

Abstract
Cancer is responsible for the deaths of millions of people worldwide each year. Once metastasized, the disease is incurable and shows resistance to all anti-cancer therapies. The already-elevated level of reactive oxygen species (ROS) in cancer cells is further increased by therapies. The oxidative stress activates the DNA damage response (DDR) and the stressed cancer cell moves towards cell cycle arrest. Once arrested, the majority of cancer cells will undergo programmed cell death in the form of apoptosis. If the cancer cell is able to exit the cell cycle prior to cell division and enter a protected G0 state, it is able to withstand and survive therapy as a polyaneuploid cancer cell (PACC) and eventually seed resistant tumor growth.
Keywords: Polyploid giant cancer cell (PGCC), polyaneuploid cancer cell (PACC), reactive oxygen species, cell cycle, therapy resistance
Introduction
Globally, 9.5 million people died as a result of cancer in 2018. [1] In 2020, it is estimated that 1.8 million people across the United States will be newly diagnosed with cancer and of all diagnosed patients, 600,000 will die as a result of the disease. [2] Metastatic disease is responsible for 90% of all cancer related deaths. [3] Cancers confined to the primary site are considered curable and can be removed through primary therapies such as surgery or radiation. Once cancer metastasizes and spreads to sites across the body it is considered incurable. [3]
Metastatic disease is treated with systemically-administered anti-cancer therapeutics, such as chemotherapy, with the goal of killing the rapidly dividing cancer cells. Eventually, however, cancer becomes resistant to all types of anti-cancer therapies. While reducing total tumor burden, therapeutic exposure may also induce the emergence of a small subset of multi-therapy-resistant cancer cells that survive and seed recurrence following therapeutic intervention. [4, 5] These cells show resistance not only to the applied therapy, but also other anti-cancer drugs and therapies applied following initial exposure. [4, 6] These resistant cells are present in small numbers in the primary tumor, having already evolved to the same lethal phenotype: resistance to external stress. It is hypothesized that they play a key role in the metastasizing potential of cancer and have been shown to activate as a response to stress, including chemotherapeutic insult. [3, 4]
The emergence – and survival – of these rare resistance-mediating cells may be attributed to a relationship between the cancer cell’s stress response pathways and reactive oxygen species (ROS). When applied, anti-cancer therapies affect the internal homeostasis of the cell by damaging nuclear content and generating ROS. [7, 8] The generation of ROS across the cell shifts the redox balance towards a state of oxidative stress. [7, 9] Genomic instability is a Hallmark of cancer, and, therefore, various pathways of the DNA damage response (DDR) are already activated even in untreated and non-stressed cancer cells. The DDR can be affected by a number of factors ranging from the cell’s cell cycle status to the type of DNA strand breakage. [10, 11] The DDR is not a single pathway, but instead is a suite of biochemical pathways and responses that sense DNA damage and determine the fate of the cell. Fates include repair during various stages of proliferation, slow cell cycling, and cell cycle arrest to allow for more extensive DNA repairs, or apoptosis in the case of damage extending past the point of repair. [10, 12] Both therapeutic DNA damage and high ROS inflict their own damage on the nuclear contents of the cell, while regulating the DDR, apoptosis, and the cell’s entry into cell cycle arrest. [7, 13]
Reactive oxygen species as a moderator of cancer cell death
ROS are primarily produced as natural byproducts of cellular respiration. The generation and removal of ROS determines the cellular redox balance, contributing to the overall homeostasis of the cell. [9, 14] In general, all cells rely on the transcription of antioxidants, such as glutathione, to reduce ROS when levels increase past signaling thresholds. If ROS generation outpaces the reduction capabilities of the cell’s antioxidants, the cell will experience oxidative stress and the mitochondria and nucleus will be damaged. [7, 14] ROS molecules such as superoxide (O−2), hydroxyl radicals (OH−), and hydrogen peroxide (H2O2,) are present in the cytosol and organelles of all cells. [14] Tightly-regulated levels of ROS play a critical role in both normal and cancer cells as initiators and regulators of various cellular signaling pathways. [9, 14, 15] In cancer cells, it is well documented that both ROS and antioxidant levels are increased compared to normal cell populations (Figure 1A). [16, 17] The elevated, yet balanced, ROS levels can activate signaling pathways that contribute to cancer’s ability to metastasize. [7, 15, 18–20] For example, ROS promotes metastasis by aiding in EMT transformation pathways via TGF-β1. [18]
Figure 1.
