

Inherited platelets disorders can be severe, especially after trauma or surgical procedures in some monogenic disorders, as in Bernard–Soulier syndrome [BSS; Mendelian Inheritance in Man (MIM) #231200]. BSS is a rare autosomal recessive macrothrombocytopenia (incidence of about one per million). Its hallmark is a defective adhesion of platelets to the sub-endothelium, resulting from quantitative or qualitative defects in the glycoprotein Ib (GPIb)-IX-V complex, a platelet receptor for von Willebrand Factor (VWF), which is composed of four subunits: GPIbα, GPIbβ, GPIX and GPV.1 Laboratory diagnosis is based on prolonged bleeding time, moderate-to-severe thrombocytopenia (platelet count typically ranges from 20 to 100 × 109/l), giant platelets and deficient ristocetin-dependent platelet agglutination.2 Very little is known about the biochemical and clinical features of heterozygous carriers of the mutations causing BSS, and about the impact in general population individuals of variation in genes encoding the GPIb-IX-V complex when present in heterozygosity. In fact, family members with only one mutated allele are generally asymptomatic, with sub-normal platelet count, slightly enlarged platelets and marginally reduced levels of glycoproteins expression.
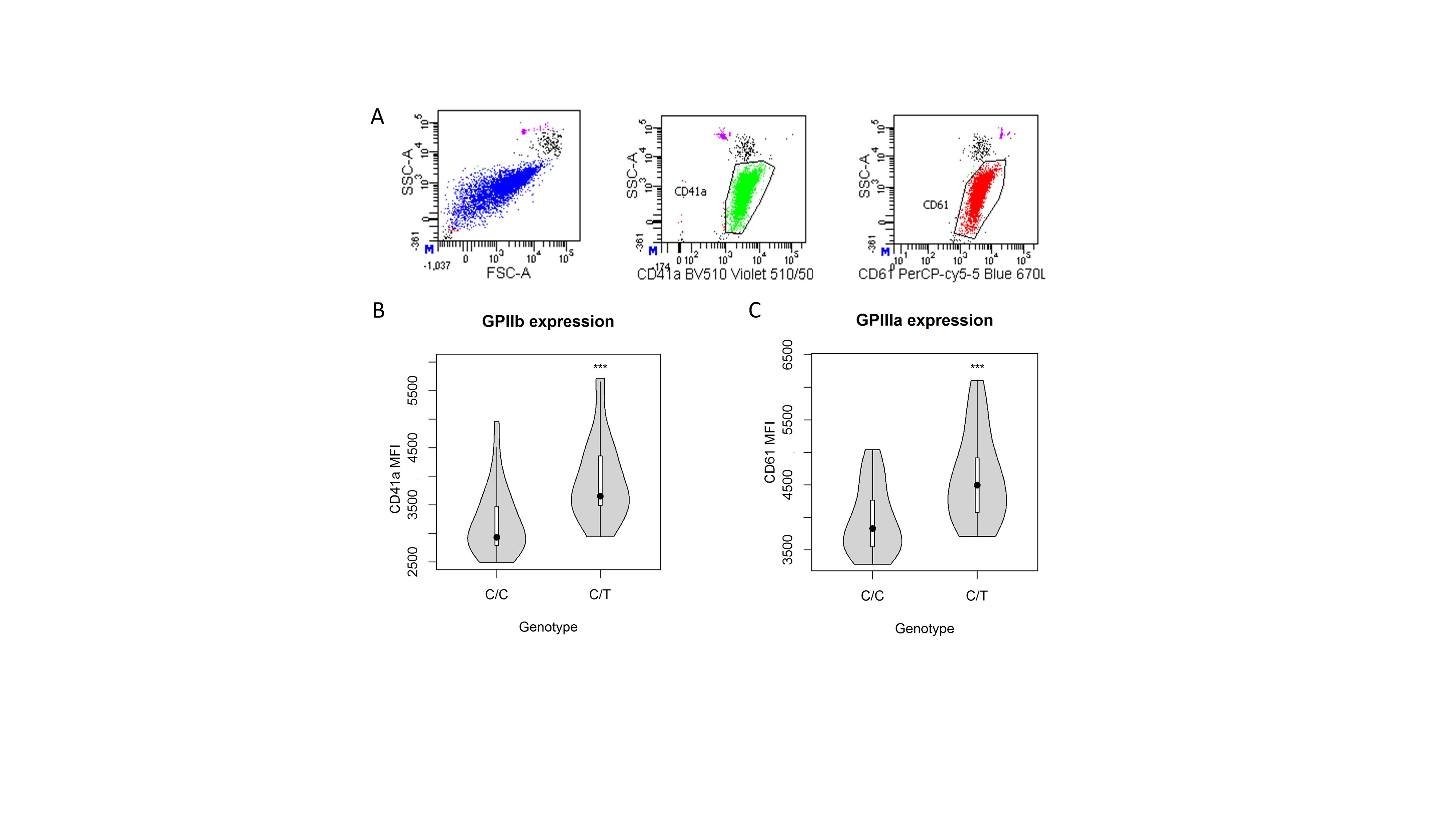
Here, to dissect the impact of genetic variability on platelet count, a sequencing-based whole-genome association study was performed in 6528 volunteers included in the SardiNIA general population cohort.3 Six signals were identified (Table SI, Data S1 for description), including a novel nonsynonymous variant [22:19711445:C/T; minor allele frequency (MAF) = 0·0045; P = 1·172 × 10−16], mapping in the second exon (c.C79T, p.P27S) of the GP1BB gene (Figure S1). Completely independent of previously reported associations in the same genomic region (Data S1), p.P27S is Sardinian-specific, being completely missing in large sequencing datasets such as 1000 