

Abstract
The pancreas is a vital organ with digestive and endocrine roles, and diseases of the pancreas affect millions of people yearly. A better understanding of the pancreas proteome and its dynamic post-translational modifications (PTMs) is necessary to engineer higher fidelity tissue analogues for use in transplantation. The extracellular matrix (ECM) has major roles in binding and signaling essential to the viability of insulin-producing islets of Langerhans. To characterize PTMs in the pancreas, native and decellularized tissues from four donors were analyzed. N-glycosylated and phosphorylated peptides were simultaneously enriched via electrostatic repulsion-hydrophilic interaction chromatography and analyzed with mass spectrometry, maximizing PTM information from one workflow. A modified surfactant and chaotropic agent assisted sequential extraction/on-pellet digestion was used to maximize solubility of the ECM. The analysis resulted in the confident identification of 3,650 proteins, including 517 N-glycoproteins and 148 phosphoproteins. We identified 214 ECM proteins, of which 99 were N-glycosylated, 18 were phosphorylated, and 9 were found to have both modifications. Collagens, a major component of the ECM, were the most highly glycosylated of the ECM proteins and several were also heavily phosphorylated, raising the possibility of structural and thus functional changes resulting from these modifications. To our knowledge, this work represents the first characterization of PTMs in pancreatic ECM proteins. This work provides a basal profile of PTMs in the healthy human pancreatic ECM, laying the foundation for future investigations to determine disease-specific changes such as in diabetes and pancreatic cancer, and potentially helping to guide the development of tissue replacement constructs. Data are available via ProteomeXchange with identifier PXD025048.
Keywords: electrostatic repulsion-hydrophilic interaction chromatography (ERLIC), post-translational modifications (PTM), phosphopeptide, glycopeptide, mass spectrometry, pancreas, extracellular matrix
Graphical Abstract
INTRODUCTION
Protein function is deeply tied to its structure.1 Though there are only approximately 20,000 human genes, the molecular complexity of the human proteome is exponentially increased through post-translational modifications (PTMs), resulting in over ~6 million proteoforms.2, 3 PTMs play many critical biological roles, from regulating transcription via methylation of histones to signaling degradation of substrate proteins via ubiquitination.4, 5 PTMs, regardless of moiety size, can alter protein structure and may greatly affect downstream biological processes.6
The profiling of PTMs has been greatly enhanced through the use of mass spectrometry (MS)-based analytical tools. When coupled with liquid chromatography (LC), thousands of analytes may be identified and quantified in a single LC-MS experiment. The two most extensively studied PTMs with MS are glycosylation and phosphorylation (most often at Ser, Thr, and Tyr). It is believed that at least half of the human proteome is glycosylated and phosphorylated. These PTMs, furthermore, are the two most abundant and most commonly studied PTMs.7 Aberrant regulation of both glycosylation and phosphorylation may also be implicated in numerous diseases, including pancreatic cancer and Alzheimer disease.8–11
Often, only one PTM is the target for enrichment during sample preparation in many analyses. Most analyses of PTMs via MS utilize a bottom-up workflow where proteins are digested into peptides, often using trypsin. Due to their low stoichiometry, an enrichment step is often needed to have adequate analyte signal for confident identifications of these PTM-containing peptides. Enrichment strategies for glyco- and phosphopeptides have been reviewed previously.12, 13 It is the actions of many protein interactions and PTMs together in vivo, however, that drive biological processes, particularly in the extracellular matrix (ECM). The ECM, also called the matrisome, that surrounds cells not only provides structural support, but also binds membrane receptors, such as integrins, and regulates growth factor diffusion and binding, influencing cell signaling.14, 15 To fully understand in vivo interactions, characterizing multiple PTMs simultaneously can maximize biological insights.16 Several studies have separated and analyzed multiple PTMs at once using gradient elution from materials retaining peptides with both glycosylation and phosphorylation.17–21 Simultaneous analyses can also prove useful in determining cross-talk interactions of proteins through PTMs.22 An example of PTM cross-talk is between O-GlcNAc and phosphorylation.23, 24 This type of cross-talk involves both competition in modifying the same or proximal residues and in regulation of enzymatic function due to structural changes caused by PTMs. Thus, PTM cross-talk has major implications for protein function.
An important consideration for analyses is that the ECM is also unique between tissue types, including pancreatic tissue. The pancreas is an essential organ with both exocrine and endocrine functions. Most of the mass of the organ is comprised of acinar cells which produce digestive enzymes. The islets of Langerhans, which compose 1–2% of total organ volume,25 are comprised of numerous endocrine cell types that produce hormones like insulin. Dysfunction or absence of insulin function characterizes diabetes, which affects millions each year. Pancreas or islet transplantation are the only treatment options that can provide continuous control of blood glucose levels, although isolated islets have impaired survival and function in part due to removal from the surrounding pancreatic microenvironment and loss of cell-ECM interactions.26–29 Characterization of PTMs in the pancreatic ECM may provide clues into cell signaling and other processes that help promote islet viability and proper function.
This present work analyzes N-glycosylation and phosphorylation in the pancreas proteome and matrisome. These two PTMs are heavily implicated in signaling processes and may be involved in cross-talk interactions. To our knowledge, this is the first study characterizing PTMs in the human pancreatic ECM. Further investigation of the modified proteins is needed to elucidate how protein function is altered as a result of PTM-induced structural changes. Analysis of these proteins in pancreatic diseases may also identify disease biomarkers and potential targets for therapeutic intervention.
EXPERIMENTAL
Materials and reagents:
PolySAX LP™ (12 μm, pore size 300 Å) and TopTips™ for the spin-tip enrichment were from PolyLC and Glygen Corp, respectively (Columbia, MD, USA). Bond Elut OMIX C18 pipette tips (10–100 μL) were from Agilent (Santa Clara, CA).
Sodium dodecyl sulfate solution (SDS; 10% in water), iodoacetamide (IAA), Roche cOmplete™, mini, EDTA-free protease inhibitor tablets, and Roche PhosSTOP™ phosphatase inhibitor tablets were from MilliporeSigma (St. Louis, MO). Ammonium acetate, tris base, and urea were from Fisher Scientific (Pittsburgh, PA). C18 Sep-Pak cartridges (1 cc, 50 mg sorbent each) were from Waters (Milford, MA). Dithiothreitol (DTT), lyophilized LysC/trypsin, and frozen sequencing grade trypsin were from Promega (Madison, WI). Pierce™ protein and peptide BCA assays were from Thermo Scientific (Rockford, IL). Optima™ LC/MS grade acetonitrile (ACN), water, and trifluoracetic acid (TFA) were from Fisher Scientific (Waltham, MA).
All solution concentrations refer to concentrations in water.
Organ tissue procurement and processing:
Pancreas tissues were procured by the University of Wisconsin Organ and Tissue Donation Services from donors with no indication of diabetes or pancreatitis, with consent obtained for research from next of kin and authorization by the University of Wisconsin-Madison Health Sciences Institutional Review Board. IRB oversight of the project is not required because it does not involve human subjects as recognized by 45 CFR 46.102(f). The organs were received within 24 h of recovery and trimmed of extra-pancreatic connective tissues, including duodenum, large arteries, and veins. The parenchyma was cut into 1 cm3 cubes and frozen at −80 °C for future use. One piece of frozen pancreas per donor was thawed and rinsed with 1× phosphate-buffered saline (PBS) followed by sterile water, and then manually chopped into small pieces. The pieces were immersed in sterile water and homogenized for 3 s using a kitchen immersion blender, then pelleted (16,100 × g, 5 min). Any floating lipids and fats were removed, and the translucent supernatant was discarded. The pellet was flash frozen and stored at −80 °C.
For decellularization, thawed pancreas pieces were homogenized and treated with 2.5 mM deoxycholate and lipase over an 18-h period and the resulting pancreatic ECM was washed, lyophilized, and stored at −80 °C until analysis, as previously described.30, 31
Tissue lysis and protein extraction:
Sample preparation was based on the surfactant and chaotropic agent assisted sequential extraction/on-pellet digestion (SCAD) method.30 Lyophilized pancreatic tissues (four donors, with a native and decellularized aliquot for each donor) were thawed from −80 °C, added to 200 μL of 4% SDS in 50 mM Tris buffer (pH=8) containing one tablet each of protease and phosphatase inhibitor, then incubated at 95 °C with moderate shaking for 10 min. The tissues were then lysed at 4 °C using a probe sonicator (Thermo Fisher Scientific, 60 W, 20kHz) at 50% amplitude for 45 seconds, with a 30 second rest every 15 seconds. The lysates were then pelleted via centrifugation at 16,000 ×g for 15 min at 4 °C. The supernatants were recovered, and the protein concentrations measured via protein BCA assay according to the manufacturer protocol.
Protein precipitation and digestion:
Approximately 0.5 mg of protein from each sample was then transferred to a separate tube, where proteins were reduced with 5 mM DTT at room temperature for 1 h. Alkylation was done with 15 mM IAA for 30 min in the dark. Alkylation was then quenched by adding DTT to 5 mM. Protein precipitation was done by adding chilled acetone at 8X sample volume and incubating overnight at −20 °C. The precipitate was pelleted, and the supernatant was removed. The pellet was then washed with chilled 80% (v/v) acetone and incubated for 2 h at −20 °C. The precipitate was pelleted, and the supernatant disposed again, followed by air-drying at room temperature. The protein pellets were resuspended in 150 μL of 8 M urea in 50 mM Tris buffer (pH=8) followed by addition of LysC/trypsin (1:100 w/w). Samples were incubated for 4 h at 37 °C for this initial digestion. Samples were then diluted with 50 mM tris to <1 M urea. Trypsin was then added to each sample (1:100 w/w), followed by overnight incubation at 37 °C. Digestion was quenched by addition of TFA to each sample to a concentration of 0.3%.
