

Abstract
The neuroprotective role of human adipose-derived stems cells (hASCs) has raised great interest in regenerative medicine due to their ability to modulate their surrounding environment. Our group has demonstrated that exosomes derived from hASC (hASCexo) are a cell-free regenerative approach to long term recovery following traumatic brain injury (TBI). Previously, we demonstrated the efficacy of exosome treatment with intravenous delivery at 3 h post TBI in rats. Here, we show efficacy of exosomes through intranasal delivery at 48 h post TBI in mice lengthening the therapeutic window of treatment and therefore increasing possible translation to clinical studies.
Our findings demonstrate significant recovery of motor impairment assessed by an elevated body swing test in mice treated with exosomes containing MALAT1 compared to both TBI mice without exosomes and exosomes depleted of MALAT1. Significant cognitive improvement was seen in the reversal trial of 8 arm radial arm water maze in mice treated with exosomes containing MALAT1. Furthermore, cortical damage was significantly reduced in mice treated with exosomes containing MALAT1 as well as decreased MHCII+ staining of microglial cells. Mice without exosomes or treated with exosomes depleted of MALAT1 did not show similar recovery. Results demonstrate both inflammation related genes and NRTK3 (TrkC) are target genes modulated by hASC exosomes and further that MALAT1 in hASC exosomes regulates expression of full length TrkC thereby activating the MAPK pathway and promoting recovery.
Exosomes are a promising therapeutic approach following TBI with a therapeutic window of at least 48 h and contain long noncoding RNA’s, specifically MALAT1 that play a vital role in the mechanism of action.
Keywords: Exosomes, Adipose derived stem cells, MALAT1, Inflammation, NRTK3, TrkC, TBI
1. Introduction
Traumatic brain injury (TBI) is a leading cause of death and associated disability in the US. From 2006 to 2014, there was a 53% increase in the number of TBI-related emergency department visits, hospitalizations, and deaths. Approximately 155 people die each day in the US from injuries that include a TBI (Prevention, 2019). Currently, treatment options are limited to rehabilitation and symptom management even though damage has the potential to occur for years after insult. A focus on preventing or slowing this secondary cell death therefore is a priority with an urgent need for therapeutic treatment. Therapeutic window was underscored, in a recent review, as an essential aspect of pre-clinical studies as many TBI subjects either do not seek or have limited access to care, however only 16% of the drugs in pre-clinical studies surveyed demonstrated therapeutic windows beyond 24 h (Mohamadpour et al., 2019). Thus, one goal of our studies was to explore extended therapeutic windows.
Neuroinflammation has been widely investigated experimentally as a target for prospective TBI therapeutics due to its contribution to secondary cell death that can potentially occur for years after the initial insult (Acosta et al., 2013; McKee and Daneshvar, 2015; Ferguson et al., 2017; Mouzon et al., 2019). There is an acute peripheral myeloid, microglial and astroglial response to injury within 24 h in controlled cortical impact (CCI) model (Acabchuk et al., 2016). For example, in aged mice there is a chronic and exaggerated infiltration of peripherally derived monocytes that is closely linked to chronic cognitive deficits (Morganti et al., 2016, 2019; Chou et al., 2018; Bachstetter et al., 2020). Our previously published data using RNAseq show the hASC exosomes clearly reduce inflammatory pathways following CCI and down regulate expression of MHC, IBA1 and GFAP in young rodents (Patel et al., 2018).
Neurotrophin 3 (NT3) regulates neuronal survival, morphology, excitability, axon conduction velocity, migration, neurite outgrowth through activation of its receptor TrkC. TrkC is a membrane-bound receptor that, upon neurotrophin NT3 binding, undergoes autophosphorylation and then phosphorylates members of the mitogen-activated protein kinases (MAPK aka ERK) pathway. MAPK signaling leads to cell differentiation and proliferation. As a result of alternative splicing, the NRTK3 gene has two isoforms: the TrkC full length (TrkC_FL) which has the tyrosine kinase domain and TrkC truncated (Trk_T1) which lacks the intracellular domain but transmembrane and extracellular domains are identical to TrkC_FL. Hence, TrkC_T1 can bind to NT3 robustly but is unable to transduce signals associated with its tyrosine kinase activity.
Exosomes are a critical part of the hASC secretome, containing cargo that limits the secondary cell death following TBI by modulating neuroinflammation. MALAT1, a long noncoding RNA (lncRNA), is a significant component of the cargo and controls key biological processes that includes inflammation. MALAT1 is shown to regulate gene expression and splicing in several systems (Eissmann et al., 2012; Gutschner et al., 2013; Liu et al., 2015). MALAT1 is of particular interest as it is highly expressed in hASC exosomes (Patel et al., 2016), confirmed by RNAseq of hASC exosomes, and it mediates alternative splicing of genes to influence the genomic landscape.
Exosomes are an ideal vector for delivery due to their ability to evade immune rejection, modify immune responses and their unique capability of honing to injured tissue. Previously, we demonstrated the efficacy of exosome treatment with intravenous delivery at 3 h post TBI in rats. Here, we show efficacy of exosomes through intranasal delivery at 48 h post TBI in mice lengthening the window of treatment and therefore allowing for a more applicable therapeutic approach.
2. Materials and methods
2.1. Exosome isolation and collection from ASCs
hASC (Zenbio, catalog #ASC-F) were trypsinized and cell pellets were transfected with or mCherry (2 μg, Addgene, 128744) (Shi et al., 2019) or pMAX GFP using Nucleofector® kit (Lonza, catalog #VPE-1001). The cell/DNA solution was transferred to a cuvette and the program initiated (0.34 kV, 960 μF). Medium (500 μL) was added immediately and cells were gently transferred to 100 mm plates and allowed to grow for 48 h. To deplete MALAT from hASC exosomes, we used MALAT1 antisense oligonucleotide (ASO; ID: 39524 ASO from Ionis Pharmaceuticals), validated for specificity and designed for efficient uptake by cells. The ASOs were added to hASC and incubated for 48 h. The expression levels of MALAT1 were verified in the exosomes using human MALAT1 primers in qPCR. Exosomes were isolated from conditioned media (CM) and verified that exosomes contain GFP as previously described by our lab (Patel et al., 2016; El Bassit et al., 2017). Briefly, CM derived from hASC was collected after 48h and centrifuged at 3000 g for 15 min to remove dead cells. ExoSpin™ (Cell Guidance system; Catalog EX05) reagent was added to the CM and incubated for 20 min at room temperature along with XenoLight 1,1-dioctadecyl-3,3, 3,3-tetramethylindotricbocyanine iodide (DiR) (catalog #125964; Caliper Life Sciences). Following centrifugation at 1500g for 30 min to remove cellular debris, the supernatant was applied to top of ExoSpin columns and centrifuged at 50×g for 60 s. Exosomes were eluted in PBS by centrifugation at 50×g for 60 s. Nanoparticle tracking analysis from NanoSight (NTA3.1, Build 3.1.46 RRID SCR-014239) was used to analyze peak diameter and concentration of exosomes obtained from 106 hASC. Analysis showed exosomes size to be 89±7 nm.
2.2. Animal model and surgical procedures
The University of South Florida Institutional Animal Care and Use Committee (IACUC) approved all experimental procedures with animals. All mice were housed under normal conditions (20 °C, 50% relative humidity, and a 12 h light/dark cycle). All studies were performed by personnel blinded to the treatment condition.
A total of C57BL6 male mice were subjected to either TBI by controlled cortical impact or sham surgery of a craniectomy only. The mice were randomly distributed into the following groups: Surgery with no TBI (Sham control SHAM; N = 15), TBI no treatment (TBI; N = 30), TBI with exosomes (TBI+EXO; N = 21), and TBI with exosomes depleted of MALAT1 (TBI+EXO-M; N = 21). Deep anesthesia was administered to mice in a stereotaxic frame (David Kopf Instruments) using 1–2% isoflurane in nitrous oxide/oxygen (60/30%). Animals were secured on a stereotaxic frame, eye ointment was administered and the skull was exposed by a midline excision. From bregma, a center with the coordinates of −0.2 mm anteroposterior and −0.2 mm mediolateral was identified and an approximate 4 mm diameter craniectomy performed using a handheld drill. The resultant bone flap was discarded. The brain was impacted with a 3 mm diameter impactor tip rotated 20° on the vertical axis to adjust for curvature of the brain at the cerebral cortex with a velocity of 4.0 m/s reaching a depth of 0.95 mm (mild TBI) and a dwell time of 300 ms. Body temperature of the animals was regulated during surgery with thermal heating pad. Following surgery, scalps were sutured closed and mice were transferred to a recovery cage on top of a heating pad until fully ambulatory. All animals were closely monitored postoperatively. Mice were maintained on a regular rodent diet throughout experiment.
2.3. Intranasal delivery of exosomes
48 h following TBI surgery, mice were lightly anesthetized with 1–2% isoflurane in nitrous oxide/oxygen (60/30%) and intranasal delivery of treatment was given in equal volumes bilaterally between nostrils. Sham mice and TBI no treatment received PBS intranasally, whereas TBI+EXO and TBI+EXO-M groups received exosomes with and without MALAT1, respectively. Volume administered was determined based on the concentration of exosomes so that each mouse would receive a total of 10 μg exosomes, with no more than 5 μl administered per nostril and 10 min of recovery between intranasal injections before moving to the opposite nostril.
2.4. Behavioral testing
2.4.1. Elevated body swing test (EBST)
The elevated body swing test measures asymmetric motor behavior seen in unilateral brain injuries (Borlongan and Sanberg, 1995). Briefly, a mouse is placed into an empty cage and allowed to habituate for 2 min. To begin, the mouse was in a neutral position with all four paws on the ground and held about 1 inch from the base of its tail. The mouse was lifted from the surface and a swing was recorded when the mouse moved more than 10° from the vertical axis to either side. A total of 20 swings were recorded for each animal. An animal with a score of 10 exhibit no bias, whereas a score of 14 or higher indicates asymmetry and an associated motor deficit. The total number of swings made to the higher side were summed and divided by the total animals per group, computing an average number of biased swings per treatment group. Following traumatic brain injury, mice were tested 24 h post TBI and divided into 3 groups based on their EBST score in order to give an equal average score between groups before treatment was administered.