Genomes Project,4 Genome of the Netherlands (GoNL) project,5 Genome Aggregation Database (gnomAD),6 the Exome Sequencing Project in the National Heart, Lung, and Blood Institute’s (NHLBI’s) Trans-Omics for Precision Medicine (TOPMed) programme.7 No homozygous and 57 carriers for the rare 22:19711445-T allele were found. The platelet count in wild-type homozygous was 242·87 ± 117·05 × 109/l (mean ± 1·96 × SD), whereas in p.P27S carriers was 174·17 ± 91·51 × 109/l, corresponding to a reduction of 70·13 × 109/l for each copy of the minor allele (Fig 1A). With this large effect, the novel founder mutation explains ~1·05% of phenotypic variance for platelet count, representing the largest phenotypic effect among all the independent variants reported to date in the genome-wide association studies (GWAS) Catalogue (Tables SII and SIII).8 Moreover, in a subset of 2000 individuals, whose mean platelet volume was measured, this variant was associated with notably larger platelets (P = 2·13 × 10−10), consistently with evidence of morphologically enlarged platelets in patients with BSS (Fig 1B). To assess platelet functionality, a seven-colour flow cytometry panel (Table SIV) was set up in 24 of 57 p.P27S carriers (42·1%) and in an equal number of matched unrelated controls. Monoclonal antibodies directed against the GPIIb-IIIa complex (CD41a and CD61), and the VWF receptor complex (CD42a and CD42b) were used to investigate the basal receptor expression in resting platelets. The p.P27S carriers showed increased levels of GPIIb (CD41a, +22·36%; P = 1·61 × 10−4; N = 48) and GPIIIa (CD61, +16·20%; P = 6·61 × 104; N = 48) a typical finding in the presence of enlarged platelets (Figure S2). The expression of GPIX and GPIbα glycoproteins and their correct assembly into the GPIb-IX-V complex are known to be impaired by a defective GPIbβ peptide9. Indeed, despite carrying only one mutated allele, the p.P27S heterozygous showed appreciably lower basal