

Abstract
This work deals with the synthesis, crystal structure, computational study and antiviral potential of mixed ligand copper(II) complex [Cu(L)(phen)](1), (where, H2L = (Z)-N’-((E)-2-hydroxy-3,5-diiodobenzylidene)-N,N-dimethylcarbamohydrazonothioic acid, phen = 1,10-phenanthroline). The Schiff base ligand (H2L) is coordinated with Cu(II) ion in O, N, S-tridentate mode. The copper complex (1) crystallized in the monoclinic system of the space group P21/c with eight molecules in the unit cell and reveals a square pyramidal geometry. Furthermore, we also perform quantum chemical calculations to get insights into the structure-property relationship and functional properties of ligand (H2L) and its copper (II) complex [Cu(L)(phen)](1). Complex [Cu(L)(phen)](1) was also virtually designed in-silico evaluation by Swiss-ADME. Additionally, inspiring by recent developments to find a potential inhibitor for the COVID-19 virus, we have also performed molecular docking study of ligand and its copper complex (1) to see if our compounds shows an affinity for the main protease (Mpro) of COVID-19 spike protein (PDB ID: 7C8U). Interestingly, the results are found quite encouraging where the binding affinity and inhibition constant were found to be -7.14 kcal/mol and 5.82 μM for ligand (H2L) and -6.18 kcal/mol and 0.76 μM for complex [Cu(L)(phen)](1) with Mpro protein. This binding affinity is reasonably well as compared to recently known antiviral drugs. For instance, the binding affinity of ligand and complex was found to be better than docking results of chloroquine (-6.293 kcal/mol), hydroxychloroquine (-5.573 kcal/mol) and remdesivir (-6.352 kcal/mol) with Mpro protein. The present study may offer the technological solutions and potential inhibition to the COVID-19 virus in the ongoing and future challenges of the global community.
In the framework of synthesis and characterization of mixed ligand copper (II) complex; the major conclusions can be drawn as follow:
Keywords: Tridentate ligand, Copper complex, X-ray crystallographic study, Molecular docking, COVID-19 main protease (Mpro), Potential inhibitor COVID-19
Graphical abstract
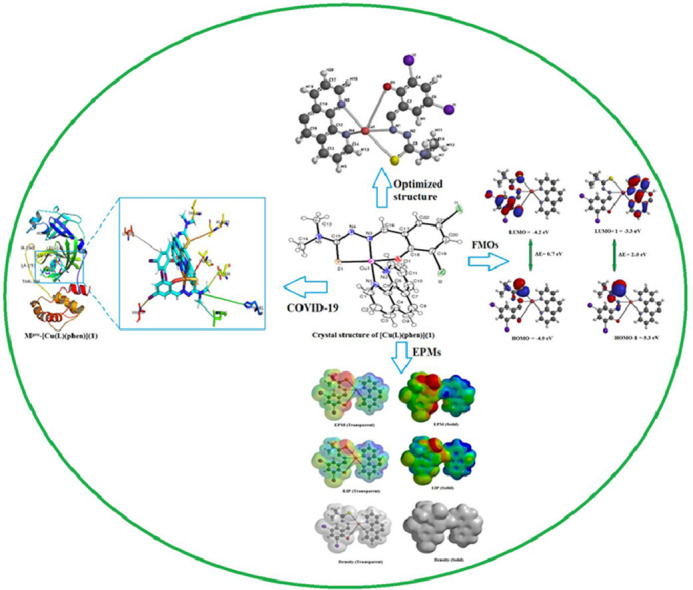
1. Introduction
The coronavirus disease (COVID- 2019) pandemic, as reported by the World Health Organization (WHO) , caused by a newly discovered coronavirus classified as severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) emerged in the city of Wuhan, Capital of Hubei Province, Central China in late 2019, which was later coined as COVID-19 [[1], [2]–3]. The SARS-CoV-2 is the seventh and newest addition to the SARS family noted for their crown-like, glycoprotein structure, Spike(S)-protein structure and ability to bind to cells easily. The bioinformatics in the genetic code hidden within the ribonucleic acid (RNA) chain of the virus itself is one of huge obstacle that researchers have had to overcome when looking for a cure or vaccine. As the world is witnessing severe damages of COVID-19 pandemic, it is a great challenge for finding a therapeutic agent to treat COVID-19 virus. Having no alternatives, Remdesivir, Chloroquine, and Hydroxychloroquine are under testing in clinical trials to access their efficacy against COVID-19 virus [[4], [5], [6], [7]–8]. One of the first antiviral drugs tested, the Ribavirin, a nucleoside analog that acts as an RNA polymerase inhibitor, was used on patients with MERS and SARS-CoV-2 protease inhibitors [[9], [10]–11].
A recent study revealed COVID-19 main protease (Mpro) is responsible for the proteolytic mutation of this virus and is essential for its life cycle [12]. Chloroquine and Hydroxychloroquine are aminoquinolines, a class of heterocyclic scaffolds with an amino group. They are able to form metal complexes with transition metal ions as an inhibitor for COVID-19 [13,14]. Schiff bases have large importance in medicinal and pharmaceutical fields. A lot of papers have been reported in this field and recently several Schiff bases and their complexes have been studied against COVID-19 [[15], [16], [17]–18]. Furthermore, recent studies have shed a light on molecular docking methodologies as powerful techniques in establishing therapeutic strategies to combat COVID-19 pandemic. In context to the recent demands, molecular docking approach has been applied for screening of organic molecules and coordination complexes as potent inhibitor of COVID-19 virus [[19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34]–35]. Metal complexes will eventually lead to the destruction of COVID-19 virus. Copper or their compounds can be a powerful weapon in the fight against COVID-19 and future pandemics [36,37]. Recently, our lab has synthesized a novel copper (II) complex [38] and studied its binding affinity and inhibition constant (-8.400 kcal/mol and 0.661 μM, respectively) for the main protease (Mpro) of COVID-19 spike protein (PDB ID: 6LU7). Unlike the previous studies, we report here, synthesis of a tridentate Schiff base ligand (H2L) and a square pyramidal copper (II) complex [Cu(L)(phen)](1) (Scheme 1 ) and its structural properties and binding affinities with main protease (Mpro) of COVID-19 spike protein (PDB ID: 7C8U) is studied by molecular docking methods to find a possible therapeutic drug candidate for COVID-19 virus.
Scheme 1.

Synthetic route for Schiff base and mixed ligand complex [Cu(L)(phen)](1).
2. Experimental
2.1. Materials used for synthesis
The chemicals used were of AnalaR grade. Cu(OAc)2.H2O, 4,4-dimethyl-3-thiosemicarbazide, 3,5-diiodosalicylaldehyde, 1,10-phenanthroline, were obtained from Sigma-Aldrich and were used without further purification.
2.2. Characterization techniques
Different spectral techniques (UV-vis, IR and HRMS) were used to analyse the prepared ligand and complex [Cu(L)(phen)](1). Elemental analytical data and quantum chemical calculations were also applied for these purposes. The FT-IR spectra of the compounds were recorded on a Shimadzu IR Affinity-1S Fourier transform infrared spectrophotometer in the range 4000–400 cm−1 range using KBr pellets and the electronic spectra of the compounds were taken on a Thermo scientific UV-Vis recording spectrophotometer Evolution-3000 in quartz cells. Melting point was measured on a Boetius micro melting point apparatus. The HRMS was made on a XEVO G2-XS QTOF High Resolution Mass Spectrometer (HRMS) equipped with dual electrospray ionization and atmospheric pressure chemical ionization (ESI/APCI) with mass range: 100–1900 m/z.
