

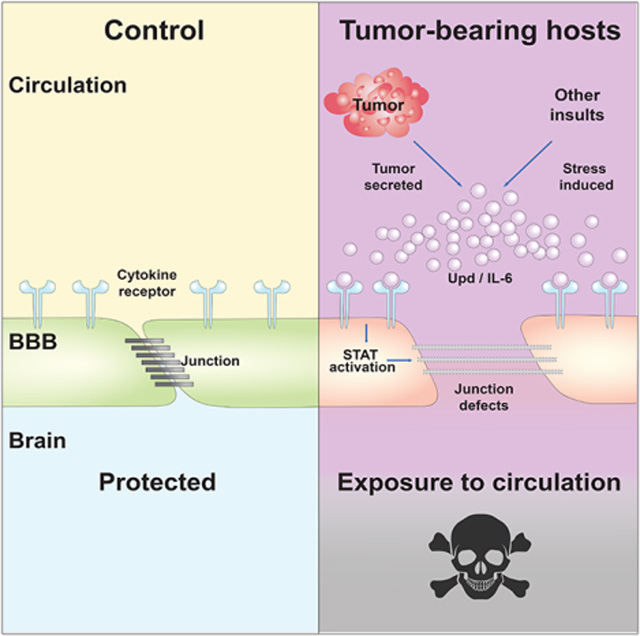
SUMMARY
Cancer patients often die from symptoms that manifest at a distance from any tumor. Mechanisms underlying these systemic physiological perturbations, called paraneoplastic syndromes, may benefit from investigation in non-mammalian systems. Using a non-metastatic Drosophila adult model, we find that malignant tumor-produced cytokines drive widespread host activation of JAK-STAT signaling and cause premature lethality. STAT activity is particularly high in cells of the blood-brain barrier (BBB), where it induces aberrant BBB permeability. Remarkably, inhibiting STAT in the BBB not only rescues barrier function but also extends the lifespan of tumor-bearing hosts. We identify BBB damage in other pathological conditions that cause elevated inflammatory signaling, including obesity and infection, where BBB permeability also regulates host survival. IL-6-dependent BBB dysfunction is further seen in a mouse tumor model, and it again promotes host morbidity. Therefore, BBB alterations constitute a conserved lethal tumor-host interaction that also underlies other physiological morbidities.
Graphical Abstract
eTOC blurb
Kim et al. use a fly cancer model to uncover a systemic effect of tumors, in which inflammatory signaling permeabilizes the blood-brain barrier. Preventing barrier permeability allows flies to live longer with the same tumor burden, and key aspects of these data are recapitulated in a mouse tumor model.
INTRODUCTION
A key feature of cancer is its ability to accelerate death of the host, and this can be caused by effects at a distance from the local site of tumor growth itself (McAllister and Weinberg, 2010). Paraneoplastic syndromes such as cancer-associated cachexia and coagulopathies have enormous impacts on patient morbidity and mortality (Baracos et al., 2018, Boccaccio and Comoglio, 2009), raising the question of whether there exist additional, unrecognized mechanisms through which tumors can remotely impact host tissues with lethal consequences. Given the complexity of tumor-host interactions in native mammalian contexts, cancer models in simpler organisms such as Drosophila can contribute potential mechanisms (Figueroa-Clarevega and Bilder, 2015, Kwon et al., 2015, Song et al., 2019, Bilder et al., 2021, Newton et al., 2020, Yeom et al., 2021).
Flies have made many important contributions to understanding pathways driving oncogenesis (Bilder, 2004, Gonzalez, 2013, Chatterjee and Deng, 2019). Most of this research focused on the imaginal discs, epithelial tissues that grow in the larval stage as primordia of adult organs. Mutations in tumor suppressor genes, overexpression of oncogenes, or a combination of the two can transform disc cells into ‘neoplastic tumors’ that overproliferate, lose epithelial organization, and show defective differentiation and other tumorous characteristics. A pioneering demonstration of the malignant capabilities of Drosophila neoplasms utilized a classical allograft assay to transplant disc tissue into the abdomen of an adult host, whose open circulatory system provides a growth-permissive environment (Beadle and Ephrussi, 1935, Gateff and Schneiderman, 1969). Whereas WT discs arrest growth at their appropriate size in this assay, tumorous discs will grow uncontrollably, killing the host rapidly and robustly (Gateff and Schneiderman, 1969, Pagliarini and Xu, 2003, Rossi and Gonzalez, 2015). Allografts have been previously used to study various tumor-autonomous phenotypes (Rossi and Gonzalez, 2015), and recently exploited to investigate tumor-host interactions (Bilder et al., 2021), but why tumors rapidly accelerate the lethality of hosts remains unknown. Here we chose to investigate these mechanisms.
RESULTS
Malignant fly tumors compromise the BBB
Allografts of malignant Drosophila epithelial tissues have long been known to induce robust and rapid lethality in hosts (Gateff and Schneiderman, 1969, Pagliarini and Xu, 2003, Rossi and Gonzalez, 2015). When transplanted into a WT adult, wing imaginal discs conditionally induced to express activated forms of the oncogenes Ras and aPKC (hereafter called ‘tumors’) reduce the median survival rate by >50% compared to non-oncogene-expressing control transplants (Figures 1A and B), in the absence of metastasis (Figures S1A–D). Flies dying of tumors did not show dramatic gut permeability (Figure S1E) which is associated with flies dying of old age (Clark et al., 2015), indicating a different mechanism driving mortality. We and others have shown that similar tumor models cause cachexia-like tissue wasting, due to production of an insulin antagonist with function analogous to mammalian IGFBPs (Figueroa-Clarevega and Bilder, 2015, Kwon et al., 2015). Surprisingly, depletion of this cachectogenic factor in malignant imaginal discs, which efficiently rescued wasting, did not extend lifespan of hosts bearing these tumors (Figures 1B and S1F–H). Additional unknown mechanisms must be responsible for the premature lethality induced by systemic effects of fly tumors.
Figure 1. Tumor-bearing fly hosts show glial Jak/STAT activation and premature death.
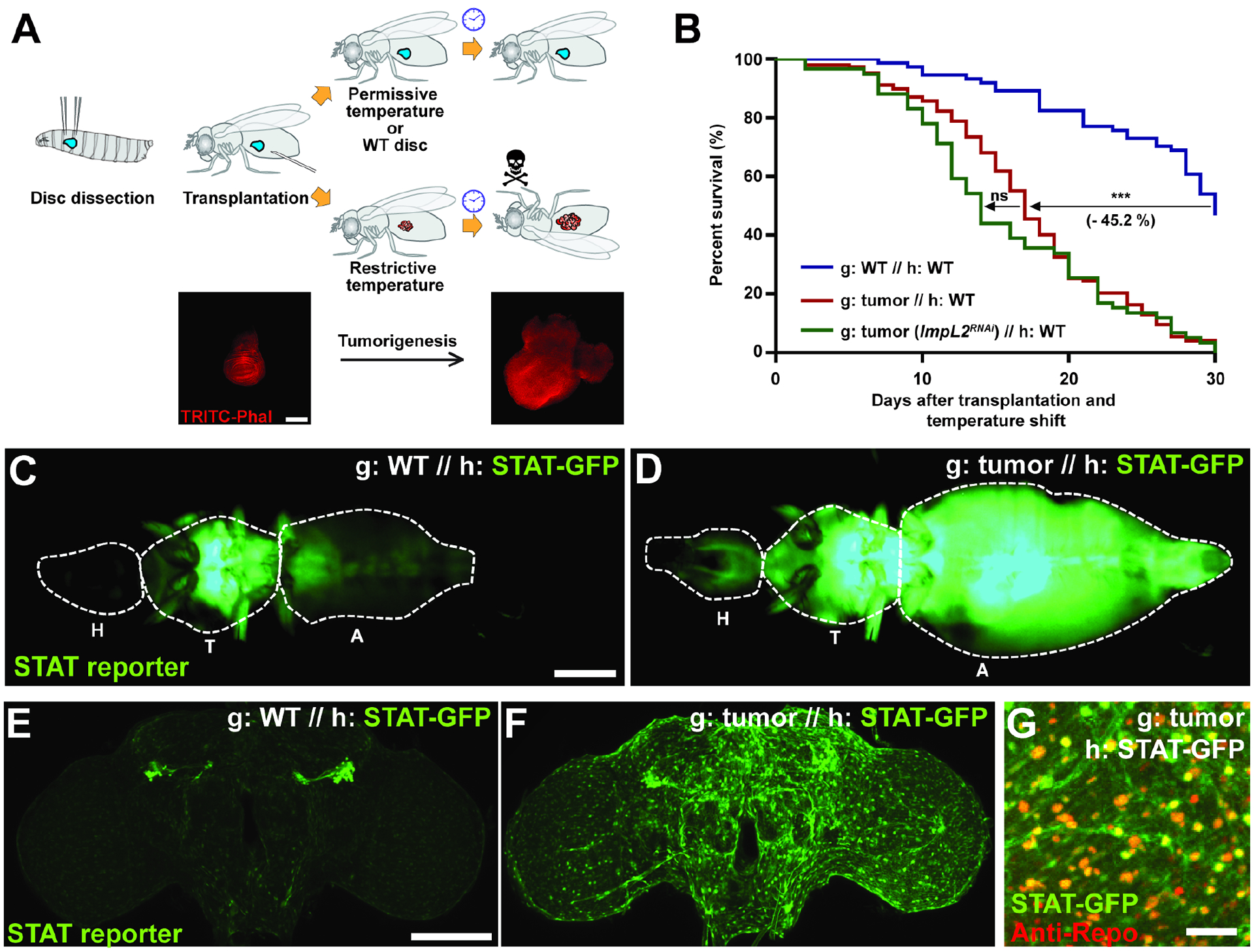
(A) Procedure to allograft tissue conditionally expressing oncogenes, with images of wing discs prior to shift to restrictive temperature and after 15 days in host following shift. Scale bar, 150 um. (B) Lifespans of hosts (h) with various graft (g) transplants. Tumor-bearing host (red, n=147) lifespan is reduced compared to hosts with control grafts (blue, n=74). Depletion of the wasting factor ImpL2 from tumor does not rescue host lifespan (green, n=59). (C, D) Tumor causes systemic activation of Jak/STAT signaling in hosts, especially in the head. H: head; T: thorax; A: abdomen. Scale bar, 500 um. (E, F) Elevated STAT activity in the brain of tumor hosts, shown by maximum intensity projection. Scale bar, 100 um. (G) STAT reporter colocalizes with Repo staining, which labels glial cells. Scale bar, 20 um.