expression levels of both GPIX (−24·69%, P = 2·66 × 10−6, N = 46; Fig 1C) and GPIbα (−26·51%, P = 3·66 × 10−8, N = 48; Fig 1D), and consequently less of the entire complex, compared to controls. This is far more than the normal expression levels of GPIX and GPIbα in carriers of other missense mutations in GP1BB, as recently reported.10 Pre-activation and reactivity changes in p.P27S platelets were investigated after exposure to the agonist adenosine diphosphate (ADP). Indeed, activated αIIbβ3 was prominently induced in the p.P27S carriers, as shown by the extent of procaspase-activating compound 1 (PAC-1) binding to resting and activated platelets (+41·94%, P = 4·84 × 10−3, N = 48, Fig 1E). Notably, no variation in the response of platelets after ADP stimulation was recently reported in patients with BSS and carriers.9 Remarkably, platelet reactivity turned out to be differentially regulated: no changes were observed in surface exposure of neo P-selectin (CD62P, +35·22%, P = 0·138, N = 48; Fig 1F) and neo granulophysin (CD63, +1·86%, P = 0·658, N = 46; Fig 1G), markers of granule content release. The unique functional effects of the p.P27S led us to examine its possible consequences on the molecular structure and conformational changes of GPIbβ by molecular modelling analysis based on the X-ray crystal structure.11 Proline–Serine substitution falls in the leucine-rich repeat N-terminal (LRRNT) domain of the 206 amino acid long protein encoded by GP1BB (Fig 2A, B). Proline residues are expected to be disruptive of structure; and indeed, in that highly conserved region and close to cysteine residues involved in the Cys26-Cys32 disulphide bridge, p.P27S could thus modify the stability and consequently the conformation of GPIbβ. To test this hypothesis, we first performed in silico molecular dynamic simulations, observing an increased conformational mobility of the amino acid backbone close to p.P27S (Figure S3), suggesting the instability of the GPIbβ glycoprotein in accordance with the observed reduction in the expression of GPIX and GPIbα. Strikingly, a greater fluctuation of the amino acids in loop 2 of the p.P27S protein was also recorded, as indicated by root-mean-square-fluctuation (Fig 2C).
