

Abstract
Chlamydia trachomatis is frequently detected in the human gastrointestinal tract despite its leading role in sexually transmitted bacterial infections in the genital tract. Chlamydia muridarum, a model pathogen for investigating C. trachomatis pathogenesis in the genital tract, can also colonize the mouse gastrointestinal tract for long periods. Genital tract mutants of C. muridarum no longer colonize the gastrointestinal tract. The mutants lacking plasmid functions are more defective in colonizing the upper gastrointestinal tract while certain chromosomal gene-deficient mutants more defective in the lower gastrointestinal tract, suggesting that Chlamydia may use the plasmid for promoting its spreading to the large intestine while using the chromosome-encoded factors for maintaining its colonization in the large intestine. The plasmid-encoded Pgp3 is critical for Chlamydia to resist the acid barrier in the stomach and to overcome a CD4+ T cell barrier in the small intestine. Once reaching the large intestine, Pgp3 is no longer required. Instead, the chromosome-encoded open reading frames TC0237/TC0668 become essential for Chlamydia to evade the group 3-like innate lymphoid cell-secreted IFNγ in the large intestine. These findings are important for exploring the medical significance of chlamydial colonization in the gut and understanding the mechanisms of chlamydial pathogenicity in the genital tract.
I. Chlamydia establishes long-lasting colonization in the gastrointestinal (GI) tracts.
Chlamydia trachomatis (CT) is a leading cause of sexually transmitted bacterial infections, which, if untreated, can trigger upper genital tract pathology, leading to long-lasting sequelae such as tubal infertility (http://www.cdc.gov/std/tg2015/chlamydia.htm). C. muridarum (CM) is frequently used for investigating CT pathogenesis since CM can infect the mouse genital tract and induce pathologies in the upper genital tract similar to those observed in CT-infected women under laparoscopy [1]. Mouse studies have revealed that the chlamydial plasmid is a critical pathogenic determinant [2–4] and the plasmid-encoded Pgp3 is largely responsible for the phenotype caused by the plasmid [5]. Some chromosomal genes have also been identified to code for chlamydial virulence factors [6–8]. Loss-of-function mutations in chromosomal genes tc0237/tc0668 significantly attenuated CM pathogenicity. Host responses may also impact CM pathogenesis. Antigen-specific CD8+ T cells [9] may promote but nonspecific CD8+ T cells [10] suppress CM pathogenesis while antigen-specific CD4+ T cells [11] can prevent chlamydial infection.
Although CT is routinely detected in human GI tracts [12–14], it is unclear whether gastrointestinal CT is beneficial or detrimental to human health. There is no significant association of non-lymphogranuloma venereum (LGV) CT with gut pathologies [15–17]. However, CT in the GI tract may affect CT pathogenicity in the genital tract either as a reservoir to seed repeated infections in the genital tract [18, 19] or induce host responses for exacerbating chlamydial induction of pathology in the genital tract [20, 21]. Revealing the mechanisms of CT colonization in the GI tract may provide new information for understanding the medical significance of the gastrointestinal CT and the pathogenic mechanisms of CT in the genital tract. The colonization by CM in the mouse GI tract is non-pathological [22]. The gastrointestinal CM may promote CM pathogenicity in the genital tract when a naïve mouse is first exposed to CM in the genital tract [20] or prevent CM infection in the genital tract when a naïve mouse is first exposed to CM in the GI tract [22]. A 2-hit model has been proposed to emphasize the contribution of the gastrointestinal CM to the pathogenicity of CM in the genital tract [21]. CM has also been tested as an oral vaccine or delivery vehicle for preventing subsequent infections in extra-gut tissues [23–25]. It is important to evaluate whether similar relationships also exist between gastrointestinal CT and genital CT during human infection. The mechanisms of CM-mouse gut interactions may be useful for designing human studies.
Orally inoculated CM can be detected in mouse rectal swabs within a few days, indicating a rapid spread of CM from the upper to the lower GI tract. Although CM is eventually cleared from the stomach within a week by gastric acid [26] and from the small intestine within a month by Th1-dominant immunity [27], CM in the large intestine persists for a long period [28]. This review will summarize recent progress on the mechanisms of chlamydial interactions with mucosal immunity using the CM-mouse gut interaction model as illustrated in Figure 1. The mechanisms by which Chlamydia overcomes mucosal barriers along the GI tract should help us understand the significance of chlamydial colonization in the GI tract and the pathogenic mechanisms of chlamydial infection in the genital tract.
Figure 1. A Chlamydia—mouse gut interaction model.