2.4.2. Radial arm water maze
At 5 weeks post TBI injury, 8 arm radial arm water (RAWM) was performed to assess cognitive function. RAWM is a hippocampal-dependent memory task that assesses spatial learning through the use of visual cues and a hidden platform. Briefly, an 8 arm RAWM (BIOSEB) was placed in 4 foot diameter water tank with water maintained at 20 °C. Two platforms were utilized: a visible platform that sat 1 inch above the water level and had a colorful flag identified it and also a hidden platform that remained 1 inch under the water line. White paint was used to cloud the water and further hide the hidden platform. A total of 15 trials was run on Days 1 and 2 in the learning phase followed by 15 trials on Day 3 of reversal testing. Each trial began with a mouse being placed in the start arm and the platform in the assigned goal arm. The goal arm remained the same for a mouse throughout the learning trials of Days 1 and 2, though the starting arm would change randomly for every trial. On Days 1 and 2, the platform alternated between hidden and visible for the first 6 trials, while the remaining trials only used the hidden platform. On Day 3, all trials only utilized the hidden platform and were placed in their “reversal” position that was directly across from their previous goal arm location. Mice were tested in two blocks of 6 trials followed by one block of 3 trials to allow for a proper resting period. On Day 3, all trials only utilized the hidden platform. Trials were run for a maximum of 60 s; if the mouse found the platform within the 60 s, they were given 30 s to remain on the platform before being returned to their home cage however, if the mouse did not find the platform at the end of 60 s, they were guided to the platform and sat on platform for 30 s. Errors were counted when a mouse entered an arm that did not contain a platform or if no arm was chosen for 15 s. RAWM performance analysis was done by analyzing the number of errors in each of the 3 blocks per day, for a total of 9 blocks over the three days. Numbers were as follows: SHAM N = 8, TBI N = 14, TBI+EXO N = 14, TBI+EXO-M N = 20, CONTROL N = 10.
2.5. Confocal imaging
To visualize localization of the exosomes post TBI, exosomes were labeled with GFP as described above. Exosomes were administered intranasally at a concentration of 10ug at 48 h after injury. Twenty-four hours later, mice were anesthetized and brains were collected. Following brains being submerged in 4% PFA solution and 30% sucrose, they were sectioned coronally at a thickness of 40 μm. Brains were stained with anti-GFP (Abcam catalog # AB13970) and imaged with confocal microscope to determine initial fate of intranasal exosomes. Further, to visualize whether exosomes co-localized with neurons, astrocytes or microglia, GFP labeled exosomes were co-labeled with anti-GFP and anti-MAP2 (neurons), anti-GFAP (astrocytes), and anti IBA-1 (microglia) and verified with confocal imaging.
2.6. Immunohistochemical analysis
2.6.1. Brain collection and sectioning
At 6 weeks post TBI, animals were anesthetized and transcardially perfused with 0.1M phosphate buffered saline (PBS) at pH 7.2 followed by 4% paraformaldehyde (PFA) in PBS at pH 7.2. Brain was collected and stored in 4% PFA solution overnight followed by 30% sucrose in PBS until brains had completely sunk. Brains were sectioned coronally at a thickness of either 40 μm with a cryostat and stored in −20 °C freezer in a cryoprotectant solution.
2.6.2. Lesion volume analysis
For lesion volume analysis, every sixth coronal section was used spanning the entire TBI insult area. Tissues were mounted onto glass slides and allowed to dry. Slides were placed in hematoxylin QS (Vector Laboratories, catalog # H-3404) and allowed to fully develop for 5–10 min, and then washed in tap water for 5 min. Slides were allowed to dry overnight before being dehydrated and cover-slipped with DPX mounting medium. To calculate lesion volume, slides were visualized under a light microscope (Nikon E600) and the impact area was outlined in the ipsilateral hemisphere and measured using Cavalieri estimator (Stereo Investigator software, MicroBrightField Inc.)
2.6.3. Major histocompatibility complex II (MHCII) staining and measurement
Immunostaining was performed on every twelfth section spanning the TBI impact area. Free-floating tissues were treated with 3% hydrogen peroxide/30% methanol in 0.1M PBS to remove endogenous peroxidase activity. Tissues were washed in PBS, and then placed in a blocking buffer solution (10% goat serum/0.3% Triton X-100/PBS) for 1 h. Tissues were then incubated in PBS-TS solution (3% goat serum/0.1% Trition X-100/PBS) with primary antibody anti-MHCII (1:2K) (BD Pharmingen catalog # 55999) overnight at 4 °C on a shaker (60 rpm). The following day, sections were washed in PBS-TS and then incubated with goat anti-rabbit secondary antibody (1:5K). Avidin-biotin substrate complex (Vector Labs, Cat# PK-6100: 1:200 dilution of stock solutions according to manufacturers instructions) was used to amplify signal for 1 h. Sections were developed with 3, 3’ – Diaminobenzidine tetra-hydrochloride (DAB) enhanced with cobalt chloride (Sigma, Cat no: D0426: 0.5 mg/ml DAB, 0.2 mg/ml Cobalt). Free-floating tissues were mounted on glass slides and after drying were dehydrated and cover-slipped using DPX mounting medium. Stained slides were scanned using Zeiss Mirax image scanner. The analysis was performed using NearCYTE image analysis software. A region of interest was drawn onto each tissue and the area of positive staining was quantified.
2.7. Primary neurons
Mouse primary neurons were plated in a 12 well plate and treated as indicated in experiments. Cells were blocked with 1% BSA for 1hr and incubated overnight with primary antibodies (1:250) against TrkC (C-terminal domain detecting TrkC_FL), ERK and p-ERK. Cells were washed with PBS three times and incubated for 1hr in Alexafluor 488 and Alexafluor 577 secondary antibody’s (1:1000, Invitrogen) at room temperature. Cells were washed once more with PBS and stained with DAPI mounting medium. Images were captured using Keyence BZX810 microscope and analyzed using Keyence Analyzer software.
2.8. PCR and quantitative real-time SYBR green qPCR
Total RNA was isolated from either brains, primary neurons or microglia as per experimental setup with TRIzol™ Reagent (Thermo-Fisher Scientific) as recommended by the manufacturer. QPCR was performed using 1.0 μl cDNA and Maxima SYBR Green/Rox qPCR master mix (Thermo Scientific). The primers used are: TrkC_FL sense primer 5′ TGATCCTCGTGGATGGACAG 3′ and anti-sense 5′ CTTCACTAGTAGATTGGCTCC3′; TrkC_T1 sense primer 5′ CCACTTCCTGAAGGAGCCCT 3′ and anti-sense 5′ CCCACTCTGGACCTCAGGTT 3′; IL1β sense primer 5′ CTCGTGGTGTCGGACCCATATGA 3′ and anti-sense 5′ TGAGGCCCAAGGCCACAGGT 3′: GAPDH sense primer 5′ TCACCACCATGGAGAAGGC 3′ and anti-sense 5′ GCTAAGCAGTTGGTGGTGCA 3′: β-actin sense primer 5′ TGTCCACCTTCCAGCAGATGT 3′ and anti-sense 5′ AGCTCAGTAACAGTCCGCCTAGA 3′. Amplification was performed on the ViiA 7 (Applied Biosystems). Real-time qPCR was then performed in triplicate on samples and standards. The plate setup included a standard series, no template control, no RNA control, no reverse transcriptase control, and no amplification control. After primer concentrations were optimized to give the desired standard curve and a single melt curve, relative quotient (RQ) was determined using the ΔΔCT method with GAPDH or HPRT as the endogenous control and control as the calibrator sample. Alternatively, the cDNA was used in PCR using a thermoblock and products were separated on 1% agarose gel. The products were visualized using ProteinSimple and AlphaView software was used for densitometric analysis.
2.9. Western blot analysis
Automated Western blot analysis using Simple WES system (ProteinSimple, Santa Clara, CA, USA) was used. The amount of lysate to antibody was optimized as per manufacturer’s instructions. A concentration of 0.4 mg/mL was found optimal to be used on all antibodies. The samples were separated on 12–230 kDa Wes Separation Module capillary cartridges of Simple Protein Wes system. Rabbit monoclonal antibody specific for phospho-ERK was used at a dilution of 1:10 while antibody specific for ERK and TrkC (C-terminal domain epitope) was used at a dilution of 1:100 in the runs. β-actin was used as a loading control (1:250 dilution of antibody). Anti-rabbit detection module kits were specific for Wes (ProteinSimple) and include Luminol-S, Peroxide, Streptavidin-HRP and anti-rabbit secondary antibody. The proteins are separated by capillary technology and analyzed based on the chemiluminescence signal peaks generated, shown as digital images representing bands as observed in traditional Western blot analysis. Using Compass software (ProteinSimple), the peak areas of TrkC_FL, pERK were estimated and normalized against β-actin.
2.10. Metabolic assay
Mouse primary neurons were plated into a poly-D-lysine coated Seahorse XFp cell culture mini-plate (Agilent Technologies, CA, USA) at a density of 4000 cells per well as determined by optimization cycles. The following day the cells were treated as indicated in experiments. The media was then changed to Seahorse XF Media (supplemented to 100 mM pyruvate, 200 mM Glutamine, and 2.5M Glucose) before being imaged on the Keyence microscope to calculate cell count for normalization. The plate was then incubated in a non-CO2 incubator at 37 °C for 1 h. Seahorse sensor cartridges were prepared and solutions loaded into ports as described for the XFp Mito Stress Test (100 μM oligomycin, 100 μM μM fluoro-carbonyl cyanide phenylhydrazone FCCP, and 50 μM Antimycin A/Rotenone are added to cells sequentially as shown in schematic of Fig. 3). Cells were run in the Seahorse XFp Analyzer. Oxygen consumption rate are measured at intervals of approximately 5–8 min. The measurements were normalized to cell counts and data was analyzed for basal respiration and proton leak using the Agilent Wave software.
Fig. 3.
