

Abstract
The 1,4-diacyloxylation of 1,3-cyclohexadiene (CHD) affords valuable stereochemically defined scaffolds for natural product and pharmaceutical synthesis. Existing cis-selective diacyloxylation protocols require superstoichiometric quantities of benzoquinone (BQ) or MnO2, which limit process sustainability and large-scale application. In this report, reaction development and mechanistic studies are described that overcome these limitations by pairing catalytic BQ with tert-butyl hydroperoxide as the stoichiometric oxidant. Catalytic quantities of bromide enable a switch from trans to cis diastereoselectivity. A catalyst with a 1:2 Pd:Br ratio supports high cis selectivity while retaining good rate and product yield. Further studies enable replacement of BQ with hydroquinone (HQ) as a source of cocatalyst, avoiding the handling of volatile and toxic BQ in large scale applications.
Keywords: homogeneous catalysis, palladium, oxidation, difunctionalization, synthetic methods
Graphical Abstract

We report an efficient and scalable cis-selective diacyloxylation protocol that employs catalytic hydroquinone with tBuOOH as a benign terminal oxidant. A bromide additive was crucial to obtaining good yields and excellent cis selectivity. The study culminates in the implementation of the optimized reaction on large scale.
Pd-catalyzed oxidations of alkenes and dienes are important industrial processes,[1] ranging from the Wacker process for production of acetaldehyde from ethylene,[2] to 1,4-diacetoxylation of 1,3-butadiene, employed in the commercial synthesis of tetrahydrofuran.[3] The latter reaction is closely related to 1,4-diacyloxylation of 1,3-cyclohexadiene (CHD) and other cyclic dienes,[4–7] which find utility in natural product and pharmaceutical synthesis.[8–12] These reactions generate cis or trans diastereomers, and their utility in synthetic routes (e.g., using Tsuji-Trost allylic substitution [13–16] or enzymatic desymmetrization [17–19]) depends on selective formation of a single diastereomer. Recently, 1,4-dibenzoyloxylation of CHD was used to access cis-1 on 150 kg scale as the first step in the synthesis of an anticancer drug candidate (Scheme 1a).[11,12] The reaction, based on a published method by Bäckvall and coworkers,[20] employs 2.2 equiv of benzoquinone (BQ), requiring the handling of nearly 500 kg of BQ in this process. BQ is toxic[21] and readily sublimes, even at room temperature,[22] and the mixture of superstoichiometric BQ and hydroquinone (HQ) byproducts formed in the reaction complicate purification efforts and lower the product yield. These challenges motivated efforts to develop improved reaction conditions. The safety hazards and operational complexities evident in this process highlight the need for a cis-selective dibenzoyloxylation method capable of using catalytic quantities of BQ, or ideally hydroquinone (HQ), as a cocatalyst.
Scheme 1.

Cis- and trans-selective diacyloxylation of 1,3-cyclohexadiene (CHD), including a (a) process-scale application leading to a drug candidate,[12] (b) trans-selective diacyloxylation using tBuOOH and catalytic benzoquinone (BQ), with a dual-function Pd catalyst,[7] and (c) goals of the present work.
BQ reoxidizes Pd0 to PdII in these reactions,[23,24] and seminal work by Bäckvall and coworkers has demonstrated that cocatalysts, such as [Co(salophen)] or [Fe(phthalocyanine)], support in situ reoxidation of HQ to BQ using O2 as the terminal oxidant.[25–28] These conditions are only compatible with formation of trans-diacyloxylation products, however. Complementary PdII/chloride catalyst systems have been identified for cis-diacyloxylation of dienes,[5] but the chloride additive inhibits the aerobic cocatalysts, even when used in catalytic quantities.[29,30] Stoichiometric MnO2 is required as a terminal oxidant in order to lower the BQ loading in these reactions.[6,31] Recently, Eastgate and coworkers reported a complementary catalytic method, using tert-butyl hydroperoxide (tBuOOH) as the oxidant.[7,32] The Pd catalyst serves a dual role in these reactions (Scheme 1b), promoting both CHD oxidation and reoxidation of HQ by tBuOOH (DA1 is a Diels-Alder adduct obtained from reaction of CHD with BQ). These conditions are appealing for large-scale application; however, they similarly access only trans-diacyloxylation product (i.e., trans-1). The present report outlines development and characterization of a new Pd(OAc)2/bromide/BQ cocatalyst system that enables large-scale cis-diacyloxylation of CHD with tBuOOH as the oxidant (Scheme 1c).
