

Abstract
The global pandemic of coronavirus disease 2019 (COVID-19), caused by severe acute respiratory syndrome coronavirus 2, has led to a dramatic loss of human life worldwide. Despite many efforts, the development of effective drugs and vaccines for this novel virus will take considerable time. Artificial intelligence (AI) and machine learning (ML) offer promising solutions that could accelerate the discovery and optimization of new antivirals. Motivated by this, in this paper, we present an extensive survey on the application of AI and ML for combating COVID-19 based on the rapidly emerging literature. Particularly, we point out the challenges and future directions associated with state-of-the-art solutions to effectively control the COVID-19 pandemic. We hope that this review provides researchers with new insights into the ways AI and ML fight and have fought the COVID-19 outbreak.
Keywords: COVID-19, SARS-CoV-2, artificial intelligence, machine learning, drug, vaccine
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus responsible for the ongoing coronavirus disease 2019 (COVID-19) global pandemic, caused mounting infections and millions of deaths, as well as incalculable devastation to the global economy [1–5]. COVID-19 is accompanied by multiple organ failure induced by increased inflammatory mediators in severe patients, which results in an increased mortality rate. Cardiac injury, such as heart failure, arrhythmias, myocarditis and sudden death, appears to be a prominent feature of multiple organ failure. Aggressive support and appropriate treatment can potentially improve recovery [6, 7]. SARS-CoV-2 along with SARS-CoV-1 and Middle East respiratory syndrome coronavirus (MERS-CoV) are positive, single-stranded RNA viruses belonging to the genus ‘Coronavirus’, the family ‘Coronaviridae’ and the order ‘Nidovirales’ [8, 9]. The SARS-CoV-2 genome can encode at least 29 proteins, including 4 structural proteins, 16 non-structural proteins and 9 accessory proteins [10, 11]. Together with their host binding sites, these proteins have become potential targets for SARS-CoV-2 antiviral drugs. For example, Remdesivir, an antiviral agent targeting RNA polymerase, has been approved by the Food and Drug Administration (FDA) as the standard of treatment for COVID-19. However, the treatments currently applied in the clinic will most likely face anticipated drug resistance arising from the evolving virus [12–14]. Therefore, a major priority has been placed on developing more therapeutic strategies and drug candidates that prevent or limit propagation and infection.
Conventional drug and vaccine discovery are daunting tasks that aim to create novel molecules with multiple desirable properties [15–17]. As a result, the process of drug and vaccine design is hugely costly and time-consuming with a low success rate. To ease this dilemma, artificial intelligence (AI) and machine learning (ML)-based models have been revolutionarily used to efficiently discover sizeable numbers of plausible, diverse and new candidate molecules in the vast molecular space [18–20]. AI/ML utilizes algorithm structures to interpret and learn the features of the input data and makes independent decisions for accomplishing specific objectives. Moreover, AI/ML can identify hit and lead compounds and provide rapid drug target inspection and optimization of drug structure design.
In this review, we focused on the AI/ML-based methods for drug discovery and vaccine design to fight against COVID-19. To this end, we surveyed a huge amount of information produced by the recent explosion of COVID-19 related studies. In addition, we highlighted the current challenges of existing approaches with potential future directions. We hope that this review could provide a strong rationale for the continued use of AI/ML-based methods in the field of drug repurposing, novel drug discovery and vaccine creation.
Search strategy
The staggering number of papers related to COVID-19 published in the form of preprints and peer-reviewed journals has posed an unprecedented challenge to the process of knowledge acquisition and information quality assessment. We undertook a systematic search in five databases, including PubMed, Google Scholar, Springer Link, Elsevier and Wiley. The search criteria consisted of the following terms: COVID-19, SARS-CoV-2, AI, ML and deep learning [21, 22]. To make the review more comprehensive, we also screened the reference list in each of the selected articles. In addition, the last literature search time was 18 June.
