

Abstract
Previous studies demonstrated that anti-hyperlipidemic drug gemfibrozil acts as NO- and heme-independent activator of NO receptor soluble guanylyl cyclase. A series of new gemfibrozil derivatives were synthesized and evaluated for sGC activation. The structure-activity relationship study identified the positions in gemfibrozil’s scaffold that are detrimental for sGC activation and those that are amendable for optimizing modifications. Compared with gemfibrozil, compounds 7c and 15b were more potent activators of cGMP-forming activity of purified sGC and exhibited enhanced relaxation of preconstricted mouse thoracic aorta rings. These studies established the overall framework needed for futher improvement of sGC activators based on gemfibrozil scaffold.
Keywords: gemfibrozil, soluble guanylyl cyclase, biochemistry, medicinal chemistry
Graphical Abstract

1. INTRODUCTION
Nitric oxide (NO) signaling is one of the fundamental pathways in mammalian physiology, which plays important roles in cardiovascular homeostasis, platelet function, gastrointestinal function, neurotransmission and other processes. A key mediator of NO signaling is soluble guanylyl cyclase (sGC), a heme protein with high affinity for NO. NO-sensitive guanylyl cyclase is a heterodimeric protein composed of one α and one β subunit. Both subunits are needed for a functional catalytic site, which uses GTP to synthesize secondary cellular messenger cGMP. Humans and mice have two functional isoforms of the α subunit (α1 and α2) and one functional β1 isoform. The heterodimer α1β1 (GC-1) is ubiquitously expressed and has a higher level of expression than the α2β1 (GC-2) counterpart, which has tissue-specific expression, and is more prevalent in brain, kidney and placenta. The binding of a NO molecule to the ferrous sGC heme induces structural changes that enhances several hundred fold the cGMP-forming activity of the enzyme. The resulting increase of intracellular cGMP level triggers cGMP-dependent processes that cause relaxation of smooth muscle cells, affects vasodilatation and GI motility [1, 2]; inhibits the adhesion of leukocyte and platelet to vascular walls, diminishes platelet aggregation; and decreases the proliferation and migration of smooth muscles cells [3]. Proper sGC function is beneficial for maintaining normal vascular plasticity, preventing atherosclerosis [4], thrombosis [5] and stroke [6]. On the contrary, deficient sGC function is associated with increased risk of myocardial infarction [7], coronary artery disease [8] [9, 10], or vascular and GI dysfunctions [11, 12]. Not surprisingly, sGC is an important therapeutic target. Pharmacological activation of sGC in cases of angina pectoris or heart failure is achieved via the use of various NO releasing organic nitrovasodilators, such as nitroglycerin, glyceryl trinitrate, isosobide dinitrate and others. Although effective, the clinical application of these NO-generating prodrugs is limited by the rapid onset of tolerance and tachyphylaxis [13]. Moreover, under inflammatory conditions these agents may generate reactive nitrogen species and cause damage to DNA, proteins and lipids [14]. Fortunately, sGC function may be stimulated NO-independently by a number of allosteric regulators. Two types of such allosteric regulators have been identified [15] - sGC stimulators and sGC activators. Allosteric sGC stimulators potentiate NO signaling by sensitizing sGC to lower doses of NO. Some of these stimulators are approved as sGC-targeting therapeutics. For example, the sGC stimulator riociguat has been approved for management of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension [16, 17], while vericiguat has been approved for heart failure [18, 19]. Although these NO-independent sGC stimulators have found clinical use, they are effective only when sGC heme is in reduced ferrous state. Many cardiovascular ailments are associated with inflammatory conditions and increased burden of oxidative stress. Under oxidative stress sGC heme may be oxidized to a ferric state [20–23], rendering sGC insensitive to NO or allosteric stimulators. Therefore, NO-independent sGC activators targeting sGC with ferric heme are of special interest. sGC activators cinaciguat (also known as BAY58-2667) and ataciguat (also known as HMR1766) have been proven effective in vitro and in vivo sGC activators [24, 25] and are considered as possible therapeutic agents [26]. Although the clinical trial of cinaciguat for the treatment of heart failure [27] showed promising results in unloading the heart, it had to be stopped due to the development of hypotension. This side effect is most likely related to the very high affinity of cinaciguat, which has an estimated EC50 in low nanomolar range [25]. Therefore, NO-independent sGC activators with lower affinity may be useful. Previous studies demonstrated that the drug gemfibrozil, which is used for treatment of type IV and V hyperlipidemias [28], also acts as a heme-independent sGC activator with vasoactive and anti-platelet properties [29]. This sGC-activating property may be responsible for superior cardiovascular protection of gemfibrozil over other fibrates [30]. The present study has been carried to obtain preliminary structure-activity information on sGC activating features of gemfibrozil, with the ultimate goal of obtaining a gemfibrozil derivative with improved sGC affinity, activation and/or vasodilating function.
2. RESULTS and DISCUSSION.
2.1. Design and synthesis.
In order to explore the development of sGC activators based upon the gemfibrozil scaffold we prepared a series of compounds. Variations of the aryl terminus, the linking heteroatom, the side-chain length and substitution, and the acid terminus were explored, as depicted in Figure 1.
Figure 1.

Gemfibrozil structure and Scaffold Modifications Sites.
We first focused our synthetic efforts on analogs with varied alkyl chain lengths and substitution. Simple nor-methyl analogs (4a-d) were readily accessible from the respective bromoacids (2a-d) using a four-step sequence (Scheme 1). Using similar chemistry, we were also able to examine the role of ether linkange as well as the substitution of the aryl ring. For this purpose, δ–chloroester 6 or δ-bromoacid 2c were employed to furnish dimethyl (7a-c) or nor-methyl analogs (8a-b) with variations on the aromatic substituents as well as ether, amine or sulfide linkages (Scheme 2).
Scheme 1.

Synthesis of nor-methyl derivatives.
Scheme 2.

Synthesis of aromatic variants.
The replacement of the gemfibrozil ether with an amine in 7a allowed us to introduce branching at the heteroatom linker (α-to the aryl group). Accordingly, amidation of amino ester 9 (an intermediate in the synthesis of 7a) with acid halides and acids, followed by saponification of the isobutyl ester, allowed us to readily prepare several compounds to explore how branching by chains of different nature affects sGC activation (Scheme 3). Several simple derivatives containing hydrophobic side-chains (10a-c) were initially prepared by this route. Similarly, substitution with additional acidic residues (10d-f) was used to assess if increased hydrogen bonding potential and/or hydrophilicity would significantly impact sGC activation. Finally, an amine-linked dimeric analog 11 was also synthesized.
Scheme 3.

Synthesis of Aniline Analogs.
We also investigated derivatives of the carboxyl terminus of gemfibrozil, including simple amide derivatives 12a-e and dimeric derivatives 13a-c (Scheme 4). Finally, ester 14 was used to prepare mono-alkyl acids 15a-b after saponification (Scheme 5).
Scheme 4.

Synthesis of amide derivatives.
Scheme 5.

Synthesis of α substituted derivatives.
2.2. Biological results.
Once the gemfibrozil derivatives described above were synthesized, we subjected them to a battery of tests to evaluate how they affect sGC activity.
2.2.1. sGC activation tests.
First, we compared the effect of 100 μM gemfibrozil or gemfibrozil derivatives on cGMP-forming activity of sGC. Previous studies demonstrated that gemfibrozil activates sGC with ferrous heme, but is more effective when sGC heme is oxidized into ferric state [29]. Therefore, the initial screening of synthesized compounds was performed using native sGC with ferrous heme (Fe2+-sGC) and sGC with oxidized ferric heme (Fe3+-sGC) (Figure 2). Based on the results of this initial screening, we selected 4c, 4d, 7a, 7c, 11 and 15b, which activated both Fe2+-sGC and Fe3+-sGC, for analysis of dose-dependent activation of Fe3+-sGC. The estimated EC50 values are reported in Table 1.
Figure 2. Upregulation of sGC activity by gemfibrozil derivatives.
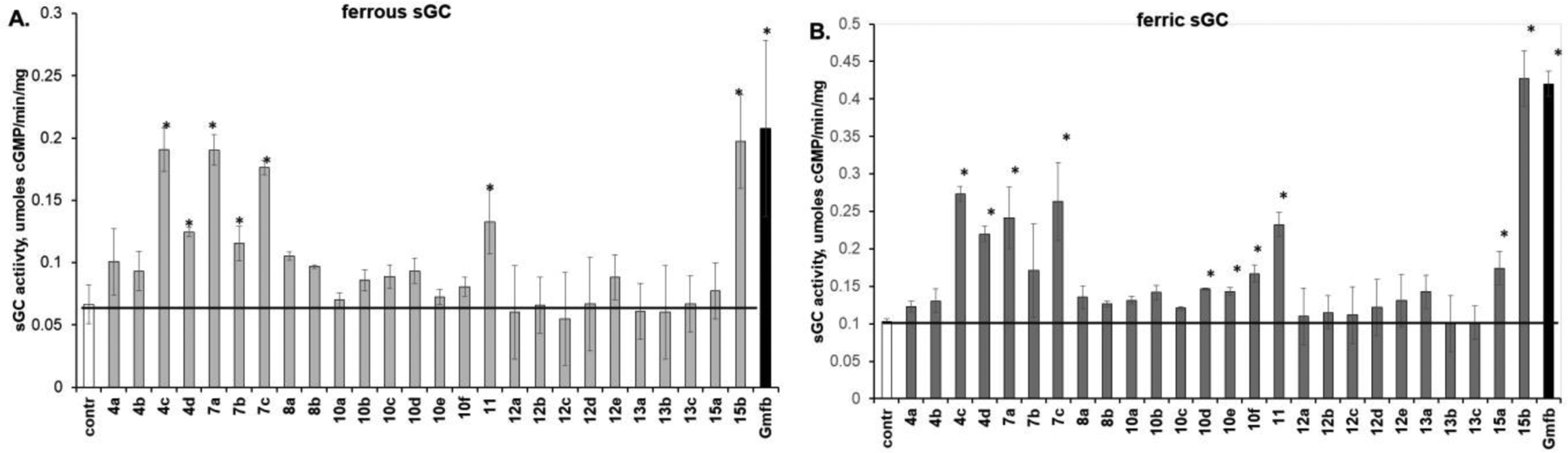
Purified human sGC containing heme in ferrous (A) or ferric (B) state was pre-incubated at room temperature for 10 minutes with 100 μM concentrations of gemfibrozil derivatives, gemfibrozil (Gem) or vehicle control (cnt) and then tested for cGMP-forming activity using the [32P]GTP assay, as described in the Method Section. Data are shown as mean ±SD from three independent tests performed in triplicate. * indicates p<0.05 vs control activity (t-test).
Table 1.
sGC-stimulating gemfibrozil derivatives
| cGMP-forming activity test | Vasorelaxation test | |||
|---|---|---|---|---|
| Compound | EC50, μM* | fold stimulation at 100 μM | IC50, μM* | MEC#, μM |
| Gemfibrozil | 624 ± 217 | 4 | 310 ± 115 | 30 |
| 4c | 707 ± 272 | 2.9 | 812 ± 453 | 100 |
| 4d | 389 ± 151 | 2.1 | >1000 | 300 |
| 7a | 50 ± 16 | 2.9 | 721 ± 161 | 100 |
| 7c | 166 ± 41 | 2.6 | 71 ± 24 | 10 |
| 11 | 119 ± 84 | 2.2 | 261 ± 77 | 10 |
| 15b | 128 ± 41 | 4.1 | 45 ± 8 | 10 |
presented as mean ± SD;
MEC – apparent minimal effective concentration, defined as lowest concentration exhibiting statistically significant difference from untreated conditions.
Analysis of sGC-activating properties of the simple nor-methyl derivatives 4a-4d revealed that the length of the carboxylic acid chain is an important factor in determining the potency of activation (Fig. 1), but that the gem-dimethyl substitution α to the carboxylic acid is less critical. Short chains in 4a and 4b were not conducive to sGC activation, while longer pentanoic and hexanoic chains of 4c and 4d, respectively, were more favorable for activation. The decreased activation by 4d vs 4c suggests that the five carbon chain (as found in the parent gemfibrozil) is optimal for sGC activation.
Analysis of these gemfibrozil derivatives also points to the importance of the benzene ring substituents. The 2,5-dimethyl substitution found in gemfibrozil provides more potent sGC activation than the mono-substituted compound 7b, which carries only the 2-methyl group (although that compound also has a sulfide linkage rather than an ether, complicating any direct comparison). Replacing the 2-methyl group with the bulkier 2-isopropyl, as seen in compound 7c, is well tolerated for activation of both Fe2+-sGC and Fe3+-sGC. Although 8b, lacking both the aryl substituents as well as the geminal dimethyl group, does not activate sGC, inclusion of the 2,5-dimethyl aryl substitution in 4d rescues the ability to activate sGC and even results in lower EC50 value (Table 1). Considering that previous work demonstrated that gemfibrozil derivatives with 3,4,5-trimethyl and (to a lesser effect) 4-fluoro aryl substituents also support sGC activation [29], these results demonstrate that substitution of the aryl core is favorable and tolerates different substitution patterns and substituents.
