Randolph TW, Franke AA, Monroe KR, Cheng I, Wilkens LR, Shepherd JA, Madeleine MM, Le Marchand L, Lim U, Lampe JW. Associations of plasma trimethylamine N-oxide, choline, carnitine, and betaine with inflammatory and cardiometabolic risk biomarkers and the fecal microbiome in the Multiethnic Cohort Adiposity Phenotype Study. Am J Clin Nutr. 2020;111:1226–1234. doi: 10.1093/ajcn/nqaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Li Y, Yang P, Liu X, Lu L, Chen Y, Zhong X, Li Z, Liu H, Ou C, Yan J, Chen M. Trimethylamine-N-oxide promotes vascular calcification through activation of NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) Inflammasome and NF-kappaB (nuclear factor kappaB) signals. Arterioscler Thromb Vasc Biol. 2020;40:751–765. doi: 10.1161/ATVBAHA.119.313414. [DOI] [PubMed] [Google Scholar]
- 42.Lynch JB, Hsiao EY. Microbiomes as sources of emergent host phenotypes. Science. 2019;365:1405–1409. doi: 10.1126/science.aay0240. [DOI] [PubMed] [Google Scholar]
- 43.Ramakrishna C, Kujawski M, Chu H, Li L, Mazmanian SK, Cantin EM. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat Commun. 2019;10:2153. doi: 10.1038/s41467-019-09884-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bromberg JS, Fricke WF, Brinkman CC, Simon T, Mongodin EF. Microbiota-implications for immunity and transplantation. Nat Rev Nephrol. 2015;11:342–353. doi: 10.1038/nrneph.2015.70. [DOI] [PubMed] [Google Scholar]
- 45.Paust HJ, Riedel JH, Krebs CF, Turner JE, Brix SR, Krohn S, Velden J, Wiech T, Kaffke A, Peters A, Bennstein SB, Kapffer S, Meyer-Schwesinger C, Wegscheid C, Tiegs G, Thaiss F, Mittrucker HW, Steinmetz OM, Stahl RA, Panzer U. CXCR3+ regulatory T cells control TH1 responses in crescentic GN. J Am Soc Nephrol. 2016;27:1933–1942. doi: 10.1681/ASN.2015020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang YH, Istomine R, Alvarez F, Al-Aubodah TA, Shi XQ, Takano T, Thornton AM, Shevach EM, Zhang J, Piccirillo CA. Salt sensing by serum/glucocorticoid-regulated Kinase 1 promotes Th17-like inflammatory adaptation of Foxp3(+) regulatory T cells. Cell Rep. 2020;30:1515–1529.e1514. doi: 10.1016/j.celrep.2020.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tabriziani H, Lipkowitz MS, Vuong N. Chronic kidney disease, kidney transplantation and oxidative stress: a new look to successful kidney transplantation. Clin Kidney J. 2018;11:130–135. doi: 10.1093/ckj/sfx091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soleimani A, Zarrati Mojarrad M, Bahmani F, Taghizadeh M, Ramezani M, Tajabadi-Ebrahimi M, Jafari P, Esmaillzadeh A, Asemi Z. Probiotic supplementation in diabetic hemodialysis patients has beneficial metabolic effects. Kidney Int. 2017;91:435–442. doi: 10.1016/j.kint.2016.09.040. [DOI] [PubMed] [Google Scholar]
- 49.Mullish BH, McDonald JAK, Pechlivanis A, Allegretti JR, Kao D, Barker GF, Kapila D, Petrof EO, Joyce SA, Gahan CGM, Glegola-Madejska I, Williams HRT, Holmes E, Clarke TB, Thursz MR, Marchesi JR. Microbial bile salt hydrolases mediate the efficacy of faecal microbiota transplant in the treatment of recurrent Clostridioides difficile infection. Gut. 2019;68:1791–1800. doi: 10.1136/gutjnl-2018-317842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pluznick JL. Gut microbiota in renal physiology: focus on short-chain fatty acids and their receptors. Kidney Int. 2016;90:1191–1198. doi: 10.1016/j.kint.2016.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marzocco S, Fazeli G, Di Micco L, Autore G, Adesso S, Dal Piaz F, Heidland A, Di Iorio B. Supplementation of short-chain fatty acid, sodium propionate, in patients on maintenance hemodialysis: beneficial effects on inflammatory parameters and gut-derived uremic toxins, a pilot study (PLAN study) J Clin Med. 2018;7:315. doi: 10.3390/jcm7100315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Behnsen J. Protectors of the neonatal gut: clostridia send pathogens packing. Cell Host Microbe. 2017;21:651–652. doi: 10.1016/j.chom.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 53.Kasahara K, Krautkramer KA, Org E, Romano KA, Kerby RL, Vivas EI, Mehrabian M, Denu JM, Backhed F, Lusis AJ, Rey FE. Interactions between Roseburia intestinalis and diet modulate atherogenesis in a murine model. Nat Microbiol. 2018;3:1461–1471. doi: 10.1038/s41564-018-0272-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franke T, Deppenmeier U. Physiology and central carbon metabolism of the gut bacterium Prevotella copri. Mol Microbiol. 2018;109:528–540. doi: 10.1111/mmi.14058. [DOI] [PubMed] [Google Scholar]
- 55.Kanbay M, Onal EM, Afsar B, Dagel T, Yerlikaya A, Covic A, Vaziri ND. The crosstalk of gut microbiota and chronic kidney disease: role of inflammation, proteinuria, hypertension, and diabetes mellitus. Int Urol Nephrol. 2018;50:1453–1466. doi: 10.1007/s11255-018-1873-2. [DOI] [PubMed] [Google Scholar]
- 56.Liu P, Wu L, Peng G, Han Y, Tang R, Ge J, Zhang L, Jia L, Yue S, Zhou K, Li L, Luo B, Wang B. Altered microbiomes distinguish Alzheimer’s disease from amnestic mild cognitive impairment and health in a Chinese cohort. Brain Behav Immun. 2019;80:633–643. doi: 10.1016/j.bbi.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 57.Peters BA, Dominianni C, Shapiro JA, Church TR, Wu J, Miller G, Yuen E, Freiman H, Lustbader I, Salik J, Friedlander C, Hayes RB, Ahn J. The gut microbiota in conventional and serrated precursors of colorectal cancer. Microbiome. 2016;4:69. doi: 10.1186/s40168-016-0218-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Iorio BR, Rocchetti MT, De Angelis M, Cosola C, Marzocco S, Di Micco L, di Bari I, Accetturo M, Vacca M, Gobbetti M, Di Iorio M, Bellasi A, Gesualdo L. Nutritional therapy modulates intestinal microbiota and reduces serum levels of total and free indoxyl sulfate and P-cresyl sulfate in chronic kidney disease (medika study) J Clin Med. 2019;8:1424. doi: 10.3390/jcm8091424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.