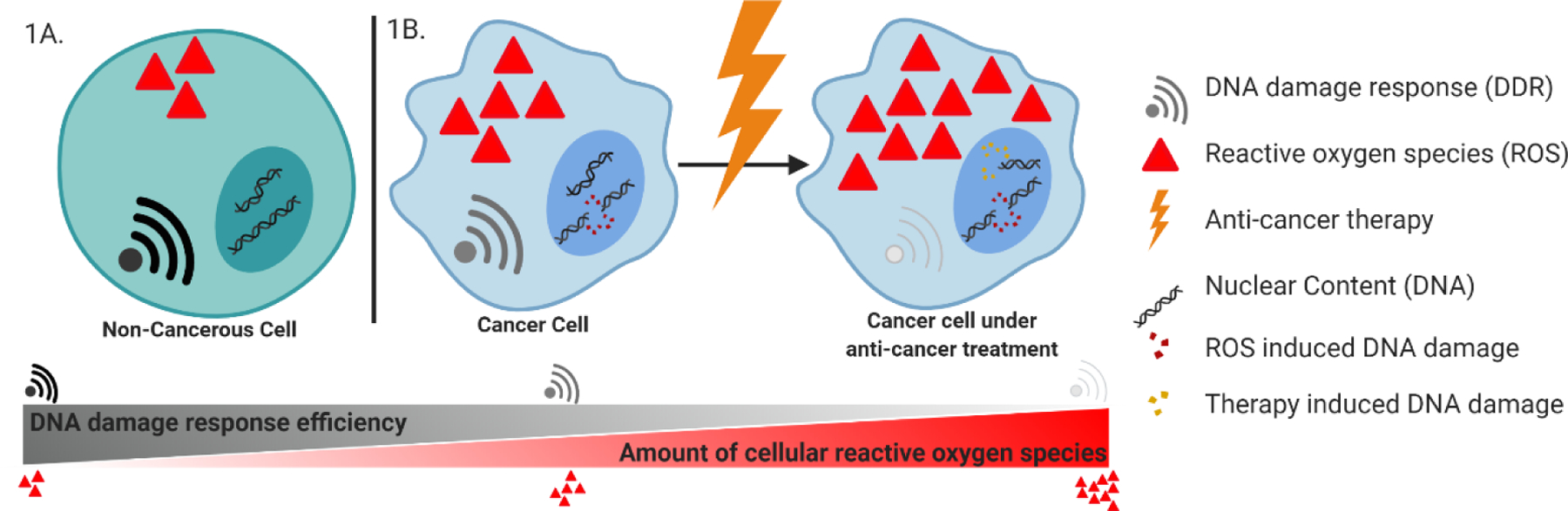
A) In non-cancerous cells, reactive oxygen species (ROS) exist at moderate to low levels to aid in the regulation of various cell signaling pathways. The DNA damage response (DDR) is capable of repairing oxidative stress-induced DNA damage and the integrity of the genome is maintained. B) Cancer cells have an increased amount of ROS, shifting the redox balance of the cell. The increased oxidative stress inflicts damage on the DNA of the cancer cell, contributing to cancer’s genomic instability. The DDR pathways, while activated from the ROS-induced DNA damage, are also hindered by the increased oxidative stress levels across the cancer cell. When anti-cancer therapy is applied to the cancer cell, the levels of ROS increase as a stress response. Both the ROS-induced and therapy-induced DNA damage activate the DDR; however, the DDR pathways are still hindered by oxidative stress and are not capable of repairing DNA at the rate it is being damaged.