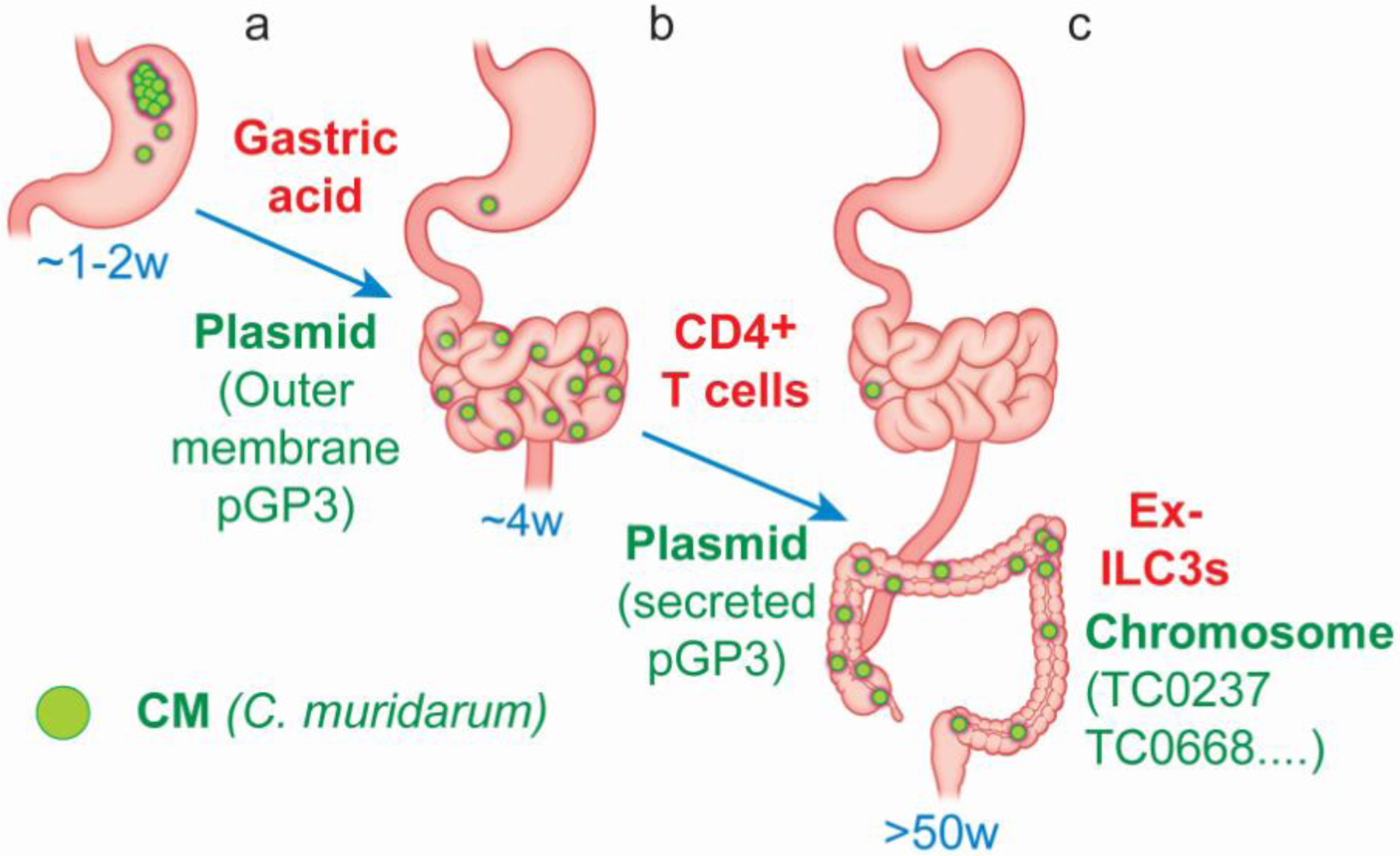
Orally inoculated Chlamydia muridarum (CM) can be detected in the rectal swabs of mice within a few days, indicating that Chlamydia can overcome multiple mucosal barriers in the gastrointestinal (GI) tract to achieve a rapid spread from the stomach into the large intestine. The residual chlamydial organisms in the stomach are eventually cleared within a week (~1w). Both the initial spread to the small intestine and the final clearance from the stomach may be regulated by gastric acid since blockade of gastric acid can significantly affect both. The chlamydial plasmid-encoded Pgp3, especially the Pgp3 that is associated with the outer membrane of chlamydial elementary bodies (EBs), is critical for promoting both the initial chlamydial spreading and the subsequent survival in the stomach (a). The small intestinal Chlamydia further spreads to the large intestine within 3 days and the spread is regulated by a CD4+ T cell-dominant immunity but the nature of the CD4+ T cells remains unclear. Nevertheless, the chlamydial plasmid-encoded Pgp3, particularly the one that is secreted into out of EBs, is also required for promoting the chlamydial spreading into the large intestine. The residual Chlamydia in the small intestine is eventually cleared in about 4 weeks (~4w). This clearance is dependent on a CD4+ Th1 response (b). Chlamydia may persist in the large intestine for hundreds of days (>50w). This long-lasting colonization is dependent on the functions of chlamydial chromosomal genes such as tc0237/tc0668 to evade IFNγ released by ILC3s. Thus, the chlamydial interactions with the IFNγ-producing ILC3s in the large intestine may ultimately determine the overall duration of chlamydial colonization in the GI tract (c).
II. Virulence factors identified in the genital tract are important for CM to colonize the GI tract.
Variants of CM with the removal of the entire plasmid or with mutations in genes encoded by the plasmid or chromosome have been identified due to the attenuation of their pathogenicity in the mouse genital tract [2, 3, 5, 7, 29, 30]. Surprisingly, these mutants were no longer able to colonize the GI tract [31–33].
II.1. CM without plasmid is more defective in colonizing the GI tract than the genital tract
CM carries a plasmid, termed pMoPn/pCM, coding for 8 putative proteins designated as glycoproteins 1 to 8 or Pgp1–8 [34]. Pgp3 [5, 35, 36] is a virulence factor while Pgp4 [37] and Pgp5 [29] regulate other chlamydial genes, including chromosomal genes involved in glycogen synthesis. CM can be depleted of the plasmid using novobiocin to create plasmid-free variants [2, 3]. Plasmid-free CM is no longer able to induce significant hydrosalpinx following intravaginal inoculation but the plasmid deficiency only has a minor impact on the CM infectivity in the genital tract. CT that lacks plasmid has been isolated from human urogenital tracts [38]. CT serovar D with or without plasmid achieved similar levels of colonization in the genital tracts of nonhuman primates [39]. Thus, the plasmid is not essential for either CT or CM to colonize the genital tract of their corresponding hosts. The next question is why Chlamydia has acquired the extra-chromosomal plasmid. Interestingly, CM was found to readily colonize the GI tract when exposed to multiple mouse mucosae [40] or to the blood stream [41]. CM in the genital tract spread to and established long-lasting colonization in the GI tract [42]. Following an oral inoculation, CM persisted in the GI tract for long periods [43] without spreading to extra-gut tissues [28]. Thus, CM is well adapted to the GI environment. When CM with or without plasmid was compared for colonizing the GI tract of mice [31], plasmid-free CM developed significantly delayed/reduced shedding of live organisms in the rectal swabs. Clearly, the plasmid is important in improving CM fitness in the mouse GI tract. It will be interesting to test whether the plasmid also improves CT fitness in the human GI tract.