2.3. Crystallographic data collection and refinement
The geometry of copper (II) coordination [Cu(L)(phen)](1) complex has also been confirmed by a single-crystal X-ray analysis. Single crystal suitable for X-ray analysis was mounted on glass fibers and used for data collection. Crystal data was collected on Enraf-Nonius MACH3 diffractometer using graphite monochromatised Mo-Kα (λ = 0.71073 Å). The crystal orientation, cell refinement and intensity measurements were made using the program CAD-4PC performing ω-scan measurements. The structure were solved by direct methods using the program SHELXS-97 [39] and refined by full-matrix least-square techniques against F2 using SHELXL-97 [40]. Anisotropic refinement was used for all non-hydrogen atoms. Hydrogen atoms were placed geometrically and held in the riding model. Data collection and structure refinement parameter along with crystallographic data of complex was summarized in Table 1 . The crystallographic data of complex have been deposited with the Cambridge Crystallographic Data Center, CCDC-2066722 for [Cu(L)(phen)] (1) as supplementary material for this paper.
Table 1.
Crystallographic Data and Refinement Parameters for complex [Cu(L)(phen)] (1).
| Crystallographic data | Complex (1) |
|---|---|
| Formula | C22H17CuI2N5OS |
| Mw (gmol−1) | 716.80 |
| temp (K) | 200(2) |
| λ (Mo Kα), (Å) | 0.71073 |
| crystal system | Monoclinic |
| space group | P 21/c |
| a (Å) | 15.1231(9) |
| b (Å) | 26.5571(15) |
| c (Å) | 12.1855(6) |
| α (°) | 90 |
| β (°) | 100.591(2) |
| γ (°) | 90 |
| V (Å3) | 4810.6(5) |
| Z | 8 |
| Dcalc (Mg/cm3) | 1.979 |
| μ (mm−1) | 3.586 |
| F(000) | 2744 |
| Crystal size(mm3) | 0.200 × 0.150 × 0.100 |
| Index ranges | -23 < = h < = 23, -40 < = k < = 40, 14 < = l < = 18 |
| Collected reflections | 208670 |
| unique reflections | 18325 |
| Absorption correction | Semi-empirical from equivalents |
| max and min trans | 0.7465 and 0.5673 |
| Refinement method | Full-matrix least-squares on F2 |
| Goodness-of-fit (GOF) on F2 | 1.067 |
| R1 [I > 2σ(I)]a | 0.0297 |
| wR2 (all data)b | 0.0683 |
| CCDC Number | 2066722 |
R1 = ∑││Fo│ – │Fc││/∑│Fo│; bwR2 = {∑ [w(Fo2 – Fc2 )2 ]/ ∑ [w(Fo2 )2 ]}1/2
2.4. Computational methods
The geometry optimization of complex [Cu(L)(phen)](1) was performed at the ωB97X-D/6-311+G** level of theory in vacuum using the Gaussian16 software package [41] and cross-validated using the Spartan 16/18 parallel suite of programs [42]. The dispersion-corrected DFT functional ωB97X-D [43] is chosen to accurately estimate the Vander walls interactions, which are expected to contribute greatly to the stability of this complex.
2.5. Molecular docking protocols
All protein docking calculations were performed using open-source AutoDock Tools [44] and AutoDock Vina (ADV) [45] programs while docking results were visualized by open-source Discovery Studio visualizer [46]. The AutoDock Vina (ADV) program is used for docking studies in the present investigation. ADV offers more accuracy in analyzing protein-ligand interactions as compared to Autodock 4.2 where the aforementioned provides more accuracy for a ligand by analyzing more than twenty rotatable bonds [45]. The spike protein of main protease (MPro) of COVID-19 was selected for molecular docking and its structure was obtained from the PDB database using (PDB ID: 7C8U; unit cell: a = 114.303 Å, b = 53.874 Å, c = 45.233 Å, α = 90°, β = 101.576(2) °, γ = 90°) and used as receptor protein [47, 48]. The Mpro is a key enzyme of SARS CoV-2 with a molecular mass of 33796.8 Da and plays a pivotal role in the processing of two polyproteins pp1a and pp1ab into 16 NSPS (non-structural proteins) [47]. The Auto Dock GUI program was used to prepare the pdbqt files of the receptor Mpro protein, ligand (H2L), and of complex [Cu(L)(phen)](1) along with the grid box settings. The grid size boundaries along X, Y, and Z axes were scaled at 30 Å, 30 Å and 30 Å, respectively, with a grid spacing of 0.508 Å to allow proper binding flexibility with a protein molecule. The output results were written into a configuration file. The receptor protein was treated as a rigid entity whereas ligand, and complex [Cu(L)(phen)](1) was kept flexible to attain the best fitting confirmation for the receptor complex. On the other hand, Kollman charges, polar hydrogen atoms were also added during the preparation of protein receptor. In addition to this, the water molecules and previously crystalized co-ligand were removed from the receptor molecule through the Auto Dock GUI program [44]. The docking protocol was validated by docking previously crystalized co-ligands with the Mpro protein. The visualization of results was performed using a free version of Discovery Studio visualizer [46].
2.6. In-silico Swiss-ADME descriptors
A large variety of in silico methods share the objective of predicting ADME (absorption, distribution, metabolism and excretion) parameters from molecular structure and their pharmacokinetic and physicochemical parameters [49]. The preliminary test in the potential application for theoretical biological activities of Schiff base ligand (H2L) and its complex [Cu(L)(phen)](1) was in-silico evaluated by Swiss-ADME through free access (http://www.swissadme.ch) to compute physicochemical descriptors as well as to predict ADME parameters, pharmacokinetic properties, drug-likeness and medicinal chemistry friendliness (among in-house proficient methods such as BOILED Egg, iLOGP and Bioavailability Radar) of one or multiple small molecules to support drug discovery [50].
2.7. Synthesis
2.7.1. Synthesis of Schiff base ligand
Schiff base (H2L) , a tridentate-ONS donor ligand was synthesized as red crystalline products by the refluxing 3,5-diiodosalicylaldehyde (20.0 mmol, 7.478 g) with 4,4-dimethyl-3-thiosemicarbazide (20.0 mmol, 2.383 g) in equimolar ratio 1:1 in MeOH for 4 h.The red crystalline product was obtained, which was filtered, washed with diethyl ether and stored in a desiccator over CaCl2. Yield: 90%. M.p.:180–190 °C. UV-Vis (MeOH) λ (nm): 255,390. Selected IR data (KBr/cm−1): 1640 (>C=N); 3109 and 1224(υAr–OH). ESI-MS: 475.31 (C10H11I2N3OS, [M+H+]). Anal. Calc. for C10H11N3OI2S (%): C, 25.28; H, 2.33; N, 8.84. Found (%): C, 25.29; H, 2.34; N, 8.85.
2.7.2. Synthesis of complex [Cu(L)(phen)](1)
To an MeOH solution (10 mL) of Cu(OAc)2.H2O (1.0 mmol, 0.199 g), a MeOH solution (10 mL) of (Z)-N’-((E)-2-hydroxy-3,5-diiodobenzylidene)-N,N-dimethylcarbamohydrazonothioic acid (1.0 mmol, 0.475 g) in the presence of triethylamine (3.0 mmol, 40 µL) as base was added with constant stirring the reaction mixture was stirred continuously for 5 h. To the above reaction mixture, the second ligand, 1, 10-phenanthroline (1.0 mmol, 0.180 g) was dissolved in a MeOH solution (10 mL) and stirring was continued for 2 h at 25 °C. After completion of the reaction, the dark blue reaction mixture was filtered to remove any solid impurities and left for slow evaporation. After 2,3 days, dark blue prismatic crystals suitable for single crystal X-ray analysis were collected, washed with MeOH and then washed with diethyl ether. The crystals were then dried in air at room temperature, and store in a desiccator over CaCl2. Yield: 70 %, M.p.:251–253 °C. UV-Vis (MeOH) λ (nm): 265,445. Selected IR data (KBr/cm−1): 1621(s), 1443(s), 1122(s) 1109(s), 740(s), 672(s). ESI-MS: 716.80 (C22H17I2N5OSCu+, [M+H+]). Anal. Calcd. for [Cu(L)(phen)](1), (%): C, 36.86; H, 2.39; N, 9.77. Found (%): C, 36.87; H, 2.40; N, 9.78.