We next evaluated the importance of the gem-dimethyl substitution alpha to the pentanoic acid chain. As shown in Fig. 2, removal of both methyl groups in compound 4d was tolerated by Fe2+-sGC, but diminished Fe3+-sGC activation. Replacement of gemfibrozil gem-dimethyl substitution by an ethyl group in compound 15a blocked the activation of Fe2+-sGC, and significantly hampered the activation of Fe3+-sGC. However, introduction of a longer hexyl chain in 15b was very well tolerated and substantially lowered the observed EC50 (Table 1).
While previous studies indicated that esterification of the carboxylic group is detrimental to sGC activating properties of gemfibrozil [29], we explored whether amide substitutions are tolerated. Activating properties of 12a-12e compounds clearly indicated that such substitution do not promote sGC activation. Even the hexyl aliphatic chain, which improved sGC activating properties when used in the nearby alpha position in 15a, had no positive effect. Connecting two gemfibrozil cores via diamide links of different length (13a-13c) also did not offer any additional benefits.
We also evaluated the importance of the atom connecting the carboxylic acid chain and the aryl moiety. Comparing gemfibrozil and aniline 7a, which has a nitrogen connecting atom, demonstrates that this modification is well tolerated, as 7a has a much better affinity based on its lower EC50 (Table 1). On the other hand, gemfibrozil provides more potent sGC activation than the sulfide-linked 7b, although that derivative has only a 2-methyl aryl substitution, complicating any direct comparison.
Considering that the aniline moiety is well tolerated, we evaluated how substitution of aniline nitrogen affects sGC activation. Acetylation, butyration, caproylation in 10a-10d were not conducive to sGC activation. The introduction of an additional carboxylic group afforded by modification with succinate (10d), glutarate (10e) or adipiate (10f) resulted in weak, yet statistically significant activation of Fe3+-sGC, but not Fe2+-sGC (Fig. 1). Therefore, we prepared an amide-linked dimer 11, which exhibited sGC activation and had a lower EC50 value, as compared to gemfibrozil.
Of interest, examination of the activation curves for the active compounds revealed anomalous behavior for two of the derivatives. As can be seen in Figure 3, the activation curves for 4c, 4d, 7a, 11 followed a typical S-shaped pattern similar to gemfibrozil, while compounds 7c and 15b exhibit a bell-shaped curve. The bell-shaped curve suggests a bi-phasic binding of these compounds to two different sites on sGC, where the first binding with a lower EC50 has an activating effect and binding to the second site (with higher EC50) has an inhibitory effect. Previous computational modeling predicted that gemfibrozil may have two binding sites within the cavity of the heme-binding pocket of sGC [29]. The bi-phasing effect of 7c and 15b is consistent with the two-binding site hypothesis. Similarly, although compound 7a had the lowest EC50 value of 50 ± 16 μM, it exhibited only a modest sGC activation at 100 μM and flattening of the curve, which could be explained by the inhibitory effect of the lower affinity binding site.
Figure 3. Dose-dependent activation of sGC by gemfibrozil derivatives.

Purified human sGC with ferric heme pre-incubated with different concentrations of indicated compounds was tested for cGMP-forming activity. Data are mean ±SD (n=9).
2.2.2. Vasodilator action of generated derivatives.
Regulation of vascular tone via promotion of vasodilation is one of the most important physiologic function of sGC. Previous studies demonstrated that activation of sGC by gemfibrozil results in vasodilation of isolated aortic rings [29]. Therefore, we tested the compounds 4c, 4d, 7a, 7c, 11 and 15b for their ability to inhibit the contraction of phenylephrine-treated isolated mouse aorta. Typical traces of changes in aortic ring tension in response to phenylephrine contraction and the vasoactive function of cumulative doses of gemfibrozil, 7c, and 15b are shown in Figure 4a. Using this approach we evaluated the dose-dependent vasodilatory effect of these compounds and estimated the minimally effective concentration (MEC) and the half-maximal inhibitory concentration IC50 (Table 1). Although the general trend that compounds with improved EC50, as compared with gemfibrozil, also exhibited a better IC50 in vasodilation tests was evident, there were some exceptions. For instance, compounds 4d and 7a exhibited improved affinity in the tests with purified sGC, but had weaker MEC and IC50 values in vasodilation experiment. This difference is most likely due to affected membrane permeability and/or bioavailability. The dose-dependent vasoactivity curves for 7c and 15b, which exhibited best vasodilatory IC50 values, are shown in Figure 4b together with gemfibrozil and 4c.
Figure 4. Vasoactivity of 7c and 15b.

(A): Representative wire myograph traces of changes in isometric tension of isolated mouse aortic rings in response to gemfibrozil, 7c or 15b or solvent (DMSO). Mouse aortic rings were isolated and prepared as described in Methods. After the contraction induced by 1 μM phenylephrine stabilized, cumulative doses of compounds were added to the organ bath and changes in isometric tension were recorded. Upticks in the traces mark when indicated amount of agonists was added. (B): Relaxation of pre-constricted mouse aortic rings in response to different concentrations of gemfibrozil, 4c, 7c or 15b. Data are mean ± SD (n=5), * - p <0.05 (one-way ANOVA followed by Tukey’s multi comparison test).
3. CONCLUSION
In summary, we described the synthesis of gemfibrozil derivatives and the analysis of their sGC activating properties. The structure-activity relationship study identified the positions in gemfibrozil’s scaffold that are detrimental for sGC activation and those that are amendable for optimizing modifications. We determined that short carboxylic acid chains, modification of gemfibrozil’s carboxyl group, and the lack of benzene ring modification are detrimental for sGC activating properties. We also demonstrated that some modification of the alpha position of the carboxylic acid, introduction of an additional carboxylic acid chain or creation of dimeric gemfibrozil derivatives with preserved carboxylic acid chains might offer additional directions for optimization of sGC activating properties in new gemfibrozil derivatives. These studies also revealed a bi-phasic response of sGC to some of gemfibrozil derivatives, providing additional support to the notion that sGC possesses two gemfibrozil-binding sites. Several compounds with improved EC50 values in sGC activity tests were generated. Two sGC-activating compounds, 7c and 15b, exhibited an enhanced vasodilatory effect, likely due to a combination of improved sGC affinity and better bioavailability. Further studies are needed to determine the effect of 7c and 15b on in vivo sGC activity and function. These SAR studies established the overall framework needed for future development of sGC activators based on gemfibrozil scaffold. Since gemfibrozil-based sGC activators are more potent activation of sGC with ferric heme, the active compounds reported here and future improved derivatives may be beneficial in clinical setting for pharmacological upregulation of sGC cardiovascular function in conditions of increased burden of oxidative stress and diminished NO-dependent sGC activation.
4. EXPERIMENTAL SECTION
4.1. Reagents and Synthesis.
Unless otherwise stated all materials were used as received and reactions were performed under anhydrous conditions under an atmosphere of N2. Reactions in vials were performed in oven-dried nitrogen-flushed vials that were either capped (most reactions) or fitted with a septum to allow nitrogen atmosphere. THF and anhydrous DMF were purchased from Sigma-Aldrich (St. Louis, MO, USA); DCM and MeCN were purchased from Fisher Scientific (Waltham, MA, USA); TEA was purchased from Fisher Scientific (Waltham, MA, USA). TEA and DIPEA were dried over CaH2, distilled and stored over 4 Å molecular sieves. THF and MeCN were dried and stored over 4 Å molecular sieves. Chloroform-d and DMSO-d6 were purchased from Cambridge Isotopes (Tewksbury, MA, USA). Reactions were monitored using normal phase thin-layer chromatography (TLC) on Millipore glass-backed 60 Å plates (indicator F-254, 250 μm). Column chromatography was perform using Silicycle SiliaFlash® P60 (230–400 mesh) silica gel as the stationary phase using eluents indicated in the experimental section. Chemical shifts (δ) are reported in parts per million (ppm) and coupling constants (J) are reported in hertz (Hz). NMR peak pattern abbreviations are as follows: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, at = apparent triplet, q = quartet, m = multiplet. NMR spectra were calibrated relative to their respective residual NMR solvent peaks, chloroform-d = 7.26 ppm (1H NMR)/77.16 ppm (13C NMR), DMSO = 2.50 ppm (1H NMR)/39.52 ppm (13C NMR). HRMS was performed in the Baylor University Mass Spectrometry Center on a Thermo Scientific LTQ Orbitrap Discovery spectrometer using +ESI or −ESI and reported for the molecular ion ([M+H]+ & [M+Na]+ or [M-H]− respectively).
4.2. General Procedure for Synthesis of 3a-d.
To a 6 dram vial was added acid 2a-d, followed by SOCl2 (2 mL). The reaction vessel was capped and heated to 75 °C for 2 h and the solvent was removed by rotary evaporation. To the crude acid chloride was added isobutanol (10 mL), and the resulting mixture was allowed to sit at 25 °C for 12 h. The solvent was removed by rotary evaporation to provide the crude product which was sufficiently pure to carry forward.
4.2.1. Isobutyl 2-bromoacetate (3a)
Isobutyl 2-bromoacetate (3a) was synthesized according to the above general procedure using 2a (1.0 g, 7.2 mmol,1 equiv.), 35% yield. 1H NMR (Chloroform-d, 600 MHz) δ 3.95 (dd, J = 6.7, 1.1 Hz, 2H), 2.84 (d, J = 1.1 Hz, 2H), 1.98 (hept, J = 7.0 Hz, 1H), 0.95 (d, J = 6.7 Hz, 6H). 13C NMR (Chloroform-d, 151 MHz) δ 167.4, 72.4, 27.8, 26.0, 19.1. +ESI-HRMS m/z: calc’d for [M+H]+ C6H12BrO2+ = 195.0015, C6H12BrO2+ found 195.0014.
4.2.2. Isobutyl 4-bromobutanoate (3b)
Isobutyl 4-bromobutanoate (3b) was synthesized according to the above general procedure using 2b (1.0 g, 5.99 mmol, 1 equiv.), 87% yield. 1H NMR (600 MHz, Chloroform-d) δ 3.86 (d, J = 6.7 Hz, 2H), 3.46 (t, J = 6.5 Hz, 2H), 2.50 (t, J = 7.2 Hz, 2H), 2.17 (p, J = 6.8 Hz, 2H), 1.92 (hept, J = 6.7 Hz, 1H), 0.92 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 172.7, 70.8, 32.9, 32.6, 27.9, 27.8, 19.2. +ESI-HRMS m/z: calc’d for [M+Na]+ C8H15BrNaO2+ = 245.0148, C8H15BrNaO2+ found 245.0150.
4.2.3. Isobutyl 5-bromopentanoate (3c)
Isobutyl 5-bromopentanoate (3c) was synthesized according to the above general procedure, using 2c (1.0 g, 5.52 mmol, 1 equiv.), 88% yield. 1H NMR (600 MHz, Chloroform-d) δ 3.85 (d, J = 6.7 Hz, 2H), 3.41 (t, J = 6.7 Hz, 2H), 2.35 (t, J = 7.3 Hz, 2H), 1.93 – 1.88 (m, 3H), 1.81 – 1.75 (m, 2H), 0.92 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 173.2, 70.6, 33.3, 33.0, 32.0, 27.7, 23.6, 19.1. +ESI-HRMS m/z: calc’d for [M+Na]+ C9H17BrNaO2+ = 259.0304, C9H17BrNaO2+ found = 259.0305.
4.2.4. Isobutyl 6-bromohexanoate (3d)
Isobutyl 6-bromohexanoate (3d) was synthesized according to the above general procedure using 2d (1.0 g, 5.12 mmol, 1 equiv.), 92% yield. 1H NMR (600 MHz, Chloroform-d) δ 3.86 (d, J = 6.7 Hz, 2H), 3.41 (t, J = 6.8 Hz, 2H), 2.34 (t, J = 7.5 Hz, 2H), 1.97–1.86 (m, 3H), 1.66 (dt, J = 15.3, 7.5 Hz, 2H), 1.48 (p, J = 7.7 Hz, 2H), 0.93 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 173.7, 70.7, 34.3, 33.6, 32.6, 27.9, 27.8, 24.3, 19.3. +ESI-HRMS m/z: calc’d for [M+H]+ C10H20BrO2+ = 251.0641, C9H11BrNaO2+ found = 251.0638.
4.3. General Procedure for Synthesis of 4a-d:
To a 6 dram vial was added esters 3a-d, followed by K2CO3 (2.5 eq) 2,5-dimethylphenol (1 eq), and MeCN (2 mL)and the reaction heated to 82 °C for 12 h. After cooling to 25 °C, 1 M NaOH (2 mL) was added and the reaction heated to 100 °C for 24 h. The resulting mixture was then partitioned between EtOAc (5 mL) and brine (10 mL) and the organic phase dried with MgSO4 and evaporated by rotary evaporation. The resulting residue was purified by silica chromatography (10–20% EtOAc in hexanes).