II.2. CM variants with mutations in chromosomal genes are more defective in colonizing the GI tract than the genital tract
Although CM variants with mutations in the chromosomal genes tc0237 and/or tc0668 are highly attenuated in inducing hydrosalpinx in the upper genital tract of mice, these variants still maintain robust infectivity in the genital tract [7]. Interestingly, these mutants can no longer colonize the GI tract [32], indicating a critical role of the chromosomal genes in improving the fitness of CM in the GI tract. The next question to address is how TC0237 or TC0668 promotes the colonization of CM in the GI tract. TC0237 is a conserved hypothetical protein that consists of 159 amino acids (AA) with no known function [44]. It is paralogous to TC0236 (172AA) and TC0235 (170AA). The three putative proteins share ~90% AA sequence identity with their CT homologs CT849, CT848, and CT847. TC0668 is also a conserved hypothetical protein with 408 AA that share 97% identity with its CT serovar D counterpart, CT389. TC0668/CT389 is localized in the chlamydial outer membrane complex [45] and shares structural homology with eukaryotic integrins such as alpha V beta 1. However, it remains unclear how these chlamydial proteins promote chlamydial colonization. Nevertheless, chlamydial mutants have been used to identify host immune effectors responsible for their inhibition.
III. Plasmid-encoded virulence factors are more important for CM to colonize the upper GI tract than the lower GI tract.
Although both plasmid- and chromosome-encoded virulence factors have been shown to improve the fitness of CM in the GI tract, their roles in different regions of the GI tract may vary. It was found that the plasmid-encoding virulence factors were more important in promoting the colonization of CM in the stomach [26] and small intestine [46–48] while those encoded in the chromosome were more important in the large intestine [47, 49, 50].
III.1. Plasmid improves the fitness of CM in distinct regions of the GI tract
The impact of the plasmid on the colonization of CM in different regions of the mouse GI tract was determined using a combination of intragastric, intrajejunal and intracolonic inoculations [46]. Following intragastric inoculation, the plasmid improved the overall colonization efficiency of CM in the GI tract by >100 folds. At the tissue level, live plasmid-free CM was not recovered from the small intestine, indicating the necessity of the plasmid for CM to undergo productive infection (or to produce infectious progenies). Nevertheless, the number of genomes of the plasmid-free CM increased in the small intestine, suggesting that CM without plasmid can still undergo persistent infection (genome proliferation without production of infectious particles). The persistent infection returned to active infection in the large intestine since infectious progenies were recovered from the large intestine after a 14-day delay. Following intrajejunal inoculation (which bypasses the gastric factors), live (infectious) plasmid-free CM was recovered from the small intestine although the recovered titer was 530-fold lower than that of the plasmid-competent CM, indicating a critical role of the plasmid in improving the infectious yields of CM in the small intestine. Together with the result from the intragastric inoculation experiment, it can be concluded that a combination of gastric and intestinal effectors may be required for inducing plasmid-free CM to undergo persistent infection in the small intestine. This may represent a relevant model for investigating the mechanisms of chlamydial persistence, a long-standing challenge faced by the Chlamydia field. It is likely that during chlamydial interactions with the stomach and small intestine, Chlamydia may have both developed the ability to undergo persistent infection for preserving strength and acquired the plasmid for increasing the efficiency of spreading to the large intestine. Finally, the plasmid still increased the infectious titer of CM in the large intestine but by only 10 folds. Thus, the plasmid is more important in improving chlamydial fitness in the small intestine than the large intestine.
III.2. Plasmid-encoded Pgp3 is essential for CM to tolerate gastric acid.
The role of plasmid-encoded Pgp3 in promoting the survival of CM was evaluated in the stomach [26]. Pgp3-deficient CM failed to colonize the GI tract following oral inoculation, but it successfully colonized the GI tract following intracolonic inoculation, suggesting that once CM reaches the colon, Pgp3 is no longer required. Thus, Pgp3 mainly helps CM overcome the barriers in the upper GI tract. Lack of Pgp3 increased the susceptibility of CM to gastric killing by >100 folds. This gastric killing was mediated mainly by gastric acid since mice deficient in gastrin restored the colonization of Pgp3-deficient CM in the stomach. The Pgp3-dependent resistance to gastric acid was recapitulated using in vitro assays. Comparing to its isogenic wild type CM, Pgp3-deficient CM was significantly more susceptible to hydrochloric acid (HCL) and pepsin and its outer membrane complexes were less stable.
Pgp3 is an immunodominant [51, 52] and trimeric protein [53] that is both secreted out of the chlamydial elementary body (EB) [35] and associated with the outer membrane of EB [54]. The secreted Pgp3 has been shown to promote chlamydial pathogenesis by neutralizing host innate immunity effectors such as the cathelicidin-related antimicrobial peptide (CRAMP) in mice or LL-37 in humans [36]. It is possible that the outer membrane-associated Pgp3 may enable CM to survive exposure to gastric acid by maintaining the integrity of the outer membrane complex. Further studies are required for testing whether the outer membrane Pgp3 can pump out proton to maintain neutral pH in chlamydial cytosolic and periplasmic regions.