AI/ML-based drug repurposing strategies for COVID-19 therapeutics
Significant efforts have been put into the use of computational methods to identify promising drug candidates for the treatment of COVID-19. In this section, we summarized the general categories of AI/ML-based drug repurposing methods for therapeutics of COVID-19, including network-based algorithms, expression-based algorithms and integrated docking simulation algorithms (Figure 1 and Table 1).
Figure 1.
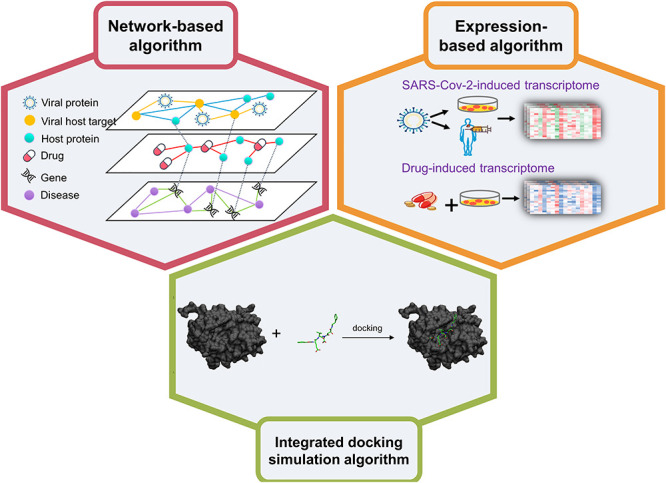
Three general categories of AI/ML-based drug repurposing methods for therapeutics of COVID-19, including network-based algorithms, expression-based algorithms and integrated docking simulation algorithms.
Table 1.
AI/ML-based studies on COVID-19 drug repositioning
| AI/ML tools | Details | Website URL | References |
|---|---|---|---|
| RL | A network label propagation to identify SARS-CoV-2 interactors | https://github.com/Murali-group/SARS-CoV-2-network-analysis | [25] |
| Dense fully convolutional neural network | Identification and ranking of protein-ligand interactions by virtual drug screening | NA | [26] |
| Natural language processing | MT-DTI to screen potential antivirals | NA | [27] |
| CNN | Identification and ranking of drug-target interactions with binding affinity | NA | [28] |
| Integrated deep learning methodology | Discovery of drug candidates by knowledge-graph-networks | https://github.com/ChengF-Lab/CoV-KGE | [30] |
| GCN | Identification and ranking of drugs by multi-rational and variational graph autoencoder | https://github.com/yejinjkim/drug-repurposing-graph | [31] |
| GCN with attentional mechanism | Discovery of drug candidates by medical knowledge graph | https://github.com/FangpingWan/NeoDTI | [32] |
| GCN | Construction of the virus-related knowledge graph | https://github.com/FangpingWan/CoV-DTI | [33] |
| GCN, network diffusion and network proximity | Identification and ranking of virus-host interactions by drug efficacy screening | https://github.com/Barabasi-Lab/COVID-19 | [34] |
| AI-based platform- InfinityPhenotype | Analysis of transcriptomic data | NA | [36] |
| Artificial neural network | Analysis of transcriptomic, proteomic, structural data and aging signatures | https://github.com/uhlerlab/covid19_repurposing | [37] |
| GCN with multi-head attention mechanism | Analysis of gene expression profiles perturbed by de novo chemicals | https://github.com/pth1993/DeepCE | [38] |
| CNN | Analysis of sequence identity and structure similarity, molecular docking | NA | [40] |
| Random forest | Prediction of docking simulation scores | NA | [41] |
| An end-to-end deep neural network | Prediction of protein ligand interaction probability, validation by drug docking algorithm | https://github.com/ekraka/SSnet | [42] |
| Naïve Bayes | Ranking based on various binding energy function, validation by docking method | NA | [43] |
Network-based algorithms
A classic way of repurpose drugs is to apply network-based algorithms [23, 24] to knowledge graphs containing relationships between different kinds of medical entities (e.g. diseases, drugs and proteins) to identify relevant host protein targets or regions of host interactome that can be targeted.