Reaction screening efforts were initiated with the previously reported tBuOOH/catalytic BQ conditions, evaluating the effect of various additives (base, DA1, etc.; see section 3 of the Supporting Information for screening data). Replacing DA1/HQ (10 mol% each; entry 1, Table 1) with 20 mol% BQ gives almost identical yield and diastereoselectivity, favoring trans-1 (entry 2). Addition of chloride to this mixture leads to higher cis-1 (entries 3 and 4), accessing 72% cis with 10 mol% Bu4NCl. Bromide and iodide lead to even higher cis selectivity (entries 5–8, also see Tables S3 and S6 in the Supporting Information), and an optimal balance of yield and cis selectivity is obtained with 5 mol% Bu4NBr (72% yield, 94% cis). Efforts to replace BQ with HQ were unsuccessful (entries 9 and 10). These reactions exhibit catalyst decomposition via Pd black formation and low product yields.
Table 1.
Reaction screening data and effect of halide additives on yield and diastereoselectivity

| ||||
|---|---|---|---|---|
| Reaction[a] | Quinone/Alkene (mol %) | Additive (mol %) | Total product (%)[b] | cis selectivity (%)[b] |
| 1 | DA1 (10 mol%) + HQ (10 mol%) | -- | 70 | 8 |
| 2 | BQ (20 mol%) | -- | 68 | 7 |
| 3 | BQ (20 mol%) | Bu4NCl (5 mol%) | 28 | 56 |
| 4 | BQ (20 mol%) | Bu4NCl (10 mol%) | 67 | 72 |
| 5 | BQ (20 mol%) | Bu 4 NBr (5 mol%) | 72 | 94 |
| 6 | BQ (20 mol%) | Bu4NBr (10 mol%) | 48 | 98 |
| 7 | BQ (20 mol%) | Bu4NI (5 mol%) | 14 | 99 |
| 8 | BQ (20 mol%) | Bu4NI (10 mol%) | 6 | 99 |
| 9 | HQ (20 mol%) | Bu4NBr (5 mol%) | 3 | 95 |
| 10 | DA1 (10 mol%) + HQ (10 mol%) | Bu4NBr (5 mol%) | 5 | 98 |
[CHD] = 420 mM, [BzOH] = 1680 mM, [Pd(OAc)2] = 10.5 mM, [Et3N] = 315 mM, [tBuOOH] = 840 mM, [biphenyl] = 42 mM (internal standard).
Yield and selectivity determined by UPLC. See Supporting Information for details.
The effect of halide:Pd ratio on initial rate and cis selectivity of the diacyloxylation was investigated more thoroughly (Figure 1). The halide loading was varied between 0–6 equiv relative to Pd and the initial rate and cis selectivity were monitored by UPLC. A 1:1 Br:Pd ratio exhibits the highest rate for cis-1 formation, but ratios between 2–3:1 Br:Pd show improved selectivity while maintaining good rates (Figure 1a). Chloride does not inhibit trans-1 formation as strongly as bromide, and, even at higher Cl:Pd ratios, only moderate diastereoselectivity is observed (Figure 1b). Iodide strongly inhibits formation of trans-1, allowing for very high cis selectivity (99% cis), but it also inhibits the rate of cis-1 formation (see Table 1 and Table S7). The relative impact of the different halide ions correlates with the relative binding affinity of the halides for PdII (I > Br > Cl),[33–36] and bromide provides the best balance of rate, selectivity, and overall catalyst performance.
Figure 1.

Effect of bromide (a) and chloride (b) on the rate and diastereoselectivity of the CHD diacetoxylation reaction. Conditions: [CHD] = 420 mM, [BzOH] = 1680 mM, [Pd(OAc)2] = 10.5 mM, [BQ] = 84 mM, [Et3N] = 315 mM, [tBuOOH] = 840 mM, [biphenyl] = 42 mM (internal standard). Reaction aliquots analyzed by UPLC. The data in gray for chloride plot reflect reactions with significant insoluble solids and they are not included in the empirical curve fits (see Supporting Information for curve fitting equations).
