

Abstract
Introduction:
Hepatocellular carcinoma (HCC) is a malignant liver tumor characterized by high molecular heterogeneity, which has hampered the development of effective targeted therapies severely. Recent experimental data have unraveled novel promising targets for HCC treatment.
Areas covered:
Eligible articles were retrieved from PubMed and Web of Science databases up to July 2021. This review summarizes the established targeted therapies for advanced HCC, focusing on the strategies to overcome drug resistance and the search for combinational treatments. In addition, conventional biomarkers holding the promises for HCC treatments and novel therapeutic targets from the research field are discussed.
Expert opinion:
HCC is a molecularly complex disease, with several and distinct pathways playing critical roles in different tumor subtypes. Experimental models recapitulating the features of each tumor subset would be highly beneficial to design novel and more effective therapies against this disease. Furthermore, a deeper understanding of combinatorial drug synergism and the role of the tumor microenvironment in HCC will lead to improved therapeutic outcomes.
Keywords: Hepatocellular carcinoma, biomarkers, DNA repair, metabolic pathways, mouse models
1. Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver cancer (PLC), accounting for ~85-90% of cases and the fourth most frequent cause of cancer-related deaths worldwide [1]. The common risk factors include hepatitis B virus (HBV) and hepatitis C virus (HCV) chronic infections, diabetes, obesity, alcoholic fatty liver disease, and nonalcoholic fatty liver disease (NAFLD). Additionally, aflatoxins, aristolochic acid, and tobacco are reported as potential pathogenetic cofactors for HCC development [2].
The molecular pathogenesis of HCC differs depending on the genetic aberrations and etiologies. Mutations in HCC tumors are only ~25% actionable; however, the lack of dominant “driver” mutations and the presence of thousands of confounding “passenger” mutations complicate the therapeutic explorations. In addition, the most frequent mutations, such as TERT promoter (~44%), TP53 (~31%), and CTNNB1 (~27%), remain undruggable [3, 4]. Moreover, the immune landscape of HCC is mostly unknown. Recently, with the increasing attention to the metabolic dysfunctions associated HCC, such as NAFLD/nonalcoholic steatohepatitis (NASH)-associated HCC, the importance of understanding the tumor microenvironment has triggered a body of new investigations.
Usually, HCC is diagnosed at a late stage, and the curative approaches, such as surgical resection and liver transplantation, can only be applied to a small portion of patients. Systemic therapies, including receptor tyrosine kinase inhibitors (RTKIs), immune-checkpoint inhibitors (ICIs), and monoclonal antibodies, have become crucial approaches to prolong the survival length of patients with inoperable HCC. In the last decade, substantial efforts to develop effective targeted treatment have been made. Although many clinical trials failed to reach their endpoints, some of these molecularly targeted approaches have been effective in specific HCC subsets. Thus, understanding the biological variations between responders and non-responders will be helpful to identify HCC patients who could benefit from a given treatment. This review discusses the approved targeted drugs against HCC and the medications that have shown translational potential in liver cancer preclinical studies.
2. Study selection
Two reviewers (H.W. and D.F.C) performed searches in PubMed and Web of Science databases to retrieve studies presenting an association between targeted therapies and HCC. The last updated search was conducted in July 2021. We conducted literature searches in the PubMed database with the following keywords: targeted therapy, hepatocellular carcinoma, HCC, liver, cancer, preclinical, mouse models, biomarkers, metabolism, signaling pathways, etc. A similar approach was applied to the Web of Science database. Determination on the inclusion and exclusion of studies was decided based on this review’s objectives, with the consensus of all the authors.
3. Established target drugs for HCC
HCC onset and progression result from the dysregulation of mechanisms regulating various cellular aspects, such as proliferation, apoptosis, motility, angiogenesis, and metabolism. The drugs used currently for HCC treatment interfere with these processes at multiple levels. In addition, with the increasing knowledge of the HCC tumor immune environment, immunotherapy either alone or in combination with targeted treatments has been developed. Overall, the current pharmacological treatments for HCC patients are classified as first-line and second-line therapies. Sorafenib, lenvatinib, and the combinational appliance of bevacizumab and atezolizumab are the first-line systemic treatments for advanced HCC. In contrast, regorafenib, cabozantinib, and ramucirumab represent the second-line systemic therapies for this disease. Furthermore, the immune checkpoint inhibitors nivolumab, pembrolizumab, and the simultaneous administration of nivolumab and ipilimumab are the drugs of choice for HCC patients who progressed on sorafenib. The pharmacological features of these drugs are summarized in Table 1, and the current algorithm for advanced HCC treatment is depicted in Figure 1.
Table 1.
Summary of approved drugs for HCC systemic treatment
| Drugs | Targets | Clinical trials number | Application | Reference |
|---|---|---|---|---|
| Sorafenib | VEGFR1-3, PDGFR, RAF kinase, KIT receptor | NCT00105443 | First line | [5] |
| Lenvatinib | VEGFR1-3, PDGFR, FGFR1-4, RET | NCT01761266 | First line | [6] |
| Atezolizumab + Bevacizumab | PDL1 + VEGF | NCT03434379 | First line | [8] |
| Regorafenib | VEGFR1-3, PDGFR, RAF kinase, FGFR1-2 | NCT01774344 | Second line | [13] |
| Cabozantinib | VEGFR1-3, MET, RET | NCT01908426 | Second line | [18] |
| Ramucirumab | VEGFR2 antibody | NCT01140347 | Second line | [10] |
| Nivolumab | PD-1 antibody | NCT01658878 | Second line | [25] |
| Nivolumab + Ipilimumab | PD-1 antibody; CTLA-4 antibody | NCT01658878 | Second line | [23] |
| Pembrolizumab | PD-1 antibody | NCT02702401 | Second line | [24, 105] |
Figure 1.
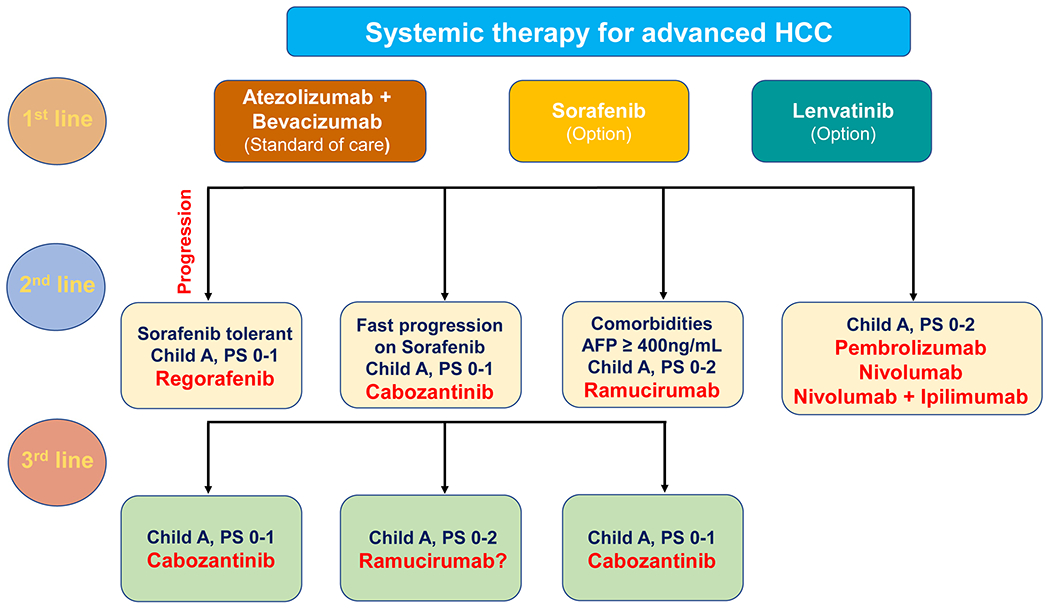
Algorithm for management of advanced HCC. Atezolizumab plus bevacizumab are offered as first-line treatment for most patients with advanced HCC, Child-Pugh class A, ECOG PS 0-1. Sorafenib and lenvatinib may be offered as optional first-line treatment of advanced HCC patients who present contraindications to atezolizumab and/or bevacizumab. Second-line therapy is an option for patients whose tumors progress while on first-line therapy and whose performance status and liver function are sufficient to tolerate it. Cabozantinib and ramucirumab are also recommended as a third-line therapy option for patients with advanced HCC. Abbreviations: PS, performance status; AFP, alpha-fetoprotein.
3.1. First-Line therapies
3.1.1. Sorafenib
Sorafenib is a multi-receptor tyrosine kinase (RTKs) inhibitor, targeting cellular processes such as angiogenesis, cell proliferation, and cell death. It is the first approved drug to treat advanced HCC patients who are not eligible for liver transplantation or surgical resection. Sorafenib targets a wide range of kinases, mainly comprising angiogenic RTKs (vascular endothelial growth factor receptors [VEGFRs] and platelet-derived growth factor receptor-β [PDGFRβ]) and cell proliferation drivers (Raf serine/threonine kinase 1 [RAF1], BRAF, and KIT). To date, no ideal biomarkers have been identified as therapeutic indicators or prognostic predictors for sorafenib treatment [5].
3.1.2. Lenvatinib
Lenvatinib was approved as a first-line treatment for unresectable HCC patients following the results from the REFLECT clinical trial (NCT01761266), where lenvatinib was non-inferior to sorafenib in terms of overall survival (OS) but showed significant improved secondary endpoints. Lenvatinib is a multi-kinase inhibitor, targeting the VEGFRs, fibroblast growth factor receptors 1-4 (FGFRs 1-4), RET, KIT, and PDGFRα75 [6].