Peptide clean-up:
Peptide samples were desalted using Sep-Pak cartridges. Cartridges were activated with ACN and 0.1% TFA, followed by sample loading and washing also in 0.1% TFA. Peptides were eluted using 50% ACN and 70% ACN, both with 0.1% FA. Eluted peptides were then dried under vacuum. Peptide concentrations were determined using the peptide BCA assay according to the manufacturer’s protocol and samples were aliquoted.
Spin-tip ERLIC enrichment:
Approximately 200 μg of peptides from each sample were resuspended in 80% ACN, 1% TFA (loading solvent). Enrichment tips were prepared by tightly packing 3 mg cotton until resistance was met. SAX beads (3 mg beads per 100 μg peptide) were resuspended in 0.1% TFA at 10 mg beads per 200 μL and activated with shaking for 15 min. The bead slurry was transferred to each tip over the cotton, with the liquid removed through centrifugation at 0.2k ×g for 2 min. Beads were conditioned with 3× 200 μL using the following solvents with mild centrifugation: ACN, 100 mM NH4Ac, 1% TFA, and loading solvent. Peptides were then loaded on the tips via slow centrifugation at 0.1k ×g for 5 min, with the flow-through reapplied to the tips. Peptides were washed with 5× 200 μL loading solvent then 1× 200 μL 80% ACN, 0.1% FA with mild centrifugation. Peptides were eluted with 200 μL of 0.1% FA and 300 mM KH2PO4 in 10% ACN with mild centrifugation. The salt elution was desalted using C18 OMIX tips according to the manufacturer’s protocol. All elutions were dried under vacuum.
UPLC-MS/MS and data analysis:
Samples were analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer coupled to a Dionex UltiMate 3000 UPLC (Thermo Fisher Scientific). Samples were either resuspended in 0.1% FA or 0.1% FA with 3% ACN. Samples were loaded onto a 15 cm capillary (75 μm i.d.) packed using 1.7 μm diameter Ethylene Bridged Hybrid (BEH) C18 material, with the integrated emitter tip in line with the instrument inlet. Each sample was analyzed in technical duplicate. More details about the UPLC-MS/MS methods can be found in Table S1.
Raw data files were searched using Byonic (Protein Metrics Inc., San Carlos, CA) embedded within Proteome Discoverer 2.1 (Thermo Fisher Scientific) against the UniProt Homo sapiens reviewed database (Aug. 2020, 20,311 sequences).32, 33 Glycan modifications were specified in Byonic with the embedded mammalian N-glycan database (309 entries) expanded with typical mannose-6-phosphate (M6P) glycans, including HexNAc(2)Hex(4–9)Phospho(1–2), HexNAc(3–4)Hex (4–9)Phospho (1–2), HexNAc(2) Hex(3–4)Phospho(1) and HexNAc(3) Hex(3–4)Phospho(1). Maximum total modifications per peptide spectral match (PSM) were fixed to one common and two rare. Identifications were filtered to 1% 2D false discovery rate (FDR).34 More details about the Proteome Discoverer 2.1 data analysis settings can be found in Table S2. The search results were later filtered to increase identification and PTM localization confidence. Further filters added were unambiguous PSM and protein assignment, Byonic score cut-off >150, |log prob|>1, and delta mod score>10.32, 35 Protein identifications were made based on the identification of at least one unique peptide that passed the filtering criteria. For the glycan-protein network, N-glycopeptides were exclusively categorized into six glycan type categories based on glycan composition: 1) M6P (containing phosphate on glycan), 2) sialic acid (containing NeuAc/NeuGc, may include fucose), 3) fucose (containing fucose but not sialic acid), 4) complex/hybrid (>2 HexNAc), 5) high mannose (2 HexNAc and >5 Hex), 6) paucimannose (2 HexNAc and <5 Hex). R 4.0.4 was used to generate figures, specifically using the tidyverse,36 igraph,37 networkD3,38 ggnetwork,39 alakazam,40 and seqinr41 libraries.
ECM proteins were determined by matching to the MatrisomeDB (available at [http://matrisomeproject.mit.edu/other-resources/human-matrisome/]). Gene ontology (GO) enrichment analysis was performed using the Protein Analysis Through Evolutionary Relationships (PANTHER) overrepresentation test (available at [http://geneontology.org/]). UniProt protein accessions were matched into a gene list against the Homo sapiens genome database. These analyses were done to determine GO biological process, cellular component, and molecular function terms. Statistical significance was determined using Fischer’s exact test with false discovery rate correction. The Sankey diagram was created using SankeyMATIC (available at [https://sankeymatic.com/build/]).
RESULTS AND DISCUSSION
The experimental workflow is shown in Fig. 1. Proteins from native and decellularized samples of pancreatic tissues from four donors were extracted and digested into peptides, followed by ERLIC spin-tip enrichment. Peptides were eluted into four fractions (flow-through, wash, and two elutions), with each fraction analyzed via MS in technical duplicate. Resulting MS spectra were searched against the UniProt Homo sapiens database and an N-glycan database in Byonic.
Fig. 1.
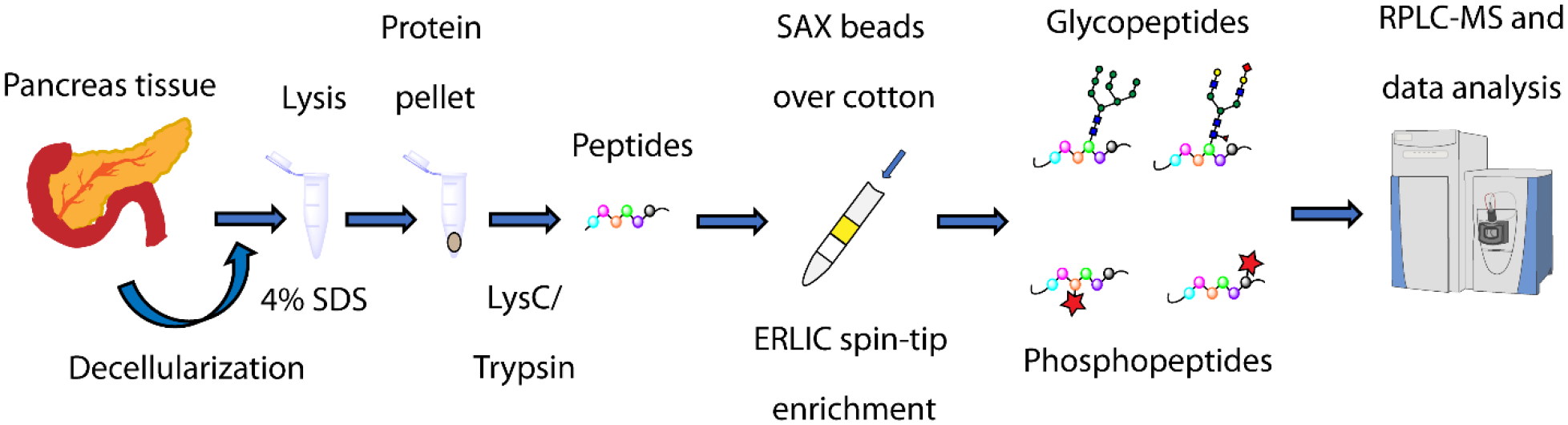
Experimental workflow. Lyophilized human pancreatic tissues were lysed via sonication in 4% SDS. Extracted proteins were precipitated with acetone before resuspension in 8 M urea followed by sequential overnight digestion with LysC and trypsin. Peptides were enriched using an ERLIC spin-tip using SAX material over cotton. The fractions were analyzed with nanoflow RPLC coupled to mass spectrometry, with identifications made using commercially available software.
The effects of tissue decellularization:
Separate samples of tissue from four donors were analyzed in the native or decellularized state. A brief, manual homogenization in water was first needed to break up tissue pieces before protein extraction. Some sample loss during this step, however, may have led to fewer protein, peptide, and PTM identifications. It is worth mentioning, however, that this step would not have led to major cell lysis, as large tissue pieces were still left after this step, which were further broken down during the lysis via sonication. On average, the protein extraction efficiency for the decellularized tissues (10%) was approximately half that of the native tissue (20%), derived by taking the mass of protein extracted as measured by the BCA assay divided by the mass of tissue. Since decellularization of pancreata should leave just the extracellular matrix behind, we sought to determine whether identifications were orthogonal in the decellularized tissues. A summary of the MS results comparing these two sample types is shown in Fig. 2.
Fig. 2.