4.3.1. 2-(2,5-dimethylphenoxy)acetic acid (4a)
2-(2,5-dimethylphenoxy)acetic acid (4a) was synthesized according to the above general procedure using K2CO3 (68 mg, 0.492 mmol, 2.51 equiv.), 2,5-dimethylphenol (24 mg, 0.196 mmol, 1 equiv.), MeCN (2 mL) followed by 3a (50 mg, 0.256 mmol, 1.3 equiv.), 32% yield. 1H NMR (Chloroform-d, 600 MHz) δ 7.04 (d, J = 7.5 Hz, 1H), 6.75 (d, J = 7.5 Hz, 1H), 6.56 (s, 1H), 4.68 (s, 2H), 2.31 (s, 3H), 2.25 (s, 3H). 13C NMR (Chloroform-d, 151 MHz) δ 173.8, 155.6, 136.9, 131.1, 124.2, 122.7, 112.5, 63.3, 21.5, 15.9. +ESI-HRMS m/z: calc’d for [M+Na]+ C10H12NaO3+= 203.0679, C9H11BrNaO2+ found = 203.0677.
4.3.2. 4-(2,5-dimethylphenoxy)butanoic acid (4b)
4-(2,5-dimethylphenoxy)butanoic acid (4b) was synthesized according to the above general procedure using K2CO3 (68 mg, 0.492 mmol, 2.5 equiv.), 2,5-dimethylphenol (24 mg, 0.196 mmol, 1 equiv.), MeCN (2 mL) followed by 3b (50 mg, 0.224 mmol, 1.1 equiv.), 32% yield. 1H NMR (Chloroform-d, 600 MHz) δ 7.04 (d, J = 7.5 Hz, 1H), 6.75 (d, J = 7.5 Hz, 1H), 6.56 (s, 1H), 4.68 (s, 2H), 2.31 (s, 3H), 2.25 (s, 3H). 13C NMR (Chloroform-d, 151 MHz) δ 173.8, 155.6, 136.9, 131.1, 124.2, 122.7, 112.5, 63.3, 21.5, 15.9. +ESI-HRMS m/z: calc’d for [M+Na]+ C12H16NaO3+ = 231.0992, C12H16NaO3+ found = 231.0990.
4.3.3. 5-(2,5-dimethylphenoxy)pentanoic acid (4c)
5-(2,5-dimethylphenoxy)pentanoic acid (4c) was synthesized according to the above general procedure, using K2CO3 (68 mg, 0.492 mmol, 2.5 equiv.), 2,5-dimethylphenol (24 mg, 0.196 mmol, 1 equiv.), MeCN (2 mL) followed by 3c (50 mg, 0.211 mmol,1.1 equiv), 33% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.00 (d, J = 7.4 Hz, 1H), 6.66 (d, J = 7.5 Hz, 1H), 6.62 (s, 1H), 3.97 (t, J = 5.7, 2H), 2.46 (t, J = 7.0 Hz, 2H), 2.31 (s, 3H), 2.17 (s, 3H), 1.90 – 1.78 (m, 4H). 13C NMR (151 MHz, Chloroform-d) δ 178.9, 157.0, 136.6, 130.5, 123.8, 120.9, 112.0, 67.3, 33.6, 28.9, 21.7, 21.6, 16.0. +ESI-HRMS m/z: calc’d for [M+Na]+ C13H18NaO3+ = 245.1148, C13H18NaO3+ found = 245.1148.
4.3.4. 6-(2,5-dimethylphenoxy)hexanoic acid (4d)
6-(2,5-dimethylphenoxy)hexanoic acid (4d) was synthesized according to the above general procedure, using K2CO3 (68 mg, 0.492 mmol, 2.5 equiv.), 2,5-dimethylphenol (24 mg, 0.196 mmol, 1 equiv.), MeCN (2 mL) followed by 3d (50 mg, 0.199 mmol, 1 equiv.), 42% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.00 (d, J = 7.4 Hz, 1H), 6.66 (d, J = 7.5 Hz, 1H), 6.62 (s, 1H), 3.95 (t, J = 6.3 Hz, 2H), 2.41 (t, J = 7.4 Hz, 2H), 2.31 (s, 3H), 2.17 (s, 3H), 1.85–1.80 (m, 2H), 1.73 (p, J = 7.5 Hz, 2H), 1.60 – 1.51 (m, 2H). 13C NMR (151 MHz, Chloroform-d) δ 178.9, 157.1, 136.6, 130.4, 123.8, 120.8, 112.1, 67.6, 34.0, 29.2, 25.9, 24.6, 21.6, 15.9. +ESI-HRMS m/z: calc’d for [M+Na]+ C14H20NaO3+ = 259.1305, C14H20NaO3+ found = 259.1302.
4.4. Synthesis of isobutyl 5-chloro-2,2-dimethylpentanoate (6):_
To a round bottom flask under an atmosphere of N2 was added diisopropylamine (2 mL, 14.5 mmol, 2.1 equiv), followed by THF (5 mL), then cooled to −78 °C for 20 min. To the above solution was added 5 M n-BuLi in hexanes (2.7 mL, 13.9 mmol, 2 equiv.) then allowed to warm to 25 °C for 1 h before being cooled again to −78 °C. To a separate round bottom flask was added isobutyl isobutyrate (1.0 g, 6.93 mmol, 1 equiv.) and THF (15 mL), then cooled to −78 °C for 30 min, after which the LDA solution was added dropwise and the mixture was stirred for 1 h. The reaction mixture was warmed to 0 °C in an ice bath, followed by the dropwise addition of 1-bromo-3-chloropropane (682 μL, 6.93 mmol). The reaction was allowed to warm to to 25 °C over 2 h, followed by removal of the solvent by rotary evaporation. The crude residue was purified by silica chromatography (10–20% EtOAc in hexanes) to provide the product as a clear oil in 82% yield. 1H NMR (Chloroform-d, 600 MHz) δ 3.85 (d, J = 6.5 Hz, 2H), 3.51 (t, J = 6.5 Hz, 2H), 1.93 (hept, J = 6.7 Hz, 1H), 1.75–1.70 (m, 2H), 1.68–1.63 (m, 2H), 1.19 (s, 6H), 0.94 (d, J = 6.7 Hz, 6H). 13C NMR (Chloroform-d, 151 MHz) δ177.5, 70.6, 45.3, 42.1, 37.9, 28.4, 27.8, 25.2, 19.1. +ESI-HRMS m/z: calc’d for [M+Na]+ C11H21ClNaO2+ = 243.1122, C11H21ClNaO2+ found = 243.1124.
4.5. Synthesis of 5-((2,5-dimethylphenyl)amino)-2,2-dimethylpentanoic acid (7a):_
To a 6 dram vial was added 9 (75 mg, 0.24 mmol, 1 equiv.), MeOH (1.0 mL), and 10 M NaOH (0.7 mL). The reaction mixture was allowed to stir at 25 °C for 22 h. To the reaction mixture was added MeOH (0.5 mL) and was heated to 80 °C for 3 d. The reaction mixture cooled to 25 °C, the volatile solvents were removed by rotary evaporation and the remaining aqueous solution was acidified to pH 5 with 3 M HCl resulting in the formation of a white precipitate. The white precipitate was then extracted with EtOAc (5 mL), the organic layer dried with Na2SO4 and the solvent removed by rotary evaporation. The product was partially purified by trituration with hexanes, then the solid material was dissolved and passed through a silica plug (60% EtOAc in hexanes) to provide the product in 70% yield. 1H NMR (600 MHz, Chloroform-d) δ 6.93 (d, J = 7.4 Hz, 1H), 6.47 (d, J = 7.3 Hz, 1H), 6.42 (s, 1H), 3.14 (t, J = 6.5 Hz, 2H), 2.29 (s, 3H), 2.09 (s, 3H), 1.70 – 1.65 (m, 4H), 1.24 (s, 6H). 13C NMR (151 MHz, Chloroform-d) δ 184.0, 146.2, 136.9, 130.0, 119.0, 117.6, 110.7, 44.4, 42.2, 37.9, 25.5, 25.2, 21.7, 17.1. −ESI-HRMS m/z: calc’d for [M-H]− C15H22NO2− = 248.1656, C15H22NO2− found = 248.1657.
4.6. Synthesis of 2,2-dimethyl-5-(o-tolyloxy)pentanoic acid (7b):_
To a 2 dram vial O-cresol (576.3 mg, 4.17 mmol, 3 equiv.) was added to a solution of 6 (305.7 mg, 1.4 mmol, 1 equiv.), NaI (675.0 mg, 4.17 mmol 3 equiv.), and K2CO3 (967.5 mg, 6.95 mmol, 5 equiv.) in DMSO (2 mL). The reaction mixture was stirred at 80 °C for 47 h. The mixture was then diluted with EtOAc (15 mL) and washed with 1 M NaOH (15 mL), then brine. The organic layer was dried with Na2SO4 and the solvent was removed by rotary evaporation to provide a 299.6 mg of a brown oil that was used without further purification. The above oil was dissolved in MeOH (2.5 mL) and added to 6 dram vial. To this solution was added 10 M NaOH (2.5 mL) and the reaction mixture was stirred at 80 °C for 48 h. The volatiles were removed from the reaction mixture by rotary evaporation. The remaining aqueous solution was washed with EtOAc (5 mL), then acidified with 3 M HCl (10 mL) and extracted with EtOAc (5 mL). Organic layer was dried with Na2SO4 and the solvent was removed by rotary evaporation. The reaction mixture was purified by silica chromatography (20% EtOAc in hexanes) to provide the product in 22% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.13 (t, J = 7.1 Hz, 2H), 6.84 (t, J = 7.4 Hz, 1H), 6.78 (d, J = 8.3 Hz, 1H), 3.95 (t, J = 6.1 Hz, 2H), 2.22 (s, 3H), 1.86 – 1.78 (m, 2H), 1.78 – 1.73 (m, 2H), 1.26 (s, 6H). 13C NMR (151 MHz, Chloroform-d) δ 183.2, 157.2, 130.7, 126.9, 126.8, 120.3, 111.0, 68.0, 42.0, 37.0, 25.3, 25.2, 16.3. +ESI-HRMS m/z: calc’d for [M+H]+ C14H20NO3+ = 259.1310, C14H20NO3+ found 259.1306.
4.7. Synthesis of 5-(2-isopropyl-5-methylphenoxy)-2,2-dimethylpentanoic acid (7c):
To a 2 dram vial was added DMSO (2 mL), 6 (305.7 mg, 1.39 mmol, 1 equiv.), NaI (625.0 mg, 4.17 mmol, 3 equiv.), K2CO3 (967.5 mg, 6.95 mmol, 5 equiv.), and thymol (626.4 mg, 4.17 mmol, 3 equiv.) The reaction mixture was heated to 80 °C for 3 d. The reaction was allowed to cool to 25 °C and was quenched with brine (50 mL), extracted with EtOAc (20 mL). The organic extract was dried with Na2SO4 and the solvent removed by rotary evaporation. The crude mixture was purified by silica chromatography (2.5 % EtOAc in hexanes) to provide the isobutyl ester of 7c in 40% yield. 1H NMR (400 MHz, Chloroform-d) δ 7.13 (d, J = 7.7 Hz, 1H), 6.77 (d, J = 7.6 Hz, 1H), 6.68 (s, 1H), 3.98 (q, J = 5.6, 5.1 Hz, 2H), 3.91 (d, J = 6.6 Hz, 2H), 3.33 (hept, J = 7.0 Hz, 1H), 2.35 (s, 3H), 1.99 (hept, J = 13.3, 6.7 Hz, 1H), 1.85 – 1.75 (m, 4H), 1.28 (s, 6H), 1.25 (d, J = 7.0 Hz, 6H), 0.99 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 189.0, 177.8, 156.1, 136.3, 134.1, 125.9, 121.0, 112.2, 70.6, 68.1, 42.3, 37.4, 27.9, 26.7, 25.3, 25.3, 22.8, 21.4, 19.2. +ESI-HRMS m/z: calc’d for [M+Na]+ C21H34NaO3+ = 357.2406, C21H34NaO3+ found 357.2409.
To a 6 dram vial was added the isobutyl ester of 7c (186.7 mg, 0.56 mmol, 1 equiv.), 10 M NaOH (2 mL), and MeOH (2 mL) and heated to 80 °C and stirred for 52 h. The volatiles were removed with rotary evaporation, reaction was quenched by the addition of 12 M HCl (2 mL). The crude mixture was extracted with EtOAc (5 mL) and the organic extract washed with brine (5 mL). The organic layer was then dried with Na2SO4 and evaporated with rotary evaporation. The crude product was purified by silica chromatography (20% EtOAc in hexanes) to provide the product in 77% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.08 (d, 1H), 6.73 (d, J = 7.1 Hz, 1H), 6.63 (s, 1H), 3.93 (t, 2H), 3.31 – 3.22 (sept, 1H), 2.31 (s, 3H), 1.84 – 1.78 (m, 2H), 1.78 – 1.72 (m, 2H), 1.26 (s, 6H), 1.20 (d, 6H). 13C NMR (151 MHz, Chloroform-d) δ 156.0, 136.3, 134.0, 125.9, 121.0, 112.2, 67.9, 41.9, 37.0, 26.7, 25.2, 25.0, 22.8, 21.4. −ESI-HRMS m/z: calc’d for [M-H]− C17H25O3− 277.1809, C17H25O3− found 277.1809.