Anti-cancer therapy increases internal ROS levels by introducing exogenous agents to the cell, as well as, by causing extensive DNA damage thus causing a response of ROS generation. [7, 21] This causes the redox balance to shift in favor of oxidative stress, subsequently damaging organelles and altering signaling pathways (Figure 1B). One damaging effect occurs in the nucleus, where DNA damage continues to accumulate from both therapeutic effects, as well as, the ROS-mediated oxidation of bases (Figure 1B). [7, 8] The increase in oxidative stress also hinders the corrective capabilities of the cell’s DDR pathways, inhibiting some, if not all, repair pathways (Figure 1B). [13, 22] The DDR, unable to repair the DNA breaks, will signal the cell to move into cell cycle arrest to prevent the replication of damaged DNA. There are several possible outcomes following cell cycle arrest. If unable to repair its severely damaged DNA, the DDR can induce apoptosis, a form of programmed cell death. [23] In a distinct mechanism, the elevated ROS levels across some cancer cells can also trigger programmed cell death through ROS-induced apoptosis. [13, 24] As a result of oxidative stress, pores along the mitochondrial membrane can be oxidized or the mitochondrial membrane may depolarize, allowing cytochrome c to be released into the cytoplasm, initiating the apoptotic cascade. [10, 13, 24, 25] Cancer cells can avoid apoptotic cell death by promoting the expression of anti-apoptotic proteins such as Bcl-2. It has been noted that an increase in Bcl-2 may also play a role in promoting antioxidant recruitment and generation during oxidative stress to manage ROS and mitochondrial membrane depolarization, thus evading apoptosis. [26] It has also been reported that cancer cells are capable of re-working and optimizing pathways responsible for the re-generation of glutathione and other antioxidants. [16] In doing so, the antioxidants are capable of reducing the oxidized membrane of the mitochondria and can help prevent the escape of cytochrome c. [27] The mitochondrial membrane stabilization by antioxidants as a response to prolonged exposure of ROS can prevent the cancer cell from succumbing to therapeutic treatments [24]; however, the hindered DDR still prevents the cancer cell from returning to the cell cycle. [10] The cancer cell remains in a steady state of cell cycle arrest.
Protective cell-cycle arrest
Exit from the cell cycle is a well-documented phenomenon in many cell types and organisms ranging from plants and yeast to animals and humans (Figure 2A). [28] Various stimuli will induce a cell to exit the cell cycle and enter a “dormant” G0 state, including overcrowding and contact inhibition [29], absence of growth factors or nutrients [30], and DNA or cellular damage. [31] Though cell cycle exit is a survival strategy observed in many cancer types [32], often as a result of DNA damage from systemic anti-cancer therapies such as chemotherapies. [33]
Figure 2.
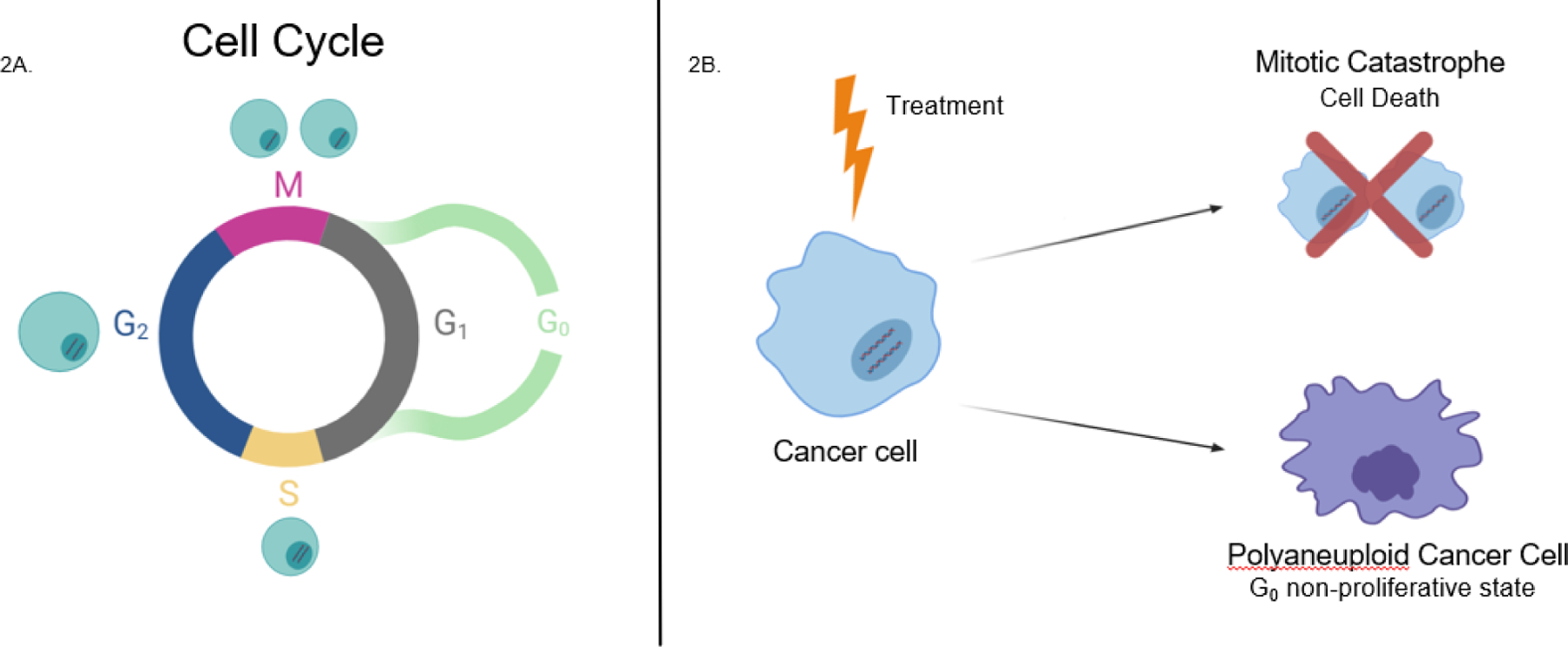
A) In a normal cell cycle, the cell will go through various stages of replication and growth in order to divide. In G1 (Growth 1), the cell will synthesize proteins and factors needed throughout the rest of the cycle, the cell will also begin to grow slightly in size. In S phase all nuclear DNA will be replicated, giving the cell two sets of genomic DNA. In G2 (Growth 2) the cell will continue to grow in size and produce new proteins that it will need in the division process. Ending in Mitosis, the cell will finally begin the process of division, splitting apart its DNA and organelles to give rise to two identical daughter cells, who continue the cycle back into G1. There is also a G0 phase outside of the traditional cycle, used as a protective non-proliferative state that the cell can use to exit the cycle to repair DNA damage, when necessary. B) A cancer treated with therapy has two possible fates: 1) attempt to undergo cell divide and apoptose due to mitotic catastrophe, or 2) escape the cell cycle and not complete mitosis or cytokinesis and enter the protective G0 state as a PACC.