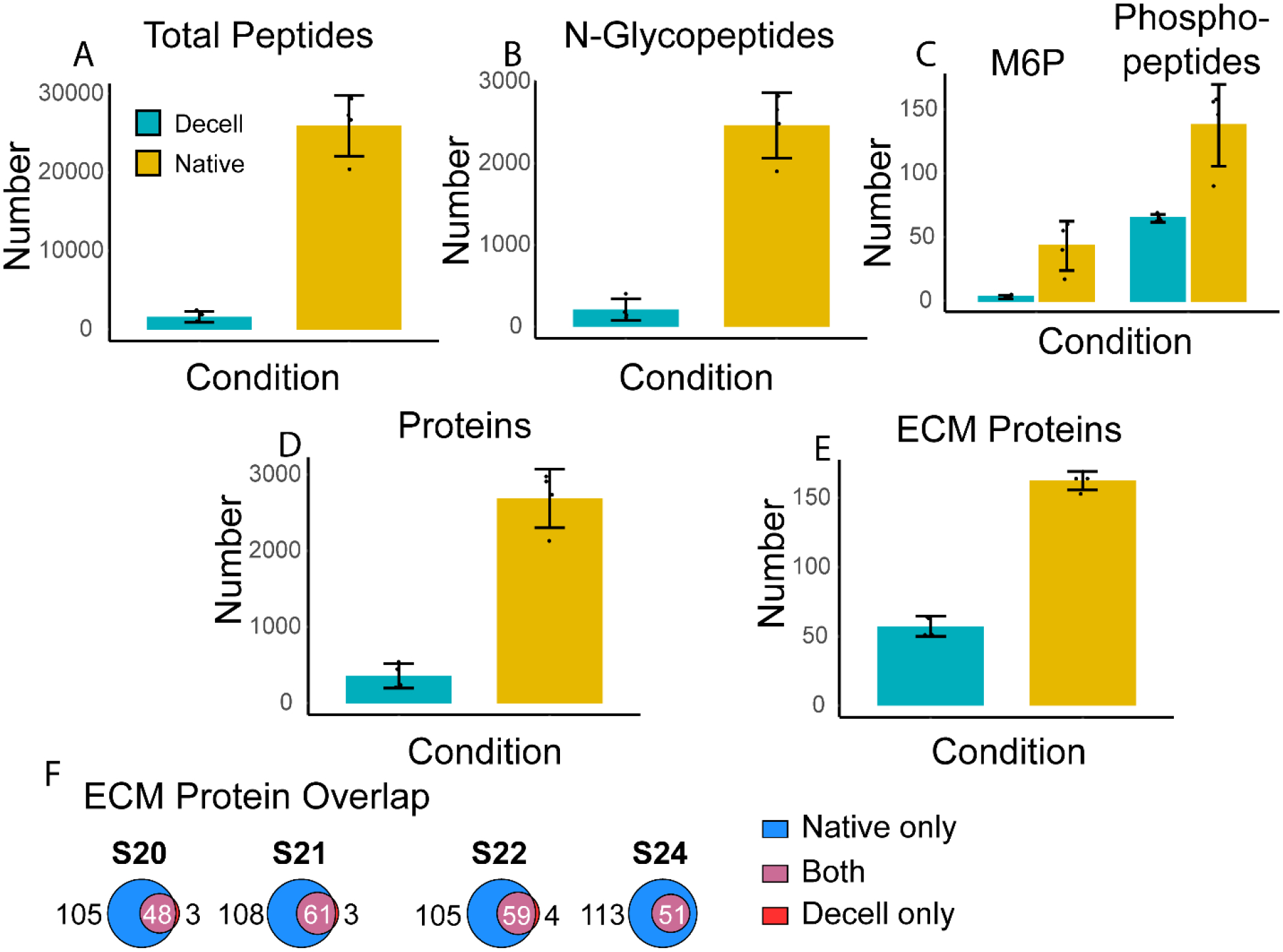
Pancreata from four patients (S20, S21, S22, and S24) were analyzed as native and decellularized samples. These two conditions were compared in the number of identified: A) total peptides, B) N-glycopeptides, C) peptides with the N-glycan mannose-6-phosphate (M6P) modification and phosphopeptides, D) total proteins, and E) ECM proteins. F) compares the overlap of ECM proteins identified from the native samples versus the decellularized samples.
As panels A-E of Fig. 2 show, more species were identified at the protein and peptide levels in the native state than in the decellularized state. Indeed, losses of ECM proteins may differ based on the decellularization protocol used. In this work, both glycosylation and phosphorylation identifications were drastically decreased in the decellularized samples, suggesting that these PTMs may have been released during decellularization or solubilized in the discarded decellularized extracts. This may be true for glycopeptide identifications, which experienced a nearly 90% decrease from native to decellularized tissues.
Proteins were defined as part of the ECM by matching to MatrisomeDB.42 Comparing the overlap of ECM proteins identified, most, if not all, species identified in the decellularized samples were also found in the matching native sample. Though decellularization leaves mainly the ECM scaffold behind from the tissues, the remaining proteins may also be identified in the native tissues. At the PTM level, this result also follows. The added digestion and washing steps needed to decellularize tissues can lead to removal of labile PTMs, including phosphorylation. This suggests that analyzing the decellularized forms of the tissue in addition to the native form using the methods demonstrated in this work results in little added ECM or PTM information. Previous studies have shown clear benefits to decellularization in improving coverage of the ECM using a bottom-up MS workflow,43, 44 though soluble ECM or ECM-associated proteins, including proteases, may be lost during decellularization.
ERLIC spin-tip enrichment:
MS-based analyses of the ECM are difficult due to lower solubility of ECM proteins. To maximize protein solubility, a modified SCAD method was used. The combination of a detergent (SDS) during lysis and a chaotrope (urea) and two enzymes (LysC and trypsin) ensures that hydrophobic ECM proteins are solubilized and digested prior to MS analysis. Intact glycopeptides were analyzed without any N-glycan release. Though deglycosylation may improve the ionization efficiency of peptides, the need to infer whether a peptide bore a glycan by identification of the N-glycosylation amino acid sequon (NXS/T, X ≠ P) was avoided. Furthermore, MS/MS fragmentation can be used to determine glycan compositions and peptide backbones in the same spectra. To enrich glycopeptides and phosphopeptides, a modified spin-tip method using electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) was performed.19 ERLIC uses electrostatic interactions to retain phosphopeptides at low pH and hydrophilic interactions to solubilize glycopeptides in an aqueous layer surrounding strong-anion exchange (SAX) stationary phase surface.45–47 Further addition of cotton to the enrichment tip also contributes to glycopeptide binding. After loading of the peptide mixture on the SAX material, a high organic solvent is used to wash non-modified peptides through the material. Eluting glycopeptides entails using higher aqueous solvent while phosphopeptides require breaking of the electrostatic interactions. In this work, a salt elution (KH2PO4) was used to accomplish this.
Besides confident identification of peptides from mass spectra, it is also important to have confident glycan composition matches and confident modification localization. To achieve higher confidence in identifications, PSMs were also filtered based on Byonic score and delta mod score, which factors in the localization of PTMs to amino acid residues.32
After collating the 64 searched MS data files, 46,190 total peptide groups were confidently identified, of which there were 5738 N-glycopeptides and 333 phosphopeptides. 3650 proteins were identified, of which 517 were N-glycoproteins and 148 were phosphoproteins. 214 of the total proteins were ECM proteins (106 core, 108 associated). PTMs were further mapped to individual residues, identifying 959 N-glycosites (with 262 unique glycan compositions) and 241 phosphosites. The breakdown of PSMs in each fraction of the enrichment is shown in Fig. S1. Representative stepped HCD (which provides sufficient fragmentation for N-glycopeptides48) MS2 spectra for a glyco- and phosphopeptide from ECM proteins are shown in Fig. 3.
Fig. 3.