Various stress response systems have been found to protect bacteria from acid stress [55], such as the arginine acid survival system used by bacteria capable of expressing functional arginine decarboxylase [56]. CM expresses functional arginine decarboxylase [57], although most CT serovars (except for serovar E) have accumulated loss-of-function mutations in this gene [58]. Certain species of Chlamydia that infect animals, including CM, may require their genomes to maintain a functional arginine decarboxylase to facilitate their oral-fecal transmissions. However, CT that has adapted to the genital tract, where acid killing is less robust, can afford to accumulate loss-of-function mutations in this gene. Since the Pgp3-dependent resistance to acid was also detected using purified EBs in in vitro assays, the pre-existing outer membrane-associated Pgp3 must be essential for this function. Since Pgp3 increased CM survival in in vitro acid killing assays by ~2 folds while in the stomach of mice by ~100 folds, it is hypothesized to both increase the chlamydial outer membrane resistance to acid and up-regulate chlamydial acid tolerance responses.
III.3. Pgp3 is more important for CM to colonize the small intestine than the large intestine
Following an intrajejunal inoculations, the infectious titer of Pgp3-deficient CM in the small intestine was significantly lower than that of its isogenic wild type CM [48]. Further, no live Pgp3-deficient CM was detected in either the large intestine or rectal swabs. Thus, Pgp3 can improve CM fitness in both the small intestine and large intestine but Pgp3 appears to be more important for CM to colonize the large intestine than the small intestine. However, following an intracolonic inoculation, Pgp3-deficient CM colonized the large intestine as robustly as its isogenic wild type CM, indicating that Pgp3 is not required for CM to colonize the large intestine. The absence of the intrajejunally inoculated Pgp3-deficent CM in the large intestine is probably caused by Pgp3-deficent CM’s failure to spread to the large intestine. Host immunity is hypothesized to block the spreading.
III.4. Plasmid-encoded Pgp3 is prevented from spreading by CD4+ T cell-mediated immunity
To determine the immune mechanisms that are involved in preventing the spread of Pgp3-deficient CM, the intrajejunally delivered Pgp3-deficient CM was monitored for its ability to spread to the large intestine in mice with or without deficiencies in different immune components [59]. Although Pgp3-deficient CM failed to spread to the large intestine in wild type mice, it spread in mice deficient in CD4, suggesting a critical role of CD4+ T cells in preventing the spread. This conclusion is further supported by the observation that adoptive transfer of wild type CD4+ T cells but not CD8+ T cells nor B cells prevented the spread of Pgp3-deficient CM from the small intestine to the large intestine in CD4-deficient mice.
IV. The chromosomal genes tc0237/tc0668 are more important for CM to colonize the large intestine than the small intestine
CM with mutations in chromosomal genes tc0237 and tc0668 (designated as chromosomal mutant or clone G28.51.1) was no longer able to maintain long-lasting colonization in the GI tract [32]. Nevertheless, at the tissue level, G28.51.1 still reached the colon following an oral delivery [47], suggesting that lack of growth by G28.51.1 in the large intestine is not due to its failure to reach the organ, but due to its inability to colonize there. Consistently, a direct intracolonic delivery still failed to restore G28.51.1’s ability to colonize the large intestine. Thus, G28.51.1 is designated as a colonization-deficient mutant in the mouse colon and has been used to identify the host factors responsible for its inhibition in the large intestine [47, 49, 50].
IV.1. The chromosomal double mutant G28.51.1 is an IFNγ-susceptible variant in the colon
Although G28.51.1 failed to colonize the large intestine of wild type mice following an intracolonic inoculation, it was able to colonize the organ of mice deficient in IFNγ, suggesting that G28.51.1 is susceptible to IFNγ. Consistently, IFNγ-deficiency restored the colonization of the orally delivered G28.51.1 in the colon while mice deficient in gastrin failed to do so. Thus, TC0237 and TC0668 are not required for CM to overcome the acid barrier in the stomach but are necessary for CM to evade IFNγ-mediated barrier in the large intestine.
IV.2. The mutant G28.51.1 is cleared by IFNγ produced by innate lymphoid cells (ILCs).
Rag1 knockout mice, lacking adaptive immune cells due to their failure to form antigen receptors on lymphocytes, still prevented the colonization of G28.51.1, indicating a critical role of innate immune cells. Further, depletion of IFNγ from Rag1 knockout mice fully restored the colonization of G28.51.1, demonstrating that innate IFNγ is responsible for the inhibition of G28.51.1. To determine the cellular source of the innate IFNγ responsible for inhibiting G28.51.1, a dual knockout mouse model was used [50]. Mice deficient in both Rag2 (hence lacking conventional lymphocytes) and IL-2Rcγ (IL-2 receptor common γ chain, hence lacking all lymphoid cells including ILCs) fully restored colonization of G28.51.1. The growth of G28.51.1 correlated with lack of IFNγ in the colon of the dual knockout mice. Thus, ILCs are likely the cellular source of the innate IFNγ. Furthermore, depletion of NK1.1+ cells from Rag1 knockout mice both prevented IFNγ production and rescued G28.51.1, suggesting that the responsible ILCs are either NK cells or the non-killer ILC1s or ILC3s. Finally, the inhibitory function of the responsible ILCs is dependent on IL-7 and IL-15, both of which have been shown to promote the development and differentiation of ILCs [60, 61].