Lifespan curves; *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; Log-rank test.
We considered alternative secreted proteins that might mediate tumor-induced death. Malignant Drosophila tumors originating in several organs and driven by distinct genetic alterations show common and strong upregulation of unpaired (upd) 1, 2, and/or 3 (Bunker et al., 2015, Kulshammer et al., 2015, Moberg et al., 2005, Pastor-Pareja et al., 2008, Song et al., 2019, Staley and Irvine, 2010, Vaccari and Bilder, 2005, Wu et al., 2010). These three genes encode cytokines that are considered fly orthologs of IL-6, and accordingly signal through an IL-6R-like receptor called Domeless (Dome) to activate the JAK-STAT pathway in target tissues (Brown et al., 2001, Amoyel et al., 2014). Flies transplanted with tumors showed widespread activation of a STAT activity reporter throughout the abdomen, indicative of systemic inflammation (Figures 1C and D). Interestingly, this reporter was also elevated in the brain (Figures 1E–F, S1I), especially at its surface (Figure S1J). Cell type-specific markers revealed that the activated STAT signal colocalized with glial cells rather than neurons (Figure 1G). Glia also expressed high levels of Dome (Figure S1K). These data reveal that tumors activate STAT-mediated inflammatory signaling in host glial cells.
A major role of Drosophila surface glia is to serve as the blood-brain barrier (BBB), which forms a sheath around the brain and maintains homeostasis by restricting permeability from the surrounding circulatory fluid (hemolymph). Although not of endothelial origin, these cells show significant conservation both structurally and functionally with the mammalian BBB (Hindle and Bainton, 2014, O’Brown et al., 2018). As in mammals, claudin-based junctions between fly BBB cells prevent almost all paracellular diffusion into the neuropil. We tested whether tumors might alter BBB function by injecting fluorescently-labelled 10 kDa dextran into the hemolymph and measuring dye penetration into the brain 15 hours later (Pinsonneault et al., 2011). This assay is not sensitive to circadian rhythms (Figure S2A) (Zhang et al., 2018). Whereas hosts transplanted with WT imaginal discs efficiently excluded dextran from the brain, hosts carrying transplanted tumors displayed a dramatic increase in BBB permeability, with dextran signal enhanced in the brain parenchyma (Figures 2A–C). An independent malignant tumor model, driven by genetic induction of oncogenic Yorkie in adult intestinal stem cells (Kwon et al., 2015), also showed this permeable BBB phenotype (Figure S2B). Increased BBB permeability in the transplant model was first evident 12 days after tumor induction (Figure 2D). This raised the possibility that BBB defects are merely secondary consequences of animal death. However, alternative lethality-inducing conditions such as starvation or oxidative stress did not alter BBB integrity (Figures S2C and D).
Figure 2. Fly tumors induce BBB disruption.
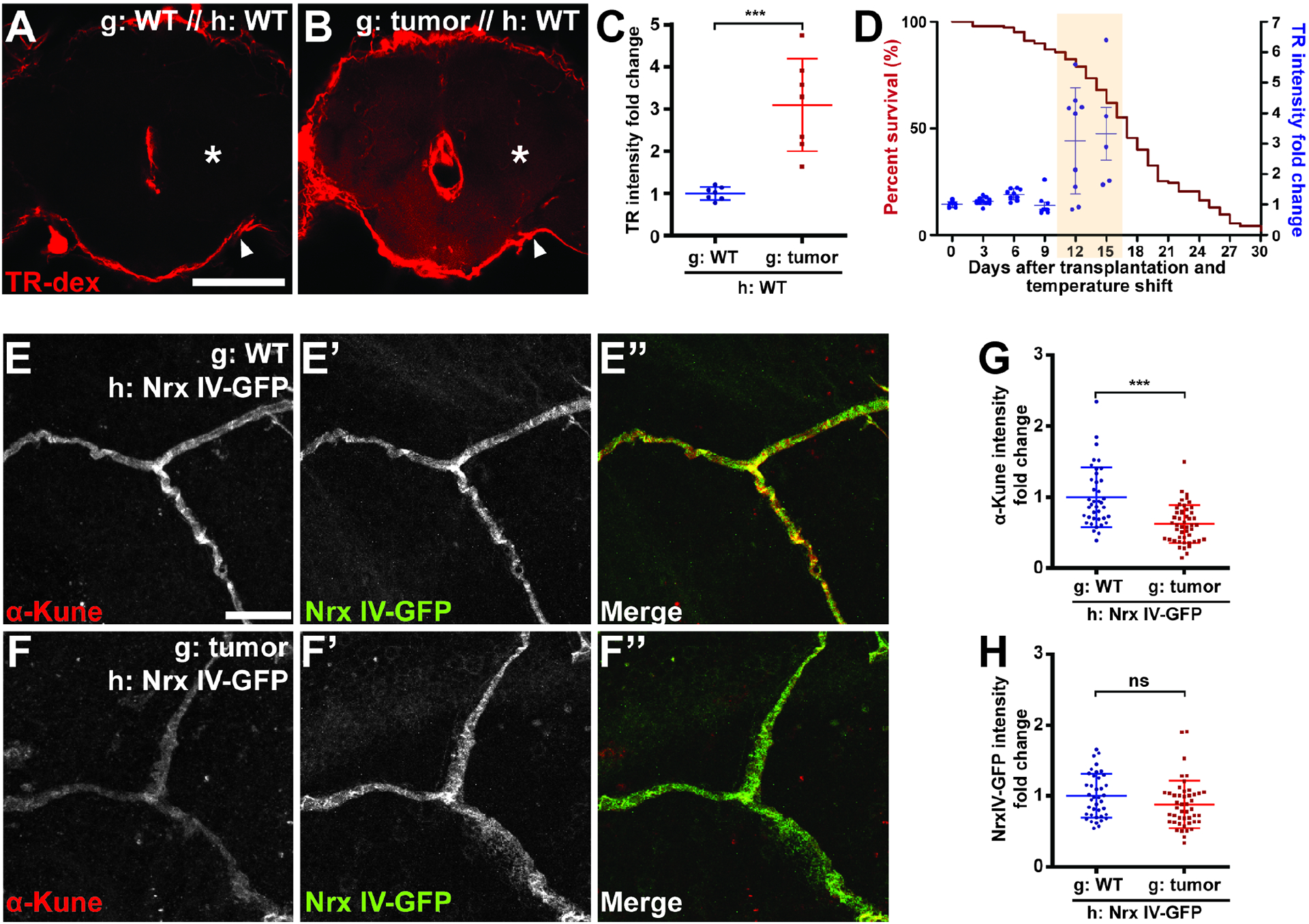
(A, B) BBB protects the brain from TR-dextran (TR-dex) penetration in control hosts, but TR-dex passes through the BBB of tumor-bearing hosts. Asterisks indicate parenchyma of brain, which is covered by BBB indicated by arrowheads. Scale bar, 100 um. (C) Quantification of TR-dex intensity in the brain (n=7 each). (D) Correlation between kinetics of BBB permeability (blue dots) and lifespan curve (red line) of tumor-bearing hosts (n≥5 each). Shaded region shows days when BBB permeability is detected. (E, F) Diffuse septate junctions of BBB cells in brains of tumor hosts compared to control. Scale bar, 10 um. (G, H) Intensity of the junctional claudin Kune but not the junctional protein NrxIV is decreased in the brain of tumor vs control hosts (n=81, 79).
Scattered plots; *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; Student’s t-test. Error bar represents mean±s.d. of normalized values to control.
The barrier function of the Drosophila BBB lies within the large, flat subperineural glia (SPG) which surround the brain (Hindle and Bainton, 2014). We examined components of the occluding intercellular junctions (septate junctions) that are formed by SPGs and limit paracellular transport. While septate junction proteins were found in narrow, dense bands between SPGs in both WT flies and hosts transplanted with control imaginal discs, septate junction proteins in tumor-transplanted hosts displayed wider and more diffuse patterns of localization (Figure 2E–F). This pattern resembles that seen in BBB-defective moody mutants, consistent with a disruption of BBB integrity (Bainton et al., 2005). In addition, levels of the Claudin homologs, Kune-kune and mega were decreased compared to other BBB junctional components (Figures 2E–H and S2E–H). These data suggest that transplanted tumors actively induce BBB disruption.