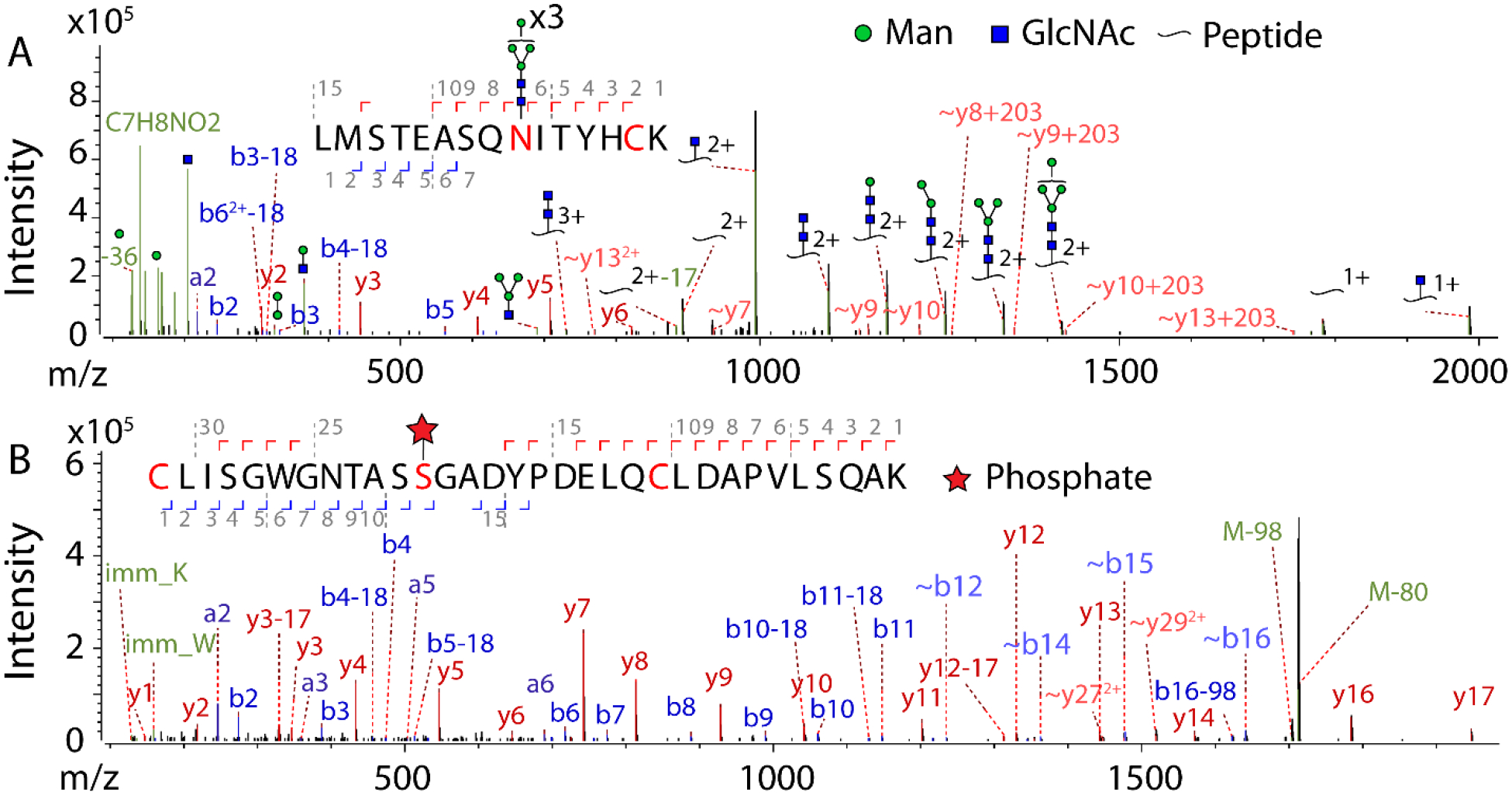
Representative single (not averaged) stepped HCD MS/MS spectra of two modified peptides from ECM proteins. Peaks were filtered at S/N > 5. A) Glycopeptide spectrum with the sequence LMSTEASQNITYHCK, with HexNAc(2)Hex(6) at N9 and carbamidomethylation at C14, from collagen alpha-1(I) chain (Gene: COL1A1, UniProt: P02452) revealing on the protein an N-glycosite at N1365. Score: 802, delta mod score: 591.6, z=3, obs. m/z: 1054.4351, mass error: 0.03 ppm. B) Phosphopeptide spectrum with the sequence CLISGWGNTASSGADYPDELQCLDAPVLSQAK, with phosphorylation at S12 and carbamidomethylation at C22, from trypsin-1 (Gene: PRSS1, UniProt: P07477) revealing on the protein a phosphosite at S150. Score=873.5, delta mod score =22.8, z=2, obs. m/z: 1752.2754, mass error: −1.73 ppm.
Pancreatic proteome profiling:
A summary of analysis of the 3650 identified proteins in this work is shown in Fig. 4.
Fig. 4.
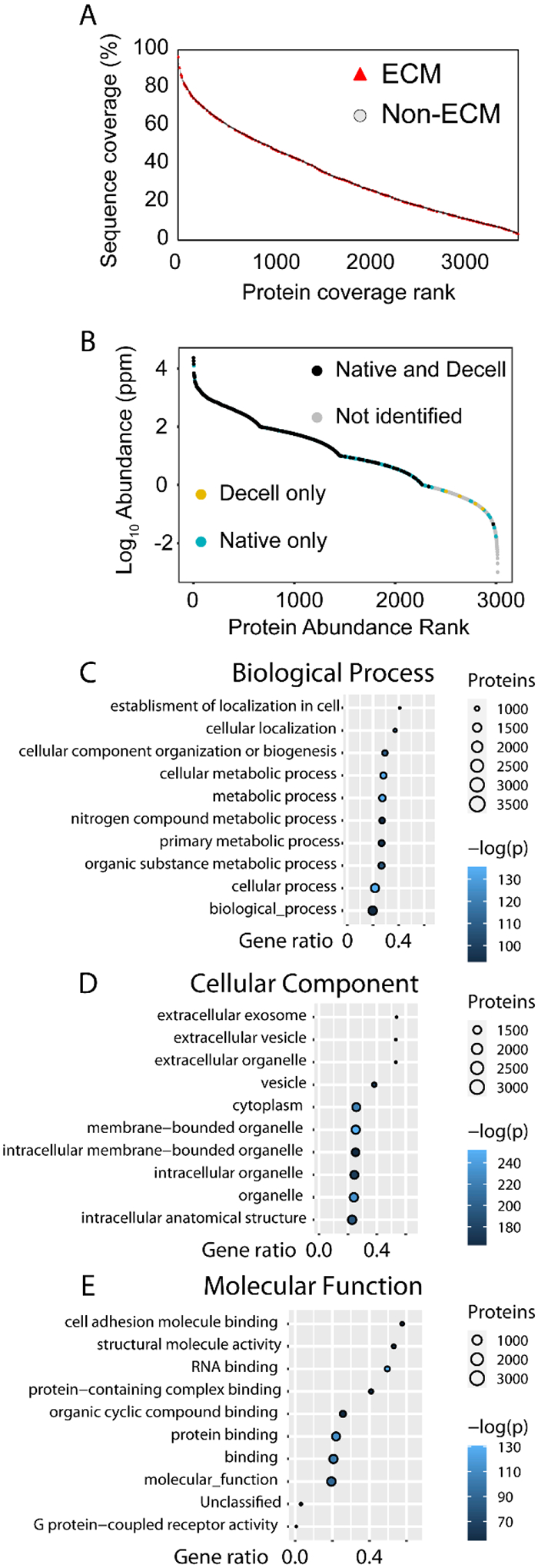
Analysis of sequence coverage and gene ontology (GO) terms of the 3650 proteins identified in this work. GO enrichment analysis was done using PANTHER with Fisher’s exact test to determine statistical significance. A) Plot of protein sequence coverage (%) versus ranking from high to low coverage. ECM proteins are highlighted with red triangles, with selected ECM proteins denoted by rank and role in the table. Non-ECM protein markers were made transparent and smaller for clarity. B) A PDAC dataset from PaxDB was plotted against protein abundance, with proteins also identified in this work in colored markers depending on in which tissue conditions the protein was identified. C-E) Plots for the top ten GO terms ranked by –log(p) for each category sorted by gene ratio. Number of proteins per GO term is denoted by circle size while –log(p) is denoted by color. Gene ratio is defined as the ratio of inputted genes over total genes per term.
Like other tissue types, the dynamic range of the pancreas proteome spans several orders of magnitude and its proteins range in size from several to hundreds of kDa. The sequence coverage of identified proteins is thus expected to vary quite widely. Coverage of all proteins identified is shown in Fig. 4A. ECM protein coverage spans the whole range, from nearly 100% protein sequence coverage of insulin down to 8% sequence coverage of collagen alpha-6(IV) chain, with lower sequence coverages also observed. The reasons for these differences in sequence coverage may vary and can be derived throughout the entirety of sample preparation to the fragmentation in the MS analysis. A likely major factor is the specific cleavage at Lys and Arg by trypsin, which are not as prevalent in more hydrophobic ECM proteins. Fewer potential sites for digestion leads to lower digestion efficiency of the protein, which may not have peptide lengths amenable to MS fragmentation. In addition, protein glycosylation may also block access by trypsin, further preventing digestion and leading to the observed lower sequence coverage of proteins. Complementary digestion with other proteases, such as GluC or AspN, may be used to increase sequence coverage of ECM proteins and decrease protein identification ambiguity.49, 50
To determine the depth of the pancreas proteome sampled during this work, the PAXdb protein abundance database was used to extract abundance values from a pancreatic ductal adenocarcinoma (PDAC) proteome dataset.51, 52 As seen in Fig. 4B, proteins from nearly three-fourths of the dynamic range (over six orders of magnitude) of this pancreatic cancer proteome were also identified in this work, demonstrating the utility of the SCAD protein extraction method at analyzing pancreatic tissue samples. Most of the overlapping proteins were identified in both the native and decellularized tissues; however, there also were proteins only identified in the native tissues. More complete coverage of the protein dynamic range could have been accomplished if more elutions were done or through larger-scale fractionation.
GO enrichment analysis using PANTHER revealed characteristics of the identified proteins.53 Results from these analyses are shown in panels Fig. 4C–E. Out of the top ten biological process GO terms, half explicitly mention metabolism, alluding to the exocrine, digestive functions of the pancreas. Some of the other terms also mention localization, likely due to proteins involved in cellular structure and movement. This is reinforced by the significant cell component GO terms, more than half of which include “extracellular.” The vesicle GO term too is significant, alluding to the intra- and intercellular transport of materials by vesicles. Enriched molecular function GO terms are further consistent with the tissue origin of the proteins, with terms involving binding and cell adhesion.
The PTM level analysis is shown in Fig. 5.
Fig. 5.