IV.3. The mutant G28.51.1 is cleared by IFNγ produced by group 3-like ILCs (or ILC3s)
NK.1.1+ ILCs that produce IFNγ include both the “killer” ILC1s (conventional NK cells) and the “helper” ILC1s [62] as well as a subset of ILC3s. Innate IFNγ produced during chlamydial infection in the airway or genital tract has been attributed to NK cells [63–65]. However, previous studies did not attempt to differentiate NK cells from other ILCs. Thus, the relative contribution of distinct ILC subsets to the production of IFNγ upon chlamydial infection remained unclear. A recent study has found that mice deficient in RORγt, a signature transcriptional factor of ILC3s, were able to fully restore colonization of G28.51.1 [50]. In contrast, deficiency in T-bet, a transcriptional factor of ILC1s, only extended G28.51.1’s colonization for one week. Thus, the colonic IFNγ responsible for inhibiting G28.51.1 is produced by ILC3s. ILC1s are not required for inhibiting G28.51.1 since G28.51.1 failed to colonize the large intestine of mice deficient in NCR1, a marker for ILC1s [66, 67]. Finally, the inhibition of G28.51.1 is independent of CCR6 [68]. Thus, the subset of ILC3s responsible for inhibiting G28.51.1 is defined as CCR6-NKp46-RORγt+ILC3s.
IV.4. IFNγ-producing ILC3s are sufficient for inhibiting G28.51.1.
Having demonstrated the necessity of IFNγ-producing ILC3s in inhibiting G28.51.1 [47, 50], the next step was addressing the sufficiency using an adoptive transfer approach [49]. The donor cells (to be transferred) were from Rag1 knockout mice (due to their ability to inhibit G28.51.1) while the recipients were mice deficient in IFNγ or IL-7R (due to their susceptibility to G28.51.1 infection). Transfer of intestinal lamina propria ILCs enriched for RORγt expression from donor mice restored the recipient mice deficient in IL-7R to inhibit G28.51.1, suggesting that the responsible ILCs are ILC3s from the intestinal lamina propria. Transfer of RORγt+ILC3s that express IFNγ ( or ex-ILC3s) from donor mice restored the recipient mice deficient in IFNγ to inhibit G28.51.1. Finally, genetically labeled RORγt+ILC3s as donor cells also prevented colonization of G28.51.1 in the large intestine of the recipient mice deficient in IFNγ. Thus, IFNγ-producing ILC3s are sufficient for regulating chlamydial colonization in the mouse large intestine.
IV.5. Can the CM-induced ILC3s contribute to the microbiota-maintained colonization resistance?
Having demonstrated both the necessity and sufficiency for IFNγ-producing ILC3s to regulate chlamydial colonization in the large intestine, the next question is to determine whether the IFNγ-producing ILC3s induced by Chlamydia can contribute to the colonization resistance maintained by gut microbiota. ILC3s can produce different cytokines in response to different signals from local tissues [69, 70]. Tissue IL-23 may induce NCR1−RORγt+ILC3s to secrete both IL-17 & IFNγ [71], potentially contributing to microbial induction of colitis [72]. T-bet co-expression is unnecessary for NCR1−RORγt+ILC3s to produce IFNγ [71] but necessary for maintaining the functionality of NCR1+RORγt+ILC3s [70]. The ILC3s responsible for interacting with CM express neither CCR6 nor NCR1 and their function is independent of T-bet [50]. Since chlamydial colonization in the GI tract is non-pathological [22], microbiota are hypothesized to also interact with the CCR6-NCR1-IFNγ-producing RORγt+ILC3s. Microbiota are known to confer colonization resistance to pathogen infections via mechanisms such as maintaining the production of IFNγ in the mucosal tissues [73–75]. However, it is not clear whether microbiota use the IFNγ-producing ILC3s for establishing IFNγ-mediated colonization resistance although microbiota can interact with regulatory ILCs for attenuating gut inflammation [76]. Besides G28.51.1 [6, 7], there are also other CM chromosomal mutants [8, 30]. It will be worth testing whether these CM variants are also defective in colonizing the large intestine and susceptible to the IFNγ secreted by the CCR6-NCR1-RORγt+ILC3s.
V. The strategies Chlamydia has acquired in the GI tract may make it a successful sexually transmitted agent in the genital tract
CM has been shown to acquire plasmid genes to promote its survival in the stomach and small intestine and chromosomal genes to maintain long-lasting colonization in the large intestine of mice. It is hypothesized that the strategies CM has acquired from its interactions with the GI tract may render itself a successful agent for invading the genital tract, including colonizing the lower genital tract, crossing the cervical and uterotubal barriers and infecting tubal epithelial cells, which may lead to long-term sequelae. The hypothesis is supported by the followings:
First, the oral-fecal route is more efficient than genital contact in transmitting CM among mice, providing opportunities for CM to adapt to the GI tract instead of the genital tract.
Second, CM maintains long-lasting colonization in the GI tract but not the genital tract, allowing CM to acquire the ability to overcome the barriers in the GI tract.
Third, CM variants deficient in virulence factors are highly defective in colonizing the GI tract but still maintain significant infectivity in the genital tract, suggesting that the primary purpose of these virulence factors is for dealing with the barriers in the GI tract. Coincidentally, the same factors also promote chlamydial infection in the genital tract.
Fourth, the mucosal barriers in the GI tract are more stringent than those in the genital tract. The strategies that CM has developed for evading the more stringent barriers in the GI tract may enable CM’s invasion of the genital tract.
Finally, the same concept may also be applicable to other microbes. It is hypothesized that the ability of urinary pathogenic E. coli or UPEC to establish persistent infection in the urinary tract may be obtained by UPEC during its passage in the GI tract [77].