BBB permeability is induced by tumor-derived cytokines
To determine whether BBB permeability is caused by JAK-STAT activating ligands, we removed Upds from either host or tumor tissue. Transplants of tumors into upd2/upd3 null mutant hosts, like transplants of tumors into WT hosts, caused permeation of injected dextran from the hemolymph to the brain (Figures 3A, B, D). By contrast, WT hosts transplanted with tumors depleted of upd2 and upd3 by RNAi maintained normal BBB function (Figure S3A). Because autocrine JAK-STAT signaling promotes tumor growth (Figure S3B) (Wu et al., 2010, Bunker et al., 2015), we also overexpressed a constitutively active form of JAK while depleting upd2 and upd3 in the transplanted tumor, and again found that the BBB remained intact in the presence of comparable tumor burden (Figures 3C, D, and S3C).
Figure 3. Jak/STAT inhibition rescues tumor-induced BBB permeability and extends host lifespan.
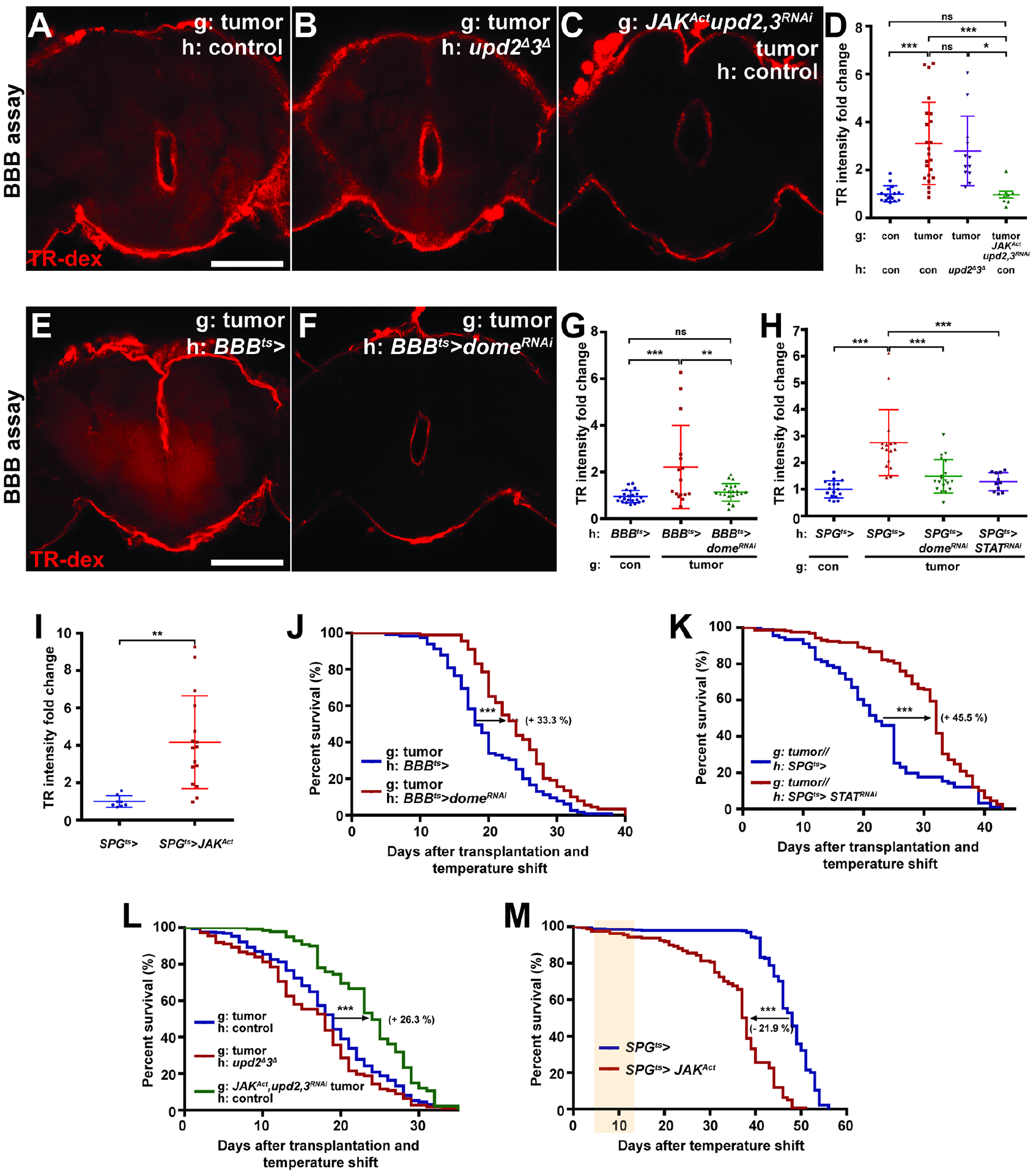
(A–D) Tumor-bearing hosts of control genotype (A) or tumor-bearing hosts that lack upd2 and upd3 (B) show permeable BBB, but depletion of upd2 and upd3 in transplanted tumors rescues the permeable BBB phenotype (C, activated JAK is coexpressed to restore autocrine growth). Scale bar, 100 um. (D) Quantification of TR-dex intensity in the brain of genotypes shown in (A) (red, n=23), (B) (purple, n=12), (C) (green, n=5), and non-tumor control (blue, n=16). (E, F) Inhibition of Jak/STAT pathway in BBB cells (via dome-RNAi) rescues permeability of BBB in tumor hosts. (G) Quantification of TR-dex intensity in the brain of genotypes shown in (E) (red, n=16), (F) (green, n=22), and non-tumor control (blue, n=22). Scale bar, 100 um. (H) Depleting Dome (green, n=19) or STAT (purple, n=11) in the SPG cells rescues permeable BBB in tumor-bearing hosts compared to tumor-bearing hosts expressing GAL4 only (red, n=16). Values are normalized to non-tumor control (blue, n=17). (I) Ectopic activation of Jak/STAT in SPG (red, n=16) is sufficient to open BBB compared to control (blue, n=8) at day 5 after temperature shift. (J) Inhibition of Jak/STAT pathway in BBB (red, n=89) extends lifespan of tumor-bearing hosts compared to control (blue, n=115). (K) Lifespan extension of tumor-bearing hosts with STAT depletion in SPG (red, n=158) compared to control (blue, n=91). (L) Control hosts bearing tumors lacking upd2 and upd3 (green, n=102) show extended lifespan compared to upd2/upd3 null hosts bearing control tumor (red, n=112) or control hosts bearing control tumor (blue, n=265). (M) Tissue-specific ectopic activation of Jak/STAT pathway in SPG (red, n=160) reduces lifespan compared to control (blue, n=358). Shaded region shows days after BBB permeability is detected (cf 3I).
Scattered plots represent mean±s.d of normalized values to control. *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; One-way anova (Tukey post test) for (D), (G) and (H), Student’s t-test for (I). Lifespan curves; *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; Log-rank test. Error bar represents mean±s.d of normalized values to control.
We further tested the necessity and sufficiency of cell-autonomous STAT activation for BBB permeabilization. Blocking STAT signaling in the BBB or SPG cells by depleting Dome or STAT with specific GAL4 drivers robustly prevented the leaky BBB phenotypes induced by tumors (Figures 3E–H). Conversely, SPG-specific overexpression of constitutively active JAK in untransplanted adults led to rapid disruption of BBB permeability (Figure 3I). Moreover, overexpression of Upd2 or Upd3 in muscle also compromised BBB permeability (Figure S3D). Taken together, the above data indicate that direct action of tumor-produced Upd cytokines on glial cells is the cause of BBB disruption.
The BBB protects fly hosts from tumor-induced death
What is the physiological consequence of tumor cytokine-induced BBB permeability? We assessed host phenotypes when STAT signaling was blocked in BBB cells and found that cachexia-like wasting was not altered (Figures S3E and F). Moreover, such manipulations had no effect on tumor size (Figure S3G). Strikingly, preventing STAT activation in BBB cells led to the extension of lifespan in tumor-bearing hosts. Median survival time was extended ~33% when Dome was depleted in all BBB cells, and by ~45% when STAT was depleted in SPG alone (Figures 3J and K). Depletion of Dome in all BBB cells or depletion of STAT in SPG cells had no positive impact on lifespan in hosts without transplants (Figures S3H and I). Consistent with this finding, WT hosts bearing upd2/3-depleted tumors with activated JAK, in which the BBB is intact, showed extended lifespan, whereas upd2/3-deficient hosts transplanted with a standard tumor still showed premature death (Figure 3L). Together, these data reveal that an intact BBB protects adult flies from lethal systemic effects of a tumor.
Is death promoted by tumors simply due to loss of core BBB functions such as ionic homeostasis? Transgenic SPG-specific STAT activation did cause premature host lethality, but death occurred long (~20 days) after the onset of BBB dysfunction (Figures 3I and M), in contrast to the synchronous kinetics seen with tumor transplants (Figure 2D). Flies depleted of the BBB junction-regulating GPCR Moody in adulthood also lived for several weeks despite brain permeability to dextran (Figures S3J and K) (Bainton et al., 2005). These results suggest that STAT-mediated BBB disruption does not alone compromise host lifespan. To verify this model, we transplanted tumors depleted of upd2 and upd3 into hosts whose BBB was compromised by STAT activation in SPG. upd2/3-depleted tumors still accelerated death of hosts in which the BBB was defective (Figure S3L). By contrast, upd2/3-depleted tumors transplanted into control hosts showed lifespans comparable to non-tumorous discs transplanted into control hosts (Figures 1B and S3L). Overall, our results demonstrate that JAK-STAT activation in BBB is necessary but not sufficient to cause rapid death of tumor-bearing hosts. They also hint that additional tumor-dependent factors may enter the brain to induce lethality.