4.8. Synthesis of 6-(phenylthio)hexanoic acid (8a):
In a 6 dram vial, 6-bromohexanoic acid (2e) (502 mg, 3.03 mmol, 1 equiv.) was added to thionyl chloride (1 mL, 13.7 mmol), which was heated to 75 °C for 1 h under nitrogen. The mixture was then allowed to cool to 25 °C before isopropanol (20 mL) was added and the reaction mixture was stirred for 2 h. The solvent was then removed by rotary evaporation and the resulting residue was purified by silica chromatography (10% EtOAc in Hexanes) to provide isopropyl 6-bromohexanoate in 82% yield. 1H NMR (DMSO-d6, 600 MHz) δ 5.00 (sept., J = 6.3 Hz, 1H), 3.40 (t, J = 6.8 Hz, 2H), 2.28 (t, J = 7.4 Hz, 2H), 1.91–1.84 (m, 2H), 1.68–1.61 (m, 2H), 1.50–1.43 (m, 2H), 1.23 (d, J = 6.3 Hz, 6H). 13C NMR (DMSO-d6, 151 MHz) δ 173.1, 67.7, 34.6, 33.7, 32.6, 27.8, 24.3, 22.0. +ESI-HRMS m/z: calc’d for [M+Na]+ C9H17BrNaO2+ 259.0306, C9H17BrNaO2+ found 259.0306.
To a 6 dram vial was added isopropyl 6-bromohexanoate (500 mg, 2.11 mmol, 1 equiv.) dissolved in THF (1 mL). Thiophenol (216 μL, 2.11 mmol, 1 equiv.) was added as a solution in 1 M NaOH (2 mL) and THF (1 mL), then the reaction mixture was stirred at 25 °C. The solution was subjected to rotary evaporation to remove the THF, quenched with 3 M HCl (5 mL) and extracted with DCM (5 mL). The organic phase was dried with MgSO4 and evaporated to provide the isopropyl ester of 8a in 87% yield. 1H NMR (DMSO-d6, 600 MHz) δ 7.32 (dd, J = 8.1, 1.4 Hz, 2H), 7.23 (t, J = 7.5, 2H), 7.16 (tt, J = 7.3, 0.8 Hz, 1H), 5.00 (sept., J = 6.3 Hz, 1H), 2.91 (t, J = 7.3 Hz, 2H), 2.26 (t, J = 7.4 Hz, 2H), 1.70–1.60 (m, 4H), 1.49–1.43 (m, 2H), 1.22 (d, J = 6.3 Hz, 6H). 13C NMR (DMSO-d6, 151 MHz) δ 173.3, 136.9, 129.1, 129.0, 125.9, 67.6, 34.6, 33.6, 28.9, 28.4, 24.7, 22.0. +ESI-HRMS m/z: calc’d for [M+H]+ C15H23SO2+ 267.1413, C15H23SO2+ found 267.1413.
In a 6 dram vial the isopropyl ester of 8a (489 mg, 1.84 mmol, 1 equiv.) dissolved in THF (2 mL) and added to of 10 M NaOH (2 mL) in a round bottom flask. The mixture was heated to 100 °C for 16 h, then allowed to cool to 25 °C. The crude material was acidified with 3 M HCl (10 mL) and cooled to 4 °C for 2 h, then filtered to afford the product in 18% yield. 1H NMR (DMSO-d6, 600 MHz) δ 7.31 (d, J = 8.1 Hz, 2H), 7.28 (t, J = 7.5 Hz, 2H), 7.17 (t, J = 7.3 Hz, 1H), 2.92 (t, J = 7.3 Hz, 2H), 2.36 (t, J = 7.4 Hz, 2H), 1.71–1.63 (m, 4H), 1.52–1.46 (m, 2H). 13C NMR (DMSO-d6, 151 MHz) δ 177.4, 136.8, 129.2, 129.0, 126.0, 33.6, 33.6, 28.9, 28.3, 24.4. +ESI-HRMS m/z: calc’d for [M+Na]+ C12H16NaO2S+ 247.0763, C12H16NaO2S+ found 247.0762.
4.9. Synthesis of 6-phenoxyhexanoic acid (8b):_
In a 6 dram vial isopropyl 6-bromohexanoate (50 mg, 0.211 mmol, 1 equiv.) was dissolved in a mixture of THF (2 mL) and 1 M NaOH (2 mL), and phenol (18.6 μL, 0.211 mmol, 1 equiv.) was added and the mixture vigorously stirred for 8 h at 80 °C. 10 M NaOH (2 mL) was then added and the mixture was heated to 100 °C for 48 h, and then 3 M HCl (10 mL) was used to quench the reaction. The mixture was extracted with EtOAc (10 mL), and the organic layer dried with MgSO4, and the solvent removed by rotary evaporation. The crude residue was then purified by silica chromatography (10% EtOAc in Hexanes) to afford the product in 46% yield. 1H NMR (Chloroform-d, 600 MHz) δ 7.27 (t, J = 8.3 Hz, 2H), 6.93 (t, J = 7.3 Hz, 1H), 6.88 (d, J = 8.0 Hz, 2H), 3.96 (t, J = 6.4 Hz, 2H), 2.40 (t, J = 7.4 Hz, 2H), 1.85–1.78 (m, 2H), 1.76–1.70 (m, 2H), 1.58–1.50 (m, 2H). 13C NMR (Chloroform-d, 151 MHz) δ 178.3, 159.1, 129.6, 120.7, 114.6, 67.6, 33.8, 29.1, 25.8, 24.6. −ESI-HRMS m/z: calc’d for [M-H]− C12H15O3− 207.1027, C12H15O3− found 207.1026.
4.10. Synthesis of isobutyl 5-((2,5-dimethylphenyl)amino)-2,2-dimethylpentanoate (9):
To a 6 dram vial was added DMF (9 mL), 6 (2.0 g, 9 mmol, 1 equiv.), NaI (4.1 g, 27 mmol, 3 equiv.), K2CO3 (3.4 g, 24.8 mmol, 2.75 equiv.), and 2,5-dimethylaniline (1.2 g, 9.9 mmol, 1.1 equiv.). The reaction mixture was heated to 62 °C and stirred for 23 h, then was allowed to cool to 25 °C, quenched with brine (50 mL), and extracted with EtOAc (20 mL). The organic extract was dried with Na2SO4 and the solvent removed by rotary evaporation. The crude mixture was partially purified by silica chromatography (5–10% EtOAc in hexanes). The partially purified product was dissolved in a 2:1 hexanes to 4 M HCl in dioxane mixture (15 mL), followed by addition of 3 M HCl in H2O (5 mL) and vigorously shaken to form a white precipitate, which was filtered and washed with EtOAc (5 mL) to provide the product in 50 % yield. 1H NMR (600 MHz, DMSO-d6) δ 6.49 (d, J = 7.8 Hz, 1H), 6.44 (d, J = 7.8 Hz, 1H), 6.37 (s, 1H), 3.03 (d, J = 6.5 Hz, 2H), 2.55 – 2.51 (m, 4H), 1.59 (s, 3H), 1.57 (s, 3H), 1.08 (hept, J = 13.4 Hz, 1H), 0.91 (ddt, J = 10.6, 7.5, 3.4 Hz, 2H), 0.86 – 0.80 (m, 2H), 0.41 (s, 6H), 0.12 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, DMSO-d6) δ 176.5, 137.3, 132.3, 131.4, 129.4, 127.0, 122.1, 69.6, 50.4, 41.0, 35.7, 26.8, 23.3, 20.8, 18.6, 17.2, 14.3. +ESI-HRMS m/z: calc’d for [M+H]+ C19H32NO2+ = 306.2428, C19H32NO2+ found = 306.2433.
4.11. Synthesis of 5-(N-(2,5-dimethylphenyl)acetamido)-2,2-dimethylpentanoic acid (10a):_
To a 1 dram vial was added 9 (50.0 mg, 0.16 mmol, 1 equiv.), THF (0.75 mL), and TEA (67.5 μL, 0.48 mmol, 3 equiv.). DCM (0.75 mL) was then added to increase homogeneity of the reaction, followed by the addition of AcCl (22.8 μL, 0.32 mmol, 2 equiv.). The reaction mixture was allowed to stir at 25 °C for 24 h and diluted with EtOAc (3 mL) and filtered. The solvent was removed with rotary evaporation and the mixture was purified with by silica chromatography (20% then 60% EtOAc in hexanes) to provide the isobutyl ester of 10a in 72% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.15 (d, J = 7.7 Hz, 1H), 7.06 (d, J = 6.9 Hz, 1H), 6.87 (s, 1H), 4.00–3.99 (m, 1H), 3.82 – 3.73 (m, 2H), 3.10–3.00 (m, 1H), 2.32 (s, 3H), 2.15 (s, 3H), 1.84 (sept J = 13.3 Hz, 1H), 1.72 (s, 3H), 1.58 – 1.38 (m, 4H), 1.15 (s, 3H), 1.15 (s, 3H), 0.86 (dd, J = 6.7, 2.3 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 177.8, 170.6, 141.6, 137.0, 132.3, 131.4, 129.6, 129.1, 70.5, 48.4, 42.4, 37.9, 27.9, 25.4, 25.2, 23.5, 22.4, 20.9, 19.2, 17.2. +ESI-HRMS m/z: calc’d for [M+Na]+ C21H33NaNO3+ = 370.2358, C21H33NaNO3+ found = 370.2358.
To a 6 dram vial was added the isobutyl ester of 10a (36.7 mg, 0.12 mmol, 1 equiv.), followed by MeOH (1.75 mL) and 10 M NaOH (1.25 mL), and the mixture stirred at 25 °C for 24 h. The organic solvent was removed by rotary evaporation and 3 M HCl was added until reaction mixture pH was 2. The solution was then extracted with EtOAc (5 mL), the organic extract dried with Na2SO4, and the solvent removed by rotary evaporation. The product was purified by trituration with hexanes to provide the product in 35% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.15 (d, J = 7.8 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 6.90 (d, J = 1.8 Hz, 1H), 4.06 (ddd, J = 13.2, 8.7, 6.7 Hz, 1H), 3.41 – 3.32 (m, 1H), 3.05 (ddd, J = 13.1, 8.4, 4.6 Hz, 1H), 2.31 (s, 3H), 2.15 (s, 3H), 1.74 (s, 3H), 1.71 – 1.40 (m, 4H), 1.17 (d, J = 10.2 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 183.0, 171.1, 141.4, 137.1, 132.2, 131.4, 129.6, 129.2, 48.3, 41.9, 37.6, 25.4, 24.9, 23.3, 22.3, 20.9, 17.1. −ESI-HRMS m/z: calc’d for [M-H]− C17H25NO3− = 290.1762, C17H25NO3− found = 290.1761.
4.12. General Procedure for Synthesis of 10b-f:
To a 1 dram vial was added the appropriate acid, HATU, TEA, and DMF and the mixture stirred for 15 min at 25 °C. To the reaction mixture was added 9 (50 mg, 0.16 mmol, 1 equiv.) and the resulting mixture stirred for the specified time at 25 °C. The reaction was quenched with 3 M HCl (2 mL) and extracted with EtOAc (2 mL). The organic layer was further washed with 1 M NaOH (2 mL) and the organic layer dried with Na2SO4. The solvent was removed with rotary evaporation. The crude material was purified by silica chromatography using the eluents indicated to provide the ester precursor of the desired product.
To a 6 dram vial was added the ester of the desired product followed by 4:1 MeOH:10 M NaOH solution (2 mL) and was stirred at 25 °C for 72 h, then the temperature was increased to 40 °C for 24 h, then to 60 °C for 48 h. The organic solvent was removed by rotary evaporation, 3 M HCl added until the reaction mixture pH was 2, the aqueous solution extracted with EtOAc (5 mL), and the organic extract dried with Na2SO4. The organic solvent was removed by rotary evaporation and the product was purified as specified.
4.12.1. Isobutyl 5-(N-(2,5-dimethylphenyl)butyramido)-2,2-dimethylpentanoate (10b isobutyl ester)
Isobutyl 5-(N-(2,5-dimethylphenyl)butyramido)-2,2-dimethylpentanoate (10b isobutyl ester) was synthesized using the above general procedure using butyric acid (16.2 μL, 0.18 mmol, 1.1 equiv), HATU (68.4 mg, 0.18 mmol, 1.1 equiv.), TEA (67.5 μL, 0.48 mmol, 3 equiv.), and DMF (1 mL). After addition of 9 (50 mg, 0.16 mmol, 1 equiv.) the reaction was stirred for 47 h followed by addition of TEA (67.5 μL, 0.48 mmol, 3 equiv.), and stirred for an additional 26 h. Silica chromatography (20-40-60% EtOAc in hexanes), provided the isobutyl ester of 10b in 34% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.15 (d, J = 7.7 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 6.85 (s, 1H), 4.08 – 4.00 (m, 1H), 3.82 – 3.73 (m, 2H), 3.05 – 2.98 (m, 1H), 2.32 (s, 3H), 2.13 (s, 3H), 1.93 (dt, J = 15.2, 7.4 Hz, 1H), 1.89 – 1.75 (m, 2H), 1.60 – 1.39 (m, 6H), 1.15 (d, J = 2.8 Hz, 6H), 0.86 (dd, J = 6.7, 2.5 Hz, 6H), 0.81 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 177.9, 173.0, 141.2, 136.9, 132.4, 131.3, 129.9, 129.0, 70.5, 48.5, 42.4, 37.9, 36.0, 27.9, 25.5, 25.2, 23.5, 20.9, 19.2, 18.7, 17.2, 14.0. +ESI-HRMS m/z: calc’d for [M+Na]+ C17H25NaNO3+ = 398.2671, C17H25NaNO3+ found = 398.2671.