VI. Concluding remarks and Caveats
Chlamydia is frequently detected in the GI tracts of both humans and animals. However, neither its significance nor the mechanisms is known. Since CT in human GI tract has been shown to be more resistant than CT in the genital tract to some antibiotic therapy, it is urgent to determine whether gastrointestinal CT is beneficial or detrimental to human health. Revealing the mechanisms of CM interactions with mouse GI tract may provide information for designing human studies. However, caution should be taken when applying information obtained from mouse studies to humans. Although CM has adapted to the GI tract of mice, it is not known whether CT has adapted to the human GI tract since oral-fecal route is more efficient for mice than humans to transmit microbes [78]. In contrast, CT is transmitted between humans via the genital tract. Thus, CT may have achieved certain level of fitness with human genital tissues. Nevertheless, non-LGV CT serovars are still frequently detected in the human rectal swabs, suggesting that CT may still maintain some level of fitness with the GI tract. The lack of association of non-LGV CT serovars with any significant pathology in the GI tract suggests that non-LGV CT serovars may also be non-pathological in the GI tract of humans. It will be interesting to reveal the survival mechanisms of the non-LGV CT serovars in the human GI tract and further determine their significance.
Outstanding Questions.
How does chlamydial plasmid-encoded pGP3 promote chlamydial resistance to gastric acid?
How does pGP3 promote chlamydial evasion of CD4+ T cell-mediated immunity in the intestine?
How does Chlamydia selectively activate ex-ILC3s to secrete IFNγ?
How do TC0237 & TC0668 mediate chlamydial resistance to IFNγ?
What is the significance of CT colonization in the human gastrointestinal tract?
Trends box.
Chlamydia mutants deficient in plasmid-encoded factors are more defective in colonizing the upper gastrointestinal tract while those deficient in chromosome-encoded factors are more defective in colonizing the lower gastrointestinal tract.
Chlamydia may have acquired the plasmid for promoting its spreading to the large intestine while using the chromosomal genes for maintaining its long-lasting colonization in the large intestine.
The plasmid-encoded pGP3 is critical for Chlamydia to resist both gastric acid in the stomach and CD4+ T lymphocyte-mediated immunity in the small intestine and it may play similar roles in aiding chlamydial infection in the female genital tract.
The chromosomal proteins TC0237 & TC0668 are essential for chlamydial evasion of ILC3-secreted IFNγ in the large intestine and they may also enable Chlamydia to evade innate IFNγ in the genital tract.
The CM-mouse gut interactions may represent a productive platform for both discovering novel mechanisms of microbe-host interactions and revealing pathogenic mechanisms of Chlamydia in the genital tract.
CM colonization in the gut is long-lasting and non-pathogenic, which may be used for both modeling microbiota-gut interactions and exploring the medical significance and utility of gastrointestinal Chlamydia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhong G et al. (2020) Animal Models of Chlamydia trachomatis Infection. In Chlamydia Biology: From Genome to Disease (Tan M et al. eds), pp. 459–482, Caister Academic Press, U.K. [Google Scholar]
- 2.O’Connell CM et al. (2007) Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J Immunol 179 (6), 4027–34. [DOI] [PubMed] [Google Scholar]
- 3.Lei L et al. (2014) Reduced live organism recovery and lack of hydrosalpinx in mice infected with plasmid-free Chlamydia muridarum. Infect Immun 82 (3), 983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong G (2017) Chlamydial Plasmid-Dependent Pathogenicity. Trends Microbiol 25 (2), 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y et al. (2014) Plasmid-encoded Pgp3 is a major virulence factor for Chlamydia muridarum to induce hydrosalpinx in mice. Infect Immun 82 (12), 5327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C et al. (2015) In vitro passage selects for Chlamydia muridarum with enhanced infectivity in cultured cells but attenuated pathogenicity in mouse upper genital tract. Infect Immun 83 (5), 1881–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conrad TA et al. (2016) The Chromosome-Encoded Hypothetical Protein TC0668 Is an Upper Genital Tract Pathogenicity Factor of Chlamydia muridarum. Infect Immun 84 (2), 467–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morrison SG et al. (2018) Chlamydia muridarum Genital and Gastrointestinal Infection Tropism Is Mediated by Distinct Chromosomal Factors. Infect Immun 86:DOI 10.1128/IAI.00141-18 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murthy AK et al. (2011) Tumor necrosis factor alpha production from CD8+ T cells mediates oviduct pathological sequelae following primary genital Chlamydia muridarum infection. Infect Immun 79 (7), 2928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xie L et al. (2020) Suppression of Chlamydial Pathogenicity by Nonspecific CD8(+) T Lymphocytes. Infect Immun 88:DOI 10.1128/IAI.00315-20 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison RP and Caldwell HD (2002) Immunity to murine chlamydial genital infection. Infect Immun 70 (6), 2741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gratrix J et al. (2015) Evidence for increased Chlamydia case finding after the introduction of rectal screening among women attending 2 Canadian sexually transmitted infection clinics. Clin Infect Dis 60 (3), 398–404. [DOI] [PubMed] [Google Scholar]
- 13.Pabbaraju K et al. (2017) Use of the APTIMA Combo 2 Assay and a Secondary Algorithm to Detect and Confirm Chlamydia trachomatis in Rectal-Only Infections. Sex Transm Dis 44 (2), 118–119. [DOI] [PubMed] [Google Scholar]
- 14.Craig AP et al. (2015) Is it time to switch to doxycycline from azithromycin for treating genital chlamydial infections in women? Modelling the impact of autoinoculation from the gastrointestinal tract to the genital tract. BMC Infect Dis 15, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee KJ et al. (2015) Chlamydial Proctitis in a Young Man Who Has Sex with Men: Misdiagnosed as Inflammatory Bowel Disease. Chonnam Med J 51 (3), 139–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mardh PA et al. (1980) Lack of evidence for an association between infection with Chlamydia trachomatis and Crohn’s disease, as indicated by micro-immunofluorescence antibody tests. Acta Pathol Microbiol Scand B 88 (1), 57–9. [DOI] [PubMed] [Google Scholar]