A protective BBB is disrupted in other chronic inflammatory conditions
The existence of a native STAT-regulated pathway to alter BBB permeability raises the question of whether physiological stimuli other than tumors might trigger it. Endocrine upregulation of Upds has been described in two other adult contexts: microbial infection and a high-fat diet (Woodcock et al., 2015, Chakrabarti et al., 2016). We found that both infection with E. faecalis and lard feeding also resulted in clear permeabilization of the BBB, with STAT reporter activation and expansion of septate junction markers in SPG cells that closely resembled those seen in tumor-bearing hosts (Figures 4A–I). Moreover, when lard-fed adults were returned to a normal diet, dextran exclusion was restored (Figure 4J), revealing that this alteration in barrier permeability is reversible and not due to permanent cellular damage.
Figure 4. BBB permeability regulates survival to chronic physiological insults.
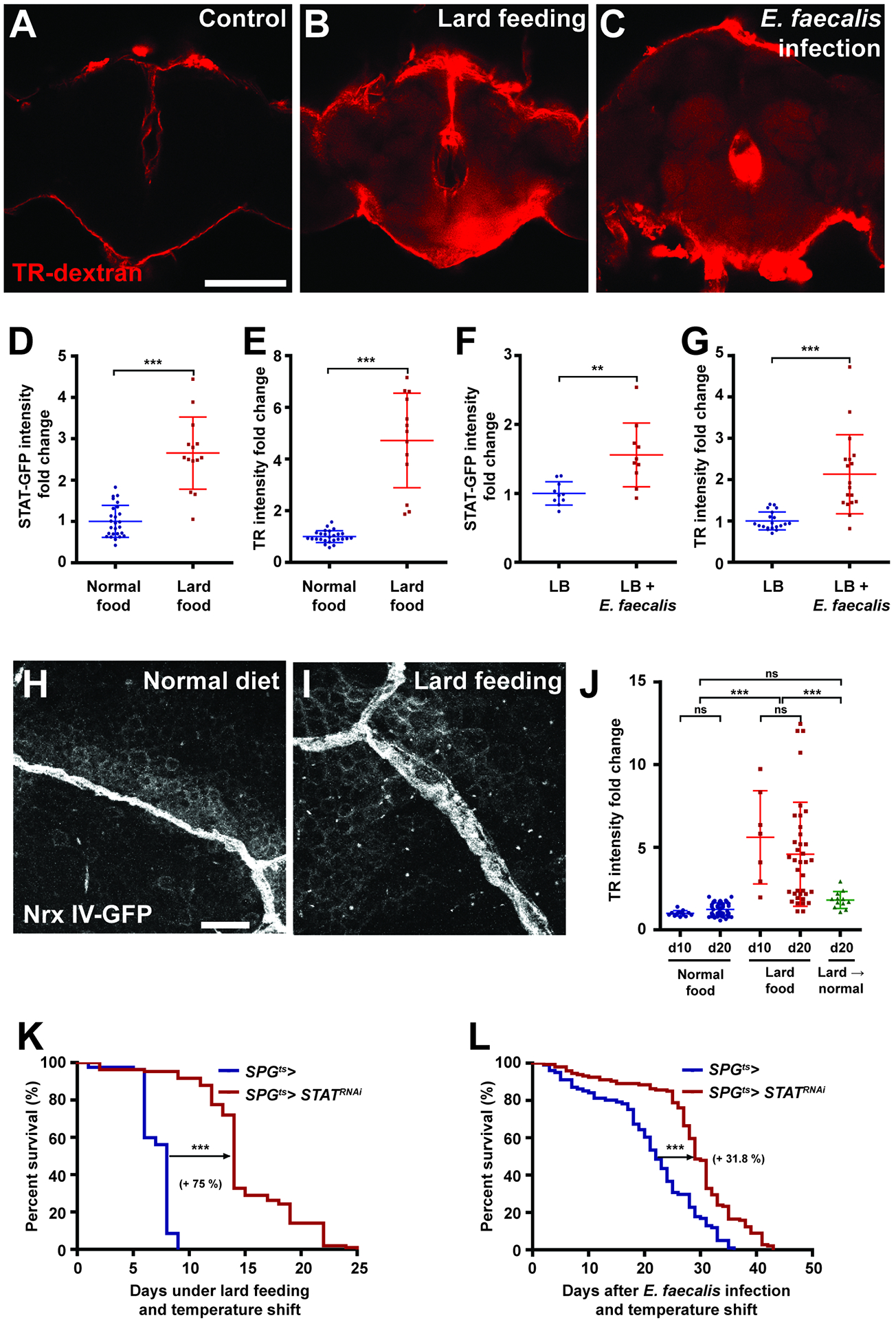
(A–C) Representative brain images of control (A), lard-fed (B), or E. faecalis infected (C) with TR-dex injection. Scale bar, 100 um. (D, E) High fat diet (red, n=14) increases brain STAT reporter intensity and TR-dex penetration compared to control (blue, n=26 each). (F, G) Injection of E. faecalis (red, n=10) increases brain STAT reporter intensity and TR-dex permeability compared to control (blue, n=10). (H, I) Septate junctions visualized by Nrx-IV-GFP shows diffuse organization under high fat diet compared to control. Scale bar, 10 um. (J) Permeable BBB under high fat diet is rescued by subsequent normal food feeding. (n>7 each). (K, L) Depleting STAT in SPG extends lifespan of flies fed high fat diet (red, n=107) (K) or infected with E. faecalis (red, n=146) (L) compared to control (blue, n=82 and n=101, respectively).
Scattered plots; *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; Student’s t-test for (D–G), one-way anova (Tukey post test) for (J). Lifespan curves; *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; Log-rank test. Error bar represents mean±s.d of normalized values to control.
We then asked whether BBB permeability plays a functional role in protecting animals from the above insults, as it does from tumors. Lard feeding and E. faecalis infection are known to reduce viability of adult flies (Woodcock et al., 2015, Chakrabarti et al., 2016). Remarkably, in both cases preventing BBB permeability through blocking STAT activation specifically in SPG cells significantly extended host lifespan (Figures 4K and L). Inflammatory opening of the BBB in these chronic conditions, as in a tumor, is therefore deleterious to the host. Together, these results demonstrate that physiological changes in BBB permeability are an important aspect of the systemic stress response, and can regulate animal survival from a variety of morbid challenges.
A mouse tumor model shows IL-6-dependent BBB permeability
To test whether tumors might systemically impact the BBB of a mammal as well, we turned to the mouse. Three weeks after implantation of B16 melanoma cells into the flank, sections through the hippocampal dentate gyrus revealed that tumor-bearing host brains showed higher levels of glial fibrillary acidic protein (GFAP) compared to controls, suggesting reactive astrocytes (Figures 5A, B, and D). Since reactive gliosis occurs in conditions including but not limited to BBB dysfunction (Yang and Wang, 2015, Liddelow and Barres, 2017), we assessed BBB function directly by tracer injection. This analysis confirmed BBB leakage: when 1 Kd NHS-Biotin was injected in the tail vein, tumor-bearing hosts showed increased extravascular levels in the brain parenchyma compared to control hosts (Figures 5E, F, and H)(O’Brown et al., 2018). Moreover, immunostaining for the junctional components Claudin-5 and Occludin revealed decreased levels in the BBB of tumor-bearing hosts (Figures 5 I, J, L and S4A, B, D).
Figure 5. Paraneoplastic BBB breakdown in an IL-6-inducing mouse tumor model.