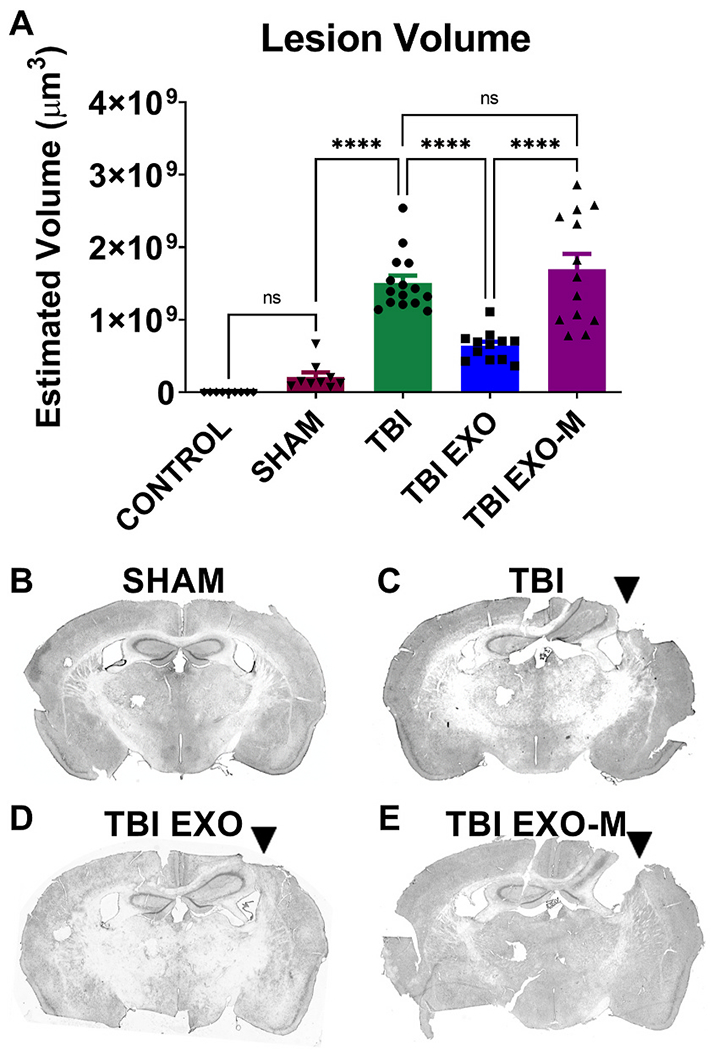
Exosomes reduce the impact lesion size following CCI.
Hematoxylin QS staining was utilized to quantify the impact area following TBI in the ipsilateral hemisphere. Using Cavalieri estimator to measure lesion volume treatment with exosomes (TBI+EXO) significantly reduced lesion volume compared to untreated TBI animals (p < 0.001 One way ANOVA F(4,52) = 36.99 followed by Tukey’s multiple comparisons) 6 weeks post TBI. However, treatment of exosomes depleted of MALAT1 (TBI+EXO-M) did not show rescue following injury when compared to untreated TBI animals. Tukeys post-hoc comparisons are indicated by asterisks on the graph ****p < 0.0001.
2.11. Data analysis
The data presented graphically depicts the group mean and standard error of the mean across treatment groups. Statistical analysis was performed using GraphPad prism software using One-Way ANOVA for most of the IHC comparisons or Two-Way repeated measures ANOVA for the behavioral testing comparison’s. Normalcy was tested using Kolmogorov-Smirnov statistic.
3. Results
3.1. Exosomes containing MALAT1 improve TBI-associated motor impairment
EBST was performed prior to CCI and then at 1 day (prior to treatment), 3, 5, and 7 days post injury. As shown in Fig. 1A EBST revealed significant asymmetry in motor activity in all injured mice at 1 day following TBI as compared to both sham mice and controls. Treatment with exosomes at 48 h after injury led to a significant improvement in EBST behavior compared to TBI only mice on days 3–7 (Two-way repeated measures ANOVA shows significant time, group and interaction effects F(16, 356) = 15.09 for the interaction. Tukey’s multiple comparison shows TBI significantly different form TBI-EXO group at 3, 5, and 7 days (p < 0.001) Control N = 20, Sham N = 8, TBI N = 21, TBI-Exo N = 25, TBI ExO-M N = 20. TBI mice show no significant recovery from either sham or control mice when tested on days 3, 5 and 7 post TBI although there is some improvement in motor behavior across days in the TBI group. TBI mice treated with exosomes depleted of MALAT1 were not different from TBI at any time point and were different from TBI exosome treatment group on Day 3 only (p < 0.001; Fig. 1A, Tukey’s multiple comparisons). These results show treatment with exosomes can significantly improve TBI-associated motor impairment in a MALAT1 dependent manner. To explore if there was a correlation between motor performance and lesion size or late phase inflammation. we ran regression analysis. As shown in Fig. 1C there was a modest correlation between motor performance on Day 3 of EBST and lesion size (R2 = 0.27, DfNDFd 1,34 F = 8.00 p = 0.009 Control N = 6, Sham N = 8, TBI N = 8, TBI-Exo N = 8, TBI EXO-M N = 11). No correlation was observed between motor performance and late phase inflammation.
Fig. 1.
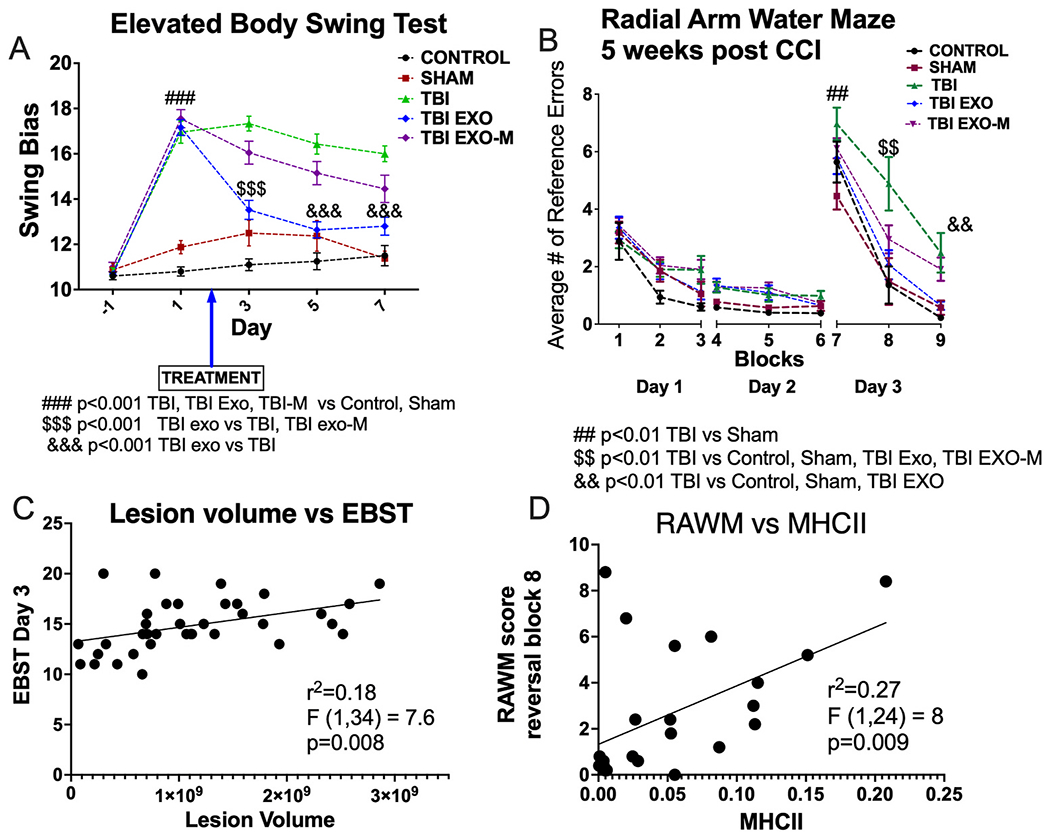
Exosomes improve both motor (EBST) and cognitive (RAWM) behavior.
A) Elevated body swing test records the number of 90° lateral swings to the right or the left and determines if an animal has a swing bias. An animal with a score of 10 exhibits no bias, whereas a score of 12 or higher indicates asymmetry and an associated motor deficit. EBST revealed significant asymmetry in motor activity following TBI when compared with control and sham mice. Following treatment, TBI mice with exosomes (TBI+EXO) significantly rescued the TBI-associated motor deficits relative to TBI. Treatment with exosomes depleted of MALAT1 (TBI+EXO-M) did not significantly improve motor performance on EBST when compared to TBI. (Two-way repeated measures ANOVA shows significant time, group and interaction effects F(16, 356) = 15.09 for the interaction. Tukey’s multiple comparison are indicated by ### (p < 0.001 TBI, TBI+EXO, TBI+EXO-M vs Control, Sham; $$$ p < 0.001 TBI+EXO vs TBI, TBI+EXO-M; &&& p < 0.001 TBI+EXO vs TBI). Animal numbers Control N = 20, Sham N = 8, TBI N = 21, TBI+EXO N = 25, TBI+EXO-M N = 20.
B) Mice were trained on an 8-arm radial arm water maze for 15 trials a day for 2 days to find a hidden platform, represented here in blocks. During training, no differences were observed between groups. One day 3, the platform was “reversed” and placed in the opposite arm the mice had previously learned. Results show the cognitive deficits associated with TBI were significantly rescued for mice treated with exosomes (Two-way repeated measures ANOVA demonstrated a treatment effect F(8, 531) = 77.5 and treatment time interaction F(32, 531) = 1.63.) Tukeys post hoc comparisons are indicated as noted on the figure. ##p < 0.01 TBI vs Sham; $$ p < 0.01 TBI vs CTRL, Sham, TBI+EXO and TBI+EXO-M; && p < 0.01 TBI vs Control, Sham and TBI+EXO. Animal numbers: Control N = 10, sham N = 7, TBI N = 12, TBI_EXO N = 15, TBI+EXO-M N = 20.
3.2. Exosomes containing MALAT1 improve cognitive impairment
Beginning at 5 weeks post injury, mice were trained in an 8 arm radial arm water maze to evaluate their spatial memory. During the acquisition of learning on days 1 and 2, there were no differences in performance between any of the 5 groups tested indicating all mice were able to learn the location of the hidden platform at a similar rate. On day 3, the platform was moved to the opposite arm (reversal). TBI mice with PBS showed a significant deficit in re-learning the platform in all 3 blocks tested compared to sham and control mice (Repeated measures 2-way ANOVA demonstrated a treatment effect F(8, 531) = 77.5 and treatment time interaction F(32, 531) = 1.63 Control N = 10, Sham N = 7, TBI N = 15, TBI-Exo N = 15, TBI Exo-M N = 20). Tukey’s multiple comparisons revealed a TBI effect on all trials on day 3 of testing (Fig. 1B). The cognitive deficits associated with TBI were significantly rescued for mice treated with hASC exosomes during blocks 8 and 9 of testing (Fig. 1B Tukey’s multiple comparisons p < 0.01). TBI mice with exosomes depleted of MALAT1 did show recovery on block 8 but not block 9 on day 3 compared to TBI without exosomes (Fig. 1B, Tukey’s multiple comparisons P < 0.01). These results show treatment with exosomes can significantly improve TBI-associated cognitive impairment in a MALAT1 dependent manner. To explore if there was a correlation between cognitive performance and lesion size or late phase inflammation, we performed a linear regression (Fig. 1D). There was no correlation between cognitive function and lesion size, there was a significant correlation with late phase inflammation (r2 = 0.27; DFn, DF, d 1,22 F = 8.00 p = 0.009 Control N = 5, Sham N = 3, TBI N = 4, TBI-EXO N = 6, TBI EXO-M N = 8).