The goal of systemic therapy is to kill cancer cells, and in particular, the rapidly proliferating cells of a growing tumor. This is achieved by inducing an overwhelming amount of DNA damage (e.g., DNA poison cisplatin and DNA polymerase II inhibitor Etoposide) or by inhibiting some aspect of normal cell cycle (e.g., microtubule stabilizer docetaxel) to force the cell into apoptosis upon cell division. [31, 34] In cancer, this DNA damage is compounded by an increase of damaging ROS. The addition of imbalanced ROS and cancer therapy-induced DNA damage overwhelms the DDR pathways within the cell [13] leading to either cell death via mitotic catastrophe or a complete exit from the cell cycle. [35] When such excessive damage occurs, the cell must exit the cycle to repair the damage, tolerate it, or succumb to it via apoptosis. [36]
To enter a protected non-proliferative state the cell must complete or abort its current cycle without triggering the cell cycle checkpoints that induce apoptosis. [31] This will lead to the cell “slipping” out of mitosis to avoid potential mitotic catastrophe(known as mitotic slippage, early or late endomitosis, or acytokinesis) [37], or bypassing mitosis altogether through endocycling [38], likely dependent on where in the cycle the DNA damage occurred. If a cell survives treatment-induced apoptosis by not completing cytokinesis, following a singular or multiple attempts at cycle completion, it will exit the cell cycle into G0 with double the standard complement of DNA: a polyaneuploid cancer cell.
Though all treatments work in distinct ways, modern systemic therapies by design only target actively proliferating cells, or cells actively engaged in the cell cycle. [39] Therefore, an exit from the process of proliferation could be a viable survival tool for cancer cells. [40] A cell within a non-proliferative state, and thus not cycling through mitosis, would not be affected by drugs designed to kill quickly dividing cells. And though the mechanism of quiescent cancer cells avoiding immune surveillance is still unclear, it is believed to be a mechanism similar to that of non-cycling stem cells that change their antigen presentation in the dormant state. [41] Hence, an exit from the proliferative state into a dormant G0 state makes these cells immunologically invisible and therapeutically untouchable. [42] These G0 cells will continue to survive in a non-proliferative state, unaffected by both cancer treatments and the immune system of the host, until a more favorable environment for proliferation returns. [43]
Polyaneuploid cancer cells (PACCs) arise from ROS induced cell-cycle arrest
Aneuploidy has been well-described in all cancer types, and is widely accepted as a distinguishing – and perhaps defining – characteristic of cancer.[44, 45] Therefore, cancer cells that exit cell cycle at any point after S phase, including following aborted mitosis, are polyaneuploid cancer cells (PACCs). These cell-cycle arrested PACCs, also referred to as polyploid giant cancer cells (PGCCs) [46], that arise following initial treatment will likely survive any subsequent rounds of therapy. It is also theorized that in this state the cell could mutate further, leading to further advantage and capability for survival. [47] When these cells eventually re-enter the cell cycle, they will have the capacity to repopulate the tumor and lead to a cancer relapse. [48]
The presence of ROS and its damaging effects on DNA in cancer cells triggers the protective response of the DDR permitting the newly-formed polyaneuploid cells to escape into a protected G0 state and avoid any further DNA damage (Figure 3). [13, 49] Shielded from therapy, these PACCs have the ability to not only survive, but re-emerge as therapeutically resistant parent cells capable of re-populating the tumor with resistant prodigy.[4]
Figure 3.