3.1.3. Atezolizumab + bevacizumab
Atezolizumab, an immune checkpoint inhibitor, is a monoclonal antibody specifically targeting programmed death-ligand 1 (PD-L1) to prevent the interaction with PD-1 and B7-1 receptors. PD-L1 interaction with PD-1 and B7-1 results in immune response suppression, especially of the T-lymphocyte compartment. PD-L1 blockade by atezolizumab can remove this inhibitory effect and thereby stimulate an anti-tumor response [7]. Bevacizumab is a monoclonal antibody that targets VEGF and inhibits angiogenesis, thus enhancing the efficacy of PD-L1 treatment by reversing VEGF-mediated immunosuppression and promoting tumor infiltration by T-cells in the tumors. The IMbrave150 clinical trial (NCT03434379) has shown significantly better overall survival and progression-free survival outcomes with atezolizumab plus bevacizumab than with sorafenib in patients with unresectable HCC who had received no previous systemic treatment [8].
3.2. Second-Line Therapies
Many clinical trials burst out in pursuit of identifying potent drugs for HCC patients who progress after sorafenib treatment but failed to reach their primary endpoints. These clinical trials include agents targeting mammalian target of rapamycin (mTOR) (NCT01035229) [9], VEGF (NCT01140347) [10] and/or FGF (NCT00825955) [11], hepatocyte growth factor (HGF) and its receptor MET (NCT01755767) [12] signaling pathways. Since 2017, only three regimens (regorafenib, cabozantinib, and ramucirumab) have been approved to treat advanced HCC after progression on sorafenib according to the guidelines. In addition, three monoclonal antibodies targeting immune checkpoints, namely nivolumab, pembrolizumab, and ipilimumab (in combination with nivolumab), have been approved by the Food and Drug Administration (FDA) after first-line treatment with sorafenib.
3.2.1. Regorafenib
Regorafenib is an RTK inhibitor targeting angiogenic VEGFR2 and angiopoietin-1 receptor (also referred to as TIE2). Regorafenib has shown more effectiveness in inhibiting tyrosine kinases and phosphatases when compared to sorafenib for the treatment of advanced HCC (NCT01774344) [13]. Regorafenib has recently been demonstrated to inhibit the phosphorylation of signal transducer and activator of transcription 3 (STAT3) through SHP1 activation and induce cell apoptosis in HCC [14]. In addition, other preclinical studies indicate that regorafenib is involved in modulating the tumor immune environment by promoting M1 macrophage polarization [15] and T-cell infiltration [16].
3.2.2. Cabozantinib
Cabozantinib inhibits various RTKs, including VEGFRs, MET, and AXL, implicated in the progression of hepatocellular carcinoma and the development of resistance to sorafenib [17]. Cabozantinib treatment resulted in more prolonged overall survival and progression-free survival than placebo in previously treated patients with advanced HCC [18].
3.2.3. Ramucirumab
Ramucirumab is an anti-VEGFR-2 monoclonal antibody that impedes the binding of the various VEGFR ligands (VEGF-A, VEGF-C, and VEGF-D), thus blocking VEGF-driven tumor angiogenesis [19]. The anti-neoplastic activity of ramucirumab has been shown in phase II and III trials (REACH-2) in patients with advanced HCC and high alpha-fetoprotein (AFP) levels (NCT01140347) [10]. This clinical trial revealed an improved overall survival in patients treated with ramucirumab compared with placebo. Importantly, ramucirumab was well tolerated and displayed a controllable safety profile.
3.2.4. Immune checkpoint inhibitors: nivolumab, nivolumab and ipilimumab, pembrolizumab
Nivolumab and pembrolizumab are human monoclonal antibodies that bind to the PD-1 receptor and block its interaction with PD-L1 and PD-L2, resulting in anti-tumor immune response re-induction and decreased tumor growth [20]. Ipilimumab is a monoclonal antibody targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), promoting anti-tumor immune responses via distinct and complementary mechanisms compared to those induced by the anti-PD1 antibody [21]. Based upon promising phase Ib/II studies [22–25], the three drugs have been approved by the FDA for patients with advanced HCC after first-line treatment with sorafenib [26].
4. Novel insights into the approved HCC drugs from the bench
With the emerging actionable drugs for advanced HCC, their underlying mechanisms remain to be delineated more precisely. In addition, the overall response rate of the HCC systemic agents is still low (~20-30%). Therefore, studies aiming to explain drug non-responsiveness or resistance and explore options for combinational treatment to improve therapeutic efficacy are highly significant. Recent genomic studies have established the landscape of molecular alterations in HCC. In particular, integrative analysis of the genomic, transcriptomic, and/or epigenomic profiles revealed that HCC is a heterogeneous disease composed of several subclasses [27, 28]. Specifically, HCC can be subdivided into two major subclasses: the proliferative and the non-proliferative subtype [29]. The proliferative HCC subset is associated with a poor prognosis, chromosomal instability, and activation of classic oncogenic signaling pathways (such as RAS- mitogen-activated protein kinase [MAPK] and protein kinase B [AKT]-mTOR). In contrast, the non-proliferative HCC subclass displays a less aggressive course with slower disease progression and frequent activation of the Wnt/β-catenin pathway [29]. However, reliable biomarkers able to stratify these tumors for tailored, targeted therapies are missing.
4.1. Prognostic biomarkers for first- and second-line systemic therapies
A wealth of biomarkers associated with response or resistance to targeted therapies have been described. However, none of them has reached the clinical stage to date. For instance, Wang et al. demonstrated that peptidase inhibitor 16 (PI16) is overexpressed in HCC tissues, and its inhibition increased the sensitivity of HCC xenografts to sorafenib by activating p38 MAPK dependent apoptosis. These findings suggest that PI16 might be a prognostic biomarker of sorafenib treatment [30]. Song et al. showed that the β-galactoside-binding protein family protein, Galectin-3, induced angiogenesis and epithelial-mesenchymal transition through phosphoinositide 3-kinase (PI3K)/AKT-glycogen synthase kinase 3β (GSK3β)/β-Catenin signaling cascade by targeting insulin-like growth factor (IGF) binding protein 3 and VIMENTIN in the HCC tumor microenvironment. Galectin-3 and β-Catenin knockdown had an additive effect on the sensitivity of HCC cells to sorafenib in a cell line xenograft model [31]. Recently, Myojin et al. established a barcoded-oncogene cDNA library and delivered it into the mouse liver to induce HCC, recapitulating the tumor diversity of genetic drivers. A genetic screen of this model revealed that HCCs expressing FGF19 were susceptible to lenvatinib treatment. Moreover, serum levels of glycosyltransferases ST6GAL were found to be positively regulated by tumor FGF19 expression, envisaging ST6GAL as a serum biomarker for predicting the outcome of lenvatinib therapy. In addition, the screening revealed that sorafenib-responsive tumors exhibited MET and NRAS activation features [32]. Also, Rodríguez-Hernández et al. found that well-differentiated HCC cells with wild-type p53 were more sensitive to sorafenib and regorafenib. In contrast, moderately- to poorly-differentiated HCC cells harboring mutated p53 and low mitochondrial respiration were more sensitive to lenvatinib and cabozantinib [33]. Recently, Teufel et al. identified the expression patterns of plasma proteins and miRNAs associated with increased HCC patients’ overall survival length following treatment with regorafenib in the RESORCE trial. Levels of these circulating biomarkers and the genetic features of tumors might be helpful to identify HCC patients who most likely would respond to regorafenib administration [34].
4.2. Drugs with synergistic anti-tumor effects to overcome therapy resistance
Drug resistance is the main limitation of the current systemic therapeutic drugs, especially for first-line drugs (sorafenib and lenvatinib). With the increasing understanding of the underlying mechanisms, novel targets have been identified as combinational therapeutic biomarkers to increase drug sensitivity and overcome drug resistance.
A myriad of mechanisms responsible for resistance to sorafenib by HCC cells have been identified; here, we summarize those in which the resistance can be therapeutically overcome. Using genome-wide clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 library screening, Wei et al. found that phosphoglycerate dehydrogenase (PHGDH), the first committed enzyme in the serine synthesis pathway, was a critical driver of sorafenib resistance. Notably, treatment with the PHGDH inhibitor NCT-503 synergized with sorafenib to abolish HCC growth in vivo [35]. Similarly, Tong et al. found that Annexin A3 (ANXA3) was enriched in sorafenib-resistant HCC cells and patient-derived xenografts. At the molecular level, ANXA3 suppressed PKCδ/p38 associated apoptosis and activated autophagy for cell survival, resulting in resistance to sorafenib. Strikingly, anti-ANXA3 monoclonal antibody therapy combined with sorafenib/regorafenib impaired tumor growth in vivo and significantly increased survival [36]. In addition, Shang et al. demonstrated that cabozantinib effectively hampered MET activity without affecting the AKT/mTOR signaling, thus providing an explanation for the unsatisfactory outcome of cabozantinib clinical trials. Moreover, combinational treatment of cabozantinib with the pan-mTOR inhibitor MLN0128 synergistically inhibited MET-activated HCCs [37]. Recently, Arechederra et al. showed that activation of the ADAMTSL5 glycoprotein was tumorigenic upon MET activation. ADAMTSL5 abrogation triggered the reduction of multiple RTKs, including MET, EGFR, PDGFRβ, FGFR4, and IGF1 receptor 1β, resulting in increased sensitivity to sorafenib, lenvatinib, and regorafenib by HCC cells [38]. Moreover, Xu et al. identified a circular RNA, circRNA-SORE, whose upregulation was associated with sorafenib resistance by HCC. Mechanistically, circRNA-SORE binds to the the oncogenic YBX1 protein and protects it from degradation, leading to resistance to sorafenib. Depletion of circRNA-SORE increased the anti-tumor effect of sorafenib in murine HCC patient-derived xenograft (PDX) models [39].