3.3. Exosomes localize to the site of injury post TBI
To visualize the location of exosomes, exosomes labeled with either mCherry or GFP were administered intranasally 48 h after TBI injury. Mice were euthanized 24 h later and brains were examined using IHC. Immunohistochemical analysis of brain sections revealed the exosomes were easily able to infiltrate the brain and were observed in the highest concentration near to the site of injury (Fig. 2A). Exosomes were also visualized in the ipsilateral corpus callosum and in the ipsilateral and contralateral hippocampus, but in lesser concentrations. Fig. 2B shows mCherry labeled exosomes in the ipsilateral hippocampus dentate gyrus. To determine location in relation to cells within the brain, GFP labeled exosomes were imaged on a confocal microscope with sections following IHC labelling with either IBA1, MAP2 along with GFAP to determine if colocalization was visualized with microglia, neurons, or astrocytes respectively. Confocal microscopy was used to determine that GFP labeled exosomes were indeed co-localized with both IBA1+ microglia (Fig. 2C), and MAP2+ neurons (Fig. 2D) within the dentate gyrus of the ipsilateral hippocampus. However, very few GFP labeled exosomes were observed to be colocalized with GFAP+ astrocytes (Fig. 2E) and instead were frequently visualized in close proximity to the astrocyte processes. Similar results were observed within the ipsilateral cortex and white matter tracts following TBI. Similar findings were observed with exosomes depleted of MALAT1, thus the presence of MALAT1 does not influence exosome localization, at least on the level examined here. In addition, we observed exosomes localized in oligodendrocytes. We also examined the spleen to determine if exosomes migrate to peripheral organs and we do observe exosomes in the spleen at this same time point (image not shown).
Fig. 2.
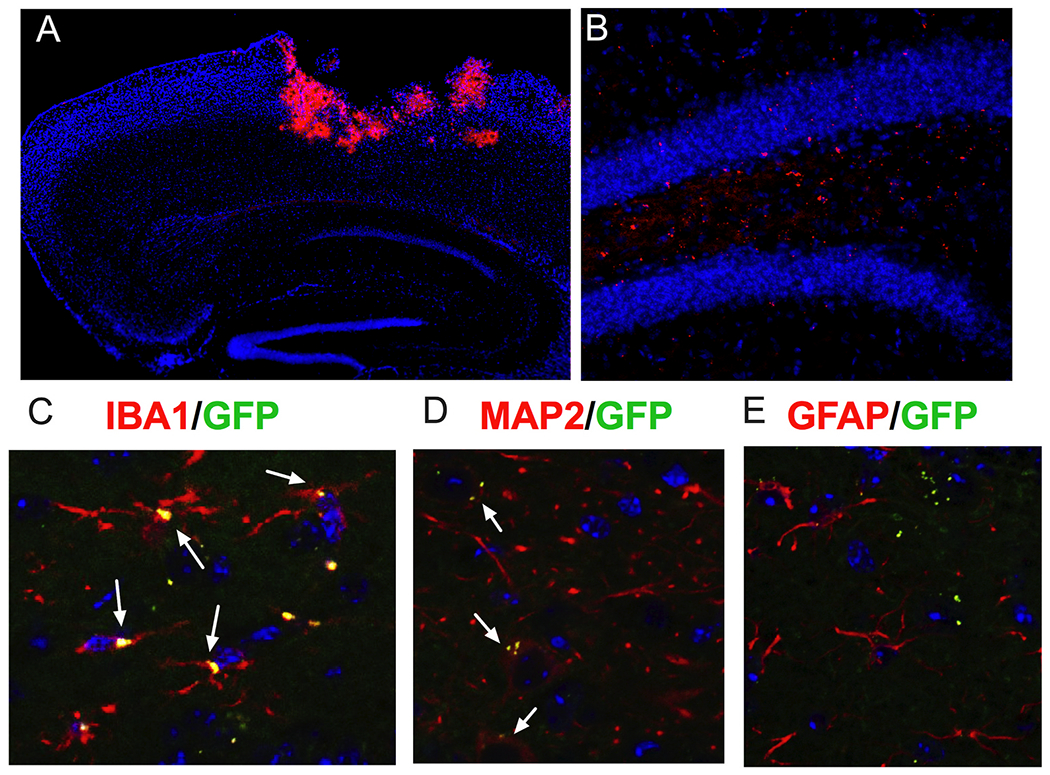
Exosomes delivered intranasally easily infiltrate the brain and are visible at the site of injuiy 24 h after treatment.
A) In this sagittal image the mCherry labelled exosomes are clearly concentrated in the area around the lesion. B) In a higher power image of the dentate gyrus you can also see mCherry labelled exosomes in a lower concentration that was not easily visible at the low power view in (A). C) To determine if exosomes were specifically observed in various cell types, merged images of exosomes co-labeled with GFP (green) and either IBA1 (microglia), MAP2 (neurons), or GFAP (astrocytes) are shown above. Exosomes were observed to be colocalized with MAP2+ neurons and IBA1+ microglia within the hippocampus (as depicted by the white arrows), however very few are seen to be co-localized with GFAP+ astrocytes and are instead frequently observed in close proximity to the astrocyte processes. Similar results were seen within the ipsilateral cortex and white matter tracts. We also observed some exosomes in oligodendrocytes in white matter tracts.
3.4. Exosomes reduce cortical brain injury and microglial activation
To further correlate the beneficial effects of exosomes treatment with behavioral improvement, hematoxylin QS staining of brain tissue sections spanning the TBI injury site was performed 6 weeks post TBI. Cortical lesion volume of the ipsilateral hemisphere was quantified using Cavalieri estimator and revealed that TBI mice treated with hASC exosomes (TBI+EXO) showed a significant reduction in cortical injury damage relative to TBI mice without treatment (Fig. 3 (p < 0.001 One way ANOVA F(4,52) = 36.99 followed by Tukey’s multiple comparisons). However, treatment of exosomes depleted of MALAT1 did not show the same rescue as there was no significant difference between TBI exosomes depleted of MALAT1 and TBI without treatment. All TBI conditions were different from both Sham and Control (P < 0.001, statistics not shown on graph). MHCII+ staining was performed to identify activated microglia following TBI. TBI alone showed an increased expression of MHCII+ microglial cells in several regions of the brain, most significantly in the thalamus but also in the fornix, corpus callosum, striatum and cortex. Quantification of the MHCII+ cells of the ipsilateral thalamus showed a significant increase in TBI mice without treatment compared to sham controls (Fig. 4A p < 0.05 One-Way ANOVA (F (4, 48) = 7.196 followed by Tukey’s multiple comparisons). Exosomes treated mice showed reduction in MHCII+ cells within the thalamus that did not reach significance from TBI alone (p = 0.054) likely due to a high variance of values in the TBI group. Treatment with exosomes depleted of MALAT1 was significantly different from the TBI group treated with exosomes (p < 0.001, Tukey’s multiple comparison test) and was not different from TBI only.
Fig. 4.
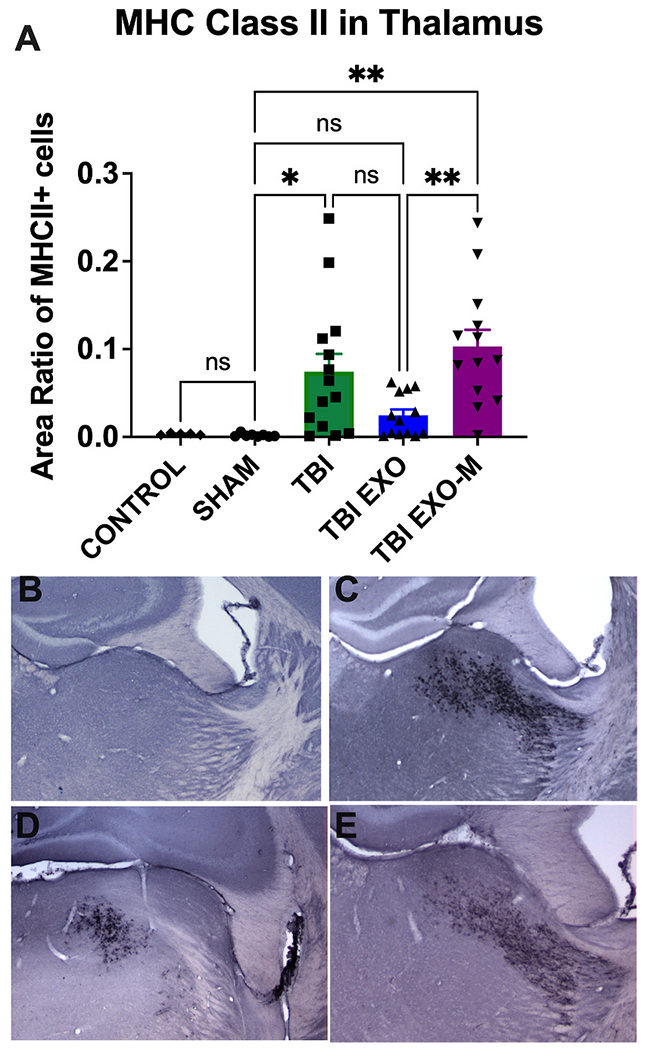
Long term upregulation of inflammation is modified by exosomes.
Six weeks following TBI, an increased expression of MHCII+ microglial cells are identified in several regions of the brain, most clearly in the thalamus but also in the fornix, corpus callosum, striatum and cortex. A) Utilizing NearCYTE image analysis software, there is a significant increase in MHCII staining in the thalamus following TBI (One-Way ANOVA (F (4, 48) = 7.196 followed by Tukey’s multiple comparisons). Treatment with exosomes reduces the MHCII microglial cells in the thalamus when compared to untreated TBI, although this did not reach significance, nor was there significance from either control or sham treatment. Treatment of exosomes depleted of MALAT1 was not different from control TBI but was different from the exosome treated group. Tukey’s post-hoc are indicated on the graph with asterisks **p < 0.01; *p < 0.05. Representative images of MHCII staining in the thalamus are shown in B) Sham, C) TBI, D) TBI+EXO, and E) TBI+EXO-M groups.