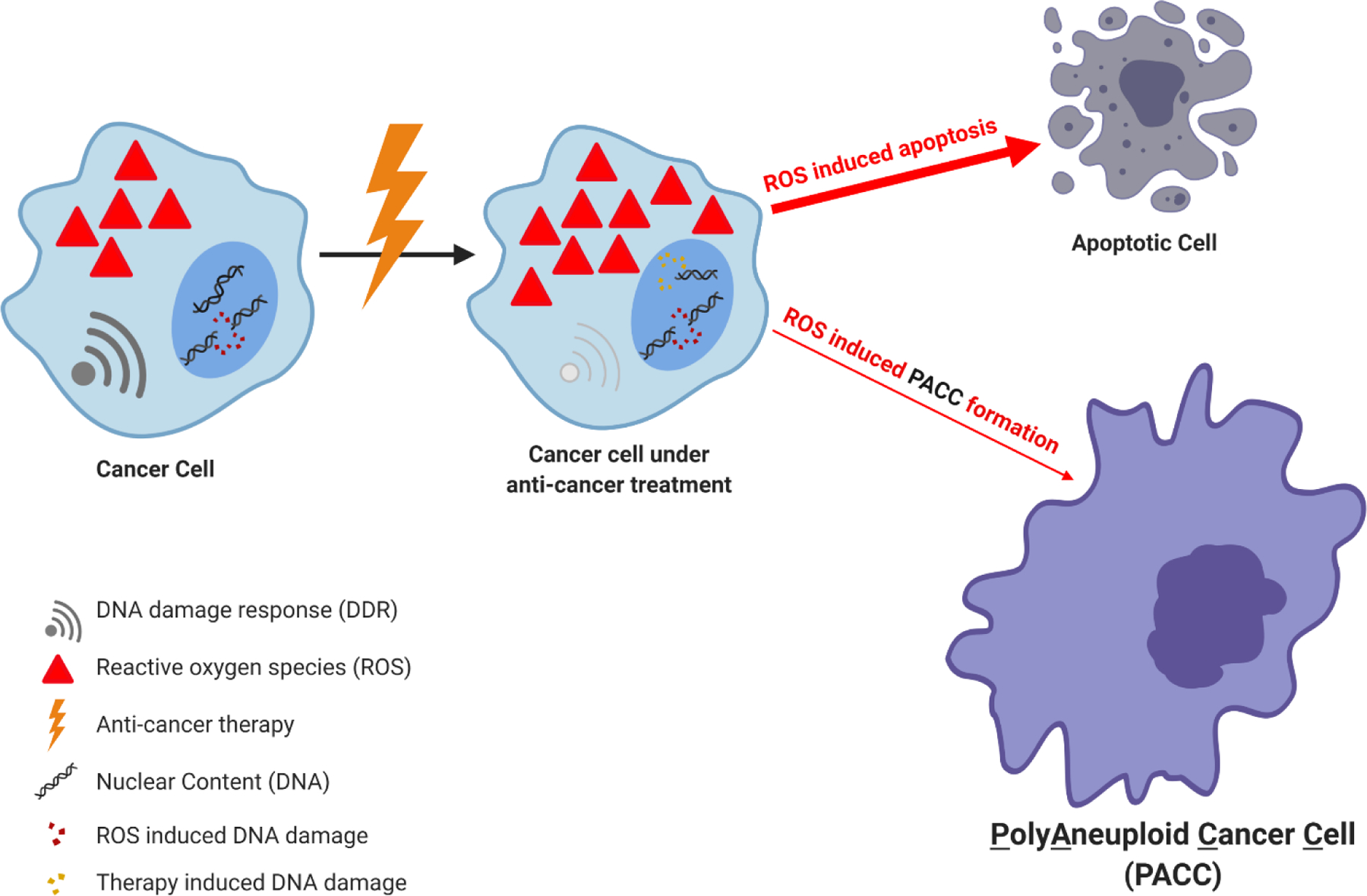
In response to anti-cancer therapy and the subsequent surge in ROS, the cancer cell is limited in its survival options. The majority of cells will be overwhelmed by the increase in nuclear damage and oxidative stress, inducing cellular death. A smaller subset of cancer cells will prematurely exit mitosis, escape the cell cycle, and enter a protective, G0 state. It is in this state that the cancer cells are protected from anti-cancer therapy and can form into polyaneuploid cancer cells (PACCs). Once a break in therapy occurs, these PACCs can re-emerge equipped with non-selective resistance to all anti-cancer therapies.