The virus-host network is based on the hypothesis that human proteins belonging to complexes or signaling pathways are closest to human interactors of viral proteins and are potentially good targets for inhibition. Law et al. [25] utilized a special network label propagation method combined with semi-supervised learning method based regularized Laplacian (RL) on a virus-host interactome to identify additional SARS-CoV-2 interactors. The RL achieved an area under the receiver-operator characteristic curve of 0.76 based on 5-fold cross-validation. In addition, the proposed methodology found the connection between endoplasmic reticulum stress, HSPA5 and anti-clotting agents. As a result, a prioritized list of human and drug targets was synthesized, which can be utilized as a reference resource for clinical research.
Several studies adopted protein–protein interactome networks-based strategies to predict drug candidates. For instance, Zhang et al. [26] implemented dense fully convolutional neural network deep learning model on protein-ligand networks using the SARS-CoV-2 3C-like (3CL) protease as the seed to identify putative drug targets for COVID-19. The proposed model virtually screened potential peptide drugs (combination of isoleucine, lysine and proline amino acids) and chemical ligands (meglumine, vidarabine, adenosine, D-sorbitol, D-mannitol, sodium gluconate, ganciclovir and chlorobutanol).
Beck et al. [27] employed a pre-trained language model named molecular transfer drug target interaction (MT-DTI) to screen FDA approved antivirals that could act on viral proteins of SARS-CoV-2. The MT-DTI model predicted that Ataznavir, an antiretroviral medication used to treat and prevent the human immunodeficiency virus (HIV), showing the best inhibitory potency against 3CL protease, followed by remdesivir, efavirenz, ritonavir and dolutegravir. In addition, Majumdar et al. [28] constructed a convolutional neural network (CNN)-based deep learning architecture to identify potential ligands for SARS-CoV-2. The protein sequence composition and ECPF4 [29] were picked to encode protein and ligands, respectively. According to the predicted drug-target interaction values, the authors presented their findings of the top 33 ligands with the highest binding affinity to the S-glycoprotein of SARS-CoV-2, which can be used to prepare antiviral drugs.
By combining multiple network-based strategies, Zeng et al. [30] developed an integrative, deep learning methodology, CoV-KGE, to identify repurposable drugs for COVID-19. After comprehensively capturing the connections among drugs, diseases, proteins/genes, pathways, expressions and then systematically analyzing the transcriptomics and proteomics data generated from SARS-CoV-2 infected human cells, the authors successfully identified 41 drug candidates (including dexamethasone, indomethacin, niclosamide and toremifene) for the potential treatment of COVID-19. Similarly, Hsieh et al. [31] built a comprehensive knowledge graph that includes multiple interactions connecting virus baits, host genes, pathways, drugs and phenotypes. Using deep graph neural embedding, they learned candidate drug’s representation of biological interactions. Rigorous experimental validation finally prioritized 22 drug candidates including azithromycin, atorvastatin, aspirin, acetaminophen and albuterol. To screen potential therapeutic drugs for COVID-19, Che et al. [32] embedded five types of entities, including drugs, genes, diseases, channels, side effects and nine relationships into medical knowledge graph. Moreover, the authors used graph convolutional networks (GCNs) with attentional mechanism to extract features from the knowledge graph and construct a prediction matrix. The experimental results indicated that the model can effectively learn the network topology around the disease, which has contributed to the precise identification of drugs for both ordinary diseases and COVID-19. In addition, five drugs (tenofovir, lopinavir, darunavir, ritonavir and ribavirin) predicted by the model have been proven effective in clinical treatment. Ge et al. [33] applied GCN algorithm and statistical analysis method to filter potential drug candidates against SARS-CoV-2. Text mining followed by in vitro assays revealed that a poly-ADP-ribose polymerase 1 inhibitor, CVL218, can reveal itself to being extremely relevant to the prevention of immunopathology induced by SARS-CoV-2 infection. CVL218 can inhibit the replication process of SARS-CoV-2 in a dose-dependent manner and can be combined with another anti-SARS-CoV-2 drug Fapilavir to enhance the efficacy. Moreover, the authors also found that CVL218 can interact with the nucleocapsid (N) protein of SARS-CoV-2 with high affinity. Therefore, CVL218 may serve as an effective therapeutic agent against COVID-19. Additionally, Gysi et al. [34] employed a multi-modal approach to the virus-host interactome integrating GCN, network diffusion and network proximity to screen drugs that can perturb the activity of host proteins connected with the COVID-19 disease module. The results of primate trials verified the true value of the proposed predictive approach, resulting in a success rate of 62% and successfully identified six drugs that reduced viral infection to treat COVID-19. In addition, the proposed pipeline offers a reduced cost and shortened timeline methodological pathway, which predicts 76 of the 77 drugs rely on network-based mechanisms that cannot beGy identified using docking-based strategies.