4.12.2. 5-(N-(2,5-dimethylphenyl)butyramido)-2,2-dimethylpentanoic acid (10b)
5-(N-(2,5-dimethylphenyl)butyramido)-2,2-dimethylpentanoic acid (10b) was synthesized using the above general procedure, using the isobutyl ester of 10b (20.4 mg, 0.054 mmol, 1 equiv.). The product was purified by dissolution in hexanes and was decanted to from insoluble impurities. The hexanes solution was evaporated to provide the product 79% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.16 (d, J = 7.9 Hz, 1H), 7.06 (d, J = 8.4 Hz, 1H), 6.88 (s, 1H), 4.14 – 4.07 (m, 1H), 3.05 – 2.97 (m, 1H), 2.31 (s, 3H), 2.13 (s, 3H), 1.99 – 1.91 (m, 1H), 1.85 – 1.77 (m, 1H), 1.66 – 1.43 (m, 7H), 1.18 (d, J = 9.3 Hz, 6H), 0.81 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 173.3, 141.1, 137.0, 132.4, 131.4, 129.9, 129.1, 48.4, 42.0, 37.6, 36.0, 25.4, 24.9, 23.3, 20.9, 18.7, 17.2, 14.0. −ESI-HRMS m/z: calc’d for [M-H]− C19H28NO3− = 318.2075, C19H28NO3− found = 318.2077.
4.12.3. Isobutyl 5-(N-(2,5-dimethylphenyl)pentanamido)-2,2-dimethylpentanoate (10c isobutyl ester)
Isobutyl 5-(N-(2,5-dimethylphenyl)pentanamido)-2,2-dimethylpentanoate (10c isobutyl ester) was synthesized using the above general procedure using caproic acid (22.5 μL, 0.18 mmol, 1.1 equiv), HATU (68.4 mg, 0.18 mmol, 1.1 equiv.), TEA (67.5 μL, 0.48 mmol, 3 equiv.), and DMF (1 mL). After addition of 9 (50 mg, 0.16 mmol, 1 equiv.) the reaction was stirred for 47 h followed by addition of TEA (67.5 μL, 0.48 mmol, 3 equiv.) and the reaction was stirred for an additional 26 h. Silica chromatography (20-40-60% EtOAc in hexanes), provided the isobutyl ester of 10c in 35% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.15 (d, J = 7.8 Hz, 1H), 7.06 (d, J = 9.9 Hz, 1H), 6.85 (s, 1H), 4.07 – 3.99 (m, 1H), 3.82 – 3.73 (m, 2H), 3.05 – 2.98 (m, 1H), 2.32 (s, 3H), 2.13 (s, 3H), 1.89 – 1.77 (m, 1H), 1.58 – 1.49 (m, 6H), 1.52 – 1.36 (m, 1H) 1.25 – 1.16 (m, 3H), 1.18 – 1.16 (m, 1H), 1.15 (d, J = 2.9 Hz, 6H), 1.14 – 1.12 (m, 1H), 0.86 (dd, J = 6.7, 2.5 Hz, 6H), 0.82 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 177.9, 173.2, 141.2, 136.9, 132.4, 131.3, 129.8, 129.0, 70.5, 48.5, 42.4, 37.9, 34.1, 31.7, 27.9, 25.5, 25.2, 25.1, 23.5, 22.6, 20.9, 19.2, 17.2, 14.0. +ESI-HRMS m/z: calc’d for [M+Na]+ C25H41NaNO3+ = 426.2984, C25H41NaNO3+ found = 426.2982.
4.12.4. 5-(N-(2,5-dimethylphenyl)pentanamido)-2,2-dimethylpentanoic acid (10c)
5-(N-(2,5-dimethylphenyl)pentanamido)-2,2-dimethylpentanoic acid (10c) was synthesized using the above general procedure, using the isobutyl ester of 10c (22.9 mg, 0.057 mmol, 1 equiv.). The product was purified by dissolution in hexanes and was decanted to from insoluble impurities. The hexanes solution was evaporated to provide the product in 76% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.16 (d, J = 7.8 Hz, 1H), 7.06 (d, J = 5.8 Hz, 1H), 6.88 (s, 1H), 4.14 – 4.06 (m, 1H), 3.01 (dd, J = 17.9, 8.3 Hz, 1H), 2.31 (s, 3H), 2.13 (s, 3H), 1.95 (dt, J = 15.2, 7.5 Hz, 1H), 1.83 (dt, J = 15.3, 7.5 Hz, 1H), 1.65 – 1.44 (m, 6H), 1.27 – 1.19 (m, 2H), 1.18 (d, J = 9.5 Hz, 6H), 1.16 – 1.09 (m, 2H), 0.82 (t, J = 7.2 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 183.5, 173.5, 141.1, 137.0, 132.4, 131.4, 129.9, 129.1, 48.3, 42.0, 37.6, 34.0, 31.6, 25.4, 25.1, 24.9, 23.3, 22.5, 20.9, 14.0. −ESI-HRMS m/z: calc’d for [M-H]− C21H32NO3− = 346.2388, C21H32NO3− found = 346.2390.
4.12.5. Isobutyl 5-(N-(2,5-dimethylphenyl)-4-methoxy-4-oxobutanamido)-2,2-dimethylpentanoate (10d diester)
Isobutyl 5-(N-(2,5-dimethylphenyl)-4-methoxy-4-oxobutanamido)-2,2-dimethylpentanoate (10d diester) was synthesized using the above general procedure using monomethyl succinic acid (24.8 mg, 0.18 mmol, 1.1 equiv), HATU (68.4 mg, 0.18 mmol, 1.1 equiv.), TEA (67.5 μL, 0.48 mmol, 3 equiv.), and DMF (1 mL). After addition of 9 (50 mg, 0.16 mmol, 1 equiv.), the reaction was stirred for 26 h followed by addition of monomethyl succinic acid (24.8 mg, 0.18 mmol, 1.1 equiv), HATU (68.4 mg, 0.18 mmol, 1.1 equiv.), TEA (67.5 μL, 0.48 mmol, 3 equiv.) was added and the reaction was stirred for an addition 24 h. Silica chromatography (20–40% EtOAc in hexanes), to provide the diester of 10d in 26% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.19 (d, J = 7.7 Hz, 1H), 7.09 (d, J = 7.0 Hz, 1H), 6.92 (s, 1H), 4.06 (ddd, J = 13.2, 9.4, 5.9 Hz, 1H), 3.84 – 3.75 (m, 2H), 3.67 (s, 3H), 3.09 – 3.01 (m, 1H), 2.66 – 2.58 (m, 1H), 2.58 – 2.50 (m, 1H), 2.34 (s, 3H), 2.32 – 2.24 (m, 1H), 2.20 (s, 3H), 2.17 – 2.07 (m, 1H), 1.87 (dt, J = 13.3, 6.7 Hz, 1H), 1.61 – 1.38 (m, 4H), 1.17 (s, 6H), 0.88 (dd, J = 6.7, 2.6 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 177.9, 173.7, 171.3, 140.6, 137.1, 132.6, 131.5, 129.8, 129.3, 70.5, 51.8, 48.6, 42.3, 37.8, 29.4, 29.2, 27.9, 25.5, 25.1, 23.4, 20.9, 19.2, 17.2. +ESI-HRMS m/z: calc’d for [M+Na]+ C24H37NaNO5+ = 442.2569, C24H37NaNO5+ found = 442.2562.
4.12.6. 5-(3-carboxy-N-(2,5-dimethylphenyl)propanamido)-2,2-dimethylpentanoic acid (10d)
5-(3-carboxy-N-(2,5-dimethylphenyl)propanamido)-2,2-dimethylpentanoic acid (10d) was synthesized using the above general procedure, using the diester of 10d (17.3 mg, 0.041 mmol, 1 equiv.). The product was partially purified with silica chromatography (80% EtOAc in hexanes plus 2 % AcOH). The product was purified was further purified by precipitation from DCM with hexanes to provide the product in 37% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.18 (d, J = 7.7 Hz, 1H), 7.09 (d, J = 7.7 Hz, 1H), 6.99 (s, 1H), 4.15 (dt, J = 14.3, 7.7 Hz, 1H), 3.02 (ddd, J = 13.1, 7.7, 5.2 Hz, 1H), 2.71 (ddd, J = 16.8, 9.1, 3.8 Hz, 1H), 2.53 (ddd, J = 17.0, 7.3, 3.8 Hz, 1H), 2.41 – 2.31 (m, 1H), 2.30 (s, 3H), 2.17 (s, 3H), 2.04 (ddd, J = 17.0, 7.2, 4.0 Hz, 1H), 1.69 (td, J = 12.3, 3.9 Hz, 1H), 1.65 – 1.44 (m, 3H), 1.19 (s, 3H), 1.16 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 183.1, 177.8, 172.3, 140.4, 137.3, 132.4, 131.6, 129.9, 129.5, 48.7, 42.0, 37.6, 29.8, 29.0, 24.6, 23.4, 20.9, 17.1. −ESI-HRMS m/z: calc’d for [M-H]− C19H26NO5− = 348.1816, C19H26NO5− found = 348.1820.
4.12.7. Isobutyl 5-(N-(2,5-dimethylphenyl)-5-methoxy-5-oxopentanamido)-2,2-dimethylpentanoate (10e diester)
Isobutyl 5-(N-(2,5-dimethylphenyl)-5-methoxy-5-oxopentanamido)-2,2-dimethylpentanoate (10e diester) was synthesized using the above general procedure using monomethyl glutaric acid (52.6 mg, 0.36 mmol, 2.2 equiv.), HATU (136.9 mg, 0.36 mmol, 2.2 equiv.), TEA (118 μL, 0.84 mmol, 6 equiv.), and DMF (1 mL). After addition of 9 (50 mg, 0.16 mmol, 1 equiv.) the reaction was stirred for 48 h at 25 °C. Silica chromatography (20–40% EtOAc in hexanes) provided the diester of 10e in 45% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.15 (d, J = 7.8 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 6.84 (s, 1H), 4.07 – 3.97 (m, 1H), 3.84 – 3.72 (m, 2H), 3.59 (s, 3H), 3.02 (ddd, J = 13.5, 8.8, 4.8 Hz, 1H), 2.31 (s, 3H), 2.27 (t, J = 6.6 Hz, 2H), 2.12 (s, 3H), 1.99 – 1.92 (m, 1H), 1.88 – 1.77 (m, 2H), 1.58 – 1.53 (m, 2H), 1.52 – 1.46 (m, 4H), 1.43 – 1.36 (m, 1H), 1.15 (s, 6H), 0.86 (dd, J = 6.7, 2.4 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 177.9, 173.8, 172.1, 140.9, 137.0, 132.4, 131.4, 129.7, 129.2, 70.5, 51.5, 48.5, 42.3, 37.9, 33.4, 33.1, 27.9, 25.5, 25.1, 23.4, 20.9, 20.6, 19.2, 17.1. +ESI-HRMS m/z: calc’d for [M+Na]+ C25H39NaNO5+ = 456.2726, C25H39NaNO5+− found = 456.2721.
4.12.8. 5-(4-carboxy-N-(2,5-dimethylphenyl)butanamido)-2,2-dimethylpentanoic acid (10e)
5-(4-carboxy-N-(2,5-dimethylphenyl)butanamido)-2,2-dimethylpentanoic acid (10e) was synthesized using the above general procedure, using the diester of 10e (22.3 mg, 0.051 mmol, 1 equiv.). The product was purified silica chromatography (80% EtOAc in hexanes plus 2 % AcOH). The product was purified was further purified by precipitation from DCM with hexanes to provide the product in 45% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.16 (d, J = 7.9 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 6.91 (s, 1H), 4.16 – 4.09 (m, 1H), 3.00 – 2.93 (m, 1H), 2.32 (s, 3H), 2.27 (t, J = 7.1 Hz, 1H), 2.13 (s, 3H), 2.05–1.97 (m, 2H), 1.90 – 1.82 (m, 1H), 1.67 – 1.56 (m, 3H), 1.56 – 1.45 (m, 4H), 1.17 (d, J = 13.6 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 183.7, 178.5, 172.8, 140.7, 137.2, 132.2, 131.5, 129.8, 129.4, 48.4, 42.0, 37.6, 33.4, 32.8, 25.8, 24.7, 23.4, 20.9, 20.4, 17.1. −ESI-HRMS m/z: calc’d for [M-H]− C20H28NO5− = 362.1973, C20H28NO5− found = 362.1974.