PACC formation can be induced in vitro by the introduction of various stressors, such as chemotherapy (Figure 4). [3, 4] Anti-cancer therapy, coupled with the relentless oxidative stress stemming from therapeutically-dependent ROS, could induce and enrich PACC populations as they are the only cells capable of surviving the damaging effects. Furthermore, the increased ROS levels as both inherent of the cancer state and as result of therapy will benefit the resistant cancer cell’s proliferation by upregulating cell signaling pathways, if able to re-enter the cell cycle. [7, 15, 18–20] If PACCs are responsible for the introduction of phenotypic resistance seen in lethal cancers, applying anti-cancer therapies to metastatic cancer may induce a fully resistant phenotype. [3, 4]
Figure 4.
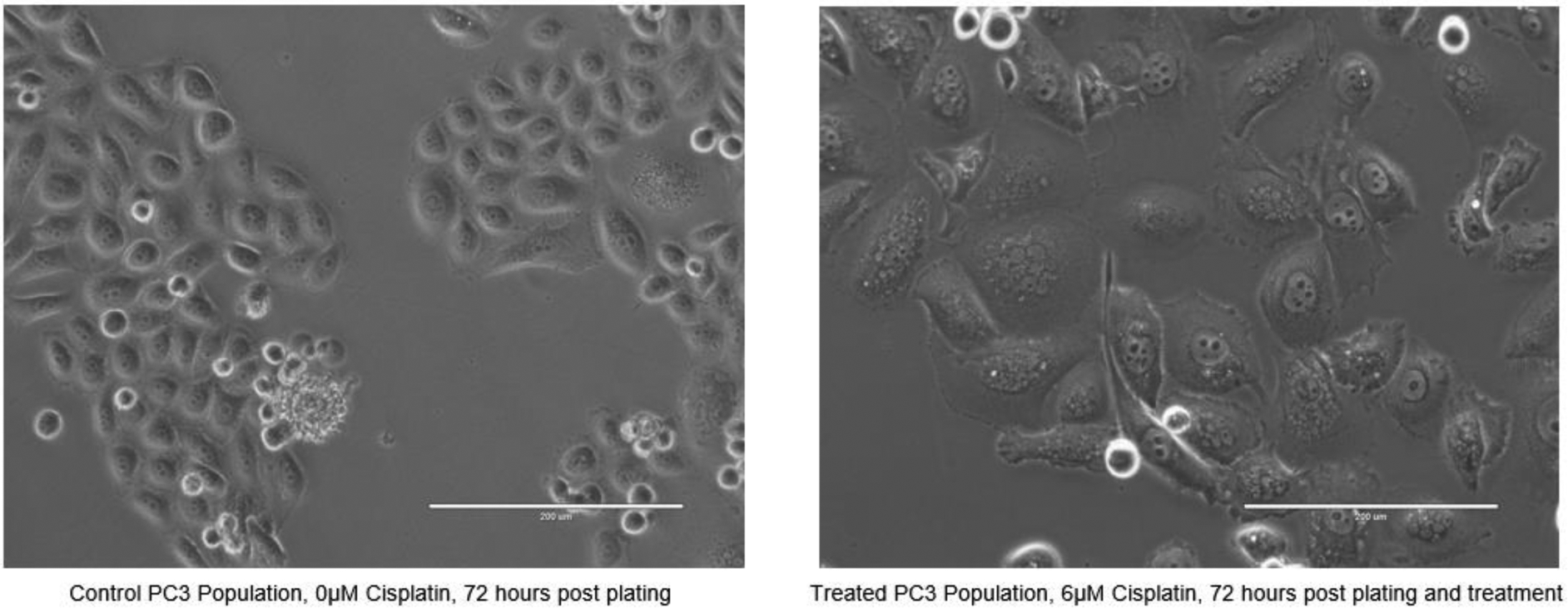
Phase contrast imaging of an untreated control PC3 population and a PC3 population treated with 6μM cisplatin 72 hours following plating and treatment. (scale = 200μm) PACCs are visibly apparent after treatment.