Fig 1.
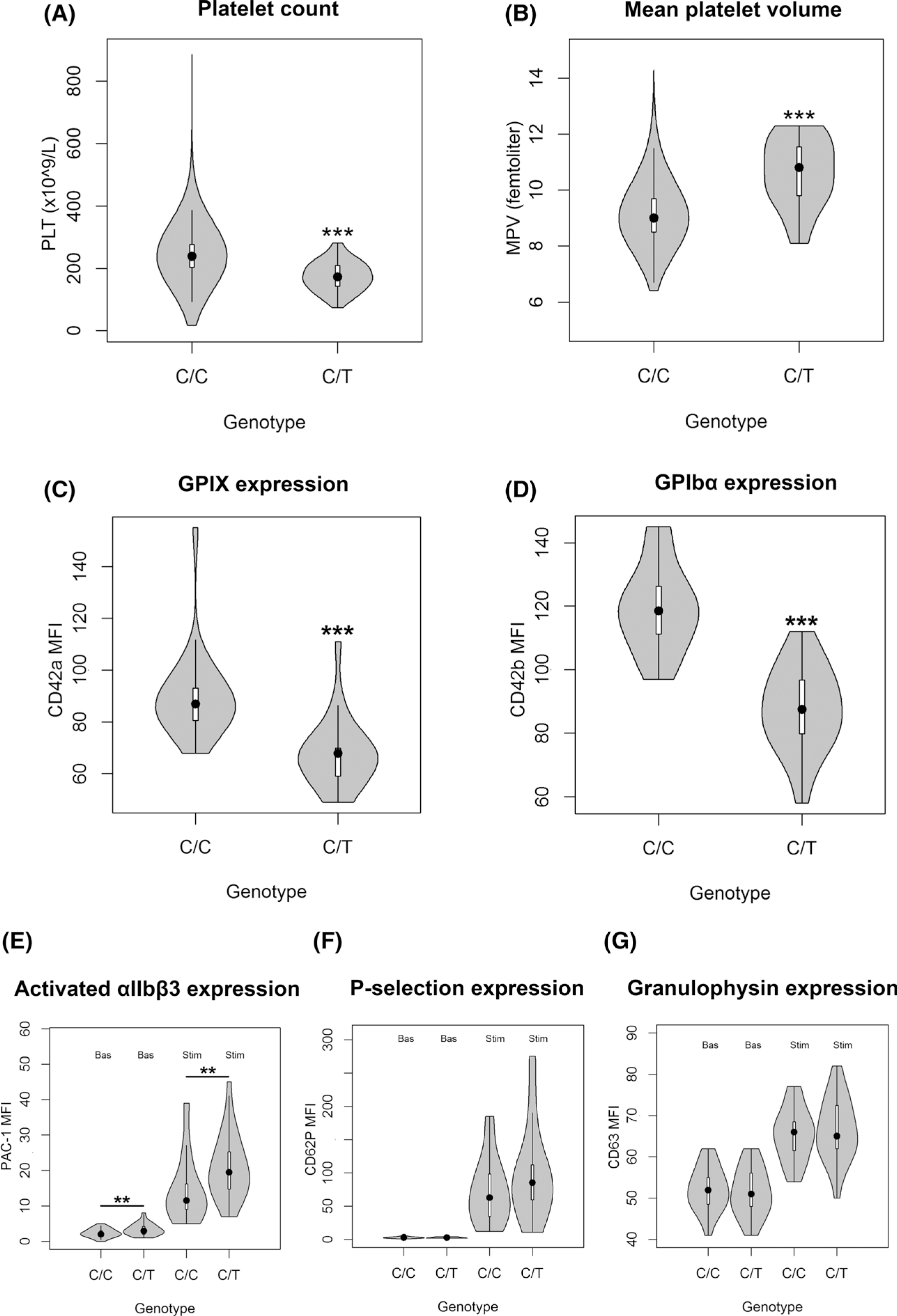
Effects of chr22:19711445 genotype on platelet-related phenotypes. (A) Platelet (PLT) count distribution stratified on 57 heterozygous carriers and 6471 homozygous wild types. (B) Mean platelet volume (MPV) distribution stratified on 28 heterozygous carriers and 1972 homozygous wild types. Basal expression levels of the main GPIb-IX-V receptor glycoproteins on resting platelets: (C) GPIX on 23 carriers and 23 controls, and (D) GPIbα on 24 carriers and 24 controls. (E–G) Expression levels of the most relevant platelet activation-dependent markers (activated αIIbβ3, P-selectin and granulophysin), in basal conditions and after stimulation with ADP. Violin plots represent the distribution of the data; the boxplots inside report the median value as a dot, the interquartile range (IQR) as a box, the 1st quartile −1·5 IQR and the 3rd quartile +1·5 IQR as whiskers. **P < 0·01, ***P < 0·001. MFI, median fluorescence intensity.
Fig 2.
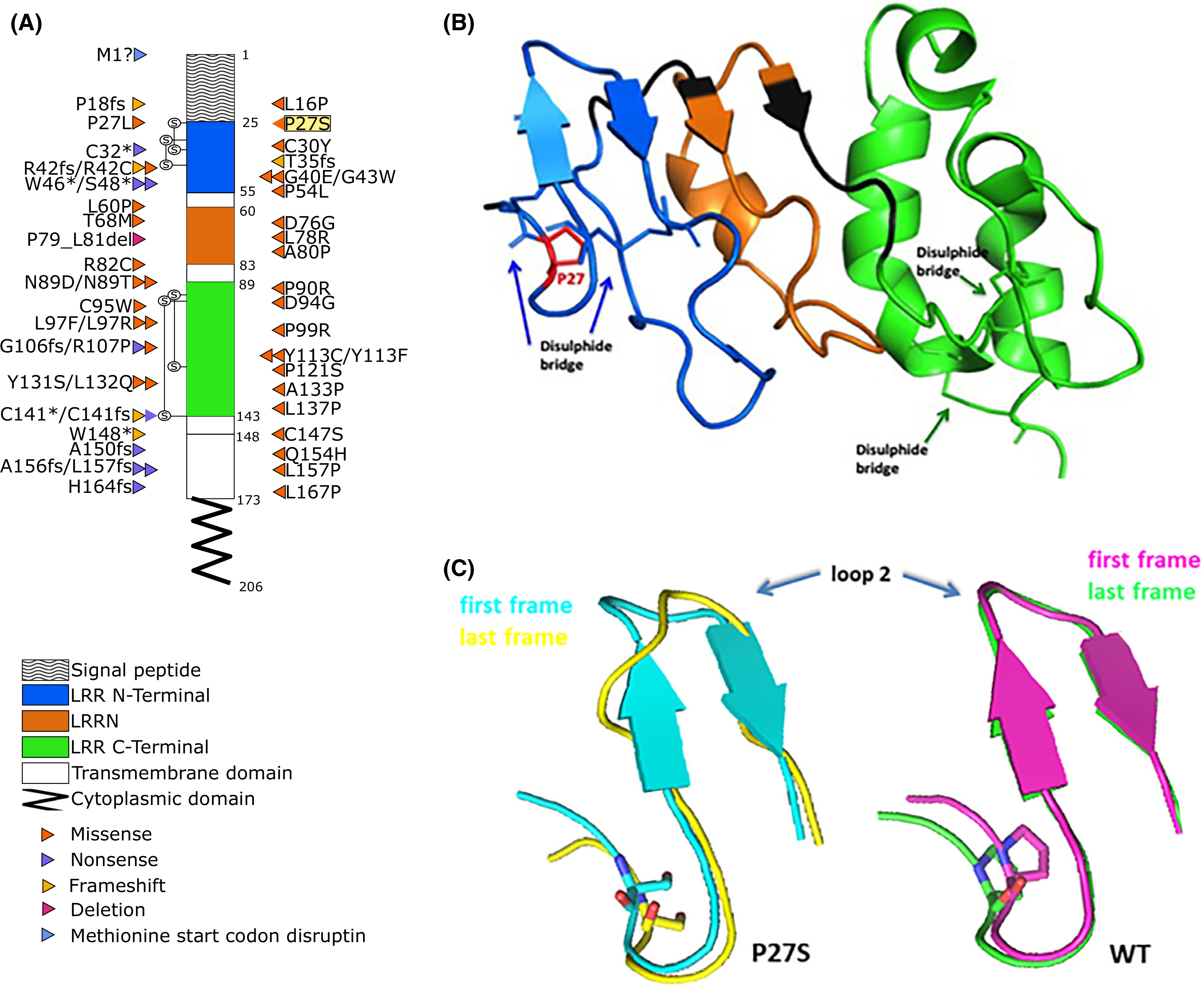
GPIbβ amino acid sequence with BSS-causing mutations and molecular modelling analyses. (A) Positions of the mutations within the coding regions of platelet glycoprotein (GP)Ibβ according to National Center for Biotechnology Information (NCBI) Reference Sequence, NP_000398.1. The different domains are indicated with different patterns. Different types of mutation are colour-coded; highlighted in yellow is the Proline27 to Serine27 substitution (P27S) described here first. Known mutations were obtained from: Savoia et al., 201416; Sivapalaratnam et al., 201717; Bragadottir et al., 201510; Qiao et al., 201518; Kunishima et al., 200119; Ferrari et al., 201820; and Bastida et al., 