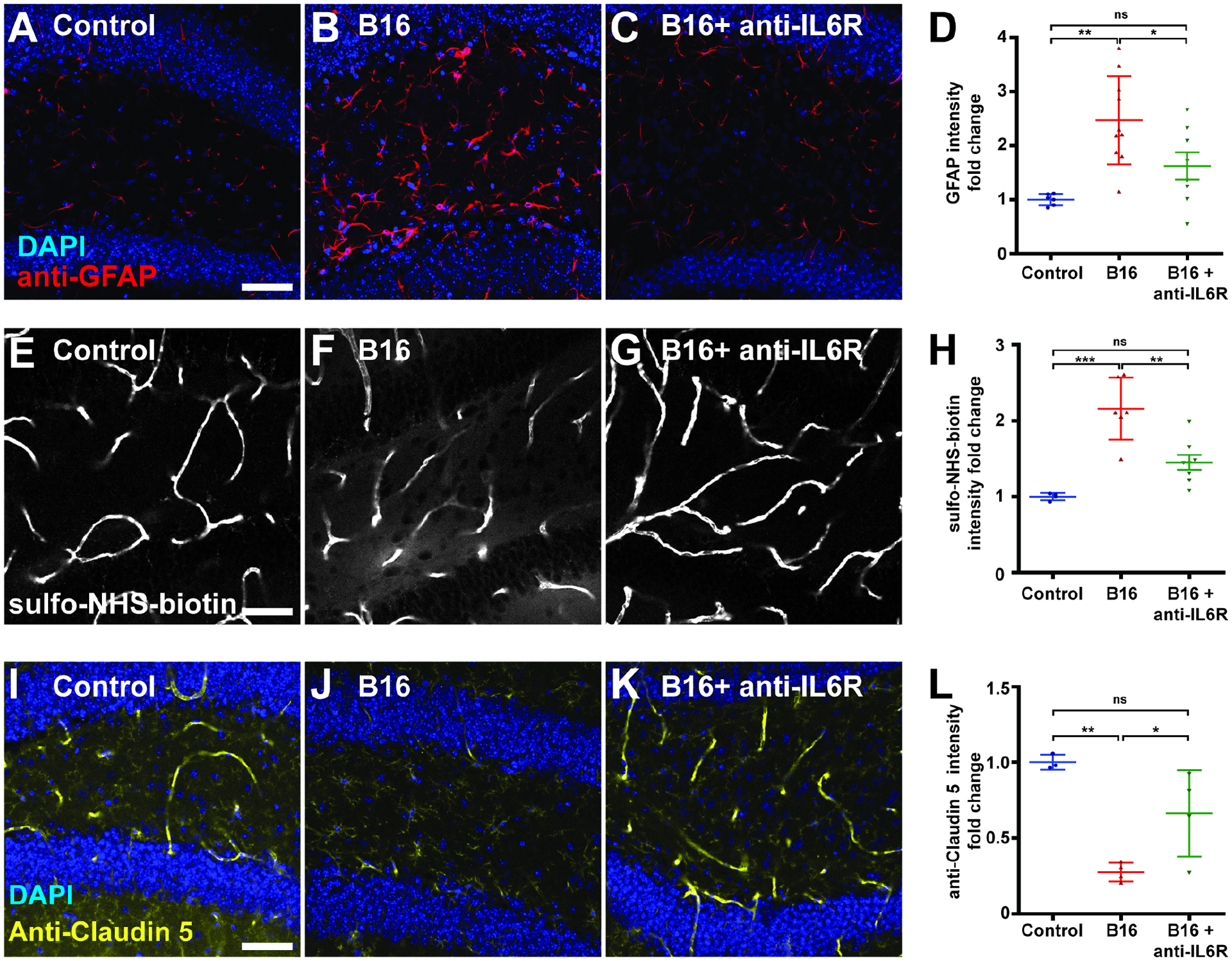
(A–D) Compared to control (A, n=6), brains of mice bearing transplanted B16-F10 melanoma cells (B, n=10) show reactive astrocytes with GFAP upregulation (red, DAPI in blue). This phenotype is reversed when tumor-bearing mice are treated with anti-IL-6R antibody (C, n=7). Scale bar, 50 um. (D) shows quantitation.
(E–H) Compared to control (E, n=4), brains of tumor-bearing mice (F, n=6) show increased BBB permeability demonstrated by extravascular presence of intravenously injected sulpho-NHS-biotin. This phenotype is reversed when tumor-bearing mice are treated with anti-IL-6R antibody (G, n=7). (H) shows quantitation.
(I–L) Compared to control (I, n=3), brains of tumor-bearing mice (J, n=4) show decreased staining of the endothelial tight junction component Claudin-5. This phenotype is reversed when tumor-bearing mice are treated with anti-IL-6R antibody (K, n=4). (L) shows quantitation.
Scattered plots; *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; one-way anova (Tukey post test). Error bar represents mean±s.d of normalized values to control.
We then explored whether this phenotype might involve IL-6-mediated inflammatory signaling, which is analogous to Upd-mediated inflammatory signaling in flies. Peripheral blood ELISAs demonstrated higher levels of circulating IL-6 in tumor-bearing hosts (Figure 6A). When these animals were subsequently injected with function-blocking antibodies against the IL-6 receptor (IL-6R), the sickness behavior and premature mortality induced by the tumor significantly improved (Figures 6C and D)(Mai et al., 2018). Comparison of hippocampal sections demonstrated that anti-IL-6R administration also caused significant decreases in both extravascular tracer intensity and reactive astrocyte induction; moreover, levels of BBB junctional components were restored (Figures 5C, D, G, H, K, L, and S4C, D). IL-6 modulates BBB permeability in other contexts (Arima et al., 2012, Rochfort and Cummins, 2015), but can also play pro-tumorigenic roles including promoting intrinsic growth and modulating the microenvironment and the anti-tumor immune response (Johnson et al., 2018, Kumari et al., 2016). Interestingly, the improved outcomes of anti-IL-6R-treated mice were not associated with a reduction of tumor burden (Figure 6B). Overall, a mammalian tumor can also compromise BBB function in an IL-6-dependent manner that promotes host morbidity.
Figure 6. IL-6 inhibition ameliorates mouse tumor morbidity associated with BBB dysfunction.
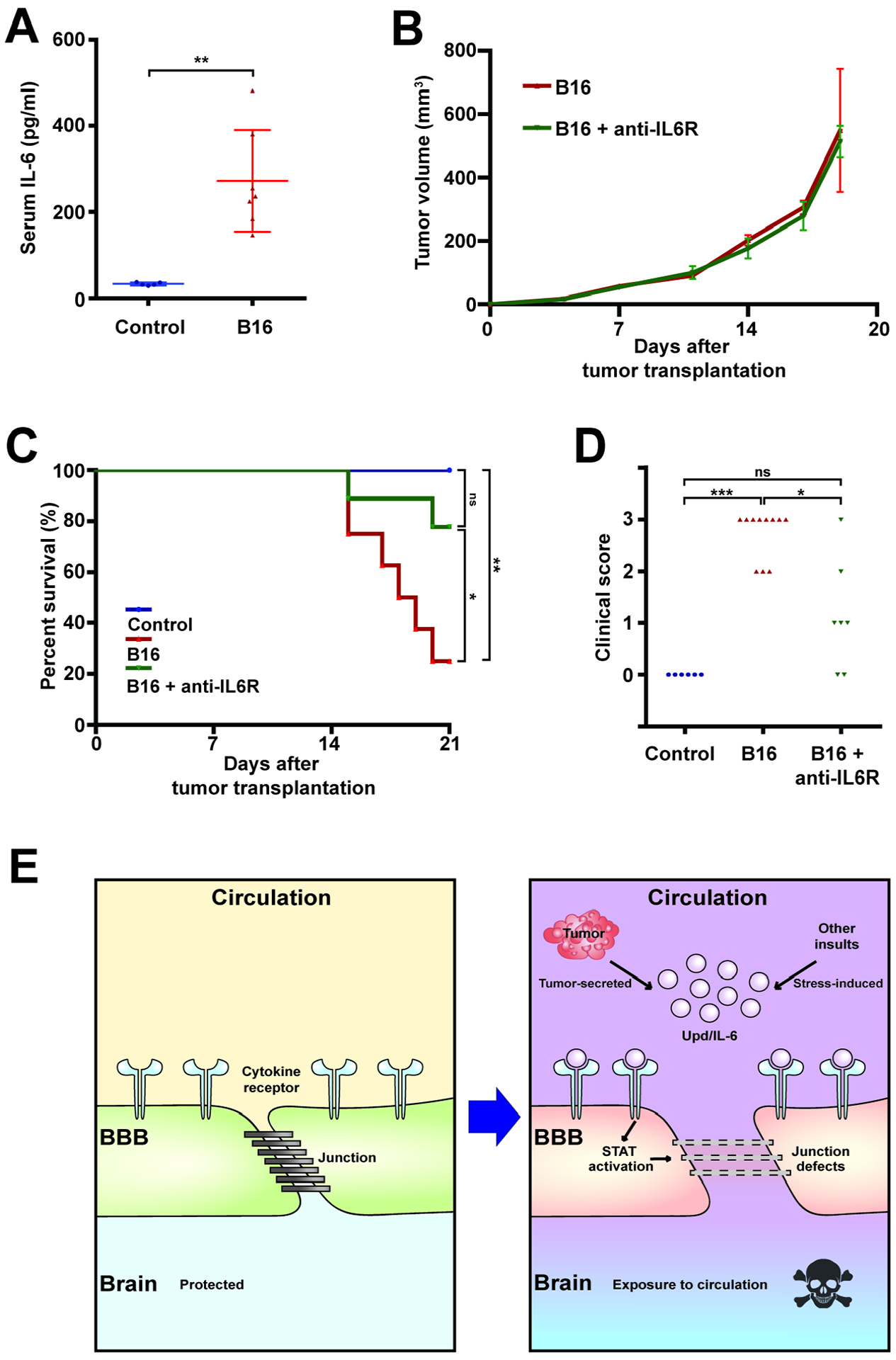
(A) Elevated levels of serum IL-6 in tumor-transplanted (red, n=7) vs control (blue, n=5) mice after 3 weeks.
(B) Tumor volume is unchanged by anti-IL-6R antibody administration (green, n=8) compared to control (red, n=8).
(C, D) Anti-IL-6R antibody administration significantly rescues both survival (C) and sickness behavior (D, clinical score of 0 indicates healthy animal) of tumor-bearing mice. Control; blue, n=6, B16; red, n=10, B16+anti-IL6R; green, n=7.
(E) Model of death elicited by inflammation-mediated BBB breakdown induced by tumors, infection, or high-fat diet.