3. Result and discussion
3.1. Chemistry
A Schiff base (H2L) ligand has synthesized by condensing 3, 5-diiodosalicylaldehyde with 4, 4-dimethyl-3-thiosemicarbazide in equimolar ratio 1:1 in MeOH. Reaction of Schiff base (H2L) and 1, 10-phenathroline with Cu(OAc)2.H2O led to the formation of mononuclear copper(II) coordination complex (Scheme 1). The Schiff base ligand is coordinated with Cu(II) ion in O, N, S-tridentate mode. The ligand can undergo tautomerism and form thiol (H2L, Form-I) or thione (HL, Form-II) as rigid Schiff base (Scheme 1).The ligand and complex gave satisfactory elemental analysis. They are stable in solid state, soluble in water and other common organic solvents. The molecular structure of ligand (H2L) and complex [Cu(L)(phen)](1) is displayed in Fig. 1. A solid state structure is planed to perform X-ray crystallography (Fig. 2). The copper complex (1) crystallized in the monoclinic system of the space group P21/c with eight molecules in the unit cell (a = 15.1231(9) Å, b = 26.5571(15) Å, c = 12.1855(6) Å, α = 90°, β = 100.591(2) °, γ = 90° and Z = 8) and reveals a square pyramidal geometry. Crystallographic and experimental data is provided in Table 1. Selected bond lengths (Å) and angles (°) is illustrated in Table 2 . UV, IR spectroscopy and ESI-MS spectra [Fig. S1, ESI-MS: 716.80 (C22H17I2N2OSCu+, (M+H+)] was consistent with the assigned structure. Copper complex of formula C22H17CuI2N5OS was prepared from mixed-ligand; a dibasic tridentate-ONS ligand (H2L) and a 1, 10-phenathroline molecule as auxiliary ligand, as described in the experimental section, in good yield (70%). Optimized structure (Fig. S2) was obtained from quantum chemical calculations supported the formation of complex [Cu(L)(phen)](1). IR spectra of copper complex (KBr pellets) display an intense absorption at ca 1635 cm−1 attributed to (>C=N), shifted ca 5 cm−1 to lower wavenumbers compared with 1640 cm−1 for H2L. UV-Vis spectra of the copper complex display intense absorption at 265 nm (π -π*) and 445 nm (n-π*).
Fig. 1.

Molecular structure of Schiff base (H2L) and copper (II) complex [Cu(L)(phen)](1).
Fig. 2.

ORTEP diagrams (a & b) of copper (II) complex [Cu(L)(phen)](1) with 50% thermal ellipsoid. Hydrogen atoms are omitted for clarity.
Table 2.
Bond distances (Å) and bond angles (°) around Cu2+ centres found in complex [Cu(L)(phen)](1).
| Bond lengths (Å) | |||
|---|---|---|---|
| Cu(1)-N(3) | 1.9507(17) | Cu(1)-S(1) | 2.2524(6) |
| Cu(1)-O(1) | 1.9542(14) | Cu(1)-N(2) | 2.2904(18) |
| Cu(1)-N(1) | 2.0300(17) | Cu(2)-N(8) | 1.9478(18) |
| Cu(2)-O(2) | 1.9576(15) | Cu(2)-N(6) | 2.0156(18) |
| Cu(2)-S(2) | 2.2616(6) | Cu(2)-N(7) | 2.2820(18) |
| Bond angles (°) | |||
|---|---|---|---|
| N(3)-Cu(1)-O(1) | 92.46(6) | N(8)-Cu(2)-O(2) | 91.91(7) |
| N(3)-Cu(1)-N(1) | 176.19(7) | N(8)-Cu(2)-N(6) | 177.68(7) |
| O(1)-Cu(1)-N(1) | 83.89(7) | O(2)-Cu(2)-N(6) | 90.06(7) |
| N(3)-Cu(1)-S(1) | 85.84(5) | N(8)-Cu(2)-S(2) | 85.66(5) |
| O(1)-Cu(1)-S(1) | 166.63(5) | O(2)-Cu(2)-S(2) | 157.24(5) |
| N(1)-Cu(1)-S(1) | 97.46(5) | N(6)-Cu(2)-S(2) | 92.99(5) |
| N(3)-Cu(1)-N(2) | 103.11(7) | N(8)-Cu(2)-N(7) | 100.84(7) |
| O(1)-Cu(1)-N(2) | 86.26(6) | O(2)-Cu(2)-N(7) | 93.03(7) |
| N(1)-Cu(1)-N(2) | 77.79(7) | N(6)-Cu(2)-N(7) | 77.81(7) |
| S(1)-Cu(1)-N(2) | 107.06(5) | S(2)-Cu(2)-N(7) | 109.67(5) |
| N(4)-Cu(1)-N(3) | 121.50(12) | N(9)-Cu(2)-N(8) | 121.56(14) |
3.2. Crystal structures of [Cu(L)(phen)](1)
The geometry of the copper (II) complex [Cu(L)(phen)](1) has also been confirmed by an X-ray crystallographic study. The ORTEP representation with the atom numbering scheme is shown in Fig. 2. Crystallographic data and molecular structure refinement important interatomic parameters are given in Table 1. Selected bond lengths (Å) and angles (°) is illustrated in Table 2. The copper (II) complex [Cu(L)(phen)](1) crystallized in the monoclinic system of the space groups P21/c with eight molecules in the unit cell (a = 15.1231(9) Å, b = 26.5571(15) Å, c = 12.1855(6) Å, α = 90°, β = 100.591(2) °, γ = 90° and Z = 8). Copper possesses a distorted square pyramidal geometry ascertained by the observed τ value 0.6 (the structural index is defined as τ = (β-α)/60 where α and β are the largest coordination angles). For perfect square pyramidal and trigonal bipyramidal geometries, τ is zero and unity, respectively [51]. Such type of distorted square pyramidal geometry was observed in similar type of copper (II) complexes (τ = 0.09–0.58) [52]. The crystal structure of the copper (II) consists of a Cu(II) ions, ONS donor atoms of a Schiff base (H2L) and N, N donor atoms of 1, 10-phenanthroline , thus forming two five membered chelate rings [S(1)-C(15)-N(4)-N(3)-Cu(1) and N(2)-C(6)-C(5)-N(1)-Cu(1) or N(6)-C(27)-C(28)-N(7)-Cu(2) and S(2)-C(37)-C(38)-N(8)-Cu(2)] and one six membered chelate ring [O(1)-C(18)-C(17)-C(16)-N(3)-Cu(1) or N(8)-C(38)-C(39)-C(34)-O(2)-Cu(2)]. The Schiff base (H2L) is tridentate, binding through O(1), N(3), S(1) and/or O(2), N(8), S(2). The second ligand, 1,10-phenathroline is bidentate , and combines through N(1) and N(2) and/or N(6) and N(7) to have five coordination at the copper centre. The Cu-N/Cu-O /Cu-S bond lengths are comparable with similar copper(II) complexes [53,54]. The unit cell packing diagrams is presented in Fig. 3. An H-bonding and intermolecular interaction for copper complex is tabulated in Table 3 . As seen in packing diagrams, complex show H-bonding and intermolecular interactions , build from N-H…O and O-H…O; acting as bridges connecting neighboring dimeric units, which stabilize the crystal structure of this complex.