4.12.9. Methyl 6-((2,5-dimethylphenyl)(5-isobutoxy-4,4-dimethyl-5-oxopentyl)amino)-6-oxohexanoate (10f diester)
Methyl 6-((2,5-dimethylphenyl)(5-isobutoxy-4,4-dimethyl-5-oxopentyl)amino)-6-oxohexanoate (10f diester) was synthesized using the above general procedure using monomethyl adipic acid (28.8 mg, 0.18 mmol, 1.1 equiv), HATU (68.4 mg, 0.18 mmol, 1.1 equiv.), TEA (67.5 μL, 0.48 mmol, 3 equiv.), and DMF (1 mL). After addition of 9 (50 mg, 0.16 mmol, 1 equiv.), the reaction was stirred for 26 h, followed by addition of monomethyl succinic acid (24.8 mg, 0.18 mmol, 1.1 equiv), HATU (68.4 mg, 0.18 mmol, 1.1 equiv.), TEA (67.5 μL, 0.48 mmol, 3 equiv.). The reaction was stirred for an additional 24 h. After evaporation of the solvent silica chromatography (20–40% EtOAc in hexanes), provided the diester of 10f in 45% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.14 (d, J = 7.7 Hz, 1H), 7.05 (d, J = 9.6 Hz, 1H), 6.83 (s, 1H), 4.00 (ddd, J = 13.8, 9.4, 5.5 Hz, 1H), 3.81 – 3.72 (m, 2H), 3.60 (s, 3H), 3.01 (ddd, J = 13.5, 8.7, 4.9 Hz, 1H), 2.31 (s, 3H), 2.21 (t, J = 7.3 Hz, 2H), 2.11 (s, 3H), 1.96 (dt, J = 15.1, 7.3 Hz, 1H), 1.88 – 1.77 (m, 3H), 1.58 – 1.52 (m, 2H), 1.52 – 1.46 (m, 4H), 1.40 (ddt, J = 16.3, 9.0, 4.5 Hz, 1H), 1.14 (d, J = 2.8 Hz, 6H), 0.85 (dd, J = 6.7, 2.5 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 177.8, 174.0, 172.5, 141.0, 137.0, 132.3, 131.4, 129.7, 129.1, 70.5, 51.5, 48.5, 42.3, 37.9, 33.9, 33.7, 27.8, 25.5, 25.1, 24.8, 24.7, 23.4, 20.9, 19.1, 17.2. +ESI-HRMS m/z: calc’d for [M+Na]+ C26H41NaNO5+ = 470.2882, C26H41NaNO5+ found = 470.2869.
4.12.10. 6-((4-carboxy-4-methylpentyl)(2,5-dimethylphenyl)amino)-6-oxohexanoic acid (10f)
6-((4-carboxy-4-methylpentyl)(2,5-dimethylphenyl)amino)-6-oxohexanoic acid (10f) was synthesized using the above general procedure, using the diester of 10f (32.0 mg, 0.07 mmol, 1 equiv.) The product was purified by silica chromatography (80% EtOAc in hexanes plus 2 % AcOH). The product was purified was further purified by precipitation from DCM with hexanes to provide the product in a 59% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.16 (d, J = 7.9 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 6.91 (s, 1H), 4.16 – 4.09 (m, 1H), 3.00 – 2.93 (m, 1H), 2.32 (s, 3H), 2.27 (t, J = 7.1 Hz, 2H), 2.13 (s, 3H), 2.05 – 1.97 (m, 1H), 1.90 – 1.82 (m, 1H), 1.67 – 1.56 (m, 4H), 1.56 – 1.43 (m, 4H).1.17 (d, J = 13.6 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 183.4, 178.8, 173.2, 141.0, 137.2, 132.3, 131.5, 129.8, 129.3, 48.7, 42.1, 37.7, 34.1, 33.5, 25.7, 24.6, 24.5, 23.5, 20.9, 17.2. −ESI-HRMS m/z: calc’d for [M-H]− C21H30NO5− = 376.2129, C21H30NO5− found = 376.2124.
4.13. Synthesis of 5,5’-(adipoylbis((2,5-dimethylphenyl)azanediyl))bis(2,2-dimethyl-pentanoic acid) (11):_
To a 1 dram vial was added SOCl2 (0.5 mL) followed by adipic acid (32.2 mg, 0.22 mmol, 1 equiv.) and the resulting mixture was heated to 75 °C for 24 h. SOCl2 was removed by rotary evaporation and the acid chloride was used without further purification. The crude acid chloride was dissolved in DCM (1 mL) and added to a 6 dram vial containing 9 (135 mg, 0.44 mmol, 2 equiv.) and DIPEA (307.4 μL, 1.76 mmol, 8 equiv.). The reaction mixture was allowed to stir at 25 °C for 3 days and the solvents were removed by rotary evaporation. To the reaction mixture was added 5 mL 3 M HCl, which was extracted with EtOAc (5 mL) and the organic extract dried with Na2SO4. The organic solvent was removed by rotary evaporation and the product was purified silica chromatography (20% to 40% EtOAc in hexanes) to provide the isobutyl ester of 11 in a 57% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.12 (d, J = 7.7 Hz, 2H), 7.04 (d, J = 7.8 Hz, 2H), 6.79 (s, 2H), 3.99 – 3.93 (m, 2H), 3.82 – 3.71 (m, 4H), 3.03 – 2.95 (m, 2H), 2.30 (s, 6H), 2.08 (d, J = 4.6 Hz, 6H), 1.85 (dtd, J = 26.9, 13.4, 11.6, 6.5 Hz, 4H), 1.75 – 1.68 (m, 2H), 1.51 – 1.44 (m, 6H), 1.42 – 1.34 (m, 6H), 1.13 (s, 12H), 0.84 (dd, J = 6.7, 2.6 Hz, 12H). 13C NMR (151 MHz, Chloroform-d) δ 177.9, 172.7, 141.0, 136.9, 131.3, 129.7, 129.1, 70.5, 48.7, 42.3, 37.8, 33.9, 33.9, 27.8, 25.5, 25.1, 25.0, 25.0, 23.4, 20.9, 19.1, 17.2. +ESI-HRMS m/z: calc’d for [M+Na]+ C44H68N2NaO6+ = 743.4975, C44H68N2NaO6+ found = 743.4967.
To a 6 dram vial was added the isobutyl ester of 11 (91.1 mg, 0.13 mmol, 1 equiv.), 10 M NaOH (0.5 mL), and MeOH (1.5 mL) and was heated to 80 °C. The reaction was stirred for 2 days and organic solvents were removed by rotary evaporation. To the reaction mixture was added 3 M HCl until reaction mixture pH was 2, extracted with EtOAc (5 mL), and dried with Na2SO4. The organic solvent was removed by rotary evaporation and the product was purified silica chromatography (60% to 100% EtOAc in hexanes + 2% AcOH) to provide the product in a 76% yield. Rotamer A: 1H NMR (600 MHz, Chloroform-d) δ 7.13 (d, J = 4.5 Hz, 2H), 7.07 (s, 2H), 6.95 (s, 2H), 4.22 (dt, J = 13.4, 7.3 Hz, 2H), 2.83 – 2.76 (m, 2H), 2.35 (s, 6H), 2.09 (s, 6H), 1.99 – 1.87 (m, 4H), 1.65 – 1.54 (m, 8H), 1.53 – 1.45 (m, 4H), 1.20 (s, 6H), 1.16 (s, 6H). 13C NMR (151 MHz, Chloroform-d) δ 183.6, 173.0, 141.3, 137.2, 132.2, 131.3, 130.1, 129.1, 48.4, 42.2, 37.9, 33.7, 25.6, 25.1, 24.7, 23.8, 21.0, 17.1. Rotamer B:1H NMR (600 MHz, Chloroform-d) δ 7.14 (d, J = 4.5 Hz, 2H), 7.05 (s, 2H), 6.88 (s, 2H), 4.02 (dt, J = 7.2, 6.4 Hz, 2H), 3.04 – 2.98 (m, 2H), 2.32 (s, 6H), 2.11 (s, 6H), 1.75 (dt, J = 13.6, 6.6 Hz, 4H), 1.65 – 1.54 (m, 8H), 1.53 – 1.45 (m, 4H), 1.18 (s, 6H), 1.17 (s, 6H). 13C NMR (151 MHz, Chloroform-d) δ 183.3, 173.1, 141.4, 137.0, 132.4, 131.4, 129.8, 129.1, 48.8, 42.2, 37.9, 33.7, 25.4, 25.0, 24.7, 23.8, 20.9, 17.3. −ESI-HRMS m/z: calc’d for [M-H]− C36H51N2O6− = 607.3753, C36H51N2O6− found = 607.3750.
4.14. General Procedure for Synthesis of 12a-e, 13a-c:
To a 2 dram vial was added gemfibrozil (1), HATU, then DMF followed by either the desired amine or TEA as indicated. The reaction mixture was stirred for 15 min at 25 °C, followed by addition of desired amine or TEA as indicated. The reaction was stirred at 25 °C for the specified amount of time. The reaction was diluted with EtOAc (5 mL) and washed sequentially with water (5 mL), 1 M HCl (5 mL), and brine (5 mL). The organic phase was then dried over MgSO4 and the solvent removed by rotary evaporation. Product was purified as specified.
4.14.1. 5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide (12a)
5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide (12a) was synthesized by the above general procedure using 7 N ammonia in MeOH (150 μL, 1.05 mmol, 5.25 equiv.), Gemfibrozil (1) (50 mg, 0.2 mmol, 1 equiv.), HATU (83.6 mg, 0.22 mmol, 1.1 equiv.), and DMF (1 mL). After the addition of TEA (27.9 μL, 0.2 mmol, 1 equiv.) the mixture was stirred for 2 h. Centrifugal evaporation provided the product in 95% yield. 1H NMR (Chloroform-d, 600 MHz) δ 7.00 (d, J = 7.5 Hz, 1H), 6.66 (d, J = 7.4 Hz, 1H), 6.61 (s, 1H), 5.68 (brs, 1H), 5.45 (brs, 1H), 3.93 (t, J = 6.1 Hz, 2H), 2.30 (s, 3H), 2.17 (s, 3H), 1.83–1.78 (m, 2H), 1.73–1.70 (m, 2H), 1.24 (s, 6H). 13C NMR (Chloroform-d, 151 MHz) δ 180.3, 157.0, 136.7, 130.5, 123.6, 120.9, 112.2, 68.0, 42.1, 37.7, 25.8, 25.3, 21.6, 16.0. +ESI-HRMS m/z: calc’d for [M+Na]+ C15H23NNaO2+ 273.1620, C15H23NNaO2+ found 273.1620.
4.14.2. 5-(2,5-dimethylphenoxy)-N,2,2-trimethylpentanamide (12b)
5-(2,5-dimethylphenoxy)-N,2,2-trimethylpentanamide (12b) was synthesized by the above general procedure using 2 M methylamine in MeOH (500 μL, 1.0 mmol, 5 equiv.), Gemfibrozil (1) (50 mg, 0.2 mmol, 1 equiv.), HATU (83.6 mg, 0.22 mmol, 1.1 equiv.) and DMF (1 mL). After the addition of TEA (27.9 μL, 0.2 mmol, 1 equiv.) the mixture was stirred for 2 h. Centrifugal evaporation provided the product in 94% yield. 1H NMR (Chloroform-d, 600 MHz) δ 7.00 (d, J = 7.4 Hz, 1H), 6.66 (d, J = 7.4 Hz, 1H), 6.60 (s, 1H), 5.72 (brs, 1H), 3.91 (t, J = 5.8 Hz, 2H), 2.80 (d, J = 4.7 Hz, 3H), 2.30 (s, 3H), 2.17 (s, 3H), 1.76–1.67 (m, 4H), 1.21 (s, 6H). 13C NMR (Chloroform-d, 151 MHz) δ 178.2, 157.0, 136.7, 130.4, 123.6, 120.9, 112.2, 68.1, 42.0, 37.6, 26.6, 25.7, 25.3, 21.6, 16.0. +ESI-HRMS m/z: calc’d for [M+Na]+ C16H25NNaO2+ 286.1778, C16H25NNaO2+ found 286.1778.
4.14.3. 5-(2,5-dimethylphenoxy)-N-ethyl-2,2-dimethylpentanamide (12c)
5-(2,5-dimethylphenoxy)-N-ethyl-2,2-dimethylpentanamide (12c) was synthesized by the above general procedure using 2 M ethylamine in THF (500 μL, 1.0 mmol, 5 equiv.), Gemfibrozil (1) (50 mg, 0.2 mmol, 1 equiv.), HATU (83.6 mg, 0.22 mmol, 1.1 equiv.), and DMF (1 mL). After the addition of TEA (27.9 μL, 0.2 mmol, 1 equiv.) the reaction mixture was stirred for 2 h. Centrifugal evaporation provided the product in 90% yield. 1H NMR (Chloroform-d, 600 MHz) δ 7.00 (d, J = 7.5 Hz, 1H), 6.66 (d, J = 7.5 Hz, 1H), 6.60 (s, 1H), 5.66 (brs, 1H), 3.91 (t, J = 5.9 Hz, 2H), 3.31–3.26 (m, 2H), 2.30 (s, 3H), 2.17 (s, 3H), 1.76–1.73 (m, 2H), 1.70–1.67 (m, 2H), 1.20 (s, 6H), 1.13 (t, J = 7.3 Hz, 3H). 13C NMR (Chloroform-d, 151 MHz) δ 177.7, 157.4, 137.0, 130.8, 124.0, 121.2, 112.6, 68.5, 42.3, 38.1, 34.9, 26.1, 25.6, 21.9, 16.3, 15.5. +ESI-HRMS m/z: calc’d for [M+Na]+ C17H27NNaO2+ 300.1934, C17H27NNaO2+ found 300.1934.