201813. (B) Three-dimensional structure of GPIbβ sequence, colour-coded according to the schematic representation in (A). (C) X-ray structure of protein (26–143 aa code 3RFE) showing the impact of p.P27S on GPIbβ glycoprotein conformation; in particular, the superposition between the first (teal) and last (yellow) frame of the molecular dynamics for the p.P27S protein (left), and the superposition between the first (pink) and last (green) frame for the wild-type (WT) protein (right) are reported.
In summary, all typical findings of macrothrombocytopenias (i.e. BSS) were observed in the p.P27S obligate carriers characterised in the present study: low levels of large platelets and low expression of GPIX and GPIbα glycoproteins, as shown by flow cytometry. As one might anticipate, the most severe cases are caused by deletions and nonsense mutations, but some missense mutations are disabling enough to be clinically significant. In one of the reported cases,12 a charge difference is introduced (p.Asn89Asp); in the other,13 as in this case, the Proline residue is replaced (p.Pro27Leu), which is expected to disrupt secondary structure in the protein. That p.P27S influences conformational changes and stability of GPIbβ, in turn affecting GPIb-IX-V complex function, is further clearly supported by the in silico molecular dynamic analyses. Noteworthy, a critical interaction of GPIbβ with GPIX involves N-terminal residues 15 through 32 of GPIbβ, precisely including Proline 27.14 According to Hardy–Weinberg expectation, at least four p.P27S homozygous individuals, most likely with BSS, are expected in Sardinia, but none have been reported to date: this may suggest that BSS is likely underdiagnosed in Sardinia, consistent with other reports.15 Thus, clinicians should be aware of the novel p.P27S mutation in the molecular characterisation of Sardinian-origin patients with a clinical picture of platelet macrocytosis and platelet count of <100 × 109/l.