For large-scale applications, it would be ideal to use HQ as the source of cocatalyst to avoid handling of large quantities of BQ;[37] however, the data in Table 1 show that poor results are obtained with HQ as the cocatalyst (entries 9–10). In order to probe the origin of this effect, 1,4-diacetoxylation of CHD was analyzed by 1H NMR spectroscopy throughout the reaction time course to track formation of the product cis-2 and quinone speciation (Figure 2a and Figure S7). Cis-2 forms steadily throughout the reaction, while the quinone speciation never reaches steady state. BQ is slowly consumed during the first 18 h, together with the appearance of HQ and DA1. Formation of DA1 is irreversible and consumes approximately 50% of the original BQ, lowering the amount of BQ/HQ available to support catalytic turnover (Figure S7). At late stages of the reaction, the rate of HQ oxidation is competitive with BQ consumption, and a small increase in [BQ] is evident.
Figure 2.

(a) Full reaction 1H NMR time course showing cis-2 formation and quinone speciation. Conditions: [CHD] = 420 mM, [AcOH-d4] = 1680 mM, [Pd(OAc)2] = 10.5 mM, [Bu4NBr] = 21 mM, [BQ] = 84 mM, [Et3N] = 315 mM, [tBuOOH] = 840 mM, [methyl 3,5-dinitrobenzoate] = 21 mM (internal standard). Reaction conducted inside an NMR spectrometer at 24 °C using a 3:1 mixture of iPrOD-d8/acetone-d6 employing AcOD-d4 as the nucleophile to ensure solubility throughout the entire 24-hour time course.39 (b) HQ oxidation studies under catalytically relevant conditions. Conditions: [HQ] = 84 mM, [AcOH] = 1680 mM, [Pd(OAc)2] = 8.4 mM, [Bu4NBr] = 16.8 mM, [Et3N] = 315 mM, [tBuOOH] = 840 mM, [methyl 3,5-dinitrobenzoate] = 8.4 mM (internal standard). (c) Simplified mechanistic cycle showing trade-off between HQ oxidation and product formation.
The rate of HQ oxidation evident during catalysis is much slower than expected from the previously reported study of Pd(OAc)2-catalyzed oxidation of HQ by tBuOOH.[7] This behavior was traced to the inhibitory effect of catalytic additives (Bu4NBr, AcOH and/or TEA) on HQ oxidation. As shown in Figure 2b, HQ undergoes efficient Pd(OAc)2-catalyzed oxidation to BQ in the absence of these additives, as expected,[7] while each of the individual additives slow or inhibit the reaction. When all three additives are combined, slow HQ oxidation is observed together with Pd catalyst decomposition (Pd black formation). Pd decomposition under these conditions can arise from the reversible reaction of PdII with HQ to form Pd0 (see ref. [38] and dashed arrows in Figure 2c), followed by irreversible decomposition of Pd0 into Pd black. Kinetically competent oxidation of HQ is observed, however, if 1 equiv of BQ is included in the reaction mixture, owing to improved Pd0 stability. Complete conversion of HQ to BQ under these conditions is complete in 16 h, commensurate with the catalytic time course shown in Figure 2a (see section 7b of the Supporting Information for details).
The above observations rationalize the ineffectiveness of HQ as a source of cocatalyst (cf. Table 1, entries 9 and 10): because HQ oxidation is relatively slow, the Pd catalyst will decompose and lead to low product yields unless BQ is included at the start of the reaction. Nonetheless, the results in Figure 2b offer an alternative strategy for use of HQ as the source of cocatalyst. The process is initiated by combining the Pd(OAc)2 catalyst and tBuOOH oxidant with HQ in the absence of other additives. These conditions enable efficient oxidation of HQ to BQ. The other reaction components (NBu4Br, NEt3, BzOH and CHD) may then be added to this “pre-oxidation mixture” to achieve the desired 1,4-dibenzoyloxylation of CHD. Preliminary optimization of this concept on small scale set the stage for demonstration on larger scale (Figure 3). Minor changes identified as beneficial in this effort include use of a slightly higher Br:Pd ratio (2.25:1) to enhance the cis selectivity (deemed more important than rate or total yield of mixed isomers) and use of a higher proportion of acetone in the cosolvent mixture to ensure all components remain soluble throughout the reaction. Implementation of the optimized conditions led to a 75% in-process yield of cis-1, with 94% cis selectivity, based on HPLC analysis of product mixture. Work-up and crystallization of the reaction stream led to a significant upgrade in the diastereomeric purity (>99.9% cis, based on HPLC analysis) and delivered 137 g of the final product (67% yield).