Fig. 3.

A packing view of copper (II) complex [Cu(L)(phen)](1) (along the a-axis).
Table 3.
Hydrogen bonds for Cu-complex [Cu(L)(phen)] (1) [Å and °].
| D-H...A | d(D-H) | d(H...A) | d(D...A) | <(DHA) |
|---|---|---|---|---|
| C(11)-H(11)...O(2) | 0.95 | 2.48 | 3.308(3) | 145.4 |
| C(13)-H(13C)...I(1)#1 | 0.98 | 3.18 | 4.060(2) | 150.9 |
| C(16)-H(16)...I(1)#1 | 0.95 | 3.14 | 3.945(2) | 143.6 |
| C(22)-H(22)...I(4)#1 | 0.95 | 3.31 | 3.848(2) | 117.8 |
| C(23)-H(23)...I(3)#2 | 0.95 | 3.26 | 4.111(3) | 149.5 |
| C(24)-H(24)...S(1) | 0.95 | 2.94 | 3.822(3) | 155.2 |
| C(35)-H(35A)...I(2)#3 | 0.98 | 3.30 | 3.999(2) | 129.8 |
| C(35)-H(35C)...I(2)#4 | 0.98 | 3.31 | 3.895(3) | 119.9 |
| C(36)-H(36B)...I(2)#4 | 0.98 | 3.32 | 3.896(4) | 119.7 |
Symmetry transformations used to generate equivalent atoms: #1 -x+2,-y+1,-z+1 #2 x, y, z-1 #3 x-1,-y+3/2, z-1/2 #4 x-1, y, z
3.3. Optimized geometry of [Cu(L)(phen)](1)
The molecular structure of complex [Cu(L)(phen)](1) is also obtained computationally. The geometry optimization of complex [Cu(L)(phen)](1) was performed at the ωB97X-D/6-311+G** level of theory in vacuum using the Gaussian16 software package [41] and cross-validated using the Spartan 16/18 parallel suite of programs [42]. The optimized structure of copper (II) complex [Cu(L)(phen)](1) was displayed in Fig. S2. The bond length and bond angle of the optimized structure was also collected. As shown in Fig. S2, the experimental bond lengths of the Cu(II) ion with O(1), N(1) and S(1) atoms are 1.954, 2.030 and 2.252 Å, which are found to be 1.923, 2.010 and 2.232 Å, in the optimized geometry , respectively. The bond lengths of the Cu(II) metal ion and N(4), N(5) atoms of 1,10-phenanthroline ring was 2.043 and 2.047 Å in experimental and computed geometry, respectively.
3.4. Frontier molecular orbital (FMO) analysis
The chemical reactivity and stability of the compounds is related to the contribution of frontier orbitals containing HOMO and LUMO location on different parts of the compounds [55]. Furthermore, the differences between the energies of the HOMO and LUMO show different parameters such as activity, stability and excitability. For example a large HOMO-LUMO energy gap, means high excitation energies for many excited states, a good stability and a high chemical hardness for the compounds. So, the energy gap of HOMO and LUMO was calculated in order to investigation the stability of ligand (H2L) and complex [Cu(L)(phen)](1). The FMOs (HOMO/ LUMO and HOMO-1/ LUMO+1) energies of ligand (H2L) and complex [Cu(L)(phen)](1) were explored and their results are tabulated in Table 4 , while their pictorial description of charge densities is displayed in Figs. S3 and S4, respectively. The calculated energy gap values of HOMO/ LUMO and HOMO-1/ LUMO+1 are 3.6 and 4.1 eV for ligand (H2L); 0.7 and 2.0 eV for complex [Cu(L)(phen)](1). This energy difference (∆E) between HOMO and LUMO orbitals for ligand and its complex is found to be unequal magnitude. The highest value of ∆E revealed that ligand (H2L) has high chemical stability and low reactivity. Accompanying the energies of FMOs, its analysis also explained the ICT transitions in the ligand and copper complex. The pictograms of charge densities for HOMO/ LUMO and HOMO-1 and LUMO+1 are displayed in Figs. S3 and S4. The charge densities for HOMO are located over the (Z)-N’-((E)-2-hydroxy-3,5-diiodobenzylidene part in ligand (H2L), while those for LUMO are concentrated at N,N-dimethylcarbamohydrazonothioic acid. Similarly, the LUMO for the complex [Cu(L)(phen)](1) was located on the metal and the HOMO located on non-metal part. The HOMO and LUMO orbitals are nearly degenerate as shown by a small energy gap and a similar distribution pattern between these two orbitals in the copper complex (Fig. S4).
Table 4.
The HOMO and LUMO energies, the energy gap (∆E)a, ionization potential (IP), electro negativity (χ), electron affinity (EA), chemical potential (μ), global softness (σ), global hardness (η), and global electrophilicity (ω) for ligand (H2L) and complex [Cu(L)(phen)] (1).
| Compounds | EHOMO | ELUMO | ∆E | EHOMO-1 | ELUMO+1 | ∆E | IP | EA | χ | η | μ | ω | σ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2L | -5.6 | -2.0 | 3.6 | -6.0 | -1.9 | 4.1 | 4.23 | 0.23 | 2.67 | 2.21 | -2.0 | 1.61 | 0.24 |
| Complex (1) | -4.9 | -4.2 | 0.7 | -5.3 | -3.3 | 2.0 | 7.68 | 0.60 | 4.23 | 3.62 | -4.2 | 2.42 | 0.13 |
Energy gap (∆E) = ELUMO-EHOMO; units in eV.
3.5. Global reactivity parameters (GRPs) analysis
The reactive nature of compounds can be determined by global reactivity parameters (GRPs), ionization potential (I), electro negativity (χ), electron affinity (A), chemical potential (μ), global softness (σ), global hardness (η), and global electrophilicity (ω) using HOMO/LUMO energies [56]. The ionization potential and electron affinity might be utilized to depict the electron gaining and losing capacity of molecules, which interrelated with HOMO/LUMO energies [57]. In frontier molecular orbitals (FMOs), we have used following equations for global reactivity descriptors [58] as given below:
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
The results obtained from Eqs. (1)–(7) are tabulated in Table 4 indicate that the values of ionization potential are noticed to be higher than electron affinity, which disclosed that ligand (H2L) has better capability to accept an electron. Similarly, in the studied complex, the ionization potential was found to be much higher than electron affinity (A) values. The stability as well as reactivity of a chemical system was correlated to chemical potential and global hardness values [59]. The stability had a direct relation with global hardness, whereas it had an inverse relationship to its reactivity [60].
3.6. Molecular electrostatic potentials (MEPs) analysis
The molecular electrostatic potential is related to the electron density and can be used for describing chemical reactivity of compounds [55]. The 3-D plots of the MEPs (molecular electrostatic potentials); LIPs (local ionization potentials) and spin density maps for [Cu(L)(phen)](1) is displayed in Fig. S5. According to Fig. S5, the higher negative potentials (reddish yellow) are found on oxygen and sulfur atoms of deprotonated Schiff base ligand coordinated to copper(II) ion. There is a visible positive potential (greenish blue) on the surface of copper (II) complex [Cu(L)(phen)](1), which indicates its susceptibility towards further nucleophilic substitution reaction. The two nitrogen atoms of 1, 10-phenathroline ligand show the slight positive potential with slightly greenish blue surface as compared to the rest of the moieties in this complex. The positive and negative regions can interpret the location of interactions of this complex, when they get involved in electrophilic or nucleophilic interactions.