Scattered plots; *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; Student’s t-test for (A, B), Kruskal-Wallis test for (D). Lifespan curves; *P<0.05, **P<0.01, ***P<0.001, not significant (ns)P>0.05; Log-rank test. Error bar represents mean±s.d of normalized values to control.
DISCUSSION
Mechanisms of cancer lethality are multifarious and mysterious, but go well beyond the mass effect. Here we identify a paraneoplastic syndrome that in both flies and mice can promote death from cancer. In the fly, cytokines produced by distant epithelial tumors induce permeabilization of the BBB, and specifically preventing this BBB opening extends host lifespan (Figure 6E). The systemic inflammatory environment with IL-6 family cytokine upregulation seen in tumor-bearing flies is a common phenotype induced by mammalian cancer (Johnson et al., 2018, Kumari et al., 2016, Dolan et al., 2018, Seruga et al., 2008), and our experiments demonstrate that blocking IL-6 signaling in tumor-bearing mice concomitantly rescues both BBB permeability and host morbidity without impacting tumor growth. We emphasize that we have not tested whether the beneficial effects of IL-6R antibody administration lie in its direct action on mouse BBB cells. Nevertheless, the data raise the possibility that a conserved signaling axis mediating inflammatory weakening of the BBB could contribute to mortality of certain human cancer patients. We note that a leaky BBB alone is not sufficient in flies to cause acute death; instead our results suggest that other tumor-induced factors may cross the barrier to trigger mortality.
Our data further show that additional inflammatory conditions can open the fly BBB, including a high fat diet which in mice also stimulates IL-6 upregulation and downregulation of junctional Claudins that regulate BBB permeability (Kanoski et al., 2010, Sabio et al., 2008). Since obesity is strongly associated with poor outcomes of cancer patients (Lauby-Secretan et al., 2016, Arnold et al., 2015), this raises the possibility that systemic inflammatory weakening of the BBB could be a contributing factor. Indeed, lard-fed fly hosts bearing tumors show accelerated mortality (Figure S4E). It is appealing to consider that targeting such a BBB-dependent mechanism regulating host survival could be an orthogonal clinical approach to standard tumoricidal intervention in cancer patients. Such a ‘tolerance therapy’, which addresses ultimate causes of death rather than tumor growth itself, may be less prone to selective pressures that create therapeutic resistance, and may also be agnostic to specifics of tumor tissue-of-origin or genotypes.
Limitations of the study
For technical reasons, our studies focused on female animals, but we note that effects may be less prominent in males for unknown reasons. Mouse experiments demonstrating systemic perturbation of the BBB were limited to one tumor cell line known to generate IL-6 production. Note that anti-IL6R antibody treatment of tumor-bearing mice did not target the BBB specifically; conditional knockout of STAT signaling in BBB cells of tumor-bearing mice would provide precise information on the therapeutic site of IL-6 inhibition comparable to that shown in flies.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David Bilder (bilder@berkeley.edu).
MATERIALS AVAILABILITY
Drosophila stains generated in this study will be available upon request from the Lead contact, David Bilder (bilder@berkeley.edu).
DATA AND CODE AVAILABILITY
This study did not generate new computational code, software, or algorithm.
EXPERIMENTAL MODEL AND SUBJECTS DETAILS
Drosophila stocks and husbandry
Gal4 drivers used included 9-137-GAL4 (‘BBB-GAL4’, which expresses in both SPG and PG) and moody-GAL4 (‘SPG-GAL4’), along with tubGAL80ts to control GAL4 transactivation. Control flies were crossed to w1118 or a UAS-RNAi line targeting mCherry. Flies were raised on cornmeal, molasses, and yeast food at room temperature unless otherwise noted. For lard feeding, 15 % lard (weight/volume) and 0.5 % agar (weight/volume) was added to food above. For oxidative stress induction, only a cotton ball soaked in 5% sucrose and 5% H2O2 was provided. For complete starvation, flies were placed in vials containing only a water-soaked cotton ball. For circadian rhythm synchronization, flies were kept in 25°C incubators with 12 hr light: 12 hr dark cycle for >3 generations before testing. For complete genotypes, see Table S1.
METHODS DETAILS
Drosophila tumor transplantation, gut tumor induction and infection
Before transplantation or injection, all apparatus was cleaned with 70% ethanol to minimize bacterial infection. For transplantation, wing discs from wandering 3rd instar larvae kept at room temperature were dissected into PBS. One intact disc was transplanted into the abdomen of female adult flies using a fine glass needle (Rossi and Gonzalez, 2015). Adults were then incubated at 29°C to induce transgene expression. Tumor size, ovary size, BBB permeability, and STAT-GFP were measured 15 days after transplantation and temperature shift. To genetically induce gut tumors, female esg-Gal4, tub-Gal80ts>UAS-yki3SA flies were maintained at 29°C for 15 days. Tests showed that lifespan was extended by ~ 2 days in males. For infection, newly eclosed female flies were maintained at 29°C for 5 days to allow transgene induction. E. faecalis was cultured in LB at 37°C overnight and 50 nl of 1:10 diluted E. faecalis culture, corresponding to 400–500 colony-forming units, was injected into the abdomen of CO2-anaesthetized hosts using a Picospritzer II microinjector.
Drosophila Lifespan assays
Newly eclosed (0–24 hour) WT flies were kept at room temperature for 2 days prior to tumor transplantation, E. faecalis injection, or lard feeding. For lifespans involving flies with temperature-sensitive transgenes, newly eclosed adults were kept at 29°C for >4 days. Females were segregated in groups <30 individuals/vial, except for lard feeding which used only male flies to prevent larval activity from increasing food liquification. Deaths were counted daily and flies were transferred without anesthesia to new food every 2–3 days. For smurf assays, flies were raised on standard food with 2.5 % (w/v) of FD&C blue dye #1, or provided with a cotton ball soaked in 5% sucrose and 3% dextran sodium sulfate (DSS) to disrupt the gut (Rera et al., 2012).
Measurement of BBB permeability and brain imaging in Drosophila
Using a fine glass needle, ~100 nl of 25 mg/ml 10,000 MW TR-dextran was injected in the abdomen of adult females. If tumor-bearing hosts were bloated, excess hemolymph was removed using a glass needle. 15 hours after dextran injection, flies were fixed in 4% paraformaldehyde for 80 minutes and washed in 0.1% PBST before brain dissection. Brains were imaged using a Zeiss LSM 700 confocal microscope on the same day of fixation to minimize diffusion of dextran. Average intensity was measured in a confocal section from the central plane of the brain at two regions using ImageJ. STAT-GFP intensity was quantitated in the optic lobes via a maximum intensity projection taken at 2 um intervals. Immunohistochemistry followed standard protocols. To quantify anti-Kune and anti-Mega stainings and NrxIV-GFP, and Nrg-GFP, the intensity plot profile of lines (10 um) across each bicellular segment of the septate junctions was measured and the area below the profile was calculated using ImageJ.
Preparation and handling of tumor-bearing mice
C57BL/6J mice were purchased from the Jackson Laboratory. All mice used were aged 10 to 15 weeks. B16-F10 cells were cultured under standard conditions (Marcus et al., 2018). Cells were washed and resuspended in PBS, and 100 ul containing 1 × 106 cells was injected subcutaneously. Mice used as controls received only PBS subcutaneously. For anti-IL-6R antibody treatment, mice received either PBS or anti-IL-6R (200 ug, Intraperitoneal injection) starting on day 9 and repeated every three days until mice were sacrificed. Sickness behavior was assayed according to (Mai et al., 2018). Tests suggested that female mice showed greater sickness behavior than male mice, so females were subsequently analyzed. Tumor growth was measured using calipers, and tumor volume was estimated using the ellipsoid formula: V=(π/6)ABC. All experiments were approved by the University of California Berkeley Animal Care and Use Committee.
Mouse brain imaging and measurement of IL-6 levels
Mice were sacrificed at 21 days or earlier when required by IACUC guidelines. For immunohistochemistry alone, mice were anesthetized and transcardially perfused with 10 ml of PBS and 20 ml of 4% paraformaldehyde (PFA) at a rate of 2 ml/min. The brains were dissected and postfixed with 4% PFA at 4°C overnight. For tracers, mice were injected intravenously with 0.2 mg/g body weight Sulfo-NHS-Biotin dissolved in 0.9% sterile saline in the lateral tail vein. Fifteen minutes after injection, brains were dissected and drop-fixed in 4% paraformaldehyde (PFA) overnight at 4 °C. All brains were cryoprotected in 30% sucrose in PBS for 48 hr, embedded in tissue freezing medium, and then stored at −80 °C. Frozen brains were cryostat sectioned at the thickness of 30 um, mounted on slides and stored at −20°C until processed. Immunohistochemistry, Biotin detection and mounting followed standard protocols and samples were imaged on a Zeiss 700 confocal microscope. For each mouse, >5 sections of the dentate gyrus of the hippocampus were analyzed; values shown are intensity averages of sections analyzed in the hilar region. Leakage of Sulfo-NHS-Biotin into brain parenchyma was quantified using FIJI ImageJ software to determine the integrated fluorescence intensity outside of CD31-labelled vasculature. A staining failure in CD31/Biotin staining on sections from one tumor-bearing mouse led to this sample being discarded. Claudin and Occludin levels were quantified as mean intensity of hilar region signal following subtraction of background measured in non-vascular neuropil. For IL-6 level measurements, blood was collected by cardiac puncture from control and tumor-bearing mice. IL-6 was quantified from harvested serum using an ELISA kit following the manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification
Quantification is described in the Methods section.