Figure 3.
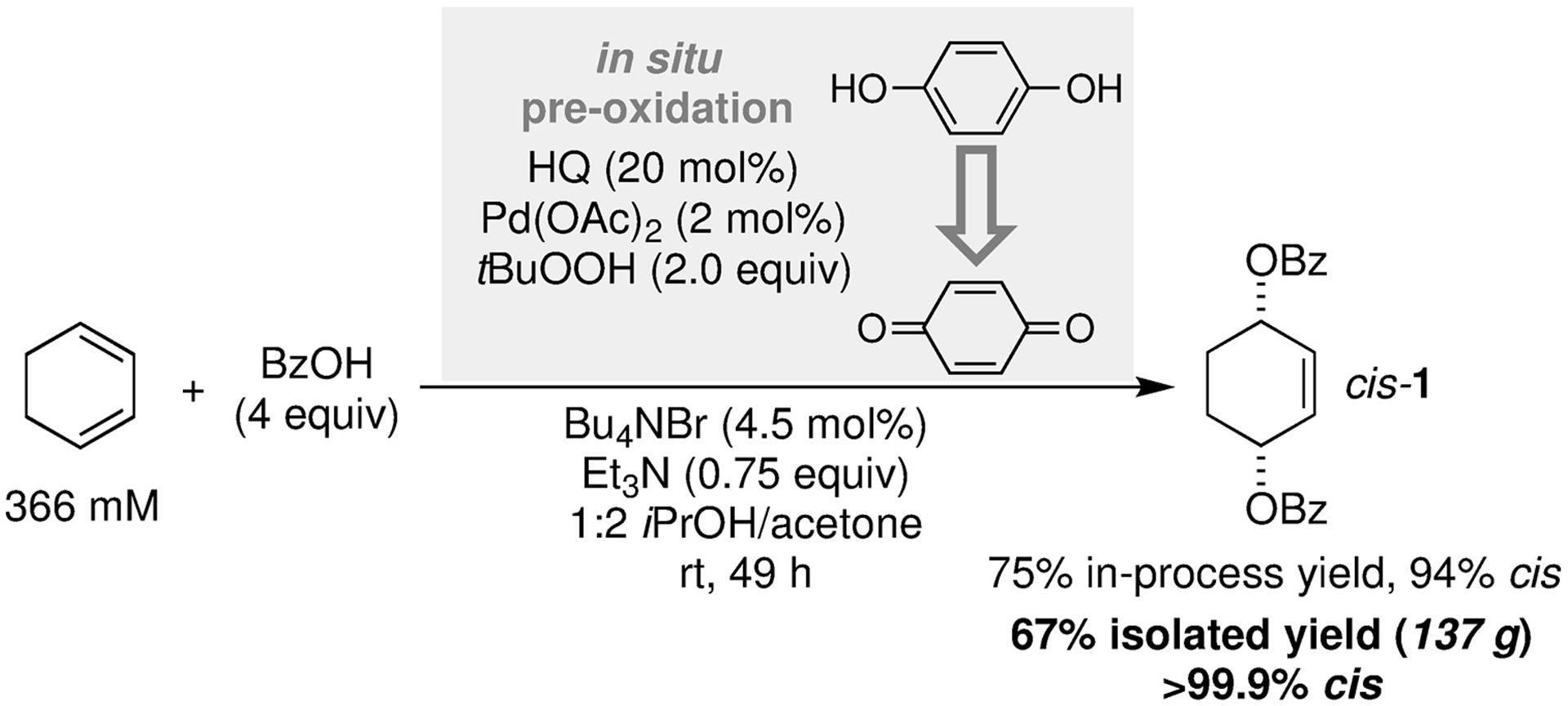
Pre-oxidation concept that enables HQ to be used as the cocatalyst (gray box), and demonstration of this sequential process to access 137 g of product, with >99.9% cis selectivity, following isolation.