3.7. Molecular docking with Mpro of COVID-19
There are several Schiff bases and copper(II) complexes which showed good inhibition properties against COVID-19. For example, Johnson et al. reported [61] that the copper(II) complex of salicylaldehyde benzoyl hydrazone exhibits significantly better inhibitory activity than does salicylaldehyde benzoyl hydrazone itself and also found relatively non-toxic to mice cells. Adamantanamine hydrochloride is a well-known antiviral drug which a mutation makes influenza virus as amantadine (AdNH2) resistant so a new formalism with copper(II) adamantanamines was found to be effective [62]. Very recently, Andreou et al. [47] have also studied the role of copper and N-acetylcysteine (NAC) in conjugation with antiviral treatments for SARS-CoV-2. In order to find potent SARS-CoV-2 inhibitor, we have docked the ligand (H2L) and copper(II) complex [Cu(L)(phen)](1) with main protease (MPro) of spike protein (PDB ID: 7C8U) of SARS-COV-2, which is very vital for attaching the virus to its host cell [63]. The stronger binding was predicted between ligand and receptor protein through negative value of binding affinity [64]. As the molecular docking was done to predict the binding affinity and pose of ligand against SARS-COV-2 main protease (PDB ID: 7C8U), we have collected the molecular docking results tabulated in Table 5 . These include the most important parameters including the binding affinity, inhibition constant and different amino acid residues of Mpro that interacts with the ligand (H2L) and copper (II) complex [Cu(L)(phen)](1). The docked ligand (H2L) and copper (II) complex [Cu(L)(phen)](1) inside the Mpro protein with its focussed view for interacting residues are displayed in Figs. 4 and 5 , respectively. Selected amino acid residues between ligand, complex and COVID-19-Mpro (PDB ID: 7C8U) are ; HIS-163, HIS-41, SER-144, LEU-141, MET-165, GLU-166, HIS-172, THR-190 for docked ligand (H2L); LEU-262, HIS-41,VAL-297,LEU-141,GLU-166, VAL-171,ALA-193, THR-196 for docked complex [Cu(L)(phen)](1).The binding affinity and inhibition constant were found to be -7.14 kcal/mol and 5.82 µM, -6.18 kcal/mol and 0.76 µM, respectively, for the best-docked confirmation of ligand (H2L) and copper (II) complex [Cu(L)(phen)](1) with Mpro protein at inhibition bounding site of receptor protein (PDB ID: 7C8U). This binding affinity is reasonably well as compared to recently known antiviral drugs. For instance, the binding affinity of the ligand (H2L) and copper (II) complex [Cu(L)(phen)](1) was found to be better than recently docked results of chloroquine (-6.293 kcal/mol), hydroxychloroquine (-5.573 kcal/mol) and remdesivir (-6.352 kcal/mol) with Mpro protein [65]. The inhibitory activity results is comparable with our previous work on copper(II) coordination complex [38].
Table 5.
The molecular docking results for Schiff base ligand (H2L), complex [Cu(L)(phen)] (1), and Mpro including the binding affinity, inhibition constant and different amino acid residues of Mpro that interact with the ligands.
| Molecule | Binding affinity (kcal/mol) | Inhibition constant, Ki (µM) | Interacting protein residues |
||
|---|---|---|---|---|---|
| H-bond | Electrostatic | Hydrophobic | |||
| H2L | -7.14 | 5.82 | HIS-163 (5.44Å) |
HIS-41(4.59Å), SER-144(2.88Å), LEU-141(3.20Å) |
MET-165(2.56Å), GLU-166(1.89Å), HIS-172 (5.69Å), THR-190(3.28 Å). |
| [Cu(L)(phen)](1) | -6.18 | 0.76 | LEU-262 (4.23Å) |
HIS-41(3.28Å), VAL-297(4.42Å), LEU-141(1.96Å), |
GLU-166(2.37Å), VAL-171 (2.78Å), ALA-193(3.09Å), THR-196(3.05Å) |
Fig. 4.

The docked Schiff base ligand (H2L) inside the Mpro protein (PDB ID: 7C8U) with its focused view for interacting residues around the docked ligand.
Fig. 5.

The docked copper (II) complex [Cu(L)(phen)](1) inside the Mpro protein (PDB ID: 7C8U) with its focused view for interacting residues around the docked ligand.
3.8. Intermolecular interactions with Mpro of COVID-19
Apart from the binding affinity and inhibition constant values, it is also very important to visualize the hydrogen bond interactions, and intermolecular interactions between ligand (H2L), copper (II) complex [Cu(L)(phen)](1) and Mpro protein. A representation of docked ligand (H2L) inside the Mpro protein (PDB ID: 7C8U) with its focused view for interacting residues along with H-bond and intermolecular interactions is displayed in Fig. 6 . There is observed one π-hydrogen bond with a distance of 5.44 Å between NH2 of HIS-163 residue and π-electrons of ligand (H2L). Beside the π-hydrogen bond, there are also seen three π-anionic electrostatic interactions between π-orbitals of ligand (H2L) and (COO−) group of anionic HIS-41, SER-144 and LEU-141 residue. The third kind of four interactions is seen as hydrophobic, which are between the π-electrons of ligand and π-electrons of MET-165, GLU-166, HIS-172, THR-190 residues with distances of 2.56, 1.89, 5.69 and 3.28 Å, respectively. There are some important hydrogen bond and intermolecular interactions was also seen between complex [Cu(L)(phen)](1) and Mpro protein. These are summarized as: one π-hydrogen bond with a distance of 4.23 Å between NH2 of LEU-262 residue and π-electrons of ligand (H2L) inside the complex and Mpro; three π-anionic electrostatic interactions between π-orbitals of ligand (H2L) and (COO−) group of anionic HIS-41, VAL-297 and LEU-141 residue with distances of 3.28, 4.42 and 1.96 Å, respectively; four hydrophobic interactions, which are between the π-electrons of ligand and π-electrons of GLU-166, VAL-171, ALA-193 and THR-196 residues with distances of 2.37, 2.78, 3.09 and 3.05 Å, respectively. This type of π-π- stacked interactions is supposed to increase binding affinity in ligand (H2L). Similar observations was found for complex [Cu(L)(phen)](1). Hence, ligand (H2L) and complex [Cu(L)(phen)](1) with SARS-COV-2 main protease (7C8U) was stabilized , and ligand (H2L) was fitted within the substrate binding packet of SARS-COV-2 main protease (PDB ID: 7C8U) having different binding modes, hydrogen bond interactions, and hydrophobic interactions.
Fig. 6.

The representation of docked ligand (H2L) inside the Mpro protein (PDB ID: 7C8U) with its focused view for interacting residues along with H-bond and intermolecular interactions; (a) H-bond donor and acceptor meshes represented by pink and green colors, respectively; (b) hydrophobic pocket represented with blue and gray colors; (c) ionizability represented by blue and orange colors; (d) SAS represented by blue and green colors, respectively (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Furthermore, H-bond is considered as a significant factor for RNA structure in biological system and less than 2.3 Å increases the binding affinity. Most significant H-bonds of ligand (H2L) with HIS-163, and complex [Cu(L)(phen)](1) with LEU-262 of amino acid residues of protein in a distance 5.44 Å and 4.23 Å, respectively was identified which is closure to standard antiviral drugs [62]. Instead of conventional hydrogen bonding between ligand and main protease, the other factors (π-bonding, dipole-dipole interactions, electrostatic interactions and hydrophobic interactions) affect the effectiveness of these compounds against the COVID-19 virus. An intuitive view of the docking cavity is displayed in Figs. 7 and 8 by calculating the total density surfaces of MPro protein along with the docked ligand (H2L) and complex [Cu(L)(phen)](1) based on H-bonding and hydrophobic interactions as shown in Fig. 7(a) and (b) for ligand (H2L), Fig. 8(a) and 8(b) for complex [Cu(L)(phen)](1), respectively. The total density surface with H-bond donor is those residues which possess polar hydrogen atoms and represented with green mesh color while H-bond acceptor is those protein residues which possess electronegative atoms and represented with red mesh colors. Fig. 7 shows that docked conformations and hydrophobic interactions of ligand (H2L) inside the Mpro protein at inhibition bounding site of receptor protein of SARS-COV-2 main protease (PDB ID: 7C8U): (a) with hydrogen bond donor and acceptor meshes represented by yellow and green colors, respectively (b) Surface representation of hydrophobic pocket represented with pink and blue colors. Fig. 8 shows the total density surfaces representation for docked copper (II) complex [Cu(L)(phen)](1) inside the Mpro protein (PDB ID: 7C8U) (a) with hydrogen bond donor and acceptor meshes represented by light green and red colors, respectively (b) Surface representation of hydrophobic pocket represented with dee pink and light blue colors. The results obtained from molecular docking indicates that there are H-bond and hydrophobic interactions between of MPro protein and docked ligand (H2L) and complex [Cu(L)(phen)] (1), which is often considered as crucial factors for potential inhabitation activity.