5. Potentially promising drug targets from experimental studies
Several signaling cascades have been proven to play a critical role in HCC initiation and progression, such as MET/HGFR, FGF19-FGF4, PI3K/AKT-mTOR, and retinoblastoma pathways. The most promising candidates and the related drugs are described below and depicted in Figure 2.
Figure 2.
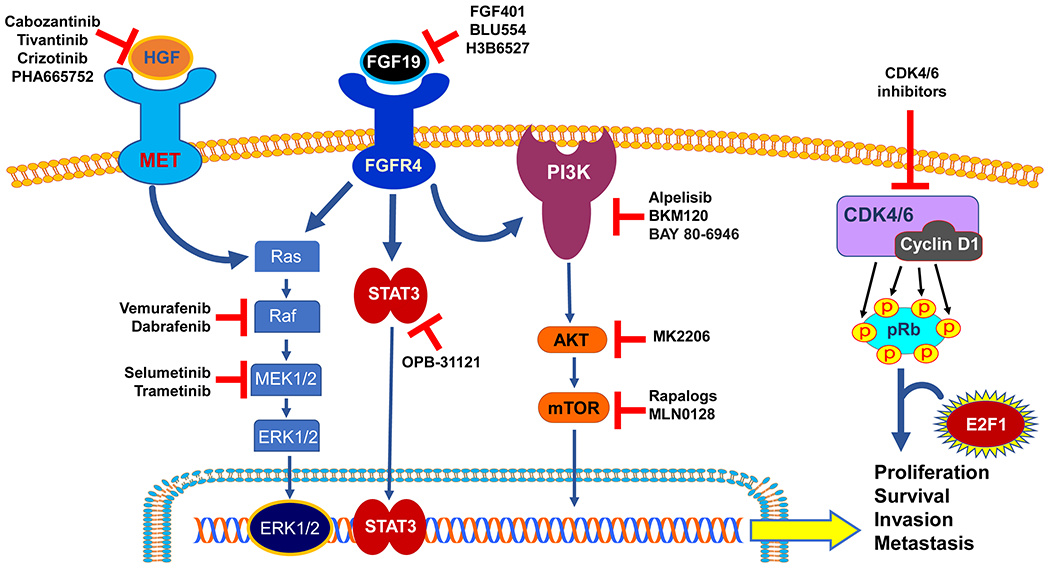
Promising drug targets from experimental studies. Multiple signaling pathways are involved in the hepatocarcinogenesis, among which the HGF/MET, FGF/FGFR, PI3K/AKT/mTOR, and CDK4/6 signaling pathways hold the promises as effective targets for HCC treatment. Targeted drugs showing efficacious anti-tumor effects from the preclinical studies are shown and summarized in the figure. Normal arrows: activation; blunted arrows: inhibition.
5.1. Oncoprotein MET
The oncoprotein MET is a well-characterized RTK implicated in hepatocarcinogenesis [40]. Elevated expression of MET and its ligand HGF are associated with poor prognosis and resistance to sorafenib treatment. Several clinical trials are ongoing to test the outcomes of MET inhibitors (cabozantinib, tivantinib [41], and capmatinib [42]) either alone or in combination with other targeted molecules or immunotherapy. Recently, the preclinical field has been focusing on the compensatory mechanisms of MET suppression to put novel insights into the combinational therapeutic strategies. MET has been reported to interact with EGFR, extracellular signal-regulated kinases 1/2 (ERK1/2), VEGFR, transforming growth factor α (TGFα), and AKT/mTOR signaling cascades in HCC [43]. Several approaches have been employed in experimental in vivo models to overcome the resistance to MET inhibition, including the combination of the MET inhibitor PHA665752 with the EGFR inhibitor gefitinib [44], the use of the dual MET/VEGFR2 inhibitor NZ001 [45], and the simultaneous administration of SU11274 (a selective MET inhibitor) and an HGF-neutralizing antibody [46]. These therapeutic approaches have shown favorable anti-tumor effects, indicating that combining the inhibition of MET with one relevant oncogenic molecule holds excellent promises for paving the road of novel anti-HCC treatments.
5.2. The FGF19-FGFR4 axis
The FGF family consists of at least 5 RTKs and many cognate ligands that have long been pursued as targets for anticancer treatments [47]. Concerning HCC, FGF19 is amplified in ~10% of the patients, is implicated in sorafenib resistance, and represents a potential predictive marker of response to FGFR kinase inhibitors [48]. Therefore, several inhibitors targeting the FGF19 receptor, which is FGFR4, have been developed. FGF401, an FGFR4 kinase inhibitor, has remarkable anti-tumor activity in mice bearing HCC tumor xenografts and patient-derived xenograft models positive for FGF19, FGFR4, and KLB [49]. In addition, BLU554 [28], H3B6527 [50], and BLU9931 [51] also demonstrated anti-tumor effect in preclinical studies. Of note, FGF401 (NCT02325739), BLU554 (NCT02508467), and H3B6527 (NCT02834780) are also undergoing phase I/II clinical trials in HCC patients.
5.3. The PI3K/AKT-mTOR cascade
The PI3K/Akt-mTOR signaling pathway is a central regulator of HCC development. This signaling cascade includes various intracellular kinases, and among them, AKT is a prominent hub downstream of many RTKs. Upstream target proteins of AKT are stimuli-induced RTKs and include PI3K, phosphoinositide-dependent protein kinase (PDK), and mTOR complex 2 (mTORC2). Activated AKT enables the phosphorylation of downstream target proteins, including FOXO1, TSC1/2, and mTORC1 [52]. The use of AKT and mTOR inhibitors in HCC studies has been thoroughly reviewed elsewhere [53, 54]. Although the applications of AKT and/or mTOR inhibitors showed an efficient tumor growth inhibition capacity in vitro and in vivo, the clinical trials so far conducted failed to reach compelling outcomes (Table 2). Current research efforts are devoted to generating more efficacious inhibitors targeting the PI3K/AKT-mTOR cascades and identifying biomarkers for patient selection as well as for combination therapies. The tuberous sclerosis complex (TSC) 1 and TSC2 are known as negative regulators of mTORC1 signaling. TSC1/2 is a GTPase-activating protein for the small GTPase Rheb, which is an essential activator of mTORC1. According to the TCGA cohort, ~4% of HCCs have TSC1 mutation, and ~5% of HCCs have TSC2 mutation. An independent study based on HBV-related HCCs documented ~16.8% of samples displaying TSC1 and/or TSC2 mutations. Of note, TSC2-mutant PDTXs are more sensitive to the treatment with the mTOR inhibitor rapamycin [55]. The PI3K/AKT/mTOR cascade is also involved in many metabolic processes in the liver, suggesting the modulation of the metabolism as an alternative therapeutic strategy against HCC. He et al. showed that loss of hepatic fructose-1, 6-bisphosphate aldolase B (Aldob) favors hepatocarcinogenesis by suppressing the interaction of AKT with its negative regulator protein phosphatase 2A (PP2A). Significantly, treatment with the PP2A activator SMAP inhibited HCC tumor growth in vitro, implying the relevance of AKT inhibition as an anti-neoplastic strategy in this tumor type [56].
Table 2.
Summary of the main experimental findings of the AKT/mTOR inhibitors
| Durgs | Mechanisms of action | Preclinical findings | Clinical Trials | Reference |
|---|---|---|---|---|
| AKT inhibitors | ||||
| MK2206 | Allosteric pan-AKT inhibitor | MK2206 inhibited HCC cellular proliferation via induction of apoptosis and cell cycle arrest. MK2206 and capmatinib (c-Met inhibitor) additively or synergistically suppressed sorafenib-resistant HCC cells in vitro and sorafenib-resistant HCC xenografts in mice. Treatment with the MK-2206 repressed liver tumorigenesis in Pten−/− Sav1−/− mice, suggesting crosstalk between AKT and YAP/TAZ through IRS2. | NCT01239355 | [106-108] |
| GDC-0068 | ATP competitive pan-AKT inhibitor | GDC0068 promoted cell apoptosis in HCC cells. GDC0068 synergized with sorafenib to suppress the growth of sorafenib-resistant HCC tumors in vivo. | NCT02465060 | [109, 110] |
| ARQ 751 | Allosteric pan-AKT inhibitor | ARQ751 showed an anti-tumor effect in a DEN-induced cirrhotic rat model of HCC by increasing apoptosis and decreasing proliferation in the tumors. | NCT02761694 1 | [111] |
| ARQ 092 | Allosteric pan-AKT inhibitor | ARQ092 showed an anti-neoplastic effect in a DEN-induced cirrhotic rat model of HCC by inducing apoptosis and decreasing proliferation in the tumors. | N/A | [112] |
| mTOR inhibitors | ||||
| Rapalogs | Rapamycin analogs 2 | Oral administration of RAD001 (everolimus) to mice HCC PDXs resulted in a dose-dependent tumor growth inhibition. RAD001-induced growth suppression was associated with the inactivation of mTOR downstream targets, reduction in VEGF expression and microvessel density, inhibition of cell proliferation, and downregulation of the cell cycle genes. | [113] | |
| MLN0128 | Pan-mTOR inhibitor | MLN0128 effectively inhibited both mTORC1 and mTORC2, leading to the suppression of cell growth and induction of apoptosis in vitro and in vivo. | NCT02575339 | [114] |
| CC-233 | Pan-mTOR inhibitor | CC-233 exhibited significant cytotoxicity and growth suppression potency against HCC cell lines and primary human HCC cells. Furthermore, CC-223 oral administration dramatically inhibited HepG2 xenograft growth. | NCT01177397 | [115] |
| NVP-BEZ235 | Dual PI3K/mTOR inhibitor | NVP-BEZ235 reverted the metabolic abnormalities and cell growth driven by the insulin signaling pathway in a rat model of insulin-induced hepatocarcinogenesis. | N/A | [116] |
ARQ 751 is under clinical trials among solid tumor patients with PIK3CA/AKT/PTEN mutations.