In summary, the results described herein introduce a new catalyst system for cis-selective 1,4-diacyloxylation of 1,3-cyclohexadiene, highlighting the efficacy of bromide to alter the stereochemical course of the reaction. The PdII catalyst serves a dual role, catalyzing both 1,4-diacyloxylation of CHD and oxidation of HQ by tBuOOH. The ability of this catalyst to operate in the presence of halides contrasts other cocatalyst systems, such as Pd(OAc)2/BQ/Co(salophen)/O2, that are poisoned by halides. This feature is crucial to achieve the desired cis diastereoselectivity with a process-friendly and sustainable oxidant amenable to large-scale application. The insights from these studies have important implications for development of other industrially relevant Pd-catalyzed oxidation methods.
Supplementary Material
Acknowledgements
We would like to thank Dr. Antonio Ramirez and Dr. Michaël Fenster for useful discussions. Financial support was provided by Bristol Myers Squibb and by the NSF (CHE-1953926). Spectroscopic instrumentation was supported by the NSF (CHE-1048642), the NIH (R01GM100143 and R01GM127545) and a generous gift from Paul J. and Margaret M. Bender.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest:
The authors declare no conflict of interest.
References.
- [1].Wang D, Jaworski JN, Stahl SS, in Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives (Eds. Stahl SS, Alsters PL), Wiley-VCH Verlag GmbH & Co., Weinheim, 2016, pp. 113–138. [Google Scholar]
- [2].Jira R, in Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives (Eds. Stahl SS, Alsters PL), Wiley-VCH Verlag GmbH & Co., Weinheim, 2016, pp. 139–158. [Google Scholar]
- [3].Izawa Y, Yokoyama T, in Liquid Phase Aerobic Oxidation Catalysis: Industrial Applications and Academic Perspectives (Eds. Stahl SS, Alsters PL), Wiley-VCH Verlag GmbH & Co., Weinheim, 2016, pp. 159–170. [Google Scholar]
- [4].Brown RG, Davidson JM, J. Chem. Soc. A, 1971, 1321–1327. [Google Scholar]
- [5].Bäckvall J-E, Nordberg RE, J. Am. Chem. Soc 1981, 103, 4959–4960. [Google Scholar]
- [6].Bäckvall J-E, Bystrom SE, Nördberg RE, J. Org. Chem 1984, 49, 4619–4631. [Google Scholar]
- [7].Zheng B, Schmidt MA, Eastgate MD, J. Org. Chem 2016, 81, 3112–3118. [DOI] [PubMed] [Google Scholar]
- [8].Trost BM, Pulley SR, J. Am. Chem. Soc 1995, 117, 10143–10144. [Google Scholar]
- [9].Trost BM, Madsen R, Guile SD, Tet. Lett 1997, 38, 1707–1710. [Google Scholar]
- [10].Hua Z, Yu W, Su M, Jin Z, Org. Lett 2005, 7, 1939–1942. [DOI] [PubMed] [Google Scholar]
- [11].Trost BM, Cook GR, Tet. Lett 1996, 37, 7485–7488. [Google Scholar]
- [12].a) Aytar BS Borovika A, Chan C, Deerberg J, Domagalski NR, Eastgate MD, Fan Y, Fenster MDB, Forest RV, Gonzalez-Bobes F, Green RA, Hickey MR, Kopp ND, La Cruz TE, Lauser K, Lee HG, Leahy DK, Luo HY, Razler TM, Savage SA, Sfouggatakis C, Soumeillant MCD, Zaretsky S, Zheng B, Zhu Y (Bristol-Myers Squibb Company) PCT Int. Appl WO2019018592 A2, 2019; [Google Scholar]; b) La Cruz TE, González-Bobes F, Eastgate MD, Sfouggatakis C, Zheng B, Kopp N, Xiao Y, Fan Y, Galindo KA, Pathirana C, Galella MA, Deerberg Joerg, J. Org. Chem 2021, DOI: 10.1021/acs.joc.1c01162. [DOI] [PubMed] [Google Scholar]
- [13].Trost BM, Van Vranken DL, Chem. Rev 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]
- [14].Tsuji J, Takahashi H, Morikawa M, Tet. Lett 1965, 49, 4387–4388. [Google Scholar]
- [15].Trost BM, Li L, Guile SD, J. Am. Chem. Soc 1992, 114, 8745–8747. [Google Scholar]
- [16].Trost BM, Van Vranken DL, Bingel C, J. Am. Chem. Soc 1992, 114, 9327–9343. [Google Scholar]
- [17].Laumen K, Schneider MP, J. Chem. Soc., Chem. Commun, 1986, 1298–1299. [Google Scholar]
- [18].Kazlauskas RJ, Weissfloch ANE, Rappaport AT, Cuccia LA, J. Org. Chem 1991, 56, 2656–2665. [Google Scholar]
- [19].Bäckvall J-E, Gatti R, Schink HE, Synthesis, 1993, 343–348. [Google Scholar]
- [20].Bäckvall J-E, Granberg KL, Hopkins RB, Acta Chem. Scand 1990, 44, 492–499. [Google Scholar]
- [21].p-Benzoquinone, MSDS No. B10358: https://www.sigmaaldrich.com/catalog/AdvancedSearchPage.do (accessed 05/21/2021).