In our previous study we performed RNAseq and using Ingenuity Pathway Analysis, we evaluated pathways that followed the pattern matching the response to injury and rescue with treatment with hASCexo and identified networks that showed MALAT1 dependent modulation (Patel et al., 2018). One of the major pathways that was affected by exosome treatment in a MALAT1 dependent manner was inflammation. To examine this further in the mice with intranasal treatment with exosomes 48 h following TBI we performed qPCR for a number of inflammation markers in the cortex ipsilateral to the injury at 7 days post injury which coincides with the timepoint examined in our previous study. As shown in Fig. 5, IL1β levels are significantly increased following TBI and reduced with treatment with hASC exosomes at 7 days after injury. Treatment with exosomes depleted of MALAT1 (EXO-M) also reduced IL1β levels compared to TBI group, but to a lower extent compared to hASC exosomes (EXO) treated group.
Fig. 5.
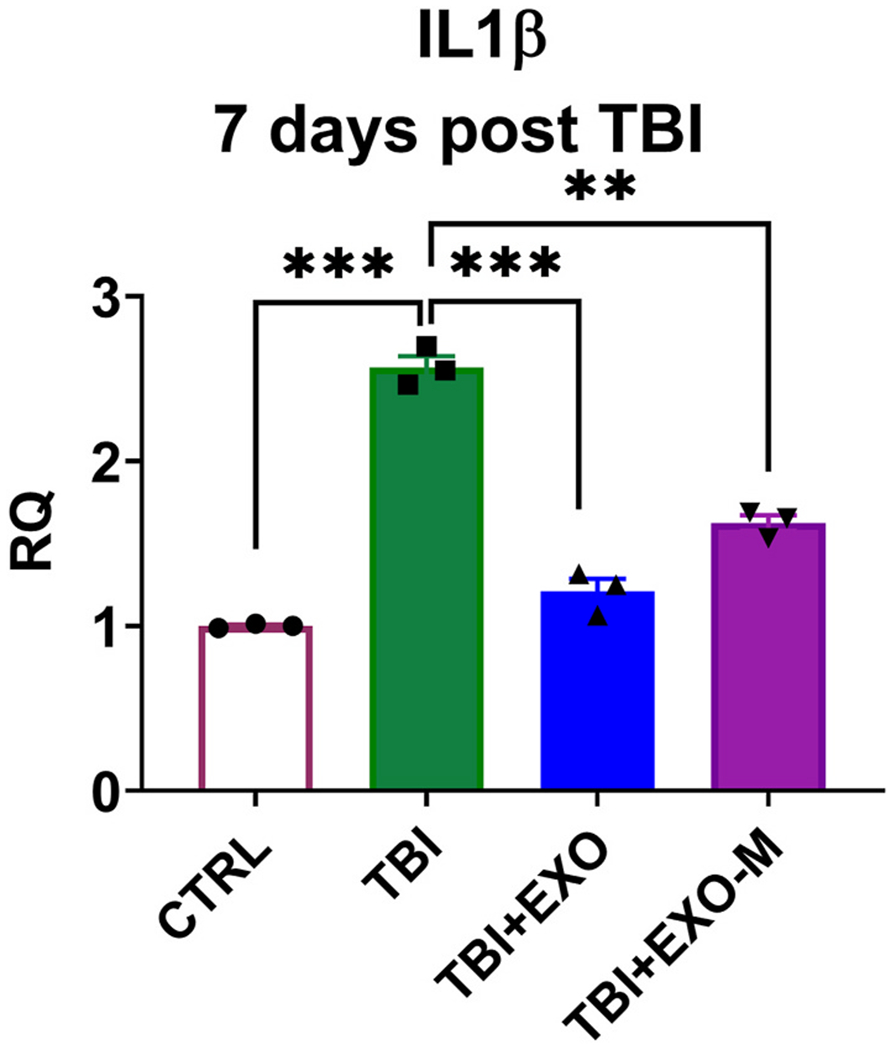
Exosomes modulate inflammation related gene expression 7 days post injury.
Total RNA was isolated and SYBR Green qPCR was performed in triplicate with primers specific for IL1β and using β-actin as internal control. The graph shows relative quantification (RQ) with no TBI (CTRL) set as reference. Statistical analysis was performed using One way ANOVA followed by Tukey’s multiple comparisons test; ***p < 0.001, ns = not significant.
3.5. TBI in mice results in decreased expression of full length (FL) TrkC which is rescued with hASCexo treatment in a MALAT1 dependent manner
MALAT1 is a known mediator of splicing and is enriched within hASC exosomes. Hence to explore potential mechanisms mediated by MALAT1 contained in hASC exosomes, we sorted our previous RNAseq dataset discussed above (Patel et al., 2018) for transcripts with the highest splicing index. The splicing index was calculated based on the formula: exon RPKM/gene RPKM. Amongst the splicing events, we focused on transcripts that followed the pattern of change with injury and rescue with hASCexo treatment in a MALAT1 dependent manner and were detected in all the mice cohorts. Our results indicated that the splicing of neurotrophic receptor tyrosine kinase 3 (NRTK3), also known as tropomysin receptor kinase C (TrkC), is substantially changed with TBI and following hASCexo treatment. The graph shows the plot of the splicing index versus exon starting position for the gene (Fig 6A).
Fig. 6.
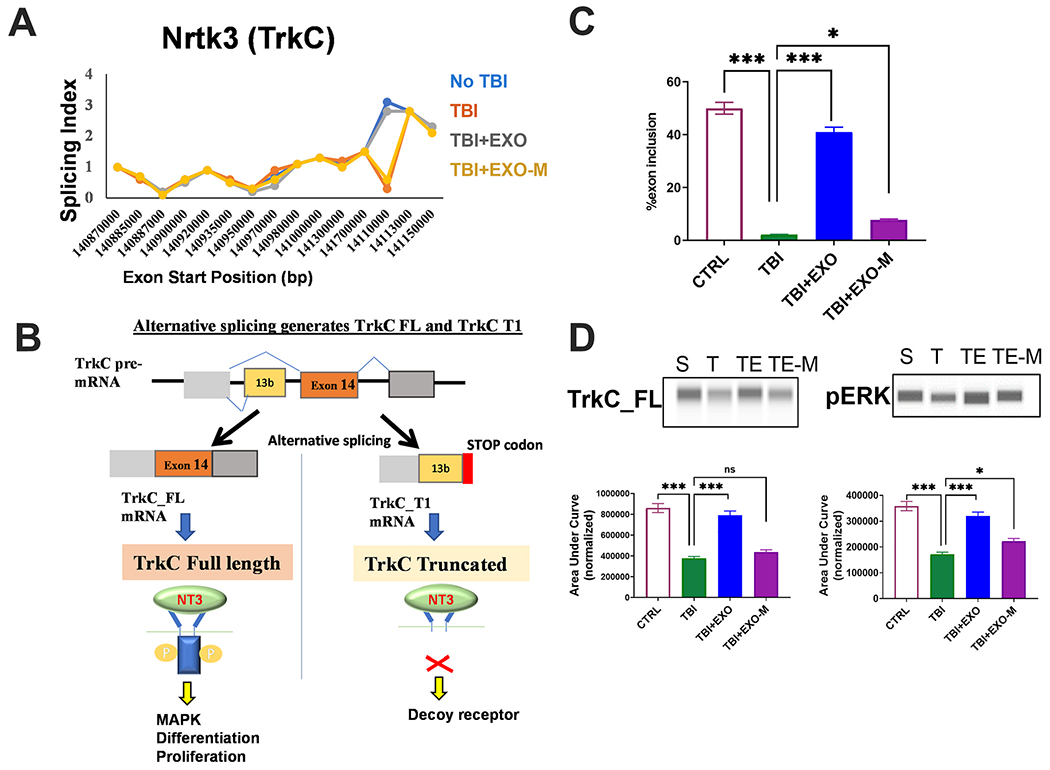
Alternative splicing of the TrkC gene is changed post TBI and exosomes treatment.
A) The splicing index was calculated based on the formula: exon RPKM/gene RPKM. The graph shows plot of the splicing index versus exon starting position for the NRTK3 (TrkC) gene in the TBI mice and treatment cohorts as indicated. (B) Schematic depicting the alternative splicing of TrkC pre-mRNA. Inclusion of exon 14 produces TrkC_FL mRNA which results in a full length receptor with tyrosine kinase activity. Inclusion of exon 13b produces TrkC_T1 mRNA which results in a truncated protein due to presence of a STOP codon. (C) 7 days post injury, total RNA was isolated from cortex of mice with sham injury, TBI, TBI treated with hASC exosomes (TBI+EXO) or TBI treated with MALAT1 depleted hASC exosomes (TBI+EXO-M); n = 5 in each group. SYBR Green qPCR was performed in triplicate with primers specific for TrkC_FL, TrkC_T1 and β-actin as internal control. The graph shows relative quantification (RQ) with no TBI (CTRL) set as reference. Percent exon 14 inclusion is calculated as (FL/(FL+T1))*100. Statistical analysis was performed using Tukey’s multiple comparisons test; ***p < 0.001 extremely significant, **p < 0.01 highly significant, ns = not significant. (D) Whole cell lysates were harvested 7 days post injury from cortex of mice with sham injury, TBI, TBI treated with hASC exosomes (TBI+EXO) or TBI treated with MALAT1 depleted hASC exosomes (TBI+EXO-M); n = 6 in each group. Automated WES (from ProteinSimple) was used for western blotting with antibodies against TrkC C-terminal domain (which detects TrkC_FL), ERK, p-ERK and β-actin (for normalization). The graph shows quantification of peaks as area under the curve (AUC) generated by Compass software (from ProteinSimple) with TrkC_FL levels normalized to β-actin and p-ERK normalized to total ERK. The virtual WES blots generated by Compass are shown above the graphs.
It is shown that TrkC levels in neurons and glia are regulated by alternative splicing producing two splice variants: the full-length receptor (TrkC_FL) and the truncated receptor (TrkC_T1) which lacks the kinase domain (Fig. 6B). The expression of the truncated variant acts as a decoy receptor and substantially inhibits signaling of NT3 via TrkC (Palko et al., 1999; Dedoni et al., 2017).
Hence, 7 days post treatment, we processed a portion of the brain from the above mice cohorts: sham, TBI, TBI treated with hASCexo (TBI+EXO) and TBI treated with MALAT1 depleted hASCexo (TBI+EXO-M) for RNA isolation. Real time SYBR Green qPCR was performed using primers specific for TrkC_FL or TrkC_T1. Our results (Fig. 6C) show a significant decrease in the functional TrkC_FL with a concurrent increase in the TrkC_T1 variant following TBI. Treatment with EXO increased the expression of TrkC_FL while treatment with EXO-M did not significantly change expression of TrkC_FL compared to the TBI group. Analyzing for percent exon 14 inclusion (calculated as TrkC_FL in total TrkC), results show a significant decrease in TBI group. The EXO group dramatically increased percent exon 14 inclusion while the EXO-M group was unable to increase exon 14 inclusion compared to TBI.