Due to the limited efficacy of Rapalogs as monotherapy against HCC, multiple clinical trials have been conducted or are ongoing to evaluate the therapeutic efficacy of Rapalogs in combination therapies. Abbreviation: N/A, not applicable.
5.4. The retinoblastoma pathway
D-type cyclins and cyclin-dependent kinases 4 and 6 (CDK4 and CDK6) are critical in driving the cell cycle’s G1 to S phase transition. They achieve this scope by phosphorylating and inactivating the retinoblastoma protein (pRb) [57]. In the absence of stimuli, active (unphosphorylated) pRb sequesters E2F1 and suppresses the latter’s transcriptional activity. Following stimulation by extracellular signals, the cyclin D-CDK4/6 complex phosphorylates pRb, leading to the activation of the E2F1 transcription factor, ultimately resulting in G1 to S phase progression and cell proliferation [57]. Recently, potent CDK4/6 inhibitors have emerged as a new therapeutic tool in many tumor types; in particular, palbociclib, ribociclib, and abemaciclib have been approved by the FDA to treat hormone receptor-positive/HER2-negative advanced breast cancer [58]. In human HCC cell lines, the CDK4/6 inhibitor palbociclib suppressed cell proliferation by inducing cell cycle arrest. In vivo, palbociclib, either alone or combined with sorafenib, hindered tumor growth and increased survival of human HCC xenografts [59]. Furthermore, the combination of palbociclib with either NF-kB [60] or FGFR4 [50] inhibitors was highly detrimental to the growth of HCC cells in vitro and in vivo. Following this encouraging preclinical data, palbociclib is currently in phase 2 clinical trial for HCC treatment (NCT01356628).
6. Novel therapeutic candidates from the preclinical arena
6.1. Targeting the metabolic pathways
Deregulation of the metabolism has been shown in virtually all cancer types, including HCC [61]. This observation is not surprising, as the liver is the primary site for many metabolic processes. HCC is metabolically different from normal liver tissue in many ways, including glycolysis, the citric acid cycle, oxidative phosphorylation, lipogenesis, lipolysis, amino-acid synthesis and catabolism, and the pentose-phosphate cascade (Figure 3). In general, these include increased glycolysis and lactate production, as well as a higher lactate-to-pyruvate ratio in HCC [62]. Moreover, higher activity of the insulin receptor pathway, such as PI3K/Akt and increased lipogenesis, fatty acid oxidation and transport were also reported [63]. Since the metabolic cascades provide the necessary nutrients for tumor cells to grow, targeting the metabolism in the liver has long been pursued to interrupt hepatocarcinogenesis (Table 3). The principal scope of targeting the metabolism for the treatment of HCC is to deprive tumor cells of the necessary nutrients without affecting the essential metabolic requirements of normal liver cells.
Figure 3.
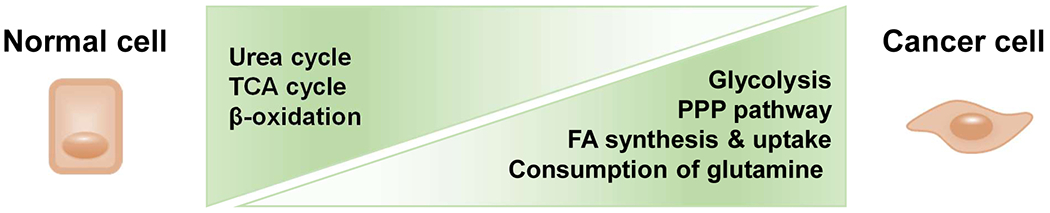
Schematic figures of metabolic alterations in HCC. Glycolysis and pentose phosphate pathway (PPP) were increased while the urea cycle, TCA cycle, and β-oxidation are decreased in the liver cancer cells. Fatty acid (FA) synthesis and uptake are gradually increased until HCC development. Glutamate produced during glutaminolysis serves as the major substrate to refuel the TCA cycle. Abbreviations: PPP, pentose phosphate pathway; FA, fatty acid; TCA, tricarboxylic acid cycle.
Table 3.
Summary of targeting metabolic processes for the treatment of HCC
| Target | Biological functions | Model | Main Findings | Drugs | Reference |
|---|---|---|---|---|---|
| Glycolysis and glycogen metabolism | |||||
| HK2 | Rate-limiting enzyme in the first glycolysis step; converting glucose into glucose-6-phosphate. | DEN-HCC model and subcutaneous xenograft models | Hepatic HK2 deletion inhibited tumor incidence in a mouse model of hepatocarcinogenesis. HK2 silencing synergizes with sorafenib to inhibit tumor growth. Upon HK2 silencing, glucose flux to pyruvate and lactate is inhibited, but TCA fluxes are maintained. HK2 silencing in combination with metformin suppressed mTORC1 in an AMPK-independent and REDD1-dependent manner. | Metformin | [64, 117] |
| PKM2 | PKM2 is involved in the conversion of phosphoenolpyruvate to pyruvate. | Subcutaneous and orthotopic xenograft models | PKM2 is upregulated in HCC; knockdown of PKM2 hampered HCC growth in both subcutaneous injection and orthotopic liver implantation models and reduced lung metastasis in vivo. | N/A | [118] |
| Lipid metabolism | |||||
| FXR | Hepatic FXR is the primary regulator of bile acid biosynthesis with essential roles in fatty-acid homeostasis. | Genetic engineered mouse model | Kainuma et al. found that FXR activation enhanced TGFβ-induced epithelial-mesenchymal transition in HCC cells. However, Yang et al. reported spontaneous development of liver tumors in the absence of the FXR. | OCA | [119, 120] |
| FASN | FASN is the master regulator of de novo lipogenesis. | Hydrodynamic transfection induced HCC | Genetic deletion of Fasn inhibited tumor development in AKT, AKT/MET, Pten loss, and c-Myc mice. Blocking cholesterol biosynthesis in combination with liver-specific Fasn knockout prevents tumor formation in mice. | C75; TVB2640 | [67, 68] |
| TCA cycle | |||||
| PDK4 | PDK4 is a key enzyme regulating the central pathway of cell metabolism by inhibiting pyruvate dehydrogenase. | Subcutaneous xenograft model | Knockdown of PDK4 increased expression of key lipogenic enzymes, FASN and stearoyl-CoA desaturase, and silencing PDK4 facilitated proliferation and migration of HCC cells | [121] | |
| SDHB | Succinate dehydrogenase converts succinate into fumarate in the TCA cycle. | Subcutaneous xenograft model | Subcutaneous implantation and tail vein injection with SDHB knockdown cells resulted in a larger tumor volume and accelerated cancer metastasis, respectively. Silencing of SDHB altered energy metabolism switched from aerobic respiration to glycolysis, resulted in the Warburg effect, and enhanced cell proliferation and motility. | [122] | |
| Oxidative phosphorylation | |||||
| NANOG | NANOG is a transcription factor and can act as a bona fide oncogene to induce carcinogenesis. | Alcohol- or obesity-HCV-induced tumor models | NANOG is induced by Toll-like receptor 4 signaling via phosphorylation of E2F1, and downregulation of Nanog slowed down HCC progression. | [123] | |
| SALL4 | SALL4 is a transcription factor and can act as a bona fide oncogene to induce carcinogenesis. | PDX HCC model | Mitochondrial oxidative phosphorylation inhibitors were shown to be particularly effective in suppressing SALL4-expressing HCC tumorigenesis in culture and in vivo. SALL4 binds approximately 50% of mitochondrial genes, including many oxidative phosphorylation genes, to activate their transcription. Oligomycin also reduced the growth of xenograft tumors grown from SALL4hi SNU-398 or HCC26.1 cells to a greater extent than sorafenib. | Oligomycin | [124] |
| Pentose-phosphate pathway | |||||
| NRF2 /KEAP1 | NRF2 is a transcriptional factor, and KEPA1 is a major negative regulator of NRF2. Activation of NRF2 increases glucose uptake and directs it to the pentose phosphate pathway. | DEN-HCC | HBV upregulates G6PD expression by HBx-mediated activation of Nrf2. In HBV-infected hepatocytes, HBx, on the one hand, results in the accumulation of p62 through inhibition of autophagic flux, and on the other hand, interacts with Keap1 through p62, enabling the formation of HBx–p62–Keap1 aggregates in the cytoplasm. These aggregates hijack Keap1 from Nrf2 leading to Nrf2 activation and G6PD expression. Nrf2 mutagenic activation drives hepatocarcinogenesis. | [71, 125, 126] | |
| Amino acid metabolism | |||||
| OGDHL | OGDHL is one of the rate-limiting components of OGDH complex, in the regulation of lipid metabolism. | Subcutaneous xenograft | The silencing of OGDHL induces lipogenesis and influences the chemosensitization effect of sorafenib in liver cancer cells by reprogramming glutamine metabolism. The reduction of reductive glutamine metabolism through OGDHL overexpression or glutaminase inhibitors sensitized tumor cells to sorafenib, a molecular-targeted therapy for HCC. | [127] | |
| p300/CBP | p300/CBP transcriptional coactivator proteins are central regulators of epigenetics. | PDX HCC model | p300/CBP epigenetically regulated the expression of glycolysis-related metabolic enzymes through modulation of histone acetylation in HCC. These data highlight the value of targeting the histone acetyltransferase activity of p300/CBP for HCC therapy. p300/CBP regulates the expression of the enzyme genes related to amino acid metabolism and nucleotide synthesis. | B029-2 | [72] |
| Purine metabolism | |||||
| IMPDH | IMPDH is a purine biosynthetic rate-limiting enzyme. | PDX HCC model | Targeted ablation of purine biosynthesis by knockdown of the IMPDH or using the drug mycophenolate mofetil (MMF) reduced HCC proliferation in vitro and decreased the tumor burden in vivo. | MMF | [128] |
Abbreviations: HK2, Hexokinase 2; PKM2, Pyruvate kinase M2; OCA, obeticholic acid; FXR, Farnesoid X receptor; FASN, Fatty acid synthase; PDK4, Pyruvate dehydrogenase kinase 4; SDHB, Succinate dehydrogenase B; NRF2, Nuclear factor erythroid 2-related factor 2; KEAP1, Kelch-like ECH-associated protein 1; OGDHL, oxoglutarate dehydrogenase-like; CBP, CREB Binding Protein; IMDPH, Inosine-5’-monophosphate dehydrogenase.