4.14.4. N-butyl-5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide (12d)
N-butyl-5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide (12d) was synthesized by the above general procedure using Gemfibrozil (1) (50 mg, 0.2 mmol, 1 equiv.), HATU (76 mg, 0.2 mmol, 1 equiv.), TEA (27.9 μL, 0.2 mmol, 1 equiv.) and DMF (1 mL). After the addition of n-butyl amine (98.6 μL, 1 mmol, 5 equiv.) the reaction was stirred for 23 h. Silica chromatography using (40% EtOAc in hexanes) provided the product in a 47% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.00 (d, J = 7.5 Hz, 1H), 6.67 – 6.65 (d, J = 7.5 Hz 1H), 6.61 (s, 1H), 5.65 (t, J = 5.8 Hz, 1H), 3.92 (t, J = 6.0 Hz, 2H), 3.25 (td, J = 7.2, 5.6 Hz, 2H), 2.30 (s, 3H), 2.17 (s, 3H), 1.78 – 1.72 (m, 2H), 1.71 – 1.67 (m, 2H), 1.50 – 1.45 (m, 2H), 1.38 – 1.31 (m, 2H), 1.21 (s, 6H), 0.92 (t, J = 7.4 Hz, 2H). 13C NMR (151 MHz, Chloroform-d) δ 177.3, 156.9, 136.5, 130.3, 123.5, 120.7, 112.1, 68.0, 41.8, 39.3, 37.6, 31.9, 25.6, 25.1, 21.4, 20.1, 15.8, 13.8. +ESI-HRMS m/z: calc’d for [M+Na]+ C19H31NNaO2+ =328.2247, found C19H31NNaO2+ =328.2247
4.14.5. 5-(2,5-dimethylphenoxy)-N-hexyl-2,2-dimethylpentanamide (12e)
5-(2,5-dimethylphenoxy)-N-hexyl-2,2-dimethylpentanamide (12e) was synthesized by the above general procedure using Gemfibrozil (1) (50 mg, 0.2 mmol, 1 equiv.), HATU (83.6 mg, 0.22 mmol, 1.1 equiv.), TEA (27.9 μL, 0.2 mmol, 1 equiv.), and DMF (1 mL). After the addition of n-hexylamine (12.84 μL, 1.0 mmol, 5 equiv.) the reaction was stirred for 2 h. Centrifugal evaporation provided the product in 92% yield. 1H NMR (Chloroform-d, 600 MHz) δ 7.00 (d, J = 7.4 Hz, 1H), 6.66 (d, J = 7.4 Hz, 1H), 6.61 (s, 1H), 5.65 (brs, 1H), 3.91 (t, J = 5.8 Hz, 2H), 3.24 (q, J = 6.6 Hz, 2H), 2.30 (s, 3H), 3.17 (s, 3H), 1.78–1.73 (m, 2H), 1.70–1.67 (m, 2H), 1.51–1.46 (m, 2H), 1.33–1.28 (m, 6H), 1.21 (s, 6H), 0.88 (t, J = 6.6 Hz, 3H). 13C NMR (Chloroform-d, 151 MHz) δ 177.4, 157.0, 136.6, 130.4, 123.6, 120.9, 112.2, 68.1, 42.0, 39.7, 37.7, 31.6, 29.8, 26.7, 25.7, 25.3, 22.7, 21.5, 15.9, 14.1 +ESI-HRMS m/z: calc’d for [M+Na]+ C15H20NaO3+ 273.1463, C15H20NaO3+ found 273.1463
4.14.6. N,N’-(ethane-1,2-diyl)bis(5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide) (13a)
N,N’-(ethane-1,2-diyl)bis(5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide) (13a) was synthesized by the above general procedure using Gemfibrozil (1) (100 mg, 0.4 mmol, 2 equiv), HATU (152.1 mg, 0.4 mmol, 2 equiv.), TEA (126.7 μL, 0.8 mmol, 4 equiv.) and DMF (1 mL) and stirred for 15 min. After addition of ethylenediamine (13.3 μL, 0.2 mmol, 1 equiv.) the reaction was stirred for 4.5 h. Silica chromatography using (40% EtOAc in hexanes) provided the product in 54 % yield. 1H NMR (600 MHz, Chloroform-d) δ 6.99 (d, J = 7.4 Hz, 2H), 6.67 (s, 2H), 6.65 (d, J = 7.6 Hz, 2H), 6.59 (d, J = 1.6 Hz, 2H), 3.89 (t, J = 5.8 Hz, 4H), 3.41 – 3.35 (m, 4H), 2.30 (s, 6H), 2.17 (s, 6H), 1.75 – 1.66 (m,8H), 1.21 (s, 12H). 13C NMR (151 MHz, Chloroform-d) δ 179.3, 157.0, 136.6, 130.4, 123.6, 120.8, 112.1, 68.0, 41.9, 40.8, 37.5, 25.6, 25.2, 21.5, 15.9. +ESI-HRMS m/z: calc’d for [M+Na]+ C32H48N2NaO4+ =547.3506, found C32H48N2NaO4 =547.3505.
4.14.7. N,N’-(butane-1,4-diyl)bis(5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide) (13b)
N,N’-(butane-1,4-diyl)bis(5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide) (13b) was synthesized by the above general procedure using Gemfibrozil (1) (100 mg, 0.4 mmol, 2 equiv.), HATU (152.1 mg, 0.4 mmol, 2 equiv.), TEA (126.7 μL, 0.8 mmol, 4 equiv.) and DMF (1 mL). After addition of putrescine (20.1 μL, 0.2 mmol, 1 equiv.) the reaction was allowed to stir at 25 °C for 4.5 h. Silica chromatography (40% EtOAc in hexanes), provided the product in 79 % yield. 1H NMR (600 MHz, Chloroform-d) δ 6.99 (d, J = 7.5 Hz, 2H), 6.65 (dd, J = 7.5, 1.5 Hz, 2H), 6.60 (d, J = 1.6 Hz, 2H), 5.93 (t, J = 5.7 Hz, 2H), 3.90 (t, J = 5.9 Hz, 4H), 3.26 (q, J = 6.4 Hz, 4H), 2.30 (s, 6H), 2.16 (s, 6H), 1.77 – 1.65 (m, 8H), 1.55 – 1.48 (m, 4H), 1.20 (s, 12H). 13C NMR (151 MHz, Chloroform-d) δ 177.7, 157.0, 136.6, 130.4, 123.5, 120.9, 112.2, 68.1, 41.9, 39.0, 37.6, 27.1, 25.6, 25.2, 21.5, 15.9. +ESI-HRMS m/z: calc’d for [M+Na]+ C34H52N2NaO4+ =575.3819, found C34H52N2NaO4+ =575.3818.
4.14.8. N,N’-(hexane-1,6-diyl)bis(5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide) (13c)
N,N’-(hexane-1,6-diyl)bis(5-(2,5-dimethylphenoxy)-2,2-dimethylpentanamide) (13c) was synthesized by the above general procedure Gemfibrozil (1) (100 mg, 0.4 mmol, 2 equiv.), HATU (152.1 mg, 0.4 mmol, 2 equiv.), TEA (126.7 μL, 0.8 mmol, 4 equiv.) and DMF (1 mL). After addition of hexamethylenediamine (23.2 mg, 0.2 mmol, 1 equiv.) the reaction was allowed to stir for 4.5 h, when an additional HATU (0.3 mmol, 0.08 equiv.) was added and allowed to stir for an additional 16 h. Silica chromatography using (40% EtOAc in hexanes), to provide the product in 28% yield. 1H NMR (600 MHz, Chloroform-d) δ 6.99 (d, J = 7.5 Hz, 2H), 6.65 (d, J = 8.0 Hz, 2H), 6.60 (d, J = 1.6 Hz, 2H), 5.79 (t, J = 5.8 Hz, 2H), 3.91 (t, J = 6.0 Hz, 4H), 3.22 (q, J = 6.7 Hz, 4H), 2.30 (s, 6H), 2.17 (s, 6H), 1.74 (td, J = 6.3, 2.4 Hz, 2H), 1.71 – 1.66 (m, 4H), 1.48 (q, J = 6.8 Hz, 6H), 1.34 – 1.31 (m, 4H), 1.21 (s, 12H). 13C NMR (151 MHz, Chloroform-d) δ 177.5, 157.0, 136.6, 130.4, 123.6, 120.9, 112.2, 68.1, 42.0, 39.2, 37.7, 29.7, 26.2, 25.7, 25.3, 21.5, 15.9. +ESI-HRMS m/z: calc’d for [M+Na]+ C36H56N2NaO4+ =603.4132, found C36H56N2NaO4+ =603.4131.
4.15. Synthesis of isobutyl 5-(2,5-dimethylphenoxy)pentanoate (14):
In a 6 dram vial 3c (50 mg, 0.211 mmol, 1.1 equiv.) was added to a solution of K2CO3 (68 mg, 0.492 mmol, 2.5 equiv.) and 2,5-dimethylphenol (24 mg, 0.196 mmol, 1 equiv.) in MeCN (2 mL). The mixture was heated to 82 °C for 14 h then extracted with EtOAc (5 mL) and the organic phase was dried with MgSO4 and evaporated by rotary evaporation. The resulting residue was purified by silica chromatography (10–25% EtOAc in hexanes) to provide the product in 78% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.02 (d, J = 7.5 Hz, 1H), 6.68 (d, J = 7.5 Hz, 1H), 6.63 (s, 1H), 3.97 (t, J = 5.7 Hz, 2H), 3.88 (d, J = 6.7 Hz, 2H), 2.43 (t, J =7.1 Hz , 2H), 2.32 (s, 3H), 2.19 (s, 3H), 1.94 (hept, J = 6.7 Hz, 1H), 1.87–1.84 (m, 4H), 0.95 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 173.8, 157.0, 136.6, 130.4, 123.7, 120.8, 112.0, 70.6, 67.3, 34.2, 29.0, 27.9, 22.0, 21.5, 19.2, 15.9. +ESI-HRMS m/z: calc’d for [M+Na]+ C17H26NaO3+ = 301.1774, C17H26NaO3+ found = 301.1775.
4.16. Synthesis of 5-(2,5-dimethylphenoxy)-2-ethylpentanoic acid (15a):
In a 6 dram vial 5 M n-BuLi in hexanes (0.9 mL, 4.5 mmol, 15 equiv.) was added to a dry solution of diisopropylamine (0.8 mL, 5.7 mmol, 19 equiv.) under nitrogen while stirring at −78 °C for 20 min in THF (3.5 mL). The reaction mixture was then allowed to warm to room temperature for 1 h before being cooled again to −78 °C. To the cold LDA solution was added ethyl bromide (0.8 mL, 10.7 mmol, 35.7 equiv.). A solution of 14 (82 mg, 0.3 mmol, 1 equiv.) was prepared in THF (15 mL) and added dropwise to the solution of LDA and ethyl bromide then stirred at −78 °C for 30 min. The reaction mixture was then allowed to warm to 0 °C in an ice bath for 1 h. The ice bath was then removed, and the reaction was allowed to warm to 25 °C over 2 h before the solvent was removed by rotary evaporation and the crude residue purified by silica chromatography (0–5% EtOAc in Hexanes) to provide the product as a clear oil in 38% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.00 (d, J = 7.4 Hz, 1H), 6.66 (d, J = 7.5 Hz, 1H), 6.61 (s, 1H), 3.94 (s, 2H), 3.88 (d, J = 6.6 Hz, 2H), 2.41–2.36 (m, 1H), 2.31 (s, 3H), 2.17 (s, 3H), 1.94 (hept, J = 6.3 Hz, 1H), 1.81–1.76 (m, 3H), 1.73–1.64 (m, 2H), 1.61–1.54 (m, 1H), 0.96–0.90 (m, 9H). 13C NMR (151 MHz, Chloroform-d) δ 176.3, 157.1, 136.6, 130.4, 123.7, 120.8, 112.1, 70.50, 67.5, 47.2, 28.7, 27.9, 27.5, 25.6, 21.6, 19.3, 15.9, 11.9. +ESI-HRMS m/z: calc’d for [M+Na]+ C19H30NaO3+ = 329.2087, C19H30NaO3+ found = 329.2090.
In a 6 dram vial the above compound, (34.9 mg, 0.114 mmol, 1 equiv.), was added to a solution of 10 M NaOH (2 mL) and toluene (2 mL) which was 110 °C for 48 h. The resulting mixture was then extracted with EtOAc (5 mL) and the organic phase dried with MgSO4 and evaporated by rotary evaporation. The resulting residue was purified by silica chromatography (10–25% EtOAc in hexanes) to provide the product in 75% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.00 (d, J = 7.5 Hz, 1H), 6.65 (d, J = 7.4 Hz, 1H), 6.62 (s, 1H), 3.98–3.92 (m, 2H), 2.44–2.40 (m, 1H), 2.30 (s, 3H), 2.17 (s, 3H), 1.88–1.80 (m, 2H), 1.76–1.68 (m, 2H), 1.64–1.57 (m, 2H), 0.97 (t, J = 7.4 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 179.5, 157.0, 136.6, 130.5, 123.8, 120.9, 112.1, 67.5, 46.4, 28.4, 27.3, 25.4, 21.6, 15.9, 11.8. +ESI-HRMS m/z: calc’d for [M+Na]+ C15H20NaO3+ = 273.1461, C15H20NaO3+ found = 273.1463.