Fig. 7.

The total density surfaces representation for docked Schiff base ligand (H2L) inside the Mpro protein (PDB ID: 7C8U) (a) with hydrogen bond donor and acceptor meshes represented by yellow and green colors, respectively (b) Surface representation of hydrophobic pocket represented with pink and blue colors (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Fig. 8.

The total density surfaces representation for docked copper (II) complex [Cu(L)(phen)](1) inside the Mpro protein (PDB ID: 7C8U) (a) with hydrogen bond donor and acceptor meshes represented by light green and red colors, respectively (b) Surface representation of hydrophobic pocket represented with dee pink and light blue colors (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
3.9. Swiss-ADME analysis
Swiss-ADME is important to analyze the pharmacokinetics and pharmacodynamics properties of the proposed molecules which could be used as drugs. Metal (II) complexes have evoked much interest for potential applications in perspective drug candidates [66,67]. The in-silico Swiss-ADME predicted that the ligand (H2L) and its copper(II) complex [Cu(L)(phen)](1) shows positive response to both blood brain barrier (BBB) and human intestinal absorption (HIA) penetration for drug discovery. The ligand was found to be non-carcinogenic and does not inhibit the P-glycoprotein. Therefore, it has protective capacity to interrupt the absorption, permeability, and retention of the drugs [68]. Swiss-ADME computed results for the ligand (H2L) and its complex [Cu(L)(phen)](1) in the different sections of the one-panel-per-molecule outputs is depicted in Table S1. The physicochemical properties like molecular weight (MW), molecular refractivity (MR), count of specific atom types and polar surface area (PSA) are compiled in bioavailability section which shows important properties for oral bioavailability. The bioavailability of molecules based on well-absorbed and poorly absorbed their lipophilicity and polarity as described by the n-octanol/water partition coefficient (log-P) and the polar surface area (PSA). The physicochemical space for oral bioavailability of the biomolecules represents the optimal range for each properties [(lipophilicity: XLOGP3 = 4.81, size: MW= 475.31 g/mol, polarity: TPSA = 32.59 Å2, solubility: logS = -6.94, saturation: Fraction Csp3 = 0.00, flexibility: number of rotatable bonds = 2 for H2L; lipophilicity: XLOGP3 = 15.58, size: MW = 716.80 g/mol, polarity: TPSA = 204.67 Å2, solubility: logS = -11.22, saturation: Fraction Csp3 = 0.00, and flexibility: number of rotatable bonds = 6 for [Cu(L)(phen)](1)]. Complex [Cu(L)(phen)](1) was predicted not orally bioavailable, because too flexible and too polar structure.
The lipophilicity and polarity of small biomolecules are collected in the BOILED-Egg section for ligand (H2L) and its complex [Cu(L)(phen)](1). The BOILED-Egg results (BBB and HIA) is given in Table S1, which can be applied to the evaluation of drug candidates for small biomolecules development [Pharmacokinetics: BBB permeant = Yes, P-gp substrate = No, CYP1A2/ CYP2C19/ CYP2C9 = Yes, CYP2D6/CYP3A4 inhibitor = No, Log Kp (skin permeation) = -4.87 cm/s, drug-likeness: bioavailability score = 0.55, medicinal chemistry: PAINS = 0, drug-likeness = No, synthetic accessibility = 2.68 for H2L; BBB permeant = No, P-gp substrate = Yes, CYP1A2/CYP2C19/ CYP2C9/CYP2D6/CYP3A4 inhibitor = No, LogKp (skin permeation) = -2.35 cm/s, drug-likeness: bioavailability score = 0.17, medicinal chemistry: PAINS = 0, drug-likeness = No, Synthetic accessibility = 6.40 for complex [Cu(L)(phen)](1)]. The complex [Cu(L)(phen)](1)] was predicted as not absorbed and not BBB penetrant (outside the Egg) as compared with standard drug (streptomycin with a TPSA of 331.43 Å2 and a WLOGP of -7.74). One major role of P-gp is to protect the central nervous system (CNS) from xenobiotics. Importantly as well, P-gp is overexpressed in some tumor cells and leads to multidrug-resistant cancers. It has been suggested that cytochromes P450 (CYP) and P-gp can process small molecules synergistically to improve protection of tissues and organisms. Schiff base ligand (H2L) and its complex [Cu(L)(phen)] (1) are substrates or inhibitors of five isoenzymes (CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4) governing important pharmacokinetic behaviors. Inhibition of these isoenzymes is certainly one major cause of pharmacokinetics-related drug-drug interactions. Similar observations were found in our previous work on copper(II) complexes [69].
4. Conclusions
-
(i)
We have successfully reported the synthesis and structural characterization of new Schiff base (H2L) and its mixed ligand copper (II) complex [Cu(L)(phen)](1).
-
(ii)
Single crystal X-ray crystallographic analysis of copper (II) complex [Cu(L)(phen)](1) is determined, which confirmed the square pyramidal geometry around Cu(II) ions.
-
(iii)
FMO findings explored the HOMO/LUMO bandgap of copper (II) complex [Cu(L)(phen)](1) in the range of 0.7–2.0 eV. GRPs explicated that [Cu(L)(phen)](1)showed greater ionization potential (IP) (7.68 eV) than the electron affinity (EA) (0.6 eV); hence, complex has greater electron-donating ability, while ligand has greater electron-accepting ability((IP: 4.23 eV and EA:0.23 eV) . Nevertheless, [Cu(L)(phen)](1) also expressed higher values of hardness (η = 3.62 eV) and a smaller value of softness (σ = 0.13 eV) compared to ligand (η = 2.21 eV and σ = 0.24 eV).\
-
(iv)
The Swiss-ADME analysis predicted that ligand and copper (II) complex [Cu(L)(phen)](1) showed positive response to blood brain barrier (BBB) and human intestinal absorption (HIA) penetration for drug discovery.
-
(v)
Additionally, a molecular docking study was performed for Schiff base (H2L) and copper (II) complex [Cu(L)(phen)](1) to see an affinity for the main protease (Mpro) of COVID-19 spike protein. Interestingly, the results are found quite encouraging where the binding affinity and inhibition constant were found to be -7.14 kcal/mol and 5.82 µM, respectively, for the best-docked confirmation of ligand as compared with complex (-6.18 kcal/mol and 0.76 µM, respectively) with Mpro protein of COVID-19. This binding affinity is reasonably well as compared to recently known and docked antiviral drugs like chloroquine (-6.293 kcal/mol), hydroxychloroquine (-5.573 kcal/mol) and remdesivir (-6.352 kcal/mol) with Mpro protein.