Statistical analysis
The Prism (Graphpad) program was used to run statistical tests. Lifespan curves were analyzed using Log-rank test, and scatter plots were analyzed using Student’s t-test (comparing 2 groups) or one-way ANOVA with Tukey post-test (comparing more than 3 groups). Kruskal-Wallis test was used to analyze nonparametric clincal score. *P<0.05, **P<0.01, ***P<0.001. Error bar represents mean ± s.d. of normalized values to control.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Repo (1:250) | DSHB | 8D12 Anti-Repo |
| Rabbit anti-GFP (1:30,000) | Molecular Probes | A6455 |
| Rabbit anti-Kune (1:250) | Gift from Dr. Beitel Northwestern University | Nelson et al. 2010 |
| Mouse anti-Mega (1:250) | Gift from Dr. Behr Leipzig University | Behr et al. 2003 |
| Rabbit anti-CD31 (1:50) | Abcam | Ab28364 |
| Goat anti-GFAP (1:1,000) | Abcam | Ab53554 |
| Mouse IL-6 ELISA MAX Standard Set | BioLegend | 431301 |
| Rat anti-IL-6R (200 ug/injection) | BioXCell | BE0047 |
| Mouse anti-Claudin-5 (10 ug/mL) | ThermoFisher Scientific | 35-2500 |
| Mouse anti-Occludin (3 ug/mL) | ThermoFisher Scientific | 33-1500 |
| Bacterial and virus strains | ||
| E. faecalis | Lazzaro lab Cornell University | N/A |
| C57BL/6J mice | Jackson Laboratory | #000664 |
| B16-F10 melanoma cell line | UC Berkeley Cell Culture Facility | B16-F10 |
| Chemicals, peptides, and recombinant proteins | ||
| TRITC-Phalloidin (1:250) | Sigma-Aldrich | P1951; RRID: AB_2315148 |
| DAPI (1:10,000) | ThermoFisher Scientific | D1306, RRID: AB_2629482 |
| 10 kDa Texas red-Dextran (25 mg/ml) | ThermoFisher Scientific | D1863 |
| FD&C Blue #1 | Spectrum Chemical MFG. Corp. | FD110 |
| Hydrogen peroxide | Fisher Chemical | H325-100 |
| Dextran sulfate sodium | MP Biomedicals | 9011-18-1 |
| Dulbecco’s modified Eagle’s medium | ThermoFisher Scientific | 11995-065 |
| Fetal bovine serum (FBS) | Omega Scientific | FB-01 |
| Glutamine | Sigma-Aldrich | G8540 |
| Penicillin-Streptomycin | ThermoFisher Scientific | 15140-122 |
| Gentamycin sulfate | Lonza | MT30-005-cR |
| β-mercaptoethanol | EMD Biosciences | 21985-023 |
| Hepes | ThermoFisher Scientific | BP310-100 |
| PBS | ThermoFisher Scientific | 14190-250 |
| Tissue freezing medium | General data healthcare | TFM-C |
| Normal donkey serum | Jackson ImmunoResearch Laboratories | 017-000-001 |
| ProLong Gold Antifade mountant | ThermoFisher Scientific | P36930 |
| EZ-Link™ Sulfo-NHS-LC-Biotin | ThermoFisher Scientific | 21335 |
| Experimental models: Organisms/strains | ||
| Nub-Gal4 | Bloomington Drosophila Stock Center | Flybase ID: FBst0086108 |
| tubGAL80ts | Bloomington Drosophila Stock Center | Flybase ID: FBst0007018 |
| UAS-Ras v12 | Bloomington Drosophila Stock Center | Flybase ID: FBst0004847 |
| UAS-aPKC ΔN | Bloomington Drosophila Stock Center | Flybase ID: FBst0051673 |
| w 1118 | Bloomington Drosophila Stock Center | Flybase ID: FBst0005905 |
| UAS-impL2RNAi (validated in PMID:27633989) | Vienna Drosophila Resource Center | Flybase ID: FBst0458731 |
| 10XStat92E-GFP | Perrimon lab Harvard University | Flybase ID: FBst0026197 |
| NrxIV-GFP | Bloomington Drosophila Stock Center | Flybase ID: FBst0050798 |
| Nrg-GFP | Bloomington Drosophila Stock Center | Flybase ID: FBst0006844 |
| upd2 Δ , upd3 Δ | Bloomington Drosophila Stock Center | Flybase ID: FBst0055729 |
| UAS-upd2RNAi (validated in PMID:23021220) | Bloomington Drosophila Stock Center | Flybase ID: FBst0033949 |
| UAS-upd3RNAi (validated in PMID:25601202) | Vienna Drosophila Resource Center | Flybase ID: FBst0478692 |
| 9-137-Gal4 (BBB-Gal4) | Bainton lab University of California-San Franscisco | Flybase ID: FBti0202073 |
| UAS-domeRNAi (validated in PMID:28045022) | Vienna Drosophila Resource Center | Flybase ID: FBti0118756 |
| Moody-Gal4 (SPG-Gal4) | Bainton lab University of California-San Franscisco | Flybase ID: FBtp0022847 |
| UAS-Hop Tum | Perrimon lab | Flybase ID: FBal0043128 |
| UAS-STATRNAi (X) (validated in PMID:17803358) | Bloomington Drosophila Stock Center | Flybase ID: FBst0026899 |
| UAS-STATRNAi (II) (validated in PMID:26690827) | Vienna Drosophila Resource Center | Flybase ID: FBst0478803 |
| 10X UAS-mCD8-GFP | Bloomington Drosophila Stock Center | Flybase ID: FBst0032184 |
| UAS-nls-GFP | Bloomington Drosophila Stock Center | Flybase ID: FBst0004775 |
| Dome-Gal4 | Bloomington Drosophila Stock Center | Flybase ID: FBst0081010 |
| Esg-Gal4 | Perrimon lab Harvard University | Flybase ID: FBti0013268 |
| UAS-yki 3SA (S111A.S168A.S250A) | Bloomington Drosophila Stock Center | Flybase ID: FBst0028817 |
| UAS-nls-GFP | Bloomington Drosophila Stock Center | Flybase ID: FBst0004776 |
| Mef2-Gal4 | Bloomington Drosophila Stock Center | Flybase ID: FBst0027390 |
| UAS-upd2 | Harrison lab University of Kentucky | Flybase ID: FBtp0110532 |
| UAS-upd3 | Harrison lab University of Kentucky | Flybase ID: FBtp0110533 |
| UAS-mCh RNAi | Bloomington Drosophila Stock Center | Flybase ID: FBst0035787 |
| UAS-moodyRNAi (validated in PMID:33307547) | Vienna Drosophila Resource Center | Flybase ID: FBst0481265 |
| Software and algorithms | ||
| Fiji | ImageJ | http://fiji.sc/ |
| Illustrator | Adobe | www.adobe.com/products/illustrator.html |
| Photoshop | Adobe | www.adobe.com/products/photoshopfamily.html |
| Acrobet DC | Adobe | acrobat.adobe.com/us/en/acrobat.html |
| Excel | Microsoft | https://products.office.com/enus/excel |
| Prism | Graphpad | www.graphpad.com/scientificsoftware/prism/ |
| Zen | Zeiss | www.zeiss.com/microscopy/us/products/microscopesoftware/zen.html |
Highlights.
Fly tumors induce paraneoplastic opening of the blood-brain barrier (BBB)
BBB permeabilization by tumor-induced Jak/STAT activation accelerates host death
BBB also protects flies from high-fat diet and non-pathogenic infections
A mouse tumor model disrupts the protective BBB in an IL-6-dependent manner
ACKNOWLEDGMENTS
We thank Roland Bainton, Chenghua Gu, Iswar Hariharan, Daniela Kaufer, Brittany Flowers, Laura Attardi and Kristin Scott for advice and discussion. We acknowledge generous gifts of reagents from Matthias Behr, Greg Beitel, Christian Klambt, Frederic Geissmann, Terry Orr-Weaver, Amita Sehgal, Laurent Seugnet, Mark Van Doren, Frederick Wolf, Tian Xu and Naoki Yamanaka and the community resources provided by TRiP at Harvard Medical School (NIH/NIGMS R01-GM084947), the Bloomington Drosophila Stock Center (NIH P40OD018537) and the Vienna Drosophila Resource Center. This work was supported by NIH grants GM090150 and GM130388 to D.B, AI113041 to D.H.R, and HD092093 to K.S., as well as an NSF Graduate Research Fellowship 1752814 to N.K.W, NIH fellowship F31CA228381 to C.J.N and a Pew Scholarship to K.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Authors declare no competing interests.