DeWaal et al. investigated glycolysis and glycogen-associated pathways in HCC development, focusing on the role of hexokinase (HK2), the essential enzyme for glucose phosphorylation. The authors found that hepatic specific deletion of HK2 inhibited tumor initiation in murine HCCs induced by diethylnitrosamine (DEN) and HK2 silencing synergized with sorafenib to inhibit tumor growth of HCC xenografts. Mechanistically, targeting HK2 inhibited the glucose flux to pyruvate and lactate. Also, HK2 depletion suppressed glycolysis and induced oxidative phosphorylation in HCC. Furthermore, HK2 depletion sensitized HCC to the anti-growth effects exerted by metformin [64]. Moreover, recent studies demonstrated the promising outcomes of targeting fatty acid synthase (FASN), a multi-enzyme that catalyzes fatty acid biosynthesis, to treat HCC. Indeed, genetic deletion of FASN in the mouse liver led to the inhibition of AKT- [65] and AKT/MET-driven [66] hepatocarcinogenesis and strongly delayed HCC development induced by phosphatase and tensin homolog (PTEN) loss [67], and c-MYC activation [68]. Of note, blocking cholesterol biosynthesis combined with FASN inhibition completely prevented HCC formation induced by PTEN deletion, indicating that simultaneous targeting of FASN and cholesterogenesis might be highly detrimental for the growth of HCC [67]. Moreover, the FASN inhibitor TVB2640 has been shown to inhibit de novo lipogenesis effectively [69] and is currently under clinical test for solid tumors (NCT02223247). Preliminary data from our group also suggest that another de novo lipogenesis inhibitor improved the effectiveness of cabozantinib in several genetically engineered mouse HCC models. Furthermore, combining FASN inhibitor with established targeted therapies and conventional chemotherapy has been envisaged for HCC treatment [70]. Overall, these in vivo data, together with the evidence of robust activity of FASN and related de novo lipogenesis in human HCC [63], point to FASN as a promising target for HCC therapy. Experimental efforts on targeting amino-acid metabolism and oxidative phosphorylation processes also reached a favorable outcome [71, 72].
6.2. Targeting the epigenome and DNA repair mechanisms
Epigenetic mechanisms modulate chromatin conformation and the accessibility of the transcriptional machinery to genes, thereby regulating their expression. Epigenetic deregulation is central to the hallmarks of cancer, leading to eventual carcinogenesis [73]. Histone deacetylation is one of the most critical epigenetic events, regulating various cellular features, such as differentiation, proliferation, and cell cycle. Histone deacetylases (HDACs), the prominent mediators of this epigenetic mechanism, are often aberrantly expressed in various tumors, including HCC [74]. The molecular mechanisms whereby HDACs contribute to hepatocarcinogenesis are complex and not completely understood. For instance, HDAC8 upregulation contributes to insulin resistance in NAFLD progression and, in coordination with the HMT KTM6 (EZH2), epigenetically represses Wnt antagonists’ expression enhancing cell proliferation in HCC [75]. Belinostat, a pan-HDAC inhibitor, has shown experimental efficacy, and it is under phase II clinical trial as a second-line systemic treatment for HCC. (NCT00321594). Intriguingly, Llopiz et al. showed that belinostat combined with immune checkpoint inhibitors (anti-CTLA-4) increased their efficacy in an experimental model of HCC [76]. Resminostat, an HDAC1/3/6 inhibitor, has been shown to induce the reversion of stem-like properties of HCC cells and significantly augmented the cytotoxic effects of sorafenib in vitro [77]. These findings provide a rationale for the ongoing clinical trial of resminostat in combination with sorafenib for HCC therapy (NCT00943449).
Furthermore, recent evidence indicates modulating different epigenetic regulators, including DNA methyltransferases and histone methyltransferases, bromodomain-containing proteins, and histone lysine demethylases, can increase immune recognition of tumor cells and synergize with immunotherapy [78]. Concerning DNA repair, Wang et al. investigated the combinational effects of targeting poly (ADP-ribose) polymerase 1 (PARP1) and DNA-dependent protein kinase catalytic subunit (DNA-PKcs) in HCC. The authors demonstrated that two DNA double-strand break repair pathways, namely homologous recombination and nonhomologous end-joining, are upregulated in an HCC mouse model, induced by overexpressing the DNA repair factors PARP1 and DNA-PKcs. Combining the PARP1 inhibitor Olaparib with the DNA-PKcs inhibitor (NU7441) suppressed HCC growth in the mouse model and HCC PDX. Because DNA-PKcs is strongly activated in human HCC [79], the results suggest the combined inhibition of both homologous recombination and nonhomologous end-joining processes is a potential therapy for liver cancer [80]. Amplification of the DNA-PKcs gene in the tumor tissue might be a reliable biomarker of the effectiveness of therapeutic strategies aimed at inhibiting DNA-PKcs in human HCC [81].
6.3. YAP/TAZ
Yes-associated protein (YAP) and its paralog, transcriptional coactivator with the PDZ-binding motif (WWTR1/TAZ), are the two transcriptional coactivators downstream of the HIPPO tumor suppressor pathway. When HIPPO is off, YAP/TAZ can be activated and translocated into the nucleus, regulating the expressions of downstream targets. It has been shown that YAP and TAZ are activated in HCC, and their induction promotes HCC development via regulating cell proliferation and survival. Studies from our group have found that conditional deletion of Yap and/or Taz led to HCC regression in mouse models induced by the overexpression of c-Myc or c-Met/sgAxin1 genes (unpublished data). The results suggest that suppressing YAP and TAZ activity might be an effective approach for HCC treatment. In addition, the tankyrase inhibitors G007LK and XAV-939 have been previously shown to be able to abolish YAP/TAZ activity in HCC cell lines [82]. However, recent studies also demonstrated that distinct oncogenic stimuli might differently activate YAP and TAZ [83]. Therefore, decisions on whether to target YAP or TAZ alone or together should be made with caution.
6.4. Additional molecules with targeted potentiality
Amplification of two-pore channel 2 (TPC2) occurs in ~6% of HCC samples based on the TCGA database. Muller et al. developed simplified analogs of the alkaloid tetrandrine as potent TPC2 inhibitors by screening a library of synthesized benzyltetrahydroisoquinoline derivatives. Notably, they found a TPC2 inhibitor with anti-tumor efficacy in mice [84]. Han et al. developed a miR122a-based approach to target telomerase reverse transcriptase (TERT) through the post-transcriptional enhancement of the ribozyme improved trans-splicing [85]. In addition, the Ser/Thr kinase polo-like kinase 1 (PLK1) plays a pivotal role in cell-cycle regulation, and targeting PLK1 with a selective PLK1 kinase inhibitor exhibited profound anti-tumor efficacy in a xenograft mouse HCC model [86]. The transcription factor Late SV40 factor (LSF) promotes hepatocarcinogenesis by regulating the major hallmarks of cancer, including cell invasion, angiogenesis, chemoresistance, and senescence [87]. The LSF inhibitors, factor quinolinone inhibitor 1 (FQI1) and FQI2, have shown compelling therapeutic effects on the HCC lesions from Alb/c-Myc transgenic mice [88]. Several microRNAs, such as miR122 [89], miR21 [90], and miR218 [91], have been identified as effective targets for HCC treatment. With the advances in the pharmacological field, targeting specific microRNAs might also be beneficial for HCC treatment.
6.5. Diagnostic biomarkers as HCC targets
Alpha-fetoprotein (AFP) and Glypican-3 (GPC3) are oncofetal glycoproteins overexpressed in HCC. The importance of AFP and GFP3 as diagnostic biomarkers for HCC is well established. From the therapeutic perspective, Hong et al. generated an epitope-optimized, high immunogenic AFP that disrupted immune tolerance and potently activated CD8 T cells to prevent mouse autochthonous HCC development [92]. Similar approaches have been evaluated in other mouse HCC models [93], supporting the idea of targeting AFP as an effective treatment for HCC. The major immune mechanisms of anti-GFP3 antibodies against HCC cells consist of antibody-dependent and complement-dependent cytotoxicity [94, 95]. Since anti-GFP3 antibodies are associated with the immune response, the combination of anti-GFP3 and immune checkpoint inhibitors is currently under intense investigation [96].
7. Conclusion
The preclinical field has unraveled several promising targets for the development of innovative treatments against HCC. RTK inhibitors and immune checkpoint inhibitors remain the most attractive options to experimental investigators. In addition, studies on targeting intracellular kinases, metabolic pathways, and epigenetic and DNA repair mechanisms add ground for novel HCC targeted therapeutic strategies. Furthermore, evaluating the anti-neoplastic potency of combinational approaches might be highly helpful to establish more effective treatments.