4.17. Synthesis of 2-(3-(2,5-dimethylphenoxy)propyl)octanoic acid (15b):
In a round bottom flask a 2 M solution LDA in THF/n-heptane/ethylbenzene (0.9 mL, 1.8 mmol, 5 equiv.) was added to a solution of dry THF (5 mL) and 1-bromohexane (0.8 mL, 5.7 mmol, 15.8 equiv.) under nitrogen while stirring at −78 °C. A solution of 14 (100 mg, 0.36 mmol, 1 equiv.) was then prepared in THF (15 mL) and added dropwise to the solution of LDA and bromohexane at −78 °C for 30 min, after which the reaction mixture was allowed to warm to 0 °C in an ice bath for 1 h. The ice bath was then removed, and the reaction was allowed to warm to 25 °C over 2 h before the solvent was removed by rotary evaporation. The crude residue was purified by silica chromatography (0–5% EtOAc in Hexanes) to provide the partially purified product as a clear oil. This material was carried forward without further purification.
In a 6 dram vial the above material was added to a solution of 10 M NaOH (2 mL) and toluene (2 mL) which was 110 °C for 48 h. The resulting mixture was then extracted with EtOAc (5 mL) and water (5 mL) and the organic phase was dried with MgSO4 and evaporated by rotary evaporation. The resulting residue was purified by silica chromatography (10–25% EtOAc in hexanes) to provide the product in 32% yield. 1H NMR (Chloroform-d, 600 MHz) δ 7.00 (d, J = 7.5 Hz, 1H), 6.66 (d, J = 7.5 Hz, 1H), 6.62 (s, 1H), 3.98–3.92 (m, 2H), 2.49–2.44 (m, 1H), 2.31 (s, 3H), 2.17 (s, 3H), 1.87–1.79 (m, 3H), 1.76–1.66 (m, 2H), 1.55–1.50 (m, 1H), 1.35–1.23 (m, 8H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, Chloroform-d) δ 180.2, 157.0, 136.6, 130.5, 123.8, 120.9, 112.1, 67.5, 44.9, 32.4, 31.8, 29.4, 28.8, 27.4, 27.3, 22.7, 21.6, 15.9, 14.2. +ESI-HRMS m/z: calc’d for [M+Na]+ C19H30NaO3+ = 329.2087, C19H30NaO3+ found = 329.2088.
4.18. Preparation of recombinant human sGC enzyme.
Full-length sGC was purified from Sf9 cells as described previously [31]. Briefly, 6 liters of Sf9 cells were harvested 72 hours after being infected with viruses expressing α1 and β1 sGC subunits. The cells disrupted by sonication and centrifuged at 100,000 × g. The supernatant was applied onto a 50 ml DEAE-FF Sepharose column and, after extensive washes, sGC was eluted with 350 mM NaCl and loaded onto a Ni-agarose column (30 ml bed volume). After washes with 40 mM imidazole, sGC was eluted with 200 mM imidazole. The fractions containing sGC-containing fractions were pooled, supplemented with 5 mM DTT and concentrated on a 10 kDa centrifugal filter concentrator (Millipore, Bedford, MA). Concentrated sGC sample was subjected to gel-filtration on Superdex 200 equilibrated with 50 mM triethanolamine pH 7.4, 250 mM NaCl, 1 mM MgCl2. For long-term storage at −80°C, the sample was supplemented with 25% glycerol.
4.19. Assay of sGC activity in vitro.
Enzymatic activity was assayed using [α−32P]GTP to [32P] cGMP conversion assay [32]. 0.5 μg sGC in 25 mM TEA, pH 7.5, 0.1 mM EGTA, 0.1 mg/ml BSA, 1 mM cGMP, 2 mM MgCl2, was incubated with indicated concentrations of gemfibrozil or its derivatives for 10 minutes at room temperature. For experiments with ferric sGC, 10 μM of sGC heme oxidizing agent 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) was added at this step. The reaction of cGMP synthesis was initiated by transferring the sample to 37°C and adding 1 mM GTP/[α−32P]GTP (~ 100000 cpm). After 15-minute incubation, the reaction was stopped by precipitation of GTP with zinc carbonate and subsequent centrifugation at 10,000×g. The supernatant containing cGMP was passed through a 2 ml alumina columns, and processed as previously reported (Sharina et. Al, BrJPharm, 2015).
4.20. Aortic ring relaxation.
All animal experiments were performed in accordance to Public Health Service’s Guide for the Care and Use of Laboratory Animals, the Foundation for Biomedical Research’s Biomedical Investigator’s Handbook for Researchers and the guidelines of the Animal Welfare Committee of the University of Texas Health Science Center (protocol AWC 18–085). C57Bl6 mice (8–10 weeks old, Jackson laboratories, Ann Harbor, MI) were euthanized by CO2. After thoracotomy, descending thoracic aorta was dissected, cut into 3–5-mm-long segments and mounted on a four-channel wire Myograph 610 (DMT, Copenhagen, Denmark) under 0.9 g of passive tension. All force measurements were recorded using Powerlab 400™ data acquisition system and LabChart software.
The rings were equilibrated for 80 min in Krebs-Henseleit solution pH 7.4, oxygenated with carbogen (95% O2, 5% CO2) with at least three buffer changes every 20 min. After equilibration the rings were contracted with a mixture of 500 nM phenylephrine and 100 nM of 5-hydroxytryptamine to achieve submaximal contraction (~1.8–2.5 grams of tension). After stabilization, gemfibrozil or gemfibrozil analogs) were added cumulatively and changes in isometric tension were recorded.
4.21. Statistical Analysis.
Statistical analysis, nonlinear regression, calculation of IC50, EC50 and was performed using GraphPad Prism 5.1 (GraphPad, La Jolla, USA). All results are expressed as mean ± SD of at least three independent experiments. Comparisons between two data groups were performed using the two tailed student t-test. Two-way ANOVA followed by Bonferroni’s multi comparison test was used for statistical evaluation of the dose-response curves. The p value <0.05 was considered statistically significant.
Supplementary Material
Highlights:
New gemfibrozil-based activators of soluble guanylyl cyclase (sGC) were synthesized and tested.
Compounds 7c and 15b have better half maximal effective concentration than gemfibrozil in sGC assay.
Compounds 7c and 15b were more effective vasorelaxants than gemfibrozil.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health (grant HL139838 to E.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE
The authors declare no competing financial interest.
REFERENCES
- 1.Ignarro LJ, Nitric oxide: a unique endogenous signaling molecule in vascular biology. Biosci Rep, 1999. 19(2): p. 51–71. doi: 10.1023/a:1020150124721 [DOI] [PubMed] [Google Scholar]
- 2.Gruetter CA, et al. , Relaxation of bovine coronary artery and activation of coronary arterial guanylate cyclase by nitric oxide, nitroprusside and a carcinogenic nitrosoamine. J Cyclic Nucleotide Res, 1979. 5(3): p. 211–24. [PubMed] [Google Scholar]
- 3.Murad F, Shattuck Lecture. Nitric oxide and cyclic GMP in cell signaling and drug development. N Engl J Med, 2006. 355(19): p. 2003–11. doi: 10.1056/NEJMsa063904 [DOI] [PubMed] [Google Scholar]
- 4.Griffith TM, et al. , Endothelium-derived relaxing factor. J Am Coll Cardiol, 1988. 12(3): p. 797–806. doi: 10.1016/0735-1097(88)90324-5 [DOI] [PubMed] [Google Scholar]
- 5.Just M, Martorana PA, and Nitz RE, [Inhibition of platelet functions by molsidomine in animals]. Pathol Biol (Paris), 1987. 35(2 Pt 2): p. 226–8. [PubMed] [Google Scholar]
- 6.Tulis DA, Salutary properties of YC-1 in the cardiovascular and hematological systems. Curr Med Chem Cardiovasc Hematol Agents, 2004. 2(4): p. 343–59. doi: 10.2174/1568016043356200 [DOI] [PubMed] [Google Scholar]
- 7.Erdmann J, et al. , Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature, 2013. 504(7480): p. 432–6. doi: 10.1038/nature12722 [DOI] [PubMed] [Google Scholar]
- 8.Wobst J, et al. , Molecular variants of soluble guanylyl cyclase affecting cardiovascular risk. Circ J, 2015. 79(3): p. 463–9. doi: 10.1253/circj.CJ-15-0025 [DOI] [PubMed] [Google Scholar]
- 9.Ehret GB, et al. , Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature, 2011. 478(7367): p. 103–9. doi: 10.1038/nature10405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu X, et al. , Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet, 2012. 44(8): p. 890–4. doi: 10.1038/s41598-017-18214-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herve D, et al. , Loss of alpha1beta1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am J Hum Genet, 2014. 94(3): p. 385–94. doi: 10.1016/j.ajhg.2014.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallace S, et al. , Disrupted Nitric Oxide Signaling due to GUCY1A3 Mutations Increases Risk for Moyamoya Disease, Achalasia and Hypertension. Clinicla Genetics, 2016. 90(4):: p. 351–60. doi: 10.1111/cge.12739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torfgard KE and Ahlner J, Mechanisms of action of nitrates. Cardiovasc Drugs Ther, 1994. 8(5): p. 701–17. doi: 10.1007/BF00877117 [DOI] [PubMed] [Google Scholar]
- 14.Forstermann U, Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat Clin Pract Cardiovasc Med, 2008. 5(6): p. 338–49. doi: 10.1038/ncpcardio1211 [DOI] [PubMed] [Google Scholar]
- 15.Sandner P, et al. , Soluble Guanylate Cyclase Stimulators and Activators. Handb Exp Pharmacol, 2019. doi: 10.1007/164_2018_197 [DOI] [PubMed] [Google Scholar]
- 16.Ghofrani HA, et al. , Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med, 2013. 369(4): p. 319–29. doi: 10.1056/NEJMoa1209657 [DOI] [PubMed] [Google Scholar]
- 17.Ghofrani HA, et al. , Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med, 2013. 369(4): p. 330–40. doi: 10.1056/NEJMoa1209655 [DOI] [PubMed] [Google Scholar]
- 18.Armstrong PW, et al. , Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N Engl J Med, 2020. 382(20): p. 1883–1893. doi: 10.1056/NEJMoa1915928 [DOI] [PubMed] [Google Scholar]
- 19.Lam CSP, et al. , Clinical Outcomes and Response to Vericiguat According to Index Heart Failure Event: Insights From the VICTORIA Trial. JAMA Cardiol, 2020. doi: 10.1001/jamacardio.2020.6455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brune B, Schmidt KU, and Ullrich V, Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. Eur J Biochem, 1990. 192(3): p. 683–8. doi: 10.1111/j.1432-1033.1990.tb19276.x [DOI] [PubMed] [Google Scholar]
- 21.Weber M, et al. , The effect of peroxynitrite on the catalytic activity of soluble guanylyl cyclase. Free Radic Biol Med, 2001. 31(11): p. 1360–7. doi: 10.1016/s0891-5849(01)00706-7 [DOI] [PubMed] [Google Scholar]
- 22.Stasch JP, et al. , Targeting the heme-oxidized nitric oxide receptor for selective vasodilatation of diseased blood vessels. J Clin Invest, 2006. 116(9): p. 2552–61. doi: 10.1172/JCI28371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerassimou C, et al. , Regulation of the expression of soluble guanylyl cyclase by reactive oxygen species. Br J Pharmacol, 2007. 150(8): p. 1084–91. doi: 10.1038/sj.bjp.0707179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schindler U, et al. , Biochemistry and pharmacology of novel anthranilic acid derivatives activating heme-oxidized soluble guanylyl cyclase. Mol Pharmacol, 2006. 69(4): p. 1260–8. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt HH, Schmidt PM, and Stasch JP, NO- and haem-independent soluble guanylate cyclase activators. Handb Exp Pharmacol, 2009(191): p. 309–39. doi: 10.1124/mol.105.018747 [DOI] [PubMed] [Google Scholar]
- 26.Evgenov OV, et al. , NO-independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nat Rev Drug Discov, 2006. 5(9): p. 755–68. doi: 10.1038/nrd2038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Erdmann E, et al. , Cinaciguat, a soluble guanylate cyclase activator, unloads the heart but also causes hypotension in acute decompensated heart failure. Eur Heart J, 2013. 34(1): p. 57–67. doi: 10.1093/eurheartj/ehs196 [DOI] [PubMed] [Google Scholar]
- 28.Quintanilla Rodriguez BS and Correa R, Gemfibrozil, in StatPearls. 2021: Treasure Island (FL). [Google Scholar]
- 29.Sharina IG, et al. , The fibrate gemfibrozil is an NO – and heme-independent activator of soluble guanylyl cyclase: in vitro studies. Br J Pharmacol, 2015. doi: 10.1111/bph.13055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jun M, et al. , Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet, 2010. 375(9729): p. 1875–84. doi: 10.1016/S0140-6736(10)60656-3 [DOI] [PubMed] [Google Scholar]
- 31.Rahaman MM, et al. , Cytochrome b5 Reductase 3 Modulates Soluble Guanylate Cyclase Redox State and cGMP Signaling. Circ Res, 2017. 121(2): p. 137–148. doi: 10.1161/CIRCRESAHA.117.310705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultz G, General principles of assays for adenylate cyclase and guanylate cyclase activity. Methods Enzymol, 1974. 38: p. 115–25. doi: 10.1016/0076-6879(74)38018-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.