REFERENCES
- AMOYEL M, ANDERSON AM & BACH EA 2014. JAK/STAT pathway dysregulation in tumors: a Drosophila perspective. Semin Cell Dev Biol, 28, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARIMA Y, HARADA M, KAMIMURA D, PARK JH, KAWANO F, YULL FE, KAWAMOTO T, IWAKURA Y, BETZ UA, MARQUEZ G, BLACKWELL TS, OHIRA Y, HIRANO T & MURAKAMI M 2012. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell, 148, 447–57. [DOI] [PubMed] [Google Scholar]
- ARNOLD M, PANDEYA N, BYRNES G, RENEHAN PAG, STEVENS GA, EZZATI PM, FERLAY J, MIRANDA JJ, ROMIEU I, DIKSHIT R, FORMAN D & SOERJOMATARAM I 2015. Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol, 16, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAINTON RJ, TSAI LT, SCHWABE T, DESALVO M, GAUL U & HEBERLEIN U 2005. moody encodes two GPCRs that regulate cocaine behaviors and blood-brain barrier permeability in Drosophila. Cell, 123, 145–56. [DOI] [PubMed] [Google Scholar]
- BARACOS VE, MARTIN L, KORC M, GUTTRIDGE DC & FEARON KCH 2018. Cancer-associated cachexia. Nat Rev Dis Primers, 4, 17105. [DOI] [PubMed] [Google Scholar]
- BEADLE GW & EPHRUSSI B 1935. Transplantation in Drosophila. Proc Natl Acad Sci U S A, 21, 642–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BILDER D 2004. Epithelial polarity and proliferation control: links from the Drosophila neoplastic tumor suppressors. Genes Dev, 18, 1909–25. [DOI] [PubMed] [Google Scholar]
- BILDER D, ONG K, HSI TC, ADIGA K & KIM J 2021. Tumor-host interactions through the lens of Drosophila. Nature Reviews Cancer, 10.1038/s41568-021-00387-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOCCACCIO C & COMOGLIO PM 2009. Genetic link between cancer and thrombosis. J Clin Oncol, 27, 4827–33. [DOI] [PubMed] [Google Scholar]
- BROWN S, HU N & HOMBRIA JC 2001. Identification of the first invertebrate interleukin JAK/STAT receptor, the Drosophila gene domeless. Curr Biol, 11, 1700–5. [DOI] [PubMed] [Google Scholar]
- BUNKER BD, NELLIMOOTTIL TT, BOILEAU RM, CLASSEN AK & BILDER D 2015. The transcriptional response to tumorigenic polarity loss in Drosophila. Elife, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAKRABARTI S, DUDZIC JP, LI X, COLLAS EJ, BOQUETE JP & LEMAITRE B 2016. Remote Control of Intestinal Stem Cell Activity by Haemocytes in Drosophila. PLoS Genet, 12, e1006089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHATTERJEE D & DENG WM 2019. Drosophila Model in Cancer: An Introduction. Adv Exp Med Biol, 1167, 1–14. [DOI] [PubMed] [Google Scholar]
- CLARK RI, SALAZAR A, YAMADA R, FITZ-GIBBON S, MORSELLI M, ALCARAZ J, RANA A, RERA M, PELLEGRINI M, JA WW & WALKER DW 2015. Distinct Shifts in Microbiota Composition during Drosophila Aging Impair Intestinal Function and Drive Mortality. Cell Rep, 12, 1656–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOLAN RD, LAIRD BJA, HORGAN PG & MCMILLAN DC 2018. The prognostic value of the systemic inflammatory response in randomised clinical trials in cancer: A systematic review. Crit Rev Oncol Hematol, 132, 130–137. [DOI] [PubMed] [Google Scholar]
- FIGUEROA-CLAREVEGA A & BILDER D 2015. Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev Cell, 33, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GATEFF E & SCHNEIDERMAN HA 1969. Neoplasms in mutant and cultured wild-tupe tissues of Drosophila. Natl Cancer Inst Monogr, 31, 365–97. [PubMed] [Google Scholar]
- GONZALEZ C 2013. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat Rev Cancer, 13, 172–83. [DOI] [PubMed] [Google Scholar]
- HINDLE SJ & BAINTON RJ 2014. Barrier mechanisms in the Drosophila blood-brain barrier. Front Neurosci, 8, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON DE, O’KEEFE RA & GRANDIS JR 2018. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol, 15, 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANOSKI SE, ZHANG Y, ZHENG W & DAVIDSON TL 2010. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J Alzheimers Dis, 21, 207–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KULSHAMMER E, MUNDORF J, KILINC M, FROMMOLT P, WAGLE P & UHLIROVA M 2015. Interplay among Drosophila transcription factors Ets21c, Fos and Ftz-F1 drives JNK-mediated tumor malignancy. Dis Model Mech, 8, 1279–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUMARI N, DWARAKANATH BS, DAS A & BHATT AN 2016. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol, 37, 11553–11572. [DOI] [PubMed] [Google Scholar]
- KWON Y, SONG W, DROUJININE IA, HU Y, ASARA JM & PERRIMON N 2015. Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev Cell, 33, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAUBY-SECRETAN B, SCOCCIANTI C, LOOMIS D, GROSSE Y, BIANCHINI F, STRAIF K & INTERNATIONAL AGENCY FOR RESEARCH ON CANCER HANDBOOK WORKING, G. 2016. Body Fatness and Cancer--Viewpoint of the IARC Working Group. N Engl J Med, 375, 794–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIDDELOW SA & BARRES BA 2017. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity, 46, 957–967. [DOI] [PubMed] [Google Scholar]
- MAI SHC, SHARMA N, KWONG AC, DWIVEDI DJ, KHAN M, GRIN PM, FOX-ROBICHAUD AE & LIAW PC 2018. Body temperature and mouse scoring systems as surrogate markers of death in cecal ligation and puncture sepsis. Intensive Care Med Exp, 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARCUS A, MAO AJ, LENSINK-VASAN M, WANG L, VANCE RE & RAULET DH 2018. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity, 49, 754–763 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCALLISTER SS & WEINBERG RA 2010. Tumor-host interactions: a far-reaching relationship. J Clin Oncol, 28, 4022–8. [DOI] [PubMed] [Google Scholar]
- MOBERG KH, SCHELBLE S, BURDICK SK & HARIHARAN IK 2005. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev Cell, 9, 699–710. [DOI] [PubMed] [Google Scholar]
- NEWTON H, WANG YF, CAMPLESE L, MOKOCHINSKI JB, KRAMER HB, BROWN AEX, FETS L & HIRABAYASHI S 2020. Systemic muscle wasting and coordinated tumour response drive tumourigenesis. Nat Commun, 11, 4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’BROWN NM, PFAU SJ & GU C 2018. Bridging barriers: a comparative look at the blood-brain barrier across organisms. Genes Dev, 32, 466–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAGLIARINI RA & XU T 2003. A genetic screen in Drosophila for metastatic behavior. Science, 302, 1227–31. [DOI] [PubMed] [Google Scholar]
- PASTOR-PAREJA JC, WU M & XU T 2008. An innate immune response of blood cells to tumors and tissue damage in Drosophila. Dis Model Mech, 1, 144–54; discussion 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PINSONNEAULT RL, MAYER N, MAYER F, TEGEGN N & BAINTON RJ 2011. Novel models for studying the blood-brain and blood-eye barriers in Drosophila. Methods Mol Biol, 686, 357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RERA M, CLARK RI & WALKER DW 2012. Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila. Proc Natl Acad Sci U S A, 109, 21528–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROCHFORT KD & CUMMINS PM 2015. The blood-brain barrier endothelium: a target for pro-inflammatory cytokines. Biochem Soc Trans, 43, 702–6. [DOI] [PubMed] [Google Scholar]
- ROSSI F & GONZALEZ C 2015. Studying tumor growth in Drosophila using the tissue allograft method. Nat Protoc, 10, 1525–34. [DOI] [PubMed] [Google Scholar]
- SABIO G, DAS M, MORA A, ZHANG Z, JUN JY, KO HJ, BARRETT T, KIM JK & DAVIS RJ 2008. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science, 322, 1539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SERUGA B, ZHANG H, BERNSTEIN LJ & TANNOCK IF 2008. Cytokines and their relationship to the symptoms and outcome of cancer. Nat Rev Cancer, 8, 887–99. [DOI] [PubMed] [Google Scholar]
- SONG W, KIR S, HONG S, HU Y, WANG X, BINARI R, TANG HW, CHUNG V, BANKS AS, SPIEGELMAN B & PERRIMON N 2019. Tumor-Derived Ligands Trigger Tumor Growth and Host Wasting via Differential MEK Activation. Dev Cell, 48, 277–286 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STALEY BK & IRVINE KD 2010. Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr Biol, 20, 1580–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VACCARI T & BILDER D 2005. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev Cell, 9, 687–98. [DOI] [PubMed] [Google Scholar]
- WOODCOCK KJ, KIERDORF K, POUCHELON CA, VIVANCOS V, DIONNE MS & GEISSMANN F 2015. Macrophage-derived upd3 cytokine causes impaired glucose homeostasis and reduced lifespan in Drosophila fed a lipid-rich diet. Immunity, 42, 133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU M, PASTOR-PAREJA JC & XU T 2010. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature, 463, 545–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Z & WANG KK 2015. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci, 38, 364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YEOM E, SHIN H, YOO W, JUN E, KIM S, HONG SH, KWON DW, RYU TH, SUH JM, KIM SC, LEE KS & YU K 2021. Tumour-derived Dilp8/INSL3 induces cancer anorexia by regulating feeding neuropeptides via Lgr3/8 in the brain. Nat Cell Biol, 23, 172–183. [DOI] [PubMed] [Google Scholar]
- ZHANG SL, YUE Z, ARNOLD DM, ARTIUSHIN G & SEHGAL A 2018. A Circadian Clock in the Blood-Brain Barrier Regulates Xenobiotic Efflux. Cell, 173, 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new computational code, software, or algorithm.