8. Expert opinion
With the development and refinement of high-throughput sequencing technologies, cancer treatment is becoming more and more targeted-orientated and individual-specific. Experimental investigations have identified numerous molecules that potentially can be targeted in HCC, including RTKs, intracellular kinases, metabolic enzymes, and epigenetic modulators. However, adequate and proper models should be established and used for reliable therapeutic predictions. Although many HCC mouse models have been generated, including chemical- or diet-induced models, genetically engineered models, and cell-line or patient-derived xenograft (PDX) models, none can perfectly recapitulate the HCC biological features in the patients. In addition, it is still challenging to establish a feasible HCC model for the study of the tumor immune environment in vivo. In this regard, it has been reported that intraperitoneal injection of carbon tetrachloride in combination with intrasplenic inoculation of oncogenic hepatocytes in the immunocompetent mice was able to establish a clinically relevant HCC model with features of fibrosis, which enabled the investigation of immune responses during HCC progression [97]. Moreover, exogenous hyper-immunogenic antigens such as LucOS [98] or polyinosinic-polycytidylic acid [99] were applied to enhance immunity in mouse HCCs. However, these models are either technically laborious or biologically unsuitable [100], adding obstacles to translating preclinical studies to the clinical setting. Therefore, investigators have to be cautious in choosing the models and interpreting the preclinical observations.
With the increasing number of non-viral-related HCCs, especially NAFLD- and NASH-related HCC, understanding the variations between each tumor subtype is imperative. For instance, NASH-related HCC are characterized by enrichment in bile and fatty acid metabolism, oxidative stress, and IFN/NFκB-related inflammation, and a decrease in DNA damage repair compared to viral/alcohol-HCC [101, 102]. A meta-analysis study of the current immunotherapy clinical trials demonstrated the lack of response to immunotherapy in human NASH-HCC [103] compared to viral/alcohol-related HCC. Similarly, in a preclinical NASH model, the mouse liver exhibited an increasing accumulation of exhausted, unconventionally activated CD8+PD1+ T cells with disease progression, which promoted HCC development and resulted in an unfavorable effect of the anti-PD1 treatment. Therefore, a better understanding of the underlying mechanism of non-viral HCC will be beneficial for the development of proper pharmacological therapies.
Combinational therapies are becoming promising strategies to improve therapeutic effectiveness in many tumor types, including HCC. In this regard, combinations of two drugs targeting different oncogenic pathways or combinations of targeted drugs with immune checkpoint inhibitors are under evaluation. The VEGF pathway promotes local immune suppression by inhibiting antigen-presenting cells and effector cells and activating suppressive elements, including Treg cells, myeloid-derived suppressor cells, and tumor-associated macrophages, providing the rationale for combining immune checkpoint inhibitors with anti-angiogenic agents. With the exciting results from the IMBrave150 trial [8], where the anti-PD1 therapy was combined with the anti-VEGF treatment, similar preclinical studies are encouraged. Therefore, a deeper understanding of the HCC immune environment in experimental models will provide more effective combinational therapeutic strategies. In addition, studies on the effectiveness of targeting the tumor stroma cells and inflammatory signals of the tumor microenvironment also hold promises from the preclinical arena.
Immune checkpoint inhibitors will inevitably provide robust support for novel anti-HCC therapies. However, the complexity of etiologies of HCC drives a significant tumor heterogeneity, leading to the challenge of the widespread appliance of the immune therapies. In addition, it has been shown by several groups that a subset of HCCs, for instance, with β-Catenin activation, is resistant to immunotherapies [104]. Therefore, the selection of drugs targeting HCCs with immunosuppressive properties remains challenging in the preclinical scenario. For this purpose, in silico high-throughput screening of chemical compound libraries followed by in vitro and in vivo testing might represent a reliable strategy.
Article Highlights.
Hepatocellular carcinoma (HCC) is a frequent liver tumor associated with high molecular heterogeneity and poor prognosis.
The combination of Atezolizumab and Bevacizumab is currently the first-line therapy against HCC. However, the benefits are somehow limited and temporary.
High-throughput technologies have recently expanded our understanding of the molecular landscape of human liver cancer and unraveled potentially actionable targets.
Targeted therapies, mainly based on RTK inhibitors and immune checkpoint inhibitors, have been mainly unsatisfactory due to the lack of specific predictive biomarkers and the rapid appearance of drug resistance.
Recent experimental data have unraveled some of the resistance mechanisms to drugs by HCC cells, and approaches to overcome this hurdle have been developed or are under investigation.
Novel therapeutic strategies from the preclinical arena, such as targeting intracellular kinases, metabolic pathways, and epigenetic and DNA repair mechanisms, are opening new viable options for treating this lethal disease.
Funding
This paper was not funded.
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Footnotes
Reviewer disclosures
One reviewer is a co-inventor on several awarded and applied patents related to small molecule inhibitors of the the transcription factor LSF. Some of the claims on the patents include treatment of various cancers for which LSF is a driver, including hepatocellular carcinoma. Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers
- 1.Villanueva A Hepatocellular carcinoma. N Engl J Med 2019;380(15):1450–62. [DOI] [PubMed] [Google Scholar]
- 2.Li S, Saviano A, Erstad DJ, et al. Risk factors, pathogenesis, and strategies for hepatocellular carcinoma prevention: Emphasis on secondary prevention and its translational challenges. J Clin Med 2020;9(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Totoki Y, Tatsuno K, Covington KR, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet 2014;46(12):1267–73. [DOI] [PubMed] [Google Scholar]
- 4.Ally A, Balasundaram M, Carlsen R, et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169(7):1327–41.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This study provides a comprehensive and integrative genomic characterization of a large cohort of HCC patients.
- 5.Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359(4):378–90. [DOI] [PubMed] [Google Scholar]
- 6.Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. The Lancet 2018;391(10126):1163–73. [DOI] [PubMed] [Google Scholar]
- 7.Butte MJ, Keir ME, Phamduy TB, et al. Programmed death-1 ligand 1 interacts specifically with the B7–1 costimulatory molecule to inhibit T cell responses. Immunity 2007;27(1):111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus Bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med 2020;382(20):1894–905. [DOI] [PubMed] [Google Scholar]
- 9.Zhu AX, Kudo M, Assenat E, et al. Effect of Everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA 2014;312(1):57–67. [DOI] [PubMed] [Google Scholar]
- 10.Zhu AX, Park JO, Ryoo B-Y, et al. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. The Lancet Oncology 2015;16(7):859–70. [DOI] [PubMed] [Google Scholar]
- 11.Llovet JM, Decaens T, Raoul J-L, et al. Brivanib in patients with advanced hepatocellular carcinoma who were intolerant to sorafenib or for whom sorafenib failed: results from the randomized phase III BRISK-PS study. J Clin Oncol 2013;31(28):3509–16. [DOI] [PubMed] [Google Scholar]
- 12.Rimassa L, Assenat E, Peck-Radosavljevic M, et al. Tivantinib for second-line treatment of MET-high, advanced hepatocellular carcinoma (METIV-HCC): a final analysis of a phase 3, randomised, placebo-controlled study. The Lancet Oncology 2018;19(5):682–93. [DOI] [PubMed] [Google Scholar]
- 13.Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet 2017;389(10064):56–66. [DOI] [PubMed] [Google Scholar]
- 14.Tai W-T, Chu P-Y, Shiau C-W, et al. STAT3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin Cancer Res 2014;20(22):5768. [DOI] [PubMed] [Google Scholar]
- 15.Ou D-L, Chen C-W, Hsu C-L, et al. Regorafenib enhances antitumor immunity via inhibition of p38 kinase/Creb1/Klf4 axis in tumor-associated macrophages. J Immunother Cancer 2021;9(3):e001657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shigeta K, Matsui A, Kikuchi H, et al. Regorafenib combined with PD1 blockade increases CD8 T-cell infiltration by inducing CXCL10 expression in hepatocellular carcinoma. J Immunother Cancer 2020;8(2):e001435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang Q, Chen W, Ren M, et al. Cabozantinib suppresses tumor growth and metastasis in hepatocellular carcinoma by a dual blockade of VEGFR2 and MET. Clin Cancer Res 2014;20(11):2959. [DOI] [PubMed] [Google Scholar]
- 18.Abou-Alfa GK, Meyer T, Cheng A-L, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med 2018;379(1):54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clarke JM, Hurwitz HI. Targeted inhibition of VEGF receptor 2: an update on ramucirumab. Expert Opin Biol Ther 2013;13(8):1187–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X, Gu Z, Chen Y, et al. Application of PD-1 blockade in cancer immunotherapy. Comput Struct Biotechnol J 2019;17:661–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rotte A Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res 2019;38(1):255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Killock D Nivolumab keeps HCC in check and opens avenues for checkmate. Nature Reviews Clinical Oncology 2017;14(7):392–92. [DOI] [PubMed] [Google Scholar]
- 23.Yau T, Kang Y-K, Kim T-Y, et al. Efficacy and safety of Nivolumab plus Ipilimumab in patients with advanced hepatocellular carcinoma previously treated with Sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncology 2020;6(11):e204564–e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. The Lancet Oncology 2018;19(7):940–52. [DOI] [PubMed] [Google Scholar]
- 25.El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. The Lancet 2017;389(10088):2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sangro B, Sarobe P, Hervás-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2021; 18:525–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee J-S, Heo J, Libbrecht L, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 2006;12(4):410–16. [DOI] [PubMed] [Google Scholar]
- 28.Hoshida Y, Nijman SMB, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Can Res 2009;69(18):7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15(10):599–616. [DOI] [PubMed] [Google Scholar]
- 30.Wang P, Jiang Z, Liu X, et al. PI16 attenuates response to sorafenib and represents a predictive biomarker in hepatocellular carcinoma. Cancer Medicine 2020;9(19):6972–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song M, Pan Q, Yang J, et al. Galectin-3 favours tumour metastasis via the activation of β-catenin signalling in hepatocellular carcinoma. Br J Cancer 2020;123(10):1521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Myojin Y, Kodama T, Maesaka K, et al. ST6GAL1 is a novel serum biomarker for lenvatinib-susceptible FGF19-driven hepatocellular carcinoma. Clin Cancer Res 2021;27(4):1150. [DOI] [PubMed] [Google Scholar]
- 33.Rodríguez-Hernández MA, Chapresto-Garzón R, Cadenas M, et al. Differential effectiveness of tyrosine kinase inhibitors in 2D/3D culture according to cell differentiation, p53 status and mitochondrial respiration in liver cancer cells. Cell Death Dis 2020;11(5):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teufel M, Seidel H, Köchert K, et al. Biomarkers associated with response to regorafenib in patients with hepatocellular carcinoma. Gastroenterology 2019;156(6):1731–41. [DOI] [PubMed] [Google Scholar]
- 35.Wei L, Lee D, Law C-T, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun 2019;10(1):4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tong M, Che N, Zhou L, et al. Efficacy of annexin A3 blockade in sensitizing hepatocellular carcinoma to sorafenib and regorafenib. J Hepatol 2018;69(4):826–39. [DOI] [PubMed] [Google Scholar]
- 37.Shang R, Song X, Wang P, et al. Cabozantinib-based combination therapy for the treatment of hepatocellular carcinoma. Gut 2020:gutjnl-2020–320716. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This article domonstated that the c-MET/ERK/p21/PKM2 cascade and VEGFR2-induced angiogenesis are the primary targets of cabozantinib in HCC treatment, and combination therapies with cabozantinib and mTOR inhibitors may be effective against human HCC.