- 17.McGarity BH et al. (1991) Deoxyribonucleic acid amplification and hybridisation in Crohn’s disease using a chlamydial plasmid probe. Gut 32 (9), 1011–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rank RG and Yeruva L (2014) Hidden in plain sight: chlamydial gastrointestinal infection and its relevance to persistence in human genital infection. Infect Immun 82 (4), 1362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bavoil PM et al. (2017) Does Active Oral Sex Contribute to Female Infertility? J Infect Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian Q et al. (2020) Gastrointestinal coinfection promotes chlamydial pathogenicity in the genital tract. Infect Immun 88 (4), e00905–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong G (2018) Chlamydia spreading from the genital tract to the gastrointestinal tract - A two-hit hypothesis. Trends Microbiol 26 (7), 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L et al. (2018) Nonpathogenic colonization with chlamydia in the gastrointestinal tract as oral vaccination for inducing transmucosal protection. Infect Immun 86 (2), e00630–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu C et al. (2018) Oral Chlamydia vaccination induces transmucosal protection in the airway. Vaccine 36 (16), 2061–2068. [DOI] [PubMed] [Google Scholar]
- 24.Zhong G, Methods and compositions for attenuated chlamydia as vaccine and vector (US Patent# 10596247), in: (USPTO), U.S.P.a.T.O. (Ed.) Patents by Inventor Guangming Zhong, Board of Regents, The University of Texas System, US, 2020. [Google Scholar]
- 25.Morrison SG et al. (2020) A Genital Infection-Attenuated Chlamydia muridarum Mutant Infects the Gastrointestinal Tract and Protects against Genital Tract Challenge. mBio 11:DOI 10.1128/mBio.02770-20 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang T et al. (2019) The plasmid-encoded pGP3 promotes Chlamydia evasion of acidic barriers in both stomach and vagina. Infect Immun 87 (5), e00844–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin H et al. (2019) Antigen-specific CD4(+) T cell-derived gamma interferon is both necessary and sufficient for clearing Chlamydia from the small intestine but not the large intestine. Infect Immun 87 (6), e00055–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L et al. (2016) The Chlamydia muridarum Organisms Fail to Auto-Inoculate the Mouse Genital Tract after Colonization in the Gastrointestinal Tract for 70 days. PLoS One 11 (5), e0155880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y et al. (2014) Transformation of Chlamydia muridarum reveals a role for Pgp5 in suppression of plasmid-dependent gene expression. J Bacteriol 196 (5), 989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giebel AM et al. (2019) Genetic Screen in Chlamydia muridarum Reveals Role for an Interferon-Induced Host Cell Death Program in Antimicrobial Inclusion Rupture. MBio 10:DOI 10.1128/mBio.00385-19 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shao L et al. (2017) The cryptic plasmid is more important for Chlamydia muridarum to colonize the mouse gastrointestinal tract than to infect the genital tract. PLoS One 12 (5), e0177691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shao L et al. (2017) Chlamydia muridarum with mutations in chromosomal genes tc0237 and/or tc0668 is deficient in colonizing the mouse gastrointestinal tract. Infect Immun 85 (8), e00321–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shao L et al. (2017) The genital tract virulence factor pGP3 is essential for Chlamydia muridarum colonization in the gastrointestinal Tract. Infect Immun 86 (1), e00429–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas NS et al. (1997) Plasmid diversity in Chlamydia. Microbiology 143 ( Pt 6), 1847–54. [DOI] [PubMed] [Google Scholar]
- 35.Li Z et al. (2008) The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect Immun 76 (8), 3415–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hou S et al. (2015) Chlamydial plasmid-encoded virulence factor Pgp3 neutralizes the antichlamydial activity of human cathelicidin LL-37. Infect Immun 83 (12), 4701–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q et al. (2020) The Repressor Function of the Chlamydia Late Regulator EUO Is Enhanced by the Plasmid-Encoded Protein Pgp4. J Bacteriol 202:DOI 10.1128/JB.00793-19 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farencena A et al. (1997) Characterization of a new isolate of Chlamydia trachomatis which lacks the common plasmid and has properties of biovar trachoma. Infect Immun 65 (7), 2965–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qu Y et al. (2015) Comparable Genital Tract Infection, Pathology, and Immunity in Rhesus Macaques Inoculated with Wild-Type or Plasmid-Deficient Chlamydia trachomatis Serovar D. Infect Immun 83 (10), 4056–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perry LL and Hughes S (1999) Chlamydial colonization of multiple mucosae following infection by any mucosal route. Infect Immun 67 (7), 3686–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai J et al. (2016) Intravenous inoculation with Chlamydia muridarum leads to a long-lasting infection restricted to the gastrointestinal tract. Infect Immun 84 (8), 2382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Q et al. (2015) In Vivo and Ex Vivo Imaging Reveals a Long-Lasting Chlamydial Infection in the Mouse Gastrointestinal Tract following Genital Tract Inoculation. Infect Immun 83 (9), 3568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yeruva L et al. (2013) Chlamydial infection of the gastrointestinal tract: a reservoir for persistent infection. Pathog Dis 68 (3), 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Read TD et al. (2000) Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res 28 (6), 1397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X et al. (2010) Identification of Chlamydia trachomatis outer membrane complex proteins by differential proteomics. J Bacteriol 192 (11), 2852–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma J et al. (2020) The cryptic plasmid improves chlamydia fitness in different regions of the gastrointestinal tract. Infect Immun 88 (3), e00860–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koprivsek JJ et