-
(vi)
Overall, the present study predicts that ligand and complex have ability to stabilized with Mpro, may have the potency to be evolved as an anti-COVID main protease drug to fight against the novel coronavirus, but before that, it must go through under the proper preclinical and clinical trials for further experimental and/or clinical scientific validation. .
CRediT authorship contribution statement
Bharti Mohan: Writing – original draft, Writing – review & editing. Mukesh Choudhary: Supervision, Conceptualization, Data curation, Formal analysis, Investigation, Methodology.
Declaration of Competing Interest
The authors declare no conflict of interest.
Acknowledgments
The author is thankful to the Sophisticated Analytical Instrument Facility (SAIF), IIT Madras, India for single crystal data collection of copper (II) complex [Cu(L)(phen)](1). The authors thank to the SAIF, IIT(ISM), India for HRMS analysis. For computer time, this research used the resources of the computational facility at Department of Chemistry, NIT Patna, India.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2021.131246.
Appendix. Supplementary materials
Supplementary material (X-ray crystallography)
Deposition number: CCDC-2066722 for [Cu(L)(phen)](1) contain the supplementary crystallographic data for this paper. This data is provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinfornationszentrum Karlstruhe Access Structures service www.ccdc.cam.ac.uk/structures.
References
- 1.Chen N., Zhou M., Dong X., Qu J., Gong F., Han Y., Qiu Y., Wang J., Liu Y., Wei Y., Xia J., Yu J., Xang X., Zhang L. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. The Lancet. 2020;395(10223):507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Q., Guan X., Wu P., Wang X., Zhou L., Tong Y., Ren R., Leung K.S.M., Lau E.H.Y., Wong J.Y., Xing X., Xiang N., Wu Y., Li C., Chen Q., Li D., Liu T., Zhao J., Liu M., Tu W., Chen C., Jin L., Yang R., Wang Q., Zhou S., Wang R., Liu H., Luo Y., Liu Y., Shao G., Li H., Tao Z., Yang Y., Deng Z., Liu B., Ma Z., Zhang Y., Shi G., Lam T.T.Y., Wu J.T., Gao G.F., Cowling B.J., Yang B., Leung G.M., Feng Z. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020;382:1199–1207. doi: 10.1056/NEJMoa2001316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) van Doremalen N., Bushmaker T., Morris D.H., Holbrook M.G., Gamble A., Williamson B.N., Tamin A., Harcourt J.L., Thornburg N.J., Gerber S.I., Smith J.O.L., de Wit E., Munster V.J. N. Engl. J. Med. 2020;382(16):1564–1567. doi: 10.1056/NEJMc2004973. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Acter T., Uddin N., Das J., Akhter A., Choudhury T.R., Kim S. Sci. Total Environ. 2020 doi: 10.1016/j.scitotenv.2020.138996. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gage A., Brunson K., Morris K., Wallen S.L., Dhau J., Gohel H., Kaushik A. Front. Nanotechnol. 2021 doi: 10.3389/frano.2021.700888. [DOI] [Google Scholar]
- 4.Mittal L., Kumari A., Srivastava M., Singh M., Asthana S. Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. J. Biomol. Struct. Dyn. 2021;39(10):3662–3680. doi: 10.1080/07391102.2020.1768151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Al-Khafaji K., Al-Duhaidahawil D., Tok T.T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dyn. 2021;39(9):3387–3395. doi: 10.1080/07391102.2020.1764392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., Yuan M.L., Zhang Y.L., Dai F.H., Liu Y., Wang O.M., Zheng J.J., Xu L., Holmes E.C., Zhang Y.Z. Nature. 2020;579:565–569. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Kozak J.J., Gray H.B., Garza-Lopez R.A. Structural stability of the SARS-CoV-2 main protease: Can metal ions affect function? J. Inorg. Biochem. 2020;211 doi: 10.1016/j.jinorgbio.2020.111179. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Li G., Clercq E.D. Therapeutic options for the 2019 novel coronavirus (2019-nCoV) Nat. Rev. Drug Discov. 2020;19:149–150. doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed] [Google Scholar]; (b) Touret F., de Lamballerie X. Of chloroquine and COVID-19. Antivir. Res. 2020;177 doi: 10.1016/j.antiviral.2020.104762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu J.T., Leung K., Bushman M., Kishore N., Niehus R., de Salazar P.M., Cowling B.J., Lipsitch M., Leung G.M. Nat. Med. 2020;26:506–510. doi: 10.1038/s41591-020-0822-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheahan T.P., Sims A.C., Leist S.R., Schäfer A., Won J., Brown A.J., Montgomery S.A., Hogg A., Babusis D., Clarke M.O., Spahn J.E., Bauer L., Sellers S., Porter D., Feng J.Y., Cihlar T., Jordan R., Denison M.R., Baric R.S. Nat. Commun. 2020;11:1–14. doi: 10.1038/s41467-019-13940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das P., Majumder R., Mandal M., Basak P. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1796799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yadav R., Imran M., Dhamija P., Chaurasia D.K., Handu S. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1796812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Razzaghi-Asl N., Ebadi A., Shahabipour S., Gholamin D. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1797536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Janabi A.S.M., Elzupir A.O., Yousef T.A. J. Mol. Struct. 2021;1228 doi: 10.1016/j.molstruc.2020.129454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Gammal O.A., Sh. Mohamed F., Rezk G.N., El-Bindary A.A. J. Mol. Liq. 2021;330 doi: 10.1016/j.molliq.2021.115522. [DOI] [Google Scholar]
- 17.Uddin M.N., Amin M.S., Rahman M.S., Khandaker S., Shumi W., Rahman M.A., Rahman S.M. Appl. Organomet. Chem. 2021;35(1) doi: 10.1002/aoc.6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mir J.M., Majid S.A., Shalla A.H. Rev. Inorg. Chem. 2021 doi: 10.1515/revic-2020-0020. [DOI] [Google Scholar]
- 19.Hussein R.K., Elkhair H.M. J. Mol. Struct. 2021;1231 doi: 10.1016/j.molstruc.2021.129979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer-Almes F.J. Comput. Biol. Chem. 2020;88 doi: 10.1016/j.compbiolchem.2020.107351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pal M., Musib D., Roy M. New J. Chem. 2021;45:1924–1933. [Google Scholar]
- 22.Gao J., Tian Z., Yang X. Biosci. Trends. 2020;14:72–73. doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 23.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sportelli M.C., Longano D., Bonerba E., Tantillo G., Torsi L., Sabbatini L., Cioffi N., Ditaranto N. Molecules. 2020;25:49. doi: 10.3390/molecules25010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkarni S.A., Nagaranjan S.K., Ramesh V., Palaniyandi V., Selvam S.P., Madhavan T. J. Mol. Struct. 2020;1221 doi: 10.1016/j.molstruc.2020.128823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venkateshan M., Muthu M., Kumar R.R. J. Mol. Struct. 2020;1220 doi: 10.1016/j.molstruc.2020.128741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altalhi T.A., Alswat K., Alsanie W.F., Ibrahim M.M., Hamdy A.A., E.Sheshtwy S. J. Mol. Struct. 2021;1228 doi: 10.1016/j.molstruc.2020.129459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baildya N., Ghosh N.N., Chattopadhyay A.P. J. Mol. Struct. 2020;1219 doi: 10.1016/j.molstruc.2020.128595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardoso W.B., Mendanha S.A. J. Mol. Struct. 2021;1225 doi: 10.1016/j.molstruc.2020.129143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghosh K., Amin S.A., Gayen S., Jha T. J. Mol. Struct. 2021;1224 doi: 10.1016/j.molstruc.2020.129026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Powelczyk A., Zaprutk L. Future Med. Chem. 2020;12(19):1743–1757. doi: 10.4155/fmc-2020-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mansour M.A., Aboulmagd A.M., Abdel-Rahman H.M. RSC Adv., 2020;10:34033–34045. doi: 10.1039/d0ra06424f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alshammari M.B., Ramadan M., Aly A.A., El-Sheref E.M., Bakht M.A., Ibrahim M.A.A., Shawky A.M. J. Mol. Struct. 2020 doi: 10.1016/j.molstruc.2020.129649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Terbouche C.A.R., Abdeldjebar H., Lakhdari H., Bachari K., Roisnel T., Hauchard D. J. Mol. Struct. 2020;1222 doi: 10.1016/j.molstruc.2020.128918. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Al-Janabi A.S.M., Eljupir A.O., Yousef T.A. J. Mol. Struct. 2021;1228 doi: 10.1016/j.molstruc.2020.129454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maldonado N., Amo-Ochoa P. Dalton Trans. 2021;50:2310–2323. doi: 10.1039/d0dt04066e. [DOI] [PubMed] [Google Scholar]
- 36.Raha S., Mallick R., Basak S., Duttaroy A.K. Med. Hypotheses. 2020;142 doi: 10.1016/j.mehy.2020.109814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cartos A.A., Jiniga J.M. Diagn. Microbiol. Infect. Dis. 2020;98(4) doi: 10.1016/j.diagmicrobio.2020.115176. [DOI] [Google Scholar]
- 38.Mohan B., Muhammad S., Al-Sehemi A.G., Bharti S., Kumar S., Choudhary M. Chem. Sel. 2021;6:738–745. [Google Scholar]
- 39.Sheldrick G.M. Acta Crystallogr. Sect. A. 1990;46:467. [Google Scholar]
- 40.Sheldrick G.M. Acta Crystallogr. Sect. A. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 41.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, Gaussian 16 Rev. A.03. Wallingford, CT, 2016.