- 38.Arechederra M, Bazai SK, Abdouni A, et al. ADAMTSL5 is an epigenetically activated gene underlying tumorigenesis and drug resistance in hepatocellular carcinoma. J Hepatol 2021;74(4):893–906. [DOI] [PubMed] [Google Scholar]
- 39.Xu J, Ji L, Liang Y, et al. CircRNA-SORE mediates sorafenib resistance in hepatocellular carcinoma by stabilizing YBX1. Signal Transduction and Targeted Therapy 2020;5(1):298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bouattour M, Raymond E, Qin S, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2018;67(3):1132–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pievsky D, Pyrsopoulos N. Profile of tivantinib and its potential in the treatment of hepatocellular carcinoma: the evidence to date. J Hepatocell Carcinoma 2016;3:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu X, Wang Q, Yang G, et al. A novel kinase inhibitor, INCB28060, blocks c-MET–dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res 2011;17(22):7127. [DOI] [PubMed] [Google Scholar]
- 43.Yu J, Chen GG, Lai PBS. Targeting hepatocyte growth factor/c-mesenchymal–epithelial transition factor axis in hepatocellular carcinoma: Rationale and therapeutic strategies. Med Res Rev 2021;41(1):507–24. [DOI] [PubMed] [Google Scholar]
- 44.Steinway SN, Dang H, You H, et al. The EGFR/ErbB3 pathway acts as a compensatory survival mechanism upon c-Met inhibition in human c-Met+ hepatocellular carcinoma. PLoS One 2015;10(5):e0128159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Gao X, Zhu Y, et al. The dual blockade of MET and VEGFR2 signaling demonstrates pronounced inhibition on tumor growth and metastasis of hepatocellular carcinoma. J Exp Clin Cancer Res 2018;37(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Firtina Karagonlar Z, Koc D, Iscan E, et al. Elevated hepatocyte growth factor expression as an autocrine c-Met activation mechanism in acquired resistance to sorafenib in hepatocellular carcinoma cells. Cancer Sci 2016;107(4):407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huynh H, Prawira A, Le TBU, et al. FGF401 and vinorelbine synergistically mediate antitumor activity and vascular normalization in FGF19-dependent hepatocellular carcinoma. Exp Mol Med 2020;52(11):1857–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hatlen MA, Schmidt-Kittler O, Sherwin CA, et al. Acquired on-target clinical resistance validates FGFR4 as a driver of hepatocellular carcinoma. Cancer Discov 2019;9(12):1686. [DOI] [PubMed] [Google Scholar]
- 49.Weiss A, Adler F, Buhles A, et al. FGF401, a first-in-class highly selective and potent FGFR4 inhibitor for the treatment of FGF19-driven hepatocellular cancer. Mol Cancer Ther 2019;18(12):2194. [DOI] [PubMed] [Google Scholar]
- 50.Joshi JJ, Coffey H, Corcoran E, et al. H3B-6527 is a potent and selective inhibitor of FGFR4 in FGF19-driven hepatocellular carcinoma. Cancer Res 2017;77(24):6999. [DOI] [PubMed] [Google Scholar]
- 51.Chen J, Du F, Dang Y, et al. Fibroblast growth factor 19–mediated up-regulation of SYR-related high-mobility group box 18 promotes hepatocellular carcinoma metastasis by transactivating fibroblast growth factor receptor 4 and Fms-related tyrosine kinase 4. Hepatology 2020;71(5):1712–31. [DOI] [PubMed] [Google Scholar]
- 52.Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin Cancer Biol 2019;59:125–32. [DOI] [PubMed] [Google Scholar]
- 53.Mroweh M, Roth G, Decaens T, et al. Targeting Akt in hepatocellular carcinoma and its tumor microenvironment. Int J Mol Sci 2021;22(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu X, Paliogiannis P, Calvisi DF, Chen X. Role of the Mammalian target of rapamycin pathway in liver cancer: From molecular genetics to targeted therapies. Hepatology 2021;73(S1):49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Comprehensive review on mTOR inhibitors for the treatment of liver cancer.
- 55.Ho DWH, Chan LK, Chiu YT, et al. TSC1/2 mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut 2017;66(8):1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He X, Li M, Yu H, et al. Loss of hepatic aldolase B activates Akt and promotes hepatocellular carcinogenesis by destabilizing the Aldob/Akt/PP2A protein complex. PLoS Biol 2020;18(12):e3000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sherr CJ. Cancer Cell Cycles. Science 1996;274(5293):1672. [DOI] [PubMed] [Google Scholar]
- 58.Janni W, Alba E, Bachelot T, et al. First-line ribociclib plus letrozole in postmenopausal women with HR+ , HER2− advanced breast cancer: Tumor response and pain reduction in the phase 3 MONALEESA-2 trial. Breast Cancer Res Treat 2018;169(3):469–79. [DOI] [PubMed] [Google Scholar]
- 59.Bollard J, Miguela V, Ruiz de Galarreta M, et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017;66(7):1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sheng J, Kohno S, Okada N, et al. Treatment of RB1-intact hepatocellular carcinoma with CDK4/6 inhibitor combination therapy. Hepatology 2021; 10.1002/hep.31872. [DOI] [PubMed] [Google Scholar]
- 61.Wang H, Lu J, Dolezal J, et al. Inhibition of hepatocellular carcinoma by metabolic normalization. PLoS One 2019;14(6):e0218186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Christofk HR, Vander Heiden MG, Harris MH, et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008;452(7184):230–33. [DOI] [PubMed] [Google Scholar]
- 63.Calvisi DF, Wang C, Ho C, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011;140(3):1071–83.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeWaal D, Nogueira V, Terry AR, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nature Commun 2018;9(1):446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li L, Pilo GM, Li X, et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J Hepatol 2016;64(2):333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu J, Che L, Li L, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep 2016;6(1):20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Che L, Chi W, Qiao Y, et al. Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans. Gut 2020;69(1):177. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This study uncovers a novel functional crosstalk between aberrant lipogenesis and cholesterol biosynthesis pathways in hepatocarcinogenesis, whose concomitant inhibition might represent a therapeutic option for HCC.
- 68.Jia J, Che L, Cigliano A, et al. Pivotal role of fatty acid synthase in c-myc driven hepatocarcinogenesis. Int J Mol Sci 2020;21(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Syed-Abdul MM, Parks EJ, Gaballah AH, et al. Fatty acid synthase inhibitor TVB-2640 reduces hepatic de novo lipogenesis in males with metabolic abnormalities. Hepatology 2020;72(1):103–18. [DOI] [PubMed] [Google Scholar]
- 70.Che L, Paliogiannis P, Cigliano A, et al. Pathogenetic, prognostic, and therapeutic role of fatty acid synthase in human hepatocellular carcinoma. Front Oncol 2019;9:1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ngo HKC, Kim D-H, Cha Y-N, et al. Nrf2 mutagenic activation drives hepatocarcinogenesis. Cancer Res 2017;77(18):4797. [DOI] [PubMed] [Google Scholar]
- 72.Cai L-Y, Chen S-J, Xiao S-H, et al. Targeting p300/CBP attenuates hepatocellular carcinoma progression through epigenetic regulation of metabolism. Cancer Research 2021;81(4):860. [DOI] [PubMed] [Google Scholar]
- 73.Baylin SB, Jones PA. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 2016;8(9):a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Audia JE, Campbell RM. Histone modifications and cancer. Cold Spring Harb Perspect Biol 2016;8(4):a019521–a21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tian Y, Wong VWS, Wong GLH, et al. Histone deacetylase HDAC8 promotes insulin resistance and β-Catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res 2015;75(22):4803. [DOI] [PubMed] [Google Scholar]
- 76.Llopiz D, Ruiz M, Villanueva L, et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol Immunother 2019;68(3):379–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soukupova J, Bertran E, Peñuelas-Haro I, et al. Resminostat induces changes in epithelial plasticity of hepatocellular carcinoma cells and sensitizes them to sorafenib-induced apoptosis. Oncotarget 2017;8(66):110367–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fernández-Barrena MG, Arechederra M, Colyn L, et al. Epigenetics in hepatocellular carcinoma development and therapy: The tip of the iceberg. JHEP Reports 2020;2(6):100167. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Comprehensive review on epigenetics in HCC development and therapy.