3.6. NT3 signaling is inhibited following TBI and is rescued with hASCexo treatment
Neurotrophin signaling is shown to mediate neuronal repair, regeneration and survival following brain injury. In particular, post-TBI, activation of NT3-TrkC signaling was previously shown to promote brain recovery, neuroprotection and cognition and attenuate neuronal injury (Cheng and Mattson, 1994; Grundy et al., 2004; Yang et al., 2005; Koo et al., 2013; Tang et al., 2014; Akyol et al., 2018). TrkC is a membrane-bound receptor that, upon neurotrophin NT3 binding, undergoes auto phosphorylation and then phosphorylates members of the mitogen-activated protein kinases (MAPK aka ERK) pathway. Hence, we evaluated the phosphorylation of ERK in the samples. Using WES, an automated western blotting, our results (Fig. 6D) show decrease in phosphorylation of ERK with TBI which is rescued with treatment with hASCexo (TBI + EXO). Treatment with MALAT1 depleted exosomes showed significant increase of pERK compared to TBI. We observe slightly decreased levels of pERK while comparing pERK between EXO-M and EXO groups, suggesting that other cargo contained in exosomes also affects the MAPK pathway.
To determine the chronic effects of hASCexo, we evaluated gene expression in the cortex after completion of behavior and motor tests (7 weeks post TBI) from the mouse cohorts: sham, TBI, TBI with hASCexo (TBI+EXO) and TBI with MALAT1 depleted hASCexo (TBI+EXO-M). RNA was isolated and Real time SYBR Green qPCR was performed using primers specific for TrkC_FL or TrkC_T1 (Fig. 7). Results do not show significant difference in TrkC_FL levels between TBI vs TBI EXO or TBI EXO-M indicating that in the long term, the TrkC_FL levels regain normal expression.
Fig. 7.
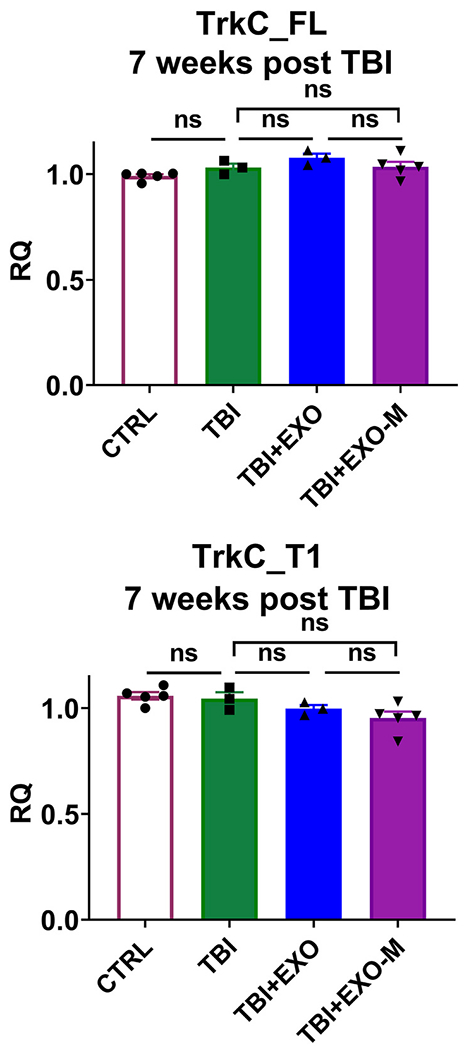
TrkC alternative splicing returns to baseline in 7 weeks post TBI.
7 weeks post injury, total RNA was isolated from cortex of mice with sham injury, TBI, TBI treated with hASC exosomes (TBI+EXO) or TBI treated with MALAT1 depleted hASC exosomes (TBI+EXO-M); n = 5 in each group. SYBR Green qPCR was performed in triplicate with primers specific for TrkC_FL, TrkC_T1 or IL1β and β-actin as internal control. The graph shows relative quantification (RQ) with sham (no TBI) set as reference. Statistical analysis was performed using Tukey’s multiple comparisons test; ns = not significant.
3.7. MALAT1 contained in hASCexo regulates splicing of TrkC and alleviates oxidative stress in primary neurons
To evaluate whether MALAT1 contained in hASCexo regulates splicing of TrkC under oxidative stress, we performed in vitro experiments using mouse primary neurons. The cortex from dams (E18) were dissected, minced into small pieces and trypsinized to remove extracellular matrix. After incubation, the tissue is dissociated by trituration and single cell suspension is obtained. The cells are checked for viability and counted and plated into T-25 flasks in neurobasal media with B27 supplement. After approximately 7 days, neurons are verified using antitubulin antibody and NeuN.
Oxidative stress induced cell damage is common in etiology of several neurobiological disorders including TBI. We induced oxidative stress with hydrogen peroxide (H2O2). Primary neuronal cells were administered 100 μM H2O2 for 1h. The cells were then treated overnight (18 h) with either 2 μg hASC exosomes (H2O2+EXO) or 2 μg MALAT1 depleted exosomes (H2O2+EXO-M). To evaluate alternative splicing in vitro, we performed real time SYBR Green qPCR to detect Trkc_FL and TrkC_T1 transcripts in primary neurons under experimental conditions of control, H2O2, H2O2+EXO, H2O2+EXO-M. Results (Fig. 8A) show a marked decrease in TrkC_FL in H2O2 treated cells. Treatment with hASCexo post H2O2, increased TrkC_FL significantly while treatment with hASCexo-MALAT1 did not increase TrkC_FL. Next, analysis for percent exon 14 inclusion (calculated as TrkC_FL in total TrkC), results show a significant decrease in H2O2 treated cells, which is reversed by treatment with hASCexo. Depleting MALAT1 from hASCexo ablated the inclusion of exon 14. These results show that alternative splicing of TrkC_FL pre-mRNA is dependent on MALAT1. Next, we performed immunocytoehemistry using antibodies for TrkC FL (antibody recognizing C-terminal containing the kinase domain) and p-ERK and ERK. Our results (Fig. 8B) show that H2O2 treatment decreases TrkC FL and activated ERK (downstream of TrkC FL signaling); treatment with hASCexo significantly increased TrkC FL expression in a MALAT1 dependent manner in concurrence with in vivo data.
Fig. 8.
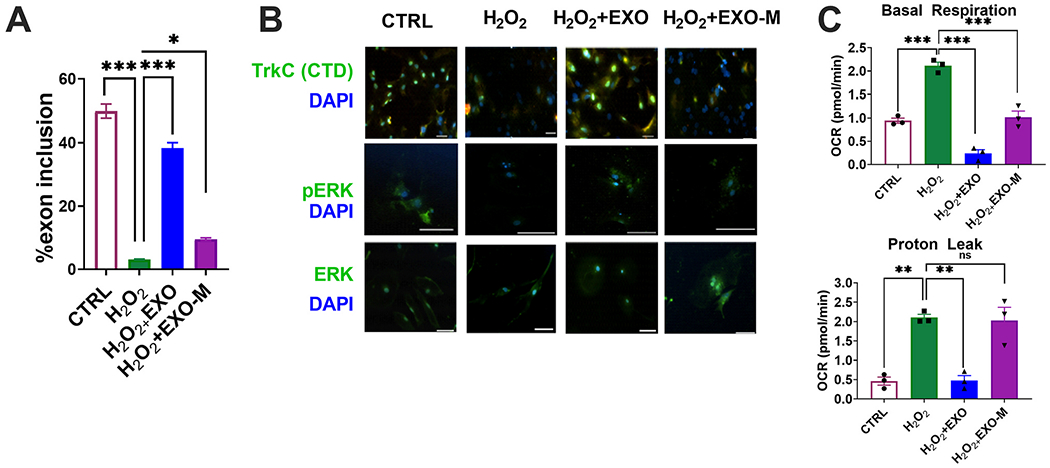
MALAT1 containing exosomes alleviates oxidative stress in primaiy neurons.
Primary neurons were treated 100 μM H2O2 for 1h followed by overnight (18 h) with either 2 μg hASC exosomes (H2O2+EXO) or 2 μg MALAT1 depleted exosomes (H2O2+EXO-M). (A) Total RNA was isolated from control, H2O2, H2O2 treated with hASC exosomes (H2O2+EXO) or H2O2 treated with MALAT1 depleted hASC exosomes (H2O2+EXO-M). The experiment was repeated 5 times independently with similar results. SYBR Green qPCR was performed in triplicate with primers specific for TrkC_FL, TrkC_T1 and β-actin as internal control. The graph shows relative quantification (RQ) with control TrkC_FL set as reference. Percent exon 14 inclusion is calculated as (FL/(FL+T1))*100. Statistical analysis was performed using One-way ANOVA’s test; ***p < 0.001, *p < 0.05. (B) Immunocytochemistry was performed using antibodies against TrkC_FL, pERK and ERK and DAPI for nuclear staining. Experiment repeated three times with similar results. Adjustment of brightness was applied uniformly to entire image. (C) Primary neurons were seeded in the Seahorse XFp miniplate in triplicate and treated as above. Mito Stress Test was performed as per manufacturer’s instructions. Measurements were normalized to cell count. Analysis generating graphs for basal respiration and proton leak were performed using Seahorse Wave software. Experiment was repeated three times and each data point represents mean ± SEM. Statistical analysis was performed by two-way ANOVA; ***p < 0.001 highly significant; **p < 0.01 significant.
A key feature of mild to severe TBI is mitochondrial dysfunction which adversely affects neuronal cell death and survival, energy metabolism and intracellular signaling. To assess the response to hASC exosome treatment with and without MALAT1 depletion, we used Agilent’s Seahorse XFp Analyzer for the Mito Stress Test in primary neurons: control, H2O2, H2O2+EXO, H2O2+EXO-M. Results (Fig. 8C) show that hASCexo treatment increased basal respiration (indicating oxygen consumption to meet cellular ATP demands) and inhibited proton leak (indicating mitochondrial damage) compared to H2O2, while depletion of MALAT1 in the hASCexo significantly attenuated this effect.