Acknowledgements:
This work was funded by US Department of Defense CDMRP/PCRP (W81XWH-20-10353), the Patrick C. Walsh Prostate Cancer Research Fund and the Prostate Cancer Foundation to S.R. Amend; and NCI grants U54CA143803, CA163124, CA093900, and CA143055, and the Prostate Cancer Foundation to K.J. Pienta. This work was also supported by the William and Carolyn Stutt Research Fund, Ronald Rose, MC Dean, Inc., William and Marjorie Springer, Mary and Dave Stevens, Louis Dorfman, the Jones Family Foundation, Timothy Hanson, and the David and June Trone Family Foundation. Figures 1, 2, and 3 were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: K.J. Pienta is a consultant for CUE Biopharma, Inc., is a founder and holds equity interest in Keystone Biopharma, Inc., and receives research support from Progenics, Inc. The other authors declare no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Ferlay JA-O, et al. , Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. (1097–0215 (Electronic)). [DOI] [PubMed]
- 2.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2020. CA: A Cancer Journal for Clinicians, 2020. 70(1): p. 7–30. [DOI] [PubMed] [Google Scholar]
- 3.Pienta KA-O, et al. , Convergent Evolution, Evolving Evolvability, and the Origins of Lethal Cancer. (1557–3125 (Electronic)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pienta KJ, et al. , Poly-aneuploid cancer cells promote evolvability, generating lethal cancer. Evolutionary Applications, 2020. 13(7): p. 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Norouzi S, et al. , Crosstalk in cancer resistance and metastasis. Critical Reviews in Oncology/Hematology, 2018. 132: p. 145–153. [DOI] [PubMed] [Google Scholar]
- 6.Eccles SA and Welch DR, Metastasis: recent discoveries and novel treatment strategies. Lancet (London, England), 2007. 369(9574): p. 1742–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perillo B, et al. , ROS in cancer therapy: the bright side of the moon. Experimental & Molecular Medicine, 2020. 52(2): p. 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lord CJ and Ashworth A, The DNA damage response and cancer therapy. Nature, 2012. 481(7381): p. 287–294. [DOI] [PubMed] [Google Scholar]
- 9.Trachootham D, et al. , Redox regulation of cell survival. Antioxidants & redox signaling, 2008. 10(8): p. 1343–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davalli P, et al. , Targeting Oxidatively Induced DNA Damage Response in Cancer: Opportunities for Novel Cancer Therapies. Oxidative medicine and cellular longevity, 2018. 2018: p. 2389523–2389523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartek J, DNA damage response, genetic instability and cancer: from mechanistic insights to personalized treatment. Molecular oncology, 2011. 5(4): p. 303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min M and Spencer SL, Spontaneously slow-cycling subpopulations of human cells originate from activation of stress-response pathways. PLOS Biology, 2019. 17(3): p. e3000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srinivas UST, Bryce WQ; Vellayappan Balamurugan A.; Jeyasekharan Anand D., ROS and the DNA damage response in cancer. Redox biology, 2019. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ray PD, Huang B-W, and Tsuji Y, Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cellular signalling, 2012. 24(5): p. 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou X, et al. , Signaling in H2O2-induced increase in cell proliferation in Barrett’s esophageal adenocarcinoma cells. (1521–0103 (Electronic)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noh J, et al. , Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nature Communications, 2015. 6(1): p. 6907. [DOI] [PubMed] [Google Scholar]
- 17.Kumari S, et al. , Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomarker insights, 2018. 13: p. 1177271918755391–1177271918755391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao Z, Chua D, and Tan NS, Reactive oxygen species: a volatile driver of field cancerization and metastasis. Molecular Cancer, 2019. 18(1): p. 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szatrowski TP and Nathan CF, Production of large amounts of hydrogen peroxide by human tumor cells. (0008–5472 (Print)). [PubMed] [Google Scholar]
- 20.Schieber M and Chandel NS, ROS function in redox signaling and oxidative stress. Current biology : CB, 2014. 24(10): p. R453–R462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rowe LA, Degtyareva N, and Doetsch PW, DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free radical biology & medicine, 2008. 45(8): p. 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curtin NJ, DNA repair dysregulation from cancer driver to therapeutic target. Nature Reviews Cancer, 2012. 12(12): p. 801–817. [DOI] [PubMed] [Google Scholar]