- 79.Evert M, Frau M, Tomasi ML, et al. Deregulation of DNA-dependent protein kinase catalytic subunit contributes to human hepatocarcinogenesis development and has a putative prognostic value. Br J Cancer 2013;109(10):2654–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang C, Tang H, Geng A, et al. Rational combination therapy for hepatocellular carcinoma with PARP1 and DNA-PK inhibitors. Proc Natl Acad Sci USA 2020;117(42):26356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cornell L, Munck JM, Alsinet C, et al. DNA-PK—a candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin Cancer Res 2015;21(4):925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jia J, Qiao Y, Pilo MG, et al. Tankyrase inhibitors suppress hepatocellular carcinoma cell growth via modulating the Hippo cascade. PLoS One 2017;12(9):e0184068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang H, Wang J, Zhang S, et al. Distinct and overlapping roles of hippo effectors YAP and TAZ during human and mouse hepatocarcinogenesis. Cell Mol Gastroenterol Hepatol 2021;11(4):1095–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Müller M, Gerndt S, Chao Y-K, et al. Gene editing and synthetically accessible inhibitors reveal role for TPC2 in HCC cell proliferation and tumor growth. Cell Chemical Biology 2021. [DOI] [PubMed] [Google Scholar]
- 85.Han SR, Lee CH, Im JY, et al. Targeted suicide gene therapy for liver cancer based on ribozyme-mediated RNA replacement through post-transcriptional regulation. Molecular Therapy - Nucleic Acids 2021;23:154–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deng Z, Chen G, Liu S, et al. Discovery of methyl 3-((2-((1-(dimethylglycyl)-5-methoxyindolin-6-yl)amino)-5-(trifluoro-methyl) pyrimidin-4-yl)amino)thiophene-2-carboxylate as a potent and selective polo-like kinase 1 (PLK1) inhibitor for combating hepatocellular carcinoma. Eur J Med Chem 2020;206:112697. [DOI] [PubMed] [Google Scholar]
- 87.Yoo BK, Emdad L, Gredler R, et al. Transcription factor Late SV40 Factor (LSF) functions as an oncogene in hepatocellular carcinoma. Proc Natl Acad Sci USA 2010;107(18):8357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rajasekaran D, Siddiq A, Willoughby JL, et al. Small molecule inhibitors of Late SV40 Factor (LSF) abrogate hepatocellular carcinoma (HCC): Evaluation using an endogenous HCC model. Oncotarget 2015;6(28):26266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bandiera S, Pfeffer S, Baumert TF, Zeisel MB. miR-122 – A key factor and therapeutic target in liver disease. J Hepatol 2015;62(2):448–57. [DOI] [PubMed] [Google Scholar]
- 90.Zhou Y, Ren H, Dai B, et al. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J Exp Clin Cancer Res 2018;37(1):324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang T, Xu L, Jia R, Wei J. MiR-218 suppresses the metastasis and EMT of HCC cells via targeting SERBP1. Acta Biochimica et Biophysica Sinica 2017;49(5):383–91. [DOI] [PubMed] [Google Scholar]
- 92.Hong Y, Peng Y, Guo ZS, et al. Epitope-optimized alpha-fetoprotein genetic vaccines prevent carcinogen-induced murine autochthonous hepatocellular carcinoma. Hepatology 2014;59(4):1448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu Z, Lu Z, Jing R, et al. Alarmin augments the antitumor immunity of lentiviral vaccine in ectopic, orthotopic and autochthonous hepatocellular carcinoma mice. Theranostics 2019;9(14):4006–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu M, Luo H, Fan M, et al. Development of GPC3-Specific chimeric antigen receptor-engineered natural killer cells for the treatment of hepatocellular carcinoma. Mol Ther 2018;26(2):366–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang Z, Jiang X, Chen S, et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front Immunol 2017;7:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nishida T, Kataoka H. Glypican 3-targeted therapy in hepatocellular carcinoma. Cancers (Basel) 2019;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li G, Liu D, Cooper TK, et al. Successful chemoimmunotherapy against hepatocellular cancer in a novel murine model. J Hepatol 2017;66(1):75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.DuPage M, Mazumdar C, Schmidt LM, et al. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012;482(7385):405–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lee J, Liao R, Wang G, et al. Preventive inhibition of liver tumorigenesis by systemic activation of innate immune functions. Cell Rep 2017;21(7):1870–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Galicia-Moreno M, Silva-Gomez JA, Lucano-Landeros S, et al. Liver cancer: therapeutic challenges and the importance of experimental models. canadian Journal of Gastroenterology and Hepatology 2021;2021:8837811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Anstee QM, Reeves HL, Kotsiliti E, et al. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16(7):411–28. [DOI] [PubMed] [Google Scholar]
- 102.Léveillé M, Estall JL. Mitochondrial Dysfunction in the Transition from NASH to HCC. Metabolites 2019;9(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pfister D, Núñez NG, Pinyol R, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021;592(7854):450–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, et al. β-Catenin activation promotes immune escape and resistance to anti–PD-1 therapy in hepatocellular carcinoma. Cancer Discov 2019;9(8):1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Finn RS, Ryoo B-Y, Merle P, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: A randomized, double-blind, phase III trial. J Clin Oncol 2019;38(3):193–202. [DOI] [PubMed] [Google Scholar]
- 106.Wilson JM, Kunnimalaiyaan S, Gamblin TC, Kunnimalaiyaan M. MK2206 inhibits hepatocellular carcinoma cellular proliferation via induction of apoptosis and cell cycle arrest. J Surg Res 2014;191(2):280–85. [DOI] [PubMed] [Google Scholar]
- 107.Han P, Li H, Jiang X, et al. Dual inhibition of Akt and c-Met as a second-line therapy following acquired resistance to sorafenib in hepatocellular carcinoma cells. Mol Oncol 2017;11(3):320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jeong S-H, Kim H-B, Kim M-C, et al. Hippo-mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J Clin Invest 2018;128(3):1010–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhai B, Hu F, Jiang X, et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther 2014;13(6):1589. [DOI] [PubMed] [Google Scholar]
- 110.Lin J, Sampath D, Nannini MA, et al. Targeting activated Akt with GDC-0068, a novel selective akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res 2013;19(7):1760. [DOI] [PubMed] [Google Scholar]
- 111.Keerthi K, Jilkova ZM, Roth GS, et al. FRI-106 - Effect of novel AKT inhibitor ARQ 751 as single agent and its combination with sorafenib on hepatocellular carcinoma in a cirrhotic rat model. J Hepatol 2017;66:S459–S60. [Google Scholar]
- 112.Roth GS, Macek Jilkova Z, Zeybek Kuyucu A, et al. Efficacy of AKT inhibitor ARQ 092 compared with sorafenib in a cirrhotic rat model with hepatocellular carcinoma. Mol Cancer Ther 2017;16(10):2157. [DOI] [PubMed] [Google Scholar]
- 113.Huynh H, Pierce Chow KH, Soo KC, et al. RAD001 (everolimus) inhibits tumour growth in xenograft models of human hepatocellular carcinoma. J Cell Mol Med 2009;13(7):1371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hsieh AC, Liu Y, Edlind MP, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012;485(7396):55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xie Z, Wang J, Liu M, et al. CC-223 blocks mTORC1/C2 activation and inhibits human hepatocellular carcinoma cells in vitro and in vivo. PLoS One 2017;12(3):e0173252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chang Z, Shi G, Jin J, et al. Dual PI3K/mTOR inhibitor NVP-BEZ235-induced apoptosis of hepatocellular carcinoma cell lines is enhanced by inhibitors of autophagy. Int J Mol Med 2013;31(6):1449–56. [DOI] [PubMed] [Google Scholar]
- 117.Dai W, Wang F, Lu J, et al. By reducing hexokinase 2, resveratrol induces apoptosis in HCC cells addicted to aerobic glycolysis and inhibits tumor growth in mice. Oncotarget 2015;6(15):13703–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wong CC-L, Au SL-K, Tse AP-W, et al. Switching of pyruvate kinase isoform L to M2 promotes metabolic reprogramming in hepatocarcinogenesis. PLoS One 2014;9(12):e115036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jia W, Xie G, Jia W. Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat Rev Gastroenterol Hepatol 2018;15(2):111–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang F, Huang X, Yi T, et al. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 2007;67(3):863. [DOI] [PubMed] [Google Scholar]
- 121.Yang C, Wang S, Ruan H, et al. Downregulation of PDK4 increases lipogenesis and associates with poor prognosis in hepatocellular carcinoma. J Cancer 2019;10(4):918–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tseng P-L, Wu W-H, Hu T-H, et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci Rep 2018;8(1):3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen C-L, Uthaya Kumar Dinesh B, Punj V, et al. NANOG metabolically reprograms tumor-initiating stem-like cells through tumorigenic changes in oxidative phosphorylation and fatty acid metabolism. Cell Metab 2016;23(1):206–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tan JL, Li F, Yeo JZ, et al. New high-throughput screening identifies compounds that reduce viability specifically in liver cancer cells that express high levels of SALL4 by inhibiting oxidative phosphorylation. Gastroenterology 2019;157(6):1615–29.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu B, Fang M, He Z, et al. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis 2015;6(11):e1980–e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kowalik MA, Guzzo G, Morandi A, et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016;7(22):32375–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dai W, Xu L, Yu X, et al. OGDHL silencing promotes hepatocellular carcinoma by reprogramming glutamine metabolism. J Hepatol 2020;72(5):909–23. [DOI] [PubMed] [Google Scholar]
- 128.Chong YC, Toh TB, Chan Z, et al. Targeted inhibition of purine metabolism is effective in suppressing hepatocellular carcinoma progression. Hepatology Commun 2020;4(9):1362–81. [DOI] [PMC free article] [PubMed] [Google Scholar]