3.8. Treatment with hASCexo reduces LPS and H2O2 induced stress in microglia
Since microglia are central to the inflammatory response to injury, we sought to evaluate the expression of TrkC splice variants in response to inflammation and oxidative stress in vitro using lipopolysaccharide (LPS) or H2O2. Immortalized microglial cells (IMG) were treated with 100 μM H2O2 for 1h or 10 ng/ml LPS for 5 h. At the end of 5 h, the cells were then treated with either 2 μg hASC exosomes (H2O2+EXO or LPS+EXO) or 2 μg MALAT1 depleted exosomes (H2O2+EXO-M or LPS_EXO-M) for 18 h. Total RNA was isolated and real time qPCR was performed using primers specific for TrkC_FL or TrkC_T1. Treatment with either LPS or H2O2 show a decrease of TrkC_FL with a concurrent increase of TrkC_T1 which was reversed by hASC exosome treatment demonstrating that hASC exosomes promoted splicing of TrkC_FL (Fig. 9). Interestingly, MALAT1 depleted exosomes were unable to increase percent exon 14 inclusion in LPS and H2O2 treated cells suggesting a cross-talk of IncRNA MALAT1 with these oxidative stress inducers to regulate TrkC_FL gene expression in microglia.
Fig. 9.
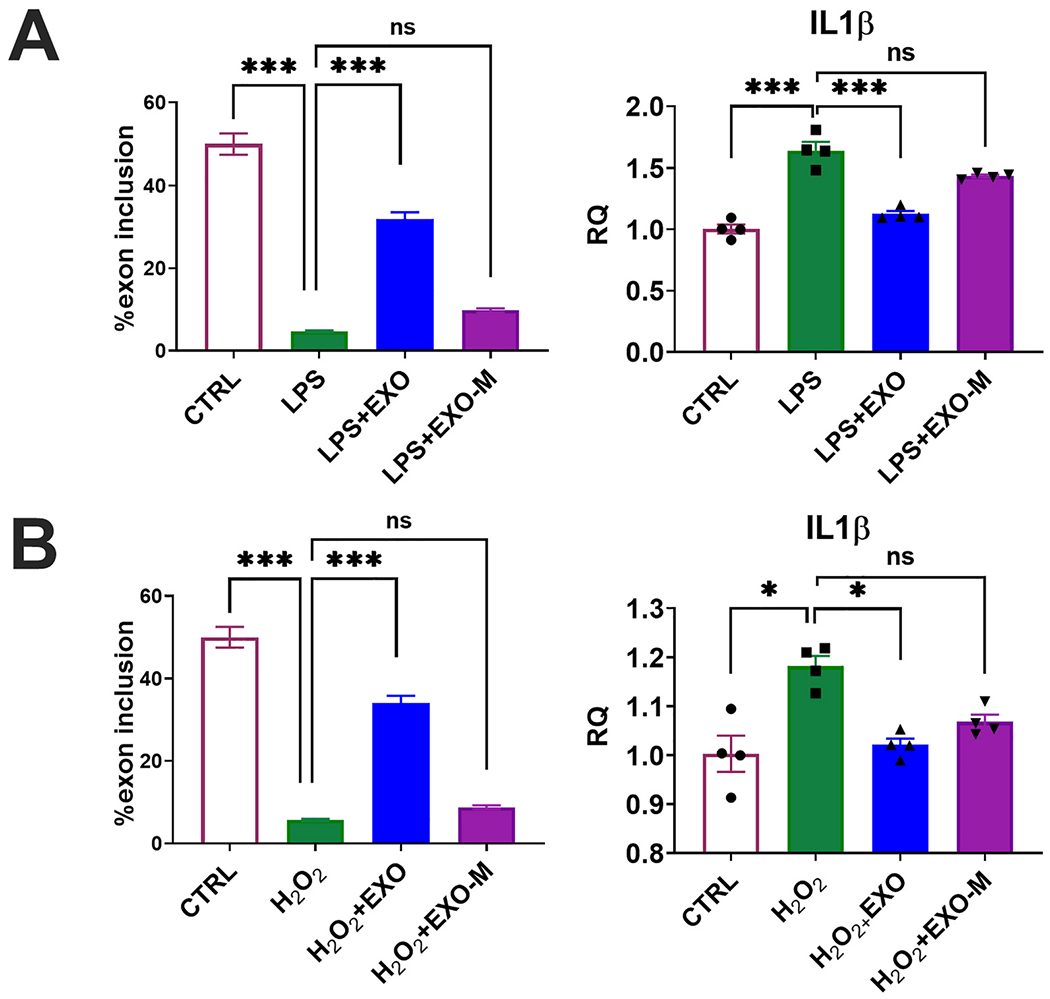
Exosomes treatment reduces inflammation and regulates TrkC alternative splicing in microglia.
Immortalized microglial cells were treated with either (A) 10 ng LPS for 5 h or (B) 100 μM H2O2 for 1h followed by overnight (18 h) with either 2 μg hASC exosomes (H2O2 +EXO) or 2 μg MALAT1 depleted exosomes (H2O2 +EXO-M). Total RNA was isolated from control, H2O2, H2O2 treated with hASC exosomes (H2O2+EXO) or H2O2 treated with MALAT1 depleted hASC exosomes (H2O2+EXO-M) LPS, LPS treated with hASC exosomes (LPS+EXO) or LPS treated with MALAT1 depleted hASC exosomes (LPS+EXO-M). The experiment was repeated four times independently with similar results. SYBR Green qPCR was performed in triplicate with primers specific for TrkC_FL, TrkC_T1 or IL1β and β-actin as internal control. The graph shows relative quantification (RQ) with control set as reference. Percent exon 14 inclusion is calculated as (FL/(FL+T1))*100. Statistical analysis was performed using Sidak’s multiple comparisons test; ***p < 0.001 extremely significant, **p < 0.01 highly significant, *p < 0.05 significant, ns = not significant.
Next, we evaluated the levels of IL1β in microglia using real time SYBR Green qPCR. IL1β levels increased in microglia with LPS treatment which was reversed by hASC exosomes treatment and depletion of MALAT1 in exosomes reversed the effect by 50% (Fig. 9).
4. Discussion
In this manuscript, we demonstrate the hASC exosomes delivered intranasally to mice 48 h following a CCI injury are able to promote recovery of the CCI induced lesion and reduce aspects of inflammation. Intranasal delivery of exosomes offers the advantage of ease of use as well as the ability of exosomes to efficiently cross the blood brain barrier and uptake by injury site and lower dose requirement compared to i.v. administration. We observed a recovery in motor function that occurs within 24 h after exosome treatment, thus demonstrating a rapid response. We also demonstrated that exosome treatment prevented one of the long term consequences of TBI that can effect functional recovery as cognitive function measured by the RAWM test was improved. We further demonstrated that these effects were observed in a MALAT1 dependent manner and explored some potential mechanistic aspects of the role of MALAT1.
It is well-established that secondary inflammation following TBI contributes to injury as outlined in the introduction. For this reason we probed markers of inflammation at both early, 7 days post injury, and late, 6 weeks post injury, time points. As observed by our previous studies with hASC exosomes, we reduced mRNA expression of pro-inflammatory cytokine IL1β in the cortex at 7 days post injury. Prevention of aspects of the secondary inflammatory response have been shown to modulate recovery (Bachstetter et al., 2015, 2016; Morganti et al., 2015, 2019; Chou et al., 2018; Rowe et al., 2018). Reduction of IL1β directly has been shown to reduce brain injury following TBI (Ozen et al., 2020). Exosomes are known to have a broad range of effects and it is likely that alterations in more than one cytokine is likely important, which we demonstrated in our previous study (Patel et al., 2018). We also examined elevated inflammatory function that continues for months following TBI (Acosta et al., 2013) and observed a continued expression of MHCII with immunohistochemistry in several regions of the brain including the corpus callosum and thalamus. This late phase chronic inflammation was also affected by our treatments in a MALAT1 dependent manner. We further explored this directly in microglia in cultures and showed that LPS treatment upregulated IL1β and that exosomes reversed this action, also in a MALAT1 dependent manner. Thus, exosomes are acting directly on microglia early in response to injury to reduce expression of cytokines and this may underlie some of our observed long-term rescue of motor and cognitive function.
Our in vivo data demonstrates that treatment with hASCexo delivered intranasally promotes repair and regeneration following TBI and improves cognition and motor capabilities. Further, in vitro treatment with hASCexo alleviates inflammation and oxidative stress in neurons and microglia. Along with our previous publications, the data presented here demonstrates that the lncRNA MALAT1 is an important mediator of the regenerative potential of hASC exosomes. The cargo of hASC exosomes contains other factors and noncoding RNA which also promote repair and healing. Hence, depletion of MALAT1 from hASC exosomes decreased the in vivo repair and regeneration by about 50% compared to hASC exosomes while the in vitro experiments elucidating the splicing mechanism of the TrkC gene show a more pronounced effect in response to depletion of MALAT1.
To further explore additional mechanistic actions of MALAT1 in hASC exosomes, we examined alternative splicing of TrkC pre-mRNA and we demonstrate that expression of TrkC_FL is decreased with TBI. Treatment with hASC exosomes promotes splicing and inclusion of exon 14 which results in increase of TrkC_FL. This is attenuated when MALAT1 is depleted from the exosomes demonstrating outcome in a MALAT1 dependent manner. Expression of TrkC_T1 acts as a decoy receptor and attenuates NT3 signaling. Other studies (Brahimi et al., 2016) have shown that aberrant neurotrophin NT3 signaling significantly affects the brain post injury. In this study, we addressed NT3 signaling via TrkC, as a possible mediator of the regenerative effects of hASC exosomes, in addition to the anti-inflammatory actions discussed above. We demonstrate that MALAT1 contained in hASCexo modulates NT3 signaling by regulating the splicing of its receptor TrkC. Importantly, we demonstrate that TrkC is a target gene modulated by hASCexo thereby activating the MAPK pathway and promoting recovery.
We also individually evaluated TrkA and TrkB levels in response to hASC exosomes and MALAT1 in primary neurons and brain samples from in vivo TBI models. Our results indicated no significant splicing changes in these receptors. We did not undertake over-expression of MALAT1 in nanoparticles as excess levels of MALAT1 may be tumor promoting; and importantly, the natural biocomponents of exosomes are not toxic and are efficiently taken up by the brain by known mechanisms thus providing significant advantage.