- 42.Lambert H., Mohan N., Lee T.C. Phys. Chem. Chem. Phys. 2019;21:14521–14529. doi: 10.1039/c9cp01762c. [DOI] [PubMed] [Google Scholar]
- 43.Chai J.D., Gordon M.H. Phys. Chem. Chem. Phys. 2008;10:6615–6620. doi: 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- 44.Morris G., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trott O., Olson A.J. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biovia D.S. Dassault Systèmes; San Diego: 2016. Discovery Studio Visualizer, v17.2.0.16349. 2016. [Google Scholar]
- 47.Andreou A., Trantza S., Filippou D., Sipsas N., Tsiodras S. COVID-19: The Potential Role of Copper and N-acetylcysteine (NAC) in a Combination of Candidate Antiviral Treatments Against SARS-CoV-2. In Vivo. 2020;34(3):1567–1588. doi: 10.21873/invivo.11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.X. Luan, W. Shang, Y. Wang, W. Yin, Y. Jiang, S. Feng, Y. Wang, M. Liu, R. Zhou, Z. Zhang, F. Wang, W. Cheng, M. Gao, H. Wang, W. Wu, R. Tian, Z. Tian, Y. Jin, H.W. Jhiang, L. Zhang, H.E. Xu, S. Jhang, RCSB PDB-7C8U: the crystal structure of COVID-19 main protease in complex with GC376, 2020, 10.2210/pdb7C8U/pdb; htttp://www.rcsb.org/structure/7C8U
- 49.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Adv. Drug. Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 50.(a) Daina A., Zoete V. Chem. Med. Chem. 2016;11:1117. doi: 10.1002/cmdc.201600182. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daina A., Michielin O., Zoete V. Sci. Rep. 2017;7:42717. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subramanian P.S., Suresh E., Dastidar P., Waghmode S., Srinivas D. Inorg. Chem. 2001;40:4291. doi: 10.1021/ic010182d. [DOI] [PubMed] [Google Scholar]
- 52.Patel R.N., Singh N., Patel D.K., Gundla V.L.N. Indian J. Chem. 2007;46A:422–427. [Google Scholar]
- 53.Tewari R., Srivastava R.C., Balundgi R.H., Chakravorty A. Inorg. Nucl. Chem. Lett. 1973;9(5):583–586. [Google Scholar]
- 54.Patel M.N., Parmar P.A., Gandhi D.S. Bioorg. Med. Chem. 2010;18(3):1227–1235. doi: 10.1016/j.bmc.2009.12.037. [DOI] [PubMed] [Google Scholar]
- 55.Mohan B., Choudhary M., Bharti S., Jana A., Das N., Muhammad S., Al-Sehemi A.G., Kumar S. J. Mol. Struct. 2019;1190:54–67. [Google Scholar]
- 56.Parr R.G., Szentpaly L.V., Liu S. J. Am. Chem. Soc. 1999;121:1922–1924. [Google Scholar]
- 57.Fukui K. Angew. Chem. Int. Ed. Engl. 1982;21:801–809. [Google Scholar]
- 58.Lawal M.M., Govender T., Maguire G.E., Kruger H.G., Honarparvar B. Int. J. Quantum Chem. 2017;118:25497. [Google Scholar]
- 59.Sert Y., Sreenivasa S., Doǧan H., Mohan N.R., Suchetan P.A., Ucun F. Spectrocrim. Acta A. 2014;130:96–104. doi: 10.1016/j.saa.2014.03.061. [DOI] [PubMed] [Google Scholar]
- 60.Murugavel S., Velan V.V., Kannan D., Bakthadoss M. J. Mol. Struct. 2016;1108:150–167. [Google Scholar]
- 61.Johnson D.K., Murphy T.B., Rose N.J., Goodwin W.H., Pickart L. Inorg. Chim. Acta. 1982;67:159–165. [Google Scholar]
- 62.Banti C., Kourkoumelis N., Hatzidimitriou A., Antoniadou I., Dimou A., Rallis M., Hoffmonn A., Schmidtke M., McGuire K., Busath D., Kolocouris A., Hadjikakou S.K. Polyhedron. 2020;185 [Google Scholar]
- 63.Boopathi S., Poma A.B., Kolandaivel P. J. Biomol. Struct. Dyn. 2020;30:1–10. [Google Scholar]
- 64.Ferreira L., dosSantos R., Oliva G., Andricolulo A. Molecules. 2015;20:13384. doi: 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang S., Krumberger M., Morris M.A., Parrocha C.M.T., Griffin J.H., Kreutzer A., Nowick J.S. Structure-Based Drug Design of an Inhibitor of the SARS-CoV-2 (COVID-19) Main Protease Using Free Software: A Tutorial for Students and Scientists. ChemRxiv. 2020 doi: 10.26434/chemrxiv.12791954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fenton R.R., Gauci R., Junk P.C., Lindoy L.F., Luckay R.C., Meehan G.V., Price J.R., Turner P., Wei G. J. Chem. Soc. Dalton Trans. 2002;10:2185–2193. [Google Scholar]
- 67.Khan T., Ahmad R., Azad I., Raza S., Joshi S., Khan A.R. Comput. Biol. Chem. 2018;75:178–195. doi: 10.1016/j.compbiolchem.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Gleich A., Kaiser B., Honscha W., Fuhrmann H., Schoeniger A. Cytotechnology. 2019;71:231. doi: 10.1007/s10616-018-0279-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mohan B., Choudhary M., Muhammad S., Das N., Singh K., Jana A., Bharti S., Algarni H., Al-Sehemi A.G., Kumar S. J. Coord. Chem. 2020;73(8):1256–1279. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material (X-ray crystallography)
Deposition number: CCDC-2066722 for [Cu(L)(phen)](1) contain the supplementary crystallographic data for this paper. This data is provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinfornationszentrum Karlstruhe Access Structures service www.ccdc.cam.ac.uk/structures.