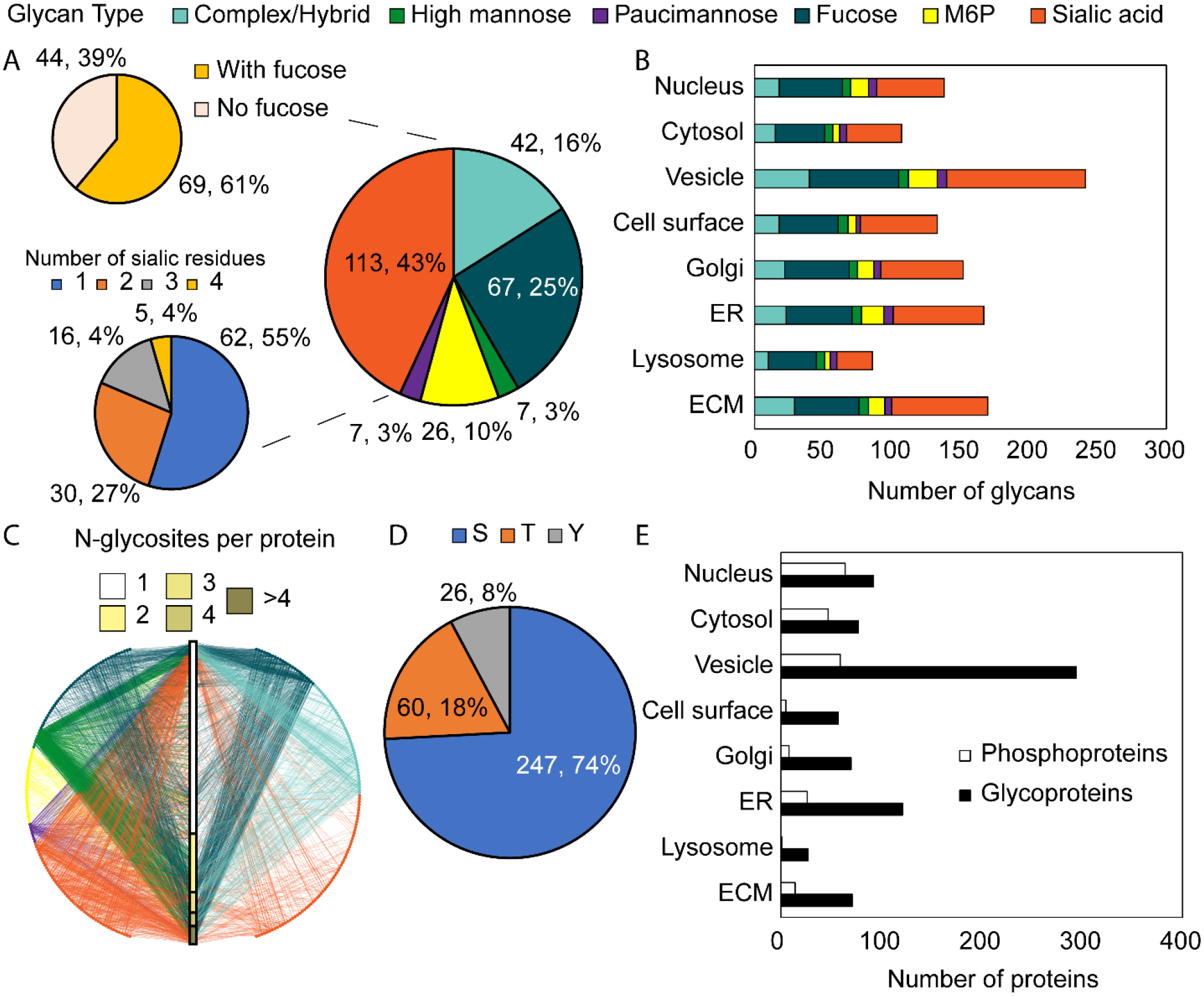
PTM level analysis. A) Breakdown of categories of glycans identified in this work. B) Glycan breakdown by GO cell component term. C) Glycan-protein interaction network showing the relationship between glycoproteins, glycans, and glycosites. Circles represent glycan compositions and intersect the central bar which represents total proteins divided by the number of glycosites. D) Breakdown of phosphorylated residue of the identified phosphosites in this work. E) Breakdown of phosphoproteins and N-glycoproteins by GO cell component term. Note that proteins may have the GO ECM term but may not have been included in MatrisomeDB.
N-glycosylation and phosphorylation (at S, T, or Y) were loaded as common, dynamic modifications during database searching. All N-glycans begin with a core structure of two GlcNAc and three mannose residues. Glycan micro-heterogeneity is further increased by branching of the glycan chain. N-glycans can be binned into six categories depending on their compositions, as described previously in the data analysis methods section. It is worth noting that complex and hybrid-type glycans here are binned together, as no linkage information is derived from glycan composition alone. As shown in Fig. 5A, a majority of the 262 glycan compositions identified were either complex/hybrid or sialic acid type. Further, most of the sialic acid type glycans also bear a fucosyl residue. Approximately half of the sialic acid type glycans bore only one sialic acid residue, while the rest had two to four residues. Increased branching, sialylation, and fucosylation of glycans have been found to mark PDAC progression and aggression.54, 55
Furthermore, M6P type glycans were also included in the N-glycan database used for peptide searching. From 126 M6P-glycopeptides we found 26 unique M6P glycan compositions. This corresponds to 58 M6P-containing glycoproteins, 15 of which are also ECM proteins. M6P glycans are high mannose structures with at least one phosphate group attached to a residue. The M6P part of the glycan tags lysosomal hydrolases for transport.56 Curiously and as shown in Fig. 5B and D, the lysosome GO cell component term did not have the highest number of proteins with M6P glycans or total glycoproteins, with the vesicle and endoplasmic reticulum (ER) having the highest numbers of annotations. The overall glycan type heterogeneity is preserved throughout the examined GO cell component terms, with the greatest number of glycans localized to the vesicle term. Fig. 5C is a glycan-protein interaction network which maps the relationships between the glycan compositions, glycosites, and glycoproteins. Each node represents a unique glycan composition, color coded by type, and maps to the individual glycoproteins with different numbers of glycosites. This is represented by the central bar. There is a vast degree of heterogeneity between the types of glycans and where they are found. Most glycoproteins have just one N-glycosite, though there were proteins found to have four or more.
Fig. 5D shows the breakdown of phosphorylated residues. Most phosphosites were at Ser (74%) or Thr (18%), with fewer sites at Tyr (8%). This is consistent with a previous report and follows from the complex fragmentation patterns of peptides containing pThr and pTyr which may interfere with confident peptide identification.18, 57
Fig. 6 examines a level further down from the PTMs, namely glycans at modified residues and numbers of modified residues on individual proteins.
Fig. 6.
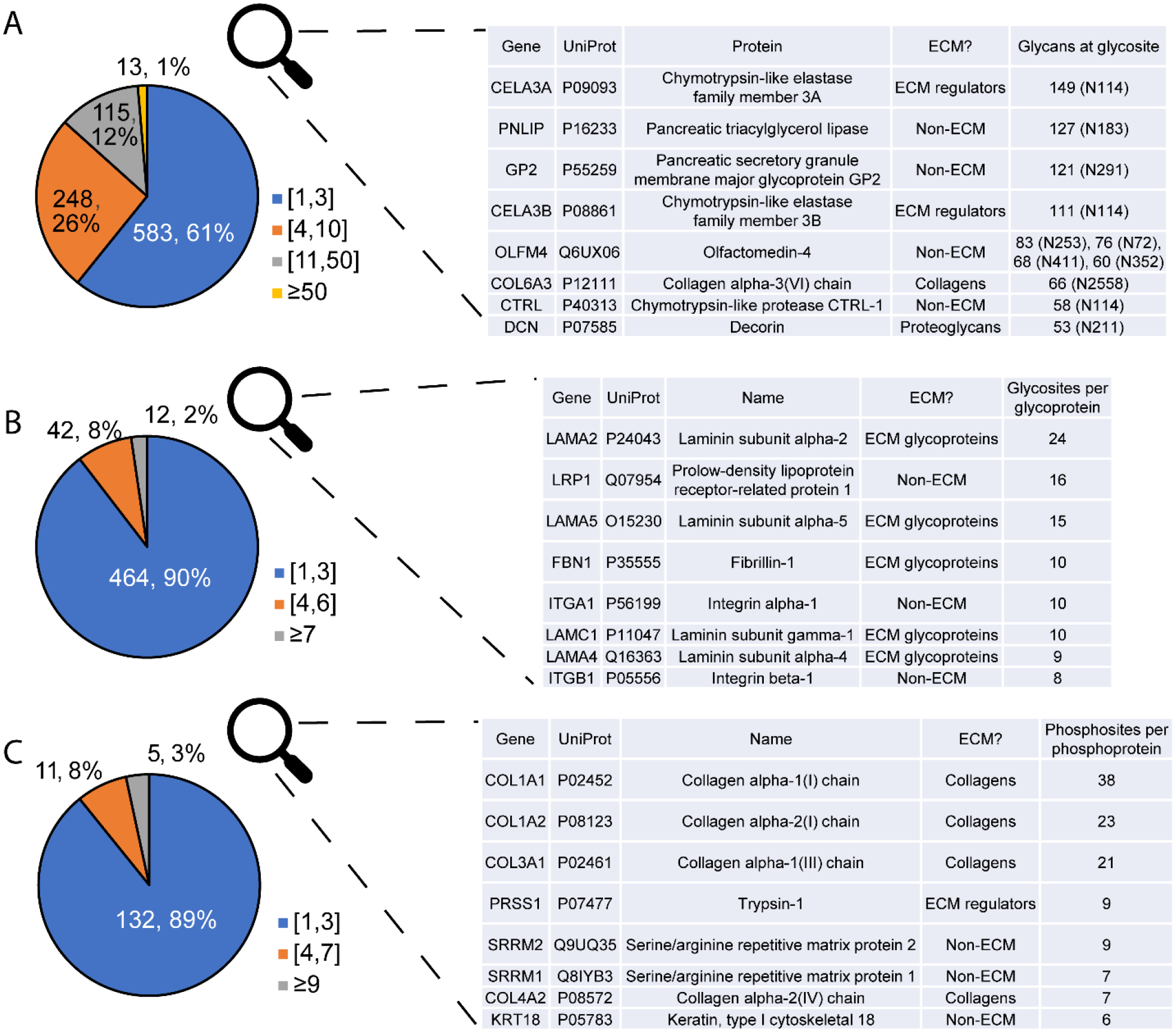
PTM site-level analysis. Top eight proteins for each category are described in the tables. A) Glycan compositions per glycosite, B) Glycosites per glycoprotein, C) Phosphosites per phosphoprotein. ECM proteins are well represented in glycosites with highest glycan microheterogeneity, most glycosites per protein, and most phosphosites per protein.