Supplementary Material
Figure S1. Manhattan plot of the genome-wide association findings.
Figure S2. Gating strategy identifying platelets.
{kind=link}
Figure S3. Molecular modelling analyses
Figure S4. Association plot for PLT at the chr3:56849749 locus.
Figure S5. Association plot for PLT at the chr6:135419631 locus.
Figure S6. Association plot for PLT at the chr15:43753426 locus.
Figure S7. Association plot for PLT at the chr1:247719769 locus.
Figure S8. Association plot for PLT at the chr19:16197320 locus.
Figure S9. Association plot for PLT at the chr22:19711445 locus.
Figure S10. Association plot for mean platelet volume (MPV) at the chr22:19711445 locus.
Figure S11. Regional association plots for PLT at the 22q11.21 locus with imputation performed using: (A) Sardinian, (B) 1000GP or (C) HRC reference panels.
Figure S12. Electrostatic surface potential calculation.
Data S1. Supplemental data.
Table SII. Associated loci from GWAS Catalogue.
Table SIII. Heritability explained in the SardiNIA study by variants reported in GWAS Catalogue.
Table SIV. Seven-colour antibody panel assessed by flow cytometry.
Table SV. Functional relevance of variants in the 95% credible set.
Supporting information A Sardinian founder mutation in GP1BB that impacts thrombocytopenia.
Table SI. List of associated regions.
Acknowledgements
We thank all the volunteers who generously participated in this study; we are grateful to Mr Mario Lovicu and Mr Nazario Olla for the logistic support provided and helpful suggestions. Supported by contracts N01-AG-1–2109 and HHSN2712011 00005C from the Intramural Research Program of the National Institute on Aging, National Institutes of Health (NIH).
Footnotes
Conflict of interest
The authors declare no competing interests.
Web resources
The URLs for data presented herein are as follows: Online Mendelian Inheritance in Man, https://www.omim.org/; SardiNIA Project, https://sardinia.irp.nia.nih.gov/; 1000 Genomes Project data repository, ftp://ftp.1000genomes.ebi.ac.uk/; GoNL, Genome of the Netherlands, http://www.nlgenome.nl/; GnomAD, http://gnomad.broadinstitute.org/; Exome Sequencing Project, https://esp.gs.washington.edu/drupal/; NHLBI TOPMed Programme, https://www.nhlbiwgs.org/; GWAS catalogue, https://www.ebi.ac.uk/gwas/.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- 1.Savoia A, Pastore A, De Rocco D, Civaschi E, Di Stazio M, Bottega R, et al. Clinical and genetic aspects of Bernard-Soulier syndrome: searching for genotype/phenotype correlations. Haematologica. 2011;96:417–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berndt MC, Andrews RK. Bernard-Soulier syndrome. Haematologica. 2011;96:355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sidore C, Busonero F, Maschio A, Porcu E, Naitza S, Zoledziewska M, et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet. 2015;47:1272–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boomsma DI, Wijmenga C, Slagboom EP, Swertz MA, Karssen LC, Abdellaoui A, et al. The Genome of the Netherlands: design, and project goals. Eur J Hum Genet. 2014;22:221–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. 2019:531210. 10.1101/531210. [DOI] [Google Scholar]
- 7.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. BioRxiv, 06 March 2019. 2019. 10.1101/563866. PPR:PPR72371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45:D896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hadjkacem B, Elleuch H, Gargouri J, Gargouri A. Bernard-Soulier syndrome: novel nonsense mutation in GPIbbeta gene affecting GPIb-IX complex expression. Ann. Hematol 2009;88:465–72. [DOI] [PubMed] [Google Scholar]
- 10.Bragadottir G, Birgisdottir ER, Gudmundsdottir BR, Hilmarsdottir B, Vidarsson B, Magnusson MK, et al. Clinical phenotype in heterozygote and biallelic Bernard-Soulier syndrome-a case control study. Am J Hematol. 2015;90:149–55. [DOI] [PubMed] [Google Scholar]