al. (2019) Distinct roles of chromosome- versus plasmid-encoded genital tract virulence factors in promoting chlamydia muridarum colonization in the gastrointestinal tract. Infect Immun 87 (8), e00265–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huo Z et al. (2020) Chlamydia deficient in plasmid-encoded pGP3 is prevented from spreading to large intestine. Infect Immun 88 (6), e00120–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Y et al. (2020) Adoptive transfer of group 3-like innate lymphoid cells restores mouse colon resistance to colonization of an IFNgamma-susceptible Chlamydia muridarum mutant. Infect Immun 89 (2), e00533–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koprivsek JJ et al. (2020) Evasion of innate lymphoid cell-regulated gamma interferon responses by Chlamydia muridarum to achieve long-lasting colonization in mouse colon. Infect Immun 88 (3), e00798–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li Z et al. (2008) Antibodies from women urogenitally infected with C. trachomatis predominantly recognized the plasmid protein pgp3 in a conformation-dependent manner. BMC Microbiol 8, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J et al. (2010) A genome-wide profiling of the humoral immune response to Chlamydia trachomatis infection reveals vaccine candidate antigens expressed in humans. J Immunol 185 (3), 1670–80. [DOI] [PubMed] [Google Scholar]
- 53.Galaleldeen A et al. (2013) Structure of the Chlamydia trachomatis immunodominant antigen Pgp3. J Biol Chem 288 (30), 22068–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen D et al. (2010) Characterization of Pgp3, a Chlamydia trachomatis plasmid-encoded immunodominant antigen. J Bacteriol 192 (22), 6017–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin J et al. (1995) Comparative analysis of extreme acid survival in Salmonella typhimurium, Shigella flexneri, and Escherichia coli. J Bacteriol 177 (14), 4097–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alvarez-Ordonez A et al. (2010) Arginine and lysine decarboxylases and the acid tolerance response of Salmonella Typhimurium. Int J Food Microbiol 136 (3), 278–82. [DOI] [PubMed] [Google Scholar]
- 57.Bliven KA et al. (2012) Characterization of the activity and expression of arginine decarboxylase in human and animal Chlamydia pathogens. FEMS Microbiol Lett 337 (2), 140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Giles TN et al. (2009) Independent inactivation of arginine decarboxylase genes by nonsense and missense mutations led to pseudogene formation in Chlamydia trachomatis serovar L2 and D strains. BMC Evol Biol 9, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He C, Xu Y, Huo Z, Wang J, Jia T, Li X, Zhong G (2021) Regulation of Chlamydia Spreading From Small Intestine to Large Intestine via an Immunological Barrier. Immunology and Cell Biology In Press. [DOI] [PubMed] [Google Scholar]
- 60.Colonna M (2018) Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 48 (6), 1104–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinette ML et al. (2017) IL-15 sustains IL-7R-independent ILC2 and ILC3 development. Nat Commun 8, 14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piersma SJ et al. (2019) Activation Receptor-Dependent IFN-gamma Production by NK Cells Is Controlled by Transcription, Translation, and the Proteasome. J Immunol 203 (7), 1981–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Onsrud M and Qvigstad E (1984) Natural killer cell activity after gynecologic infections with chlamydia. Acta Obstet Gynecol Scand 63 (7), 613–5. [DOI] [PubMed] [Google Scholar]
- 64.Rottenberg ME et al. (2000) Regulation and role of IFN-gamma in the innate resistance to infection with Chlamydia pneumoniae. J Immunol 164 (9), 4812–8. [DOI] [PubMed] [Google Scholar]
- 65.Zhao L et al. (2019) The Important Role of Dendritic Cell (DC) in iNKT-Mediated Modulation of NK Cell Function in Chlamydia pneumoniae Lung Infection. Mediators Inflamm 2019, 4742634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Narni-Mancinelli E et al. (2017) Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci Immunol 2, aam9628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sheppard S et al. (2018) The Murine Natural Cytotoxic Receptor NKp46/NCR1 Controls TRAIL Protein Expression in NK Cells and ILC1s. Cell Rep 22 (13), 3385–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin YL et al. (2017) CCR6 Deficiency Impairs IgA Production and Dysregulates Antimicrobial Peptide Production, Altering the Intestinal Flora. Front Immunol 8, 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nussbaum K et al. (2017) Tissue microenvironment dictates the fate and tumor-suppressive function of type 3 ILCs. J Exp Med 214 (8), 2331–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cella M et al. (2019) Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat Immunol 20 (8), 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buonocore S et al. (2010) Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464 (7293), 1371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Neurath MF (2019) IL-23 in inflammatory bowel diseases and colon cancer. Cytokine Growth Factor Rev 45, 1–8. [DOI] [PubMed] [Google Scholar]
- 73.Litvak Y et al. (2019) Commensal Enterobacteriaceae Protect against Salmonella Colonization through Oxygen Competition. Cell Host Microbe 25 (1), 128–139 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thiemann S et al. (2017) Enhancement of IFNgamma Production by Distinct Commensals Ameliorates Salmonella-Induced Disease. Cell Host Microbe 21 (6), 682–694 e5. [DOI] [PubMed] [Google Scholar]
- 75.Caballero S et al. (2017) Cooperating Commensals Restore Colonization Resistance to Vancomycin-Resistant Enterococcus faecium. Cell Host Microbe 21 (5), 592–602 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nakamoto N et al. (2017) Commensal Lactobacillus Controls Immune Tolerance during Acute Liver Injury in Mice. Cell Rep 21 (5), 1215–1226. [DOI] [PubMed] [Google Scholar]
- 77.Magruder M et al. (2019) Gut uropathogen abundance is a risk factor for development of bacteriuria and urinary tract infection. Nature Communications 10: 10.1038/s41467-019-13467-w (5521). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Batteiger TA et al. (2019) Detection of Rectal Chlamydia trachomatis in Heterosexual Men Who Report Cunnilingus. Sex Transm Dis 46 (7), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]