The approaches mentioned above showed that the network-based strategy applied to COVID-19 drug repurposing studies can identify effective useable drugs and drug combinations by integrating multiple types of modules, including virus-host interactions, protein–protein interactome networks, and drug-target networks.
Expression-based algorithms
One promising approach to reposition FDA approved anti-SARS-CoV-2 drugs is to observe changes in the expression of defensive genes in the disease state, which can be used as effective disease descriptor or quantitative phenotype. After that, repurposing drugs can be used to drive gene expression in the opposite direction [35]. Zhu et al. [36] inputted transcriptomic data into an AI-based platform, InfinityPhenotype, to uncover the efficacy of natural products or FDA-approved drugs. Experimental result indicated that Liquiritin exerts antiviral function by imitating type I interferon, thus it can be used as a competitive candidate for treating COVID-19. In addition, by integrating available transcriptomic, proteomic, structural data and aging signatures, Belyaeva et al. [37] proposed a computational method for drug repurposing. Given the age-dependent pathogenicity of SARS-CoV-2, the authors first identified genes that are differentially regulated by SARS-CoV-2 infection and aging based on bulk RNA-seq data. Later, the autoencoder-based architecture was used to learn the representation of gene expression profiles data and SARS-CoV-2 expression data in an unsupervised manner, thereby matching and obtaining a series of FDA-approved drugs, including clemastine, haloperidol, ribavirin and quinapril. Pham et al. [38] developed a GCN with multi-head attention mechanism to predict the differential gene expression profiles perturbed by de novo chemicals. Utilizing the newly proposed data augmentation method to extract representative features from noisy omics data, the model obtained superior performance to state-of-the-art methods. In the end, the value of the model was further proved by successfully screening chemical compounds against the clinical phenotype of COVID-19 from DrugBank.
Integrated docking simulation algorithms
Recently, remarkable improvements in docking simulation [39] coupled with advancements in AI/ML technique have been utilized to revolutionize the drug development process. Nguyen et al. [40] integrated mathematical pose (MathPose) and CNNs (MathDL) to predict spatial structure of SARS-CoV-2 3CL protease and protein-ligand binding affinities. MathPose docked the selected known complexes, and decoy complexes generated were fed into the MathDL for drug properties evaluation. According to the predicted binding affinities, the authors reported the top 15 potentially highly potent drugs to COVID-19, which provided a critical step for further drug repurposing. Similarly, Batra et al. [41] developed a powerful computational method by combining random forest regression models and ensemble docking simulations to identify promising candidates against COVID-19. The proposed model successfully screened 187 promising candidate ligands (75 of which are approved by the FDA) by cross-validation from a dataset containing nearly one million entries and a rank-ordered list of about 19 000 potential compounds. In addition, Karki et al. [42] employed an end-to-end deep neural network termed SSnet to identify protein ligand interaction probability, in combination with a drug docking algorithm Smina to evaluate the potential efficacy of clinically approved drug list. The SSnet approach was further extended to large compound libraries DrugBank and ZINC to narrow down the compounds with poor binding capacity. The truncated library can be used as a possible target for subsequent in vitro experiments. More recently, Mohapatra et al. [43] reported a Naïve Bayes classifier with inhibitors of the SARS-CoV-2 3CL protease and screened more than 2000 FDA-approved drugs. Out of the 471 drugs that were predicted by the model which can be effective for the treatment of COVID-19, the top 10 drugs were further assessed by the docking method. After which, an anti-retroviral by the name of amprenavir, a known HIV-protease inhibitor, was found to be the most effective drug.