Informal binning of glycans per glycosite in Fig. 6A shows that most glycosites have a lower level of heterogeneity, with only one to three glycan compositions found. About one-third have higher heterogeneity, with 11 to 50 glycans. A few glycosites, however, have a more extreme level of heterogeneity, with over 50 glycans found. Pancreatic triacylglycerol lipase, an enzyme that digests fats, has huge glycan micro-heterogeneity at N183, with 127 glycans found. Olfactomedin-4 had four glycosites with extreme heterogeneity. This protein is involved in cell adhesion and may play a role in promoting pancreatic cancer cell proliferation.58 Proteins with the glycosites of highest heterogeneity and highest numbers of glyco- and phosphosites (Fig. 6B and C) are described in the inset tables. Many of these proteins are part of the matrisome, with ECM glycoproteins having large numbers of glycosites per protein. Furthermore, among the proteins with the highest number of phosphosites per protein were several collagens. This is consistent with reports showing control of the collagen triple helix structure via phosphorylation.59, 60
Characterization of the pancreatic matrisome:
Out of 3650 identified proteins, 214 proteins are part of the matrisome as defined by MatrisomeDB. The core matrisome consists of collagens, glycoproteins, and proteoglycans that physically make up the ECM. The associated matrisome consists of secreted, affiliated, and regulatory proteins that closely interact with and modify the ECM.
Fig. 7 shows an analysis of ECM proteins, with the Sankey diagram in Fig. 7A showing the contributions of matrisome components to the total N-glycoproteins and phosphoproteins. Of the identified ECM proteins, 99 were N-glycosylated, 18 were phosphorylated, and 9 were found to have both modifications. Furthermore, a scatter plot of the number of glycosites versus the number of phosphosites on proteins with both modifications is shown in Fig. 7B. ECM proteins occupy the extremes of both axes, with collagen alpha-1(I) chain having the greatest number of phosphosites and laminin subunit alpha-2 having the greatest number of glycosites. Other collagens have several glycosites and numerous phosphosites as well, suggesting interactions of these PTMs with the structural roles of collagen. A previous report showed changes in collagen organization due to glycation (non-enzymatic addition of sugars to proteins), thus it would be expected that glycosylation would also have similar effects.61 This work showed both N-glycosylation and phosphorylation on collagen alpha-1(V) chain. In the pancreas specifically, type V collagen has been implicated in islet organogenesis and insulin binding.62, 63
Fig. 7.
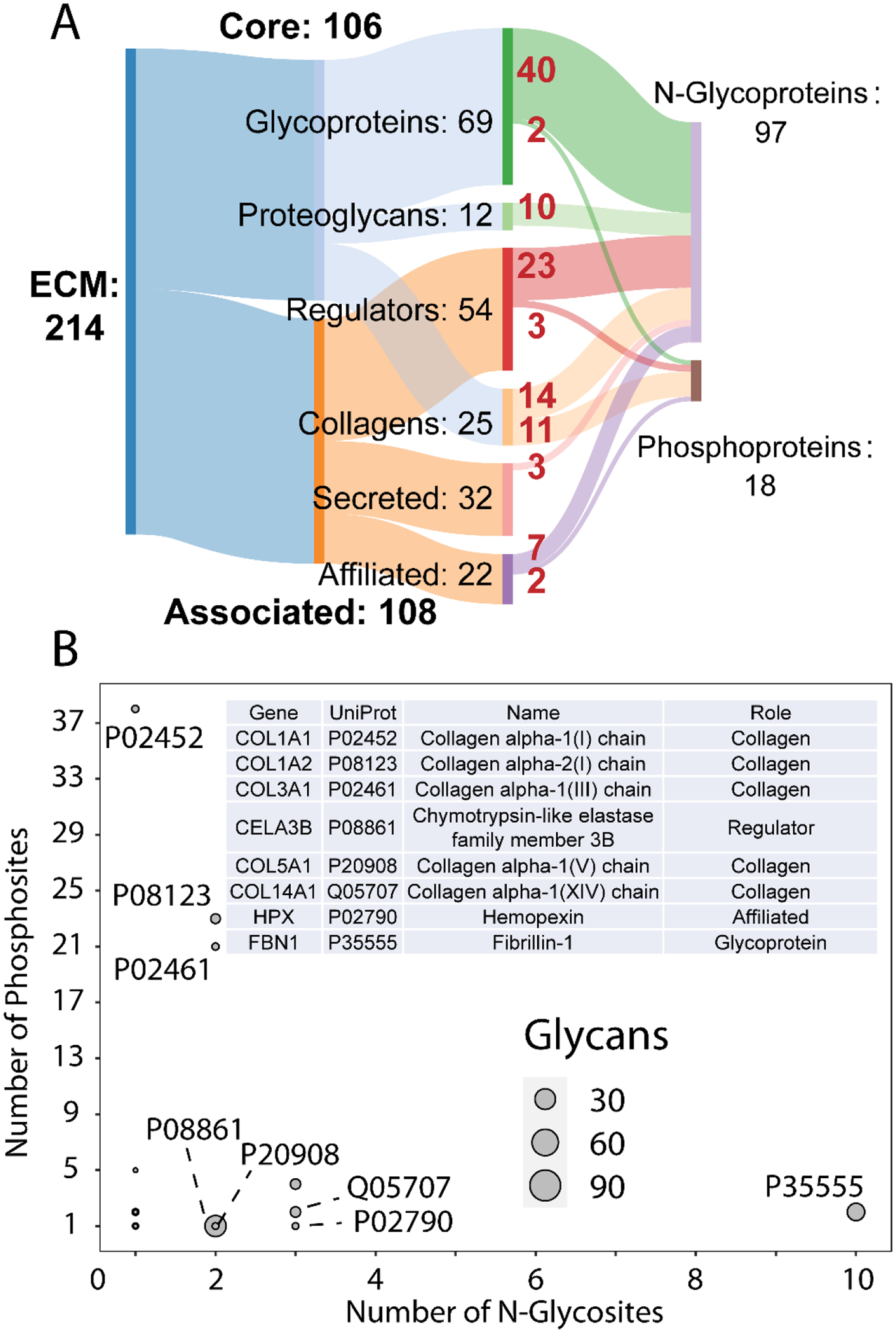
ECM protein level-analysis. A) Sankey diagram showing the breakdown of ECM protein categories. Out of 214 ECM proteins identified, 97 were found to be N-glycosylated and 18 phosphorylated. Numbers in red denote the number of proteins from each ECM protein category to the modified protein type. B) Scatterplot of proteins with at least one glycosite and phosphosite showing the number of phosphosites versus number of glycosites. The number of N-glycans on a protein is denoted by marker size. Selected ECM proteins are denoted by their UniProt accession numbers and are described in the inset table.
Although there has been much MS-based work done in analyzing the ECM proteome or overall PTMs in the pancreas,31, 44, 64–72 this is the first work specifically characterizing PTMs in the pancreatic ECM. Recent work has shown that the pancreas matrisome composition undergoes changes throughout development and maturation.73 It follows that PTMs in the matrisome would change as well, likely mirroring development of pancreas function. One limitation of this work is that while hydroxyproline is also a PTM that plays a major role in ECM protein structure,68, 74 this modification was not considered during protein database searching. The ERLIC enrichment takes advantage of the hydrophilicity of glycans. Hydroxyproline residues on their own may not contribute enough hydrophilic characteristics for the peptides they are modifying to be enriched. The importance of the ECM in diabetes and pancreatic cancer is also widely reported.75–82 Examining changes to PTMs in the pancreas and its ECM could prove useful for biomarker discovery for these diseases. Phosphorylation specifically is a key signaling PTM in regulating insulin secretion.83, 84 Further study correlating extracellular PTMs to insulin secretion mediated by intracellular phosphorylation may provide insights into the mechanisms underlying diabetes. Lastly, M6P glycosylation was also enriched in this ERLIC workflow, consistent with prior work.19, 85 M6P signaling and M6P receptors have been associated with both degradation of proteoglycans in the ECM by heparanase and potentially tumorigenesis in prior reports.86, 87 Further investigation of M6P in pancreatic cancer is thus warranted.
Underscoring this discussion of the importance of PTMs in protein function is the assertion that it is the sum and synergistic interactions of all PTM types in concert that affect protein function, not just one PTM type. Achieving this analysis will be difficult and will require advanced, top-down intact protein characterization and their PTMs via MS. A recent review by Čaval, Heck, and Reiding partially explores this in terms of “glycan meta-heterogeneity,” or glycosylation across multiple sites on an individual protein.88 Cross-talk between PTMs, whether competition for the same site or proximal modifications, increases structural complexity and results in changes to protein function that will require further investigation.