MALAT1 can regulate alternative splicing by functioning as a scaffold for splice factors which bind to the pre-mRNA of the target gene. The levels of splice factor as well as its phosphorylation state dictate the inclusion or exclusion of exons. Several pathways contribute to regulating expression of splice factors and its post-translational modifications. In our in vitro experiments using LPS and H2O2, results showed different outcome levels of MALAT1-dependent TrkC alternative splicing and this may be explained by the different pathways activated by LPS and H2O2. Elucidation of molecular mechanisms using splicing minigenes and expression studies of splice factors is underway as a separate project. Here, we demonstrate that hASC exosomes treatment activates TrkC_FL to promote repair post injury.
Although not explored in depth in this manuscript, TrkC/NT3 signaling is also important in innate immune response as monocytes secrete NT3 and express TrkC receptors (Asami et al., 2006). We have observed TrkC alternative spicing in microglia in response to LPS, as well as a direct effect of exosomes to lower LPS induced IL1β in microglia. This is an area to explore further in future studies as interactions with MAPK and Nf[ISP CHK]κB could be involved with our findings of potent actions of hASC exosomes to reduce inflammation following TBI. Further research is also underway to determine if levels of TrkC splice variants directly affect transcription of IL1β.
Acknowledgments
This work was supported by Department of Veterans’ Affairs grant I01BX003421 (PCB) and IK6BX004214 (PCB), USF internal funding (PCB), Department of Veterans’ Affairs grant I01BX003836 (NAP) and IK6BX005387 (NAP).
Abbreviations
- hASCs
human adipose derived stem cells
- hASCexo
human adipose derived stem cell exosomes
- TBI
traumatic brain injury
- RAWM
radial arm water maze
- lncRNA
long noncoding RNA
- IMG
immortalized microglial cells
- LPS
lipopolysaccharide
Footnotes
Declaration of competing interest
We have read the guidelines for Conflicts of Interest and all authors declare no conflict of interest.
PCB and NAP have an issued US Patent No. 10,857,187 Exosomes from human adipose-derived stem cells for treatment of traumatic brain injury (December 8, 2020).
References
- Acabchuk R, Briggs DI, Angoa-Perez M, Powers M, Wolferz R Jr., Soloway M, Stern M, Talbot LR, Kuhn DM, Conover JC, 2016. Repeated mild traumatic brain injury causes focal response in lateral septum and hippocampus. Concussion 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta SA, Tajiri N, Shinozuka K, Ishikawa H, Grimmig B, Diamond DM, Sanberg PR, Bickford PC, Kaneko Y, Borlongan CV, 2013. Long-term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PloS One 8, e53376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akyol O, Sherchan P, Yilmaz G, Reis C, Ho WM, Wang Y, Huang L, Solaroglu I, Zhang JH, 2018. Neurotrophin-3 provides neuroprotection via TrkC receptor dependent pErkS activation in a rat surgical brain injury model. Exp. Neurol 307, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asami T, Ito T, Fukumitsu H, Nomoto H, Furukawa Y, Furukawa S, 2006. Autocrine activation of cultured macrophages by brain-derived neurotrophic factor. Biochem. Biophys. Res. Commun 344, 941–947. [DOI] [PubMed] [Google Scholar]
- Bachstetter AD, Webster SJ, Goulding DS, Morton JE, Watterson DM, Van Eldik LJ, 2015. Attenuation of traumatic brain injury-induced cognitive impairment in mice by targeting increased cytokine levels with a small molecule experimental therapeutic. J. Neuroinflammation 12, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Zhou Z, Rowe RK, Xing B, Goulding DS, Conley AN, Sompol P, Meier S, Abisambra JF, Lifshitz J, Watterson DM, Van Eldik LJ, 2016. MW151 inhibited IL-1beta levels after traumatic brain injury with No effect on microglia physiological responses. PloS One 11, e0149451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Morganti JM, Bodnar CN, Webster SJ, Higgins EK, Roberts KN, Snider H, Meier SE, Nation GK, Goulding DS, Hamm M, Powell DK, Vandsburger M, Van Eldik LJ, Abisambra JF, 2020. The effects of mild closed head injuries on tauopathy and cognitive deficits in rodents: primary results in wild type and rTg4510 mice, and a systematic review. Exp. Neurol 326, 113180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlongan CV, Sanberg PR, 1995. Elevated body swing test: a new behavioral parameter for rats with 6-hydroxydopamine-indueed hemiparkinsonism. J. Neurosci 15, 5372–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahimi F, Maira M, Barcelona PF, Galan A, Aboulkassim T, Teske K, Rogers ML, Bertram L, Wang J, Yousefi M, Rush R, Fabian M, Cashman N, Saragovi HU, 2016. The paradoxical signals of two TrkC receptor isoforms supports a rationale for novel therapeutic strategies in ALS. PloS One 11, e0162307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prevention CfDCa, 2019. Surveillance report of traumatic brain injury-related emergency department visits, hospitalizations, and deaths–United States. In: Centers for Disease Control and Prevention. U.S. Department of Health and Human Services, 2014. [Google Scholar]
- Cheng B, Mattson MP, 1994. NT-3 and BDNF protect CNS neurons against metabolic/excitotoxic insults. Brain Res. 640, 56–67. [DOI] [PubMed] [Google Scholar]
- Chou A, Krukowski K, Morganti JM, Riparip LK, Rosi S, 2018. Persistent infiltration and impaired response of peripherally-derived monocytes after traumatic brain injury in the aged brain. Int. J. Mol. Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedoni S, Olianas MC, Ingianni A, Onali P, 2017. Interferon-beta inhibits neurotrophin 3 signalling and pro-survival activity by upregulating the expression of truncated TrkC-T1 receptor. Mol. Neurobiol 54, 1825–1843. [DOI] [PubMed] [Google Scholar]
- Eissmann M, Gutschner T, Hammerle M, Gunther S, Caudron-Herger M, Gross M, Schirmacher P, Rippe K, Braun T, Zornig M, Diederichs S, 2012. Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol. 9, 1076–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bassit G, Patel RS, Carter G, Shibu V, Patel AA, Song S, Murr M, Cooper DR, Bickford PC, Patel NA, 2017. MALAT1 in human adipose stem cells modulates survival and alternative splicing of PKCdeltaII in HT22 cells. Endocrinology 158, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S, Mouzon B, Paris D, Aponte D, Abdullah L, Stewart W, Mullan M, Crawford F, 2017. Acute or delayed treatment with anatabine improves spatial memory and reduces pathological sequelae at late time-points after repetitive mild traumatic brain injury. J. Neurotrauma 34, 1676–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy PL, Patel N, Harbuz MS, Lightman SL, Sharples PM, 2004. Adrenalectomy further suppresses the NT-3 mRNA response to traumatic brain injury but this effect is not reversed with corticosterone. Brain Res Mol Brain Res 120, 188–192. [DOI] [PubMed] [Google Scholar]
- Gutschner T, Hammerle M, Diederichs S, 2013. MALAT1 – a paradigm for long noncoding RNA function in cancer. J. Mol. Med 91, 791–801. [DOI] [PubMed] [Google Scholar]
- Koo HM, Lee SM, Kim MH, 2013. Spontaneous wheel running exercise induces brain recovery via neurotrophin-3 expression following experimental traumatic brain injury in rats. J. Phys. Ther. Sci 25, 1103–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Song L, Zeng S, Zhang L, 2015. MALAT1-miR-124-RBG2 axis is involved in growth and invasion of HR-HPV-positive cervical cancer cells. Tumour Biol. J. Int. Soc. Oncodevelop. Biol. Med 37 (3), 633–640. [DOI] [PubMed] [Google Scholar]
- McKee AC, Daneshvar DH, 2015. The neuropathology of traumatic brain injury. Handb. Clin. Neurol 127, 45–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamadpour M, Whitney K, Bergold PJ, 2019. The importance of therapeutic time window in the treatment of traumatic brain injury. Front. Neurosci 13, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, Ferguson AR, Rosi S, 2015. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J. Neurosci 35, 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti JM, Riparip LK, Chou A, Liu S, Gupta N, Rosi S, 2016. Age exacerbates the CCR2/5-mediated neuroinflammatory response to traumatic brain injury. J. Neuroinflammation 13, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti JM, Goulding DS, Van Eldik LJ, 2019. Deletion of p38alpha MAPK in microglia blunts trauma-induced inflammatory responses in mice. J. Neuroinflammation 16, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouzon B, Bachmeier C, Ojo J, Acker C, Ferguson S, Crynen G, Davies P, Mullan M, Stewart W, Crawford F, 2019. Chronic white matter degeneration, but No tau pathology at one-year post-repetitive mild traumatic brain injury in a tau transgenic model. J. Neurotrauma 36, 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozen I, Ruscher K, Nilsson R, Flygt J, Clausen F, Marklund N, 2020. Interleukin-1 beta neutralization attenuates traumatic brain injury-induced microglia activation and neuronal changes in the globus pallidus. Int. J. Mol. Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palko ME, Coppola V, Tessarollo L, 1999. Evidence for a role of truncated trkC receptor isoforms in mouse development. J. Neurosci. : Off. J. Soc. Neurosci 19, 775–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RS, Carter G, El Bassit G, Patel AA, Cooper DR, Murr M, Patel NA, 2016. Adipose-derived stem cells from lean and obese humans show depot specific differences in their stem cell markers, exosome contents and senescence: role of protein kinase C delta (PKCdelta) in adipose stem cell niche. Stem Cell Invest. 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NA, Moss LD, Lee J-Y, Tajiri N, Acosta S, Hudson C, Parag S, Cooper DR, Borlongan CV, Bickford PC, 2018. Long noncoding RNA MALAT1 in exosomes drives regenerative function and modulates inflammation-linked networks following traumatic brain injury. J. Neuroinflammation 15, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe RK, Harrison JL, Zhang H, Bachstetter AD, Hesson DP, O’Hara BF, Greene MI, Lifshitz J, 2018. Novel TNF receptor-1 inhibitors identified as potential therapeutic candidates for traumatic brain injury. J. Neuroinflammation 15, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Parag S, Patel R, Lui A, Murr M, Cai J, Patel NA, 2019. Stabilization of lncRNA GAS5 by a small molecule and its implications in diabetic adipocytes. Cell Chem Biol 26, 319–330 e316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, He H, Cheng N, Song Y, Ding W, Zhang Y, Zhang W, Zhang J, Peng H, Jiang H, 2014. PDGF, NT-3 and IGF-2 in combination induced transdifferentiation of muscle-derived stem cells into Schwann cell-like cells. PloS One 9, e73402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JT, Lee TH, Weng HH, Chang CN, Chen WC, Cheng WC, Wu JH, 2005. Dexamethasone enhances NT-3 expression in rat hippocampus after traumatic brain injury. Exp. Neurol 192, 437–443. [DOI] [PubMed] [Google Scholar]