- [22].de Kruif CG, Smit EJ, Govers HAJ, J. Chem. Phys 1981, 74, 5838–5841. [Google Scholar]
- [23].Grennberg H, Gogoll A, Bäckvall J-E, Organometallics 1993, 12, 1790–1793. [Google Scholar]
- [24].Vasseur A, Muzart J, Le Bras J, Eur. J. Org. Chem 2019, 19, 4053–4069. [Google Scholar]
- [25].Bäckvall J-E, Hopkins RB, Grennberg H, Mader MM, Awasthi AK, J. Am. Chem. Soc 1990, 112, 5160–5166. [Google Scholar]
- [26].Bäckvall J-E, Awasthi AK, Renko ZD, J. Am. Chem. Soc 1987, 109, 4750–4752. [Google Scholar]
- [27].Bergstad K, Grennberg H, Bäckvall J-E, Organometallics 1998, 17, 45–50. [Google Scholar]
- [28].For reviews on the use of catalytic quinones in Pd-catalyzed oxidation reactions, see:; a) Piera J, Bäckvall J-E, Angew. Chem. Int. Ed 2008, 19, 3506–3523; [DOI] [PubMed] [Google Scholar]; b) Liu J, Guðmundsson A, Bäckvall J-E, Angew. Chem. Int. Ed 2020, 29, 15686–15704. [Google Scholar]
- [29].Previous work has noted the incompatibility of metal macrocycle cocatalysts with chloride additives:; Grennberg H, Bäckvall J-E, J. Chem. Soc., Chem. Commun 1993, 1331–1332. [Google Scholar]
- [30]. Preliminary results in our lab reproduce the inhibitory effect of chloride noted in ref. 29.
- [31]. MnO2 is problematic and complicated to use as a stoichiometric oxidant on large scale. It generates large quantities of Mn waste, it is not soluble in typical organic solvents, and the different particle sizes and crystal polymorphs of commercially sourced MnO2 complicate reproducibility.
- [32].Eastgate MD, Buono FG, Angew. Chem. Int. Ed 2009, 48, 5958–5961. [DOI] [PubMed] [Google Scholar]
- [33].Elding LI, Inorg. Chim. Acta, 1972, 6, 647–651. [Google Scholar]
- [34].Elding LI, Inorg. Chim. Acta, 1972, 6, 683–688. [Google Scholar]
- [35].Elding LI, Olsson L-F, Inorg. Chim. Acta, 1986, 117, 9–16. [Google Scholar]
- [36].Ahrland S, Acta Chem. Scand 1956, 10, 723–726. [Google Scholar]
- [37].Compared to BQ (cf. ref. 21), HQ has lower toxicity and does not sublime under ambient conditions.; Hydroquinone, MSDS No. H9003: https://www.sigmaaldrich.com/catalog/AdvancedSearchPage.do (accessed 05/21/2021).
- [38].Bruns DL, Musaev DG, Stahl SS, J. Am. Chem. Soc 2020, 46, 19678–19688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39]. For a direct comparison of screening data with AcOH and BzOH, see Table S2 in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.