CONCLUSIONS
This work characterizes N-glycosylation and phosphorylation in healthy human pancreata and specifically the ECM using ERLIC enrichment coupled to MS. Functional studies of the PTMs identified in this work are needed to determine their ECM-specific roles, especially if these roles are essential for conferring viability to pancreatic islets. With the knowledge of the basal PTM profile of the pancreatic ECM, mimicking the in vivo pancreatic environment with high fidelity may serve as a foundation for enhancing islet viability and function after islet isolation, and following islet transplantation.
DATA REPORTING
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD025048.89 The Homo sapiens database used to search the proteomics data was from UniProt [https://www.uniprot.org/]. The human matrisome database used was from the Matrisome Project [http://matrisomeproject.mit.edu/other-resources/human-matrisome/].
Supplementary Material
Table S1. UPLC-MS/MS method details.
Table S2. Proteome Discoverer 2.1 data analysis method details.
Table S3. Donor information.
Fig. S1. ERLIC enrichment comparison of PSMs between fractions and specificities.
Figs. S2-S5. Network of statistically enriched terms from identified glycoproteins, phosphoproteins, proteins found to have both glycosylation and phosphorylation, and M6P glycoproteins.
Figs. S6-S7. Comparisons of hydrophobicity of peptide sequences identified in the native tissues versus the decellularized.
Supplemental Data S1. Excel file with sheets detailing the identified proteins and whether they are ECM, glycosylated, and phosphorylated; glycan compositions; glycosites and glycoforms; and phosphosites.
ACKNOWLEDGEMENTS
This research was supported in part by grant funding from the NIH (R21AI126419, R01DK071801, RF1AG052324, U01CA231081, and 1F31DK125021-01), and Juvenile Diabetes Research Foundation (1-PNF-2016-250-S-B and SRA-2016-168-S-B). Data presented here were also in part obtained through support from an NIH/NCATS UL1TR002373 award through the University of Wisconsin Institute for Clinical and Translational Research. The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. We would also like to acknowledge the generous support of the University of Wisconsin Organ and Tissue Donation Organization who provided human pancreas for research. Our research team would like to give special thanks to the families who donated tissues for this study. L.L. acknowledges a Vilas Distinguished Achievement Professorship and the Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
Footnotes
CONFLICTS OF INTEREST
The authors declare the following competing financial interests: J.S.O. is scientific co-founder of Regenerative Medical Solutions, Inc. He is also Chair of the Scientific Advisory Board and has stock equity. All other co-authors declare no competing interests.
REFERENCES
- 1.Uversky VN, Int. J. Mol. Sci, 2016, 17. [Google Scholar]
- 2.Smith LM and Kelleher NL, Nat. Methods, 2013, 10, 186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ponomarenko EA, Poverennaya EV, Ilgisonis EV, Pyatnitskiy MA, Kopylov AT, Zgoda VG, Lisitsa AV and Archakov AI, Int. J. Anal. Chem, 2016, 2016, 7436849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sidoli S, Lopes M, Lund PJ, Goldman N, Fasolino M, Coradin M, Kulej K, Bhanu NV, Vahedi G and Garcia BA, Sci. Rep, 2019, 9, 13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hristova V, Sun S, Zhang H and Chan DW, Clin. Proteomics, 2020, 17, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balana AT, Levine PM, Craven TW, Mukherjee S, Pedowitz NJ, Moon SP, Takahashi TT, Becker CFW, Baker D and Pratt MR, Nat. Chem, 2021, DOI: 10.1038/s41557-021-00648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khoury GA, Baliban RC and Floudas CA, Sci. Rep, 2011, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia YH, Zheng WW and Ye ZH, Eur. Rev. Med. Pharmacol. Sci, 2020, 24, 2358–2367. [DOI] [PubMed] [Google Scholar]
- 9.Choe JW, Kim HJ, Kim JS, Cha J, Joo MK, Lee BJ, Park JJ and Bak YT, Hepatobiliary Pancreat. Dis. Int, 2018, 17, 263–268. [DOI] [PubMed] [Google Scholar]
- 10.Carter AM, Tan C, Pozo K, Telange R, Molinaro R, Guo A, De Rosa E, Martinez JO, Zhang S, Kumar N, Takahashi M, Wiederhold T, Ghayee HK, Oltmann SC, Pacak K, Woltering EA, Hatanpaa KJ, Nwariaku FE, Grubbs EG, Gill AJ, Robinson B, Gillardon F, Reddy S, Jaskula-Sztul R, Mobley JA, Mukhtar MS, Tasciotti E, Chen H and Bibb JA, Proc. Natl. Acad. Sci. U. S. A, 2020, 117, 18401–18411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karikari TK, Pascoal TA, Ashton NJ, Janelidze S, Benedet AL, Rodriguez JL, Chamoun M, Savard M, Kang MS, Therriault J, Schöll M, Massarweh G, Soucy JP, Höglund K, Brinkmalm G, Mattsson N, Palmqvist S, Gauthier S, Stomrud E, Zetterberg H, Hansson O, Rosa-Neto P and Blennow K, Lancet Neurol, 2020, 19, 422–433. [DOI] [PubMed] [Google Scholar]
- 12.Riley NM, Bertozzi CR and Pitteri SJ, Mol Cell Proteomics, 2020, DOI: 10.1074/mcp.R120.002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riley NM and Coon JJ, Anal. Chem, 2016, 88, 74–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skhinas JN and Cox TR, Cell Adh Migr, 2018, 12, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hynes RO and Naba A, Cold Spring Harb. Perspect. Biol, 2012, 4, a004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glover MS, Yu Q, Chen Z, Shi X, Kent KC and Li L, International Journal of Mass Spectrometry, 2018, 427, 35–42. [Google Scholar]
- 17.Cho KC, Chen L, Hu Y, Schnaubelt M and Zhang H, ACS Chem. Biol, 2019, 14, 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Lih TM, Yang G, Chen SY, Chen L, Chan DW, Zhang H and Li QK, Anal. Chem, 2020, DOI: 10.1021/acs.analchem.9b03753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui Y, Yang K, Tabang DN, Huang J, Tang W and Li L, J. Am. Soc. Mass Spectrom, 2019, 30, 2491–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeh TT, Ho MY, Chen WY, Hsu YC, Ku WC, Tseng HW, Chen ST and Chen SF, Anal. Bioanal. Chem, 2019, 411, 3417–3424. [DOI] [PubMed] [Google Scholar]
- 21.Hong Y, Zhao H, Pu C, Zhan Q, Sheng Q and Lan M, Anal. Chem, 2018, 90, 11008–11015. [DOI] [PubMed] [Google Scholar]
- 22.Cui Y, Tabang DN, Zhang Z, Ma M, Alpert AJ and Li L, Anal. Chem, 2021, DOI: 10.1021/acs.analchem.1c00615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leney AC, El Atmioui D, Wu W, Ovaa H and Heck AJR, Proc. Natl. Acad. Sci. U. S. A, 2017, 114, E7255–E7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Z, Gucek M and Hart GW, Proc. Natl. Acad. Sci. U. S. A, 2008, 105, 13793–13798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Misawa R, Zielinski MC, Cowen P, Jo J, Periwal V, Ricordi C, Khan A, Szust J, Shen J, Millis JM, Witkowski P and Hara M, PLoS One, 2013, 8, e67454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daoud J, Petropavlovskaia M, Rosenberg L and Tabrizian M, Biomaterials, 2010, 31, 1676–1682. [DOI] [PubMed] [Google Scholar]
- 27.Llacua LA, Faas MM and de Vos P, Diabetologia, 2018, 61, 1261–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Townsend SE and Gannon M, Endocrinology, 2019, 160, 1885–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stendahl JC, Kaufman DB and Stupp SI, Cell Transplant, 2009, 18, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma F, Liu F, Xu W and Li L, J. Proteome Res, 2018, 17, 2744–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma F, Tremmel DM, Li Z, Lietz CB, Sackett SD, Odorico JS and Li L, J. Proteome Res, 2019, 18, 3156–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bern M, Kil YJ and Becker C, Curr Protoc Bioinformatics, 2012, Chapter 13, Unit13.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orsburn BC, Proteomes, 2021, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bern MW and Kil YJ, J. Proteome Res, 2011, 10, 5296–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riley NM, Hebert AS, Westphall MS and Coon JJ, Nat Commun, 2019, 10, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wickham H, Averick M, Bryan J, Chang W, McGowan LDA, François R, Grolemund G, Hayes A, Henry L and Hester J, Journal of Open Source Software, 2019, 4, 1686. [Google Scholar]
- 37.Csardi G and Nepusz T, Inter. J. Comp. Syst, 2006, 1695, 1–9. [Google Scholar]
- 38.Allaire JJ, Gandrud C, Russell K and Yetman CJ, networkD3: D3 JavaScript Network Graphs from R. R package version 0.4, https://CRAN.R-project.org/package=networkD3, (accessed June 6, 2021).
- 39.Briatte F, ggnetwork: Geometries to Plot Networks with ‘ggplot2’. R package version 0.5.9, https://CRAN.R-project.org/package=ggnetwork, (accessed June 17, 2021).