- 11.McEwan PA, Yang W, Carr KH, et al. Quaternary organization of GPIb-IX complex and insights into Bernard-Soulier syndrome revealed by the structures of GPIbβ and a GPIbβ/GPIX chimera. Blood. 2011;118:5292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiore M, De Thoré C, Ranjatoelina HR, Baas MJ, Jacquemont ML, Dreyfus M, et al. High prevalence of the natural Asn89Asp mutation in the GP1BB gene associated with Bernard-Soulier syndrome in French patients from the genetic isolate of Reunion Island. Br J Haematol. 2020;189:e67–e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bastida JM, Lozano ML, Benito R, Janusz K, Palma-Barqueros V, Del Rey M, et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica. 2018;103:148–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenny D, Morateck PA, Montgomery RR. The cysteine knot of platelet glycoprotein Ib (GPIb-beta) is critical for the interaction of GPIb-beta with GPIX. Blood. 2002;99:4428–33. [DOI] [PubMed] [Google Scholar]
- 15.Noris P, Balduini CL. Inherited thrombocytopenias in the era of personalized medicine. Haematologica. 2015;100:145–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savoia A, Kunishima S, De Rocco D, Zieger B, Rand ML, Pujol-Moix N, et al. Spectrum of the mutations in Bernard-Soulier syndrome. Hum Mutat. 2014;35:1033–45. [DOI] [PubMed] [Google Scholar]
- 17.Sivapalaratnam S, Westbury SK, Stephens JC, Greene D, Downes K, Kelly AM, et al. Rare variants in GP1BB are responsible for autosomal dominant macrothrombocytopenia. Blood. 2017;129:520–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiao J, Davis AK, Morel-Kopp MC, Ward CM, Gardiner EE, Andrews RK. Low levels of CD9 coincidental with a novel nonsense mutation in glycoprotein Ibβ in a patient with Bernard-Soulier syndrome. Ann Hematol. 2015;94:2069–71. [DOI] [PubMed] [Google Scholar]
- 19.Kunishima S, Naoe T, Kamiya T, Saito H. Novel heterozygous missense mutation in the platelet glycoprotein Ib beta gene associated with isolated giant platelet disorder. Am J Hematol. 2001;68:249–55. [DOI] [PubMed] [Google Scholar]
- 20.Ferrari S, Lombardi AM, Cortella I, Businaro MA, Bertomoro A, Di Pasquale I, et al. New heterozygous variant in GP1BB gene is responsible for an inherited form of macrothrombocytopenia. Br J Haematol. 2019;184:855–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Manhattan plot of the genome-wide association findings.
Figure S2. Gating strategy identifying platelets.
Figure S3. Molecular modelling analyses
Figure S4. Association plot for PLT at the chr3:56849749 locus.
Figure S5. Association plot for PLT at the chr6:135419631 locus.
Figure S6. Association plot for PLT at the chr15:43753426 locus.
Figure S7. Association plot for PLT at the chr1:247719769 locus.
Figure S8. Association plot for PLT at the chr19:16197320 locus.
Figure S9. Association plot for PLT at the chr22:19711445 locus.
Figure S10. Association plot for mean platelet volume (MPV) at the chr22:19711445 locus.
Figure S11. Regional association plots for PLT at the 22q11.21 locus with imputation performed using: (A) Sardinian, (B) 1000GP or (C) HRC reference panels.
Figure S12. Electrostatic surface potential calculation.
Data S1. Supplemental data.
Table SII. Associated loci from GWAS Catalogue.
Table SIII. Heritability explained in the SardiNIA study by variants reported in GWAS Catalogue.
Table SIV. Seven-colour antibody panel assessed by flow cytometry.
Table SV. Functional relevance of variants in the 95% credible set.
Supporting information A Sardinian founder mutation in GP1BB that impacts thrombocytopenia.
Table SI. List of associated regions.