- 23.Fuchs Y and Steller H, Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nature reviews. Molecular cell biology, 2015. 16(6): p. 329–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liou G-Y and Storz P, Reactive oxygen species in cancer. Free radical research, 2010. 44(5): p. 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simon HU, Haj-Yehia A, and Levi-Schaffer F, Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis, 2000. 5(5): p. 415–418. [DOI] [PubMed] [Google Scholar]
- 26.Chong SJF, Low ICC, and Pervaiz S, Mitochondrial ROS and involvement of Bcl-2 as a mitochondrial ROS regulator. Mitochondrion, 2014. 19: p. 39–48. [DOI] [PubMed] [Google Scholar]
- 27.Assi M, The differential role of reactive oxygen species in early and late stages of cancer. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 2017. 313(6): p. R646–R653. [DOI] [PubMed] [Google Scholar]
- 28.Valcourt JR, et al. , Staying alive. Cell Cycle, 2012. 11(9): p. 1680–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun P, Contact inhibition against senescence. Oncotarget, 2014. 5(17): p. 7212–7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fau Zetterberg A - Larsson O. and Larsson O, Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. (0027–8424 (Print)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barr AR, et al. , DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. (2041–1723 (Electronic)). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang B-D, et al. , A Senescence-like Phenotype Distinguishes Tumor Cells That Undergo Terminal Proliferation Arrest after Exposure to Anticancer Agents. Cancer Research, 1999. 59(15): p. 3761. [PubMed] [Google Scholar]
- 33.Roninson IB, Broude BD Fau Chang Ev, and Chang BD, If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. (1368–7646 (Print)). [DOI] [PubMed] [Google Scholar]
- 34.Lowe SW and Lin AW, Apoptosis in cancer. Carcinogenesis, 2000. 21(3): p. 485–495. [DOI] [PubMed] [Google Scholar]
- 35.Chen J, The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harbor perspectives in medicine, 2016. 6(3): p. a026104–a026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shen Z, Huhn SC, and Haffty BG, Escaping death to quiescence: avoiding mitotic catastrophe after DNA damage. Cell cycle (Georgetown, Tex.), 2013. 12(11): p. 1664–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brito DA and Rieder CL, Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Current biology : CB, 2006. 16(12): p. 1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erenpreisa J, Kalejs MS Fau Cragg M, and Cragg MS, Mitotic catastrophe and endomitosis in tumour cells: an evolutionary key to a molecular solution. (1065–6995 (Print)). [DOI] [PubMed] [Google Scholar]
- 39.Legesse-Miller A, et al. , Quiescent fibroblasts are protected from proteasome inhibition-mediated toxicity. Molecular biology of the cell, 2012. 23(18): p. 3566–3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blagosklonny MV, Cell cycle arrest is not senescence. Aging, 2011. 3(2): p. 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agudo J, et al. , Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity, 2018. 48(2): p. 271–285.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leontieva OV and Blagosklonny MV, DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging, 2010. 2(12): p. 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen W, et al. , Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells International, 2016. 2016: p. 1740936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor AM, et al. , Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer cell, 2018. 33(4): p. 676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanahan D and Robert A Weinberg, Hallmarks of Cancer: The Next Generation. Cell, 2011. 144(5): p. 646–674. [DOI] [PubMed] [Google Scholar]
- 46.Amend SR, et al. , Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. The Prostate, 2019. 79(13): p. 1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma SV, et al. , A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell, 2010. 141(1): p. 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stewart DJ, et al. , Chemotherapy dose–response relationships in non-small cell lung cancer and implied resistance mechanisms. Cancer Treatment Reviews, 2007. 33(2): p. 101–137. [DOI] [PubMed] [Google Scholar]
- 49.Ye C, et al. , Radiation-induced cellular senescence results from a slippage of long-term G2 arrested cells into G1 phase. Cell cycle (Georgetown, Tex.), 2013. 12(9): p. 1424–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]