- 40.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G and Kleinstein SH, Bioinformatics, 2015, 31, 3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Charif D and Lobry JR, in Structural Approaches to Sequence Evolution, Springer, 2007, pp. 207–232. [Google Scholar]
- 42.Naba A, Clauser KR, Ding H, Whittaker CA, Carr SA and Hynes RO, Matrix Biol, 2016, 49, 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gessel M, Spraggins JM, Voziyan P, Hudson BG and Caprioli RM, J. Mass Spectrom, 2015, 50, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knott SJ, Brown KA, Josyer H, Carr A, Inman D, Jin S, Friedl A, Ponik SM and Ge Y, Anal. Chem, 2020, 92, 15693–15698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alpert AJ, J Chromatogr, 1990, 499, 177–196. [DOI] [PubMed] [Google Scholar]
- 46.Alpert AJ, Anal. Chem, 2008, 80, 62–76. [DOI] [PubMed] [Google Scholar]
- 47.Alpert AJ, Hudecz O and Mechtler K, Anal. Chem, 2015, 87, 4704–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Riley NM, Malaker SA, Driessen MD and Bertozzi CR, J. Proteome Res, 2020, 19, 3286–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miller RM, Ibrahim K and Smith LM, J. Proteome Res, 2021, 20, 1936–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller RM, Millikin RJ, Hoffmann CV, Solntsev SK, Sheynkman GM, Shortreed MR and Smith LM, J. Proteome Res, 2019, 18, 3429–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.The Cancer Genome Atlas Research Network, Cancer Cell, 2017, 32, 185–203.e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang M, Herrmann CJ, Simonovic M, Szklarczyk D and von Mering C, Proteomics, 2015, 15, 3163–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi H, Ebert D, Muruganujan A, Mills C, Albou LP, Mushayamaha T and Thomas PD, Nucleic Acids Res, 2021, 49, D394–d403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Holst S, Belo AI, Giovannetti E, van Die I and Wuhrer M, Sci Rep, 2017, 7, 16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kontro H, Joenväärä S, Haglund C and Renkonen R, Proteomics, 2014, 14, 1713–1723. [DOI] [PubMed] [Google Scholar]
- 56.Coutinho MF, Prata MJ and Alves S, Mol Genet Metab, 2012, 105, 542–550. [DOI] [PubMed] [Google Scholar]
- 57.DeGnore J and Qin J, J. Am. Soc. Mass. Spectrom, 1998, 9, 1175–1188. [DOI] [PubMed] [Google Scholar]
- 58.Kobayashi D, Koshida S, Moriai R, Tsuji N and Watanabe N, Cancer Sci, 2007, 98, 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Acevedo-Jake AM, Ngo DH and Hartgerink JD, Biomacromolecules, 2017, 18, 1157–1161. [DOI] [PubMed] [Google Scholar]
- 60.Qiu Y, Poppleton E, Mekkat A, Yu H, Banerjee S, Wiley SE, Dixon JE, Kaplan DL, Lin YS and Brodsky B, Biophys. J, 2018, 115, 2327–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bansode S, Bashtanova U, Li R, Clark J, Müller KH, Puszkarska A, Goldberga I, Chetwood HH, Reid DG, Colwell LJ, Skepper JN, Shanahan CM, Schitter G, Mesquida P and Duer MJ, Sci. Rep, 2020, 10, 3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bi H, Ye K and Jin S, Biomaterials, 2020, 233, 119673. [DOI] [PubMed] [Google Scholar]
- 63.Yaoi Y, Hashimoto K, Takahara K and Kato I, Exp. Cell Res, 1991, 194, 180–185. [DOI] [PubMed] [Google Scholar]
- 64.Naba A, Pearce OMT, Del Rosario A, Ma D, Ding H, Rajeeve V, Cutillas PR, Balkwill FR and Hynes RO, J. Proteome Res, 2017, 16, 3083–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shortreed MR, Wenger CD, Frey BL, Sheynkman GM, Scalf M, Keller MP, Attie AD and Smith LM, J. Proteome Res, 2015, 14, 4714–4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naba A, Clauser KR, Mani DR, Carr SA and Hynes RO, Sci. Rep, 2017, 7, 40495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Petyuk VA, Qian WJ, Hinault C, Gritsenko MA, Singhal M, Monroe ME, Camp DG 2nd, Kulkarni RN and Smith RD, J. Proteome Res, 2008, 7, 3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma F, Sun R, Tremmel DM, Sackett SD, Odorico J and Li L, Anal. Chem, 2018, 90, 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sackett SD, Tremmel DM, Ma F, Feeney AK, Maguire RM, Brown ME, Zhou Y, Li X, O’Brien C, Li L, Burlingham WJ and Odorico JS, Sci. Rep, 2018, 8, 10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vigier S, Gagnon H, Bourgade K, Klarskov K, Fülöp T and Vermette P, Curr Res Transl Med, 2017, 65, 31–39. [DOI] [PubMed] [Google Scholar]
- 71.Raghunathan R, Sethi MK, Klein JA and Zaia J, Mol Cell Proteomics, 2019, 18, 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Asthana A, Tamburrini R, Chaimov D, Gazia C, Walker SJ, Van Dyke M, Tomei A, Lablanche S, Robertson J, Opara EC, Soker S and Orlando G, Biomaterials, 2021, 270, 120613. [DOI] [PubMed] [Google Scholar]
- 73.Li Z, Tremmel DM, Ma F, Yu Q, Ma M, Delafield DG, Shi Y, Wang B, Mitchell SA, Feeney AK, Jain VS, Sackett SD, Odorico JS and Li L, Nat Commun, 2021, 12, 1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rappu P, Salo AM, Myllyharju J and Heino J, Essays Biochem, 2019, 63, 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gress TM, Menke A, Bachem M, Müller-Pillasch F, Ellenrieder V, Weidenbach H, Wagner M and Adler G, Digestion, 1998, 59, 625–637. [DOI] [PubMed] [Google Scholar]
- 76.Bogdani M, Korpos E, Simeonovic CJ, Parish CR, Sorokin L and Wight TN, Curr. Diab. Rep, 2014, 14, 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hayden MR, Patel K, Habibi J, Gupta D, Tekwani SS, Whaley-Connell A and Sowers JR, J. Cardiometab. Syndr, 2008, 3, 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Filipe EC, Chitty JL and Cox TR, Int. J. Exp. Pathol, 2018, 99, 58–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holstein E, Dittmann A, Kääriäinen A, Pesola V, Koivunen J, Pihlajaniemi T, Naba A and Izzi V, Cancers (Basel), 2021, 13, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Honselmann KC, Finetti P, Birnbaum DJ, Monsalve CS, Wellner UF, Begg SKS, Nakagawa A, Hank T, Li A, Goldsworthy MA, Sharma H, Bertucci F, Birnbaum D, Tai E, Ligorio M, Ting DT, Schilling O, Biniossek ML, Bronsert P, Ferrone CR, Keck T, Mino-Kenudson M, Lillemoe KD, Warshaw AL, Fernández-Del Castillo C and Liss AS, Mol. Cancer Res, 2020, 18, 1889–1902. [DOI] [PubMed] [Google Scholar]
- 81.Tian C, Clauser KR, Öhlund D, Rickelt S, Huang Y, Gupta M, Mani DR, Carr SA, Tuveson DA and Hynes RO, Proc. Natl. Acad. Sci. U. S. A, 2019, 116, 19609–19618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zheng J, Hernandez JM, Doussot A, Bojmar L, Zambirinis CP, Costa-Silva B, van Beek E, Mark MT, Molina H, Askan G, Basturk O, Gonen M, Kingham TP, Allen PJ, D’Angelica MI, DeMatteo RP, Lyden D and Jarnagin WR, HPB (Oxford), 2018, 20, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bhatnagar S, Soni MS, Wrighton LS, Hebert AS, Zhou AS, Paul PK, Gregg T, Rabaglia ME, Keller MP, Coon JJ and Attie AD, J. Biol. Chem, 2014, 289, 25276–25286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sacco F, Seelig A, Humphrey SJ, Krahmer N, Volta F, Reggio A, Marchetti P, Gerdes J and Mann M, Cell Metab, 2019, 29, 1422–1432.e1423. [DOI] [PubMed] [Google Scholar]
- 85.Huang J, Dong J, Shi X, Chen Z, Cui Y, Liu X, Ye M and Li L, Anal. Chem, 2019, 91, 11589–11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wood RJ and Hulett MD, J. Biol. Chem, 2008, 283, 4165–4176. [DOI] [PubMed] [Google Scholar]
- 87.Brauker JH, Roff CF and Wang JL, Exp. Cell Res, 1986, 164, 115–126. [DOI] [PubMed] [Google Scholar]
- 88.Caval T, Heck AJR and Reiding KR, Mol Cell Proteomics, 2020, DOI: 10.1074/mcp.R120.002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A and Vizcaíno JA, Nucleic Acids Res, 2019, 47, D442–d450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. UPLC-MS/MS method details.
Table S2. Proteome Discoverer 2.1 data analysis method details.
Table S3. Donor information.
Fig. S1. ERLIC enrichment comparison of PSMs between fractions and specificities.
Figs. S2-S5. Network of statistically enriched terms from identified glycoproteins, phosphoproteins, proteins found to have both glycosylation and phosphorylation, and M6P glycoproteins.
Figs. S6-S7. Comparisons of hydrophobicity of peptide sequences identified in the native tissues versus the decellularized.
Supplemental Data S1. Excel file with sheets detailing the identified proteins and whether they are ECM, glycosylated, and phosphorylated; glycan compositions; glycosites and glycoforms; and phosphosites.