AI/ML-based drug discovery for COVID-19 therapeutics
During this unprecedented time due to the COVID-19 pandemic, the world has realized the necessity of novel drug development. In the past year, this process has been extensively facilitated by AI/ML [44–58]. Generally, computational drug discovery system consists of three units, including target discovery, small molecule drug discovery and predictors clinical trial outcomes (Figure 2). All studies were summarized in Table 2.
Figure 2.
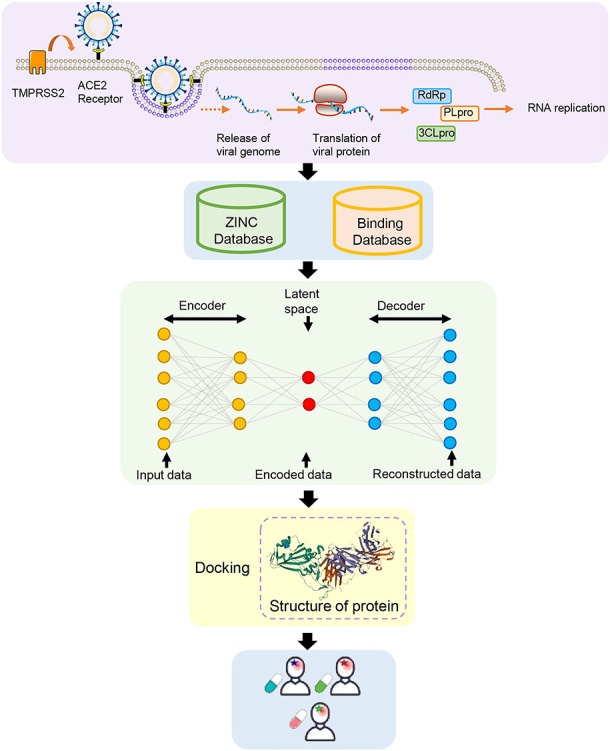
Drug discovery process based on AI/ML method.
Table 2.
AI/ML based studies on COVID-19 drug discovery
| AI/ML tools | Detail | Website URL | References |
|---|---|---|---|
| Generative chemistry pipeline | Using knowledge of crystal and homology model of target protein to design novel drugs | NA | [62] |
| Deep Q-learning network | Collection of anti-virus drugs, split structures according to chemical rules, collection of fragments, fragment libraries with medical chemistry knowledge | https://github.com/tbwxmu/2019-nCov | [63] |
| Deep docking model | Identification of potential drugs by structure-based virtual screening | NA | [64] |
| DGM | Generation of novel drugs by molecular SMILES variational autoencoder and an efficient multi-attribute controlled sampling scheme | NA | [65] |
| DGM | Generation of small molecules by transfer and reinforcement learning | NA | [66] |
| Directed message passing neural network combined with transfer learning | Identification of new drugs by virtual screening | https://github.com/pkuwangsw/COVIDVS | [67] |
| Monte Carlo tree search algorithm and multitask neural network | Discovery of new candidate ligands by iterative search and retrain strategy | NA | [68] |
Using knowledge of the crystal structure and homology model of the target protein [59–61], Zhavoronkov et al. [62] presented a generative chemistry pipeline to design novel drug-like inhibitors of COVID-19. Particularly, generative autoencoders, generative adversarial networks, genetic algorithm and language models were used to exploit the whole drug-like chemical space. Finally, several novel drug compounds were generated for further development. Tang et al. [63] developed an advanced deep Q-learning network with the fragment-based drug design called ADQN-FBDD to generate potential novel molecules targeting SARS-CoV-2 3CL protease. Inputting the initial 284 fragmented inhibitors, ADQN-FBDD identified 47 potential lead compounds and their related derivatives. Additionally, Ton et al. [64] applied a deep docking model to predict the docking scores for all the 1.3 billion compounds from ZINC 15 library and recommended the top 1000 ligands as potential SARS-CoV-2 3CL protease inhibitors. Chenthamarakshan et al. [65] developed a deep generative model (DGM), CogMol, to design drugs with low off-target activity for a given protein sequence. This AI-based platform showed its advantage that it can simultaneously process the molecule generation of a large number of target proteins without the need to retrain the model for a single target. CogMol has successfully generated 1000 drug candidates that may inhibit NSP9, 3CL protease, replicase and RBD in the S protein of SARS-CoV-2. Bung et al. [66] employed a similar generative model based on deep neural network for de novo design small molecules that inhibit SARS-CoV-2 3CL protease. The authors innovatively applied transfer and reinforcement learning to optimize the model and embedded special physicochemical property filters to ensure that the generated molecules have drug-like properties. Interestingly, the two newly proposed small molecules showed a high degree of similarity with the natural plant-derived product aurantiamide, which is significance for the production of compounds that are easy to synthesize and have comparatively fewer side effects. Recently, Wang et al. [67] used a directed massage passing neural network combined with transfer learning to build a broad-spectrum anti-SARS-CoV-2 compound prediction model. The model screened a set of prioritized compound lists from the ZINC 15 library containing 1.9 million entries. Importantly, the in vitro experimental result indicated that the selected compounds have satisfactory binding strength to SARS-CoV-2 3CL protease. More recently, by combining a Monte Carlo tree search algorithm and multitask neural network, Srinivasan et al. [68] presented a computational methodology to discover new therapeutic agents for the treatment of COVID-19. The search and retrain strategy to iteratively explore the design space and concurrently improve the accuracy of the surrogate model was shown to significantly accelerate the discovery of drug candidates.
AI/ML-based vaccine and antibody discovery for COVID-19 therapeutics
With the large volume of data and the need for automatic abstract feature learning, AI/ML has a significant contribution in areas of vaccine discovery (Figure 3 and Table 3). AL/ML models for COVID-19 vaccine development focus on the prediction of potential epitopes by using a variety of techniques, such as artificial neural network, gradient boosting decision tree and deep neural network [56, 69]. Fast et al. [70] used two artificial neural network algorithms termed MARIA and NetMHCPan4 to identify T-cell and B-cell epitopes of SARS-CoV-2. The method identified 405 T-cell epitopes with strong presentation scores for both MHC-I and MHC-II, as well as two potential neutralizing B-cell epitopes on the S protein. This finding will promote the development of potent vaccines and neutralizing antibodies for COVID-19. Using multiple bioinformatics tools, Ong et al. [71] developed Vaxign-ML, a XGBoost-based ML tool to prioritize non-structural proteins as vaccine candidates for SARS-CoV-2. The authors found that compared to the S protein, the most promising vaccine candidate for COVID-19 is nsp3, the largest non-structural protein of the “Coronaviridae” family, which scored the second highest protective antigenicity score. In addition, Prachar et al. [72] implemented a feed-forward neural network and identified 174 SARS-CoV-2 epitopes that can stably bind to 11 HLA allotypes with high prediction binding scores. Importantly, the authors assessed the current peptide-HLA prediction tools that identify epitopes relevant to SARS-CoV-2. The results suggested that several algorithms exhibit low stability and thus the predicted peptides are very likely to elicit an immune response against SARS-CoV-2. Based on this, the validated binding or non-binding peptides in this study are noted and recognized for further development of vaccines and treatment for COVID-19. Yang et al. [73] also proposed a deep neural network-based approach named DeepVacPred for prediction and design of a multi-epitope vaccine. The DeepVacPred constructed a 694aa multi-epitope vaccine containing 16 B-cell epitopes, 82 CTL epitopes and 89 HTL epitopes. Moreover, the RNA mutation of SARS-CoV-2 was tracked to ensure that the designed vaccine can manage with the mutation of the virus.
Figure 3.
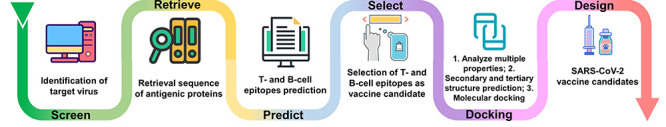
Vaccine and antibody discovery based on AI/ML method.
Table 3.
AI/ML based studies on COVID-19 vaccine design
| AI/ML tools | Details | Website URL | References |
|---|---|---|---|
| Artificial neural network | Identification of SARS-CoV-2 T-cell and B-cell epitopes based on viral protein antigen presentation and antibody binding properties | NA | [70] |
| XGBoost | Prediction of vaccine candidates from non-structural proteins | NA | [71] |
| Feed-forward neural network | Prediction of HLA-binding peptides from SARS-CoV-2 virus by binding stability | NA | [72] |
| Deep neural network | Prediction and design of multi-epitope vaccine that can manage with the mutation of the virus | https://github.com/zikunyang/DCVST | [73] |
Discussion
After the outbreak of the COVID-19 pandemic on a global scale, the question arose of how to transform research results into new effective drugs and technologies. As a result, the demand for novel drugs and faster development time has come to the forefront of research. The rise of AI/ML has greatly accelerated the often lengthy drug approval process. Faced with the ever-increasing number of computational methods for the treatment of COVID-19, properly assessing their predictive power is valuable for early action to combat the rise in cases.
Data quality
(i) One of the ways to build an effective and interpretable drug discovery model is to use drug-related entities to build neural network models. However, such algorithms have inherent limitations. For example, in order to define and obtain a larger range of drug-target interaction data, a lower affinity threshold is set, which may lead to bias in predictive performance. In addition, most studies have not evaluated the possibility that drug-target interactions may be functional associations rather than physical bindings, which also affects the quality of the data. (ii) The lack of dose-dependent profiles and virus-host interaction data may lead to unpredictable adverse reactions. An effective solution is to integrate the pharmacokinetic data of animal models and clinical trials to predict the adverse reactions of drugs to COVID-19 patients at specific doses. (iii) Different from the transcriptional characteristics related to acute infections used in expression-based algorithms, the transcriptional characteristics caused by prolonged exposure to SARS-CoV-2 can better reflect the real human response changes. Therefore, analysis of transcription characteristics at multiple time points is critical to reveal important compounds. (iv) Because of the lack of gold-standard datasets, many data-driven network-based methods use different data training models from different trial techniques, different batches and different human cell lines, in which potential data noise produces highly data-dependent results. (v) The SARS-CoV-2 infection is characterized by its highly contagious nature and inter-individual variability. Understanding the inter-individual variability has important implications for precision treatment, health care, clinical trials, vaccination and resource allocation. However, there is currently no AI/ML model that takes into account factors such as age, gender, race, complications, geographic location and social vulnerability. Therefore, collecting and processing heterogeneous data has become an urgent challenge to overcome.
Algorithm design
(i) Despite the promise of the technology, AI/ML is limited by models that are too complex and difficult to interpret, and it is unlikely to replace human test subjects anytime soon. In addition, although AI/ML provides the ability to collect, assimilate and analyze huge amounts of data, it cannot assess the value of the data for researchers. Therefore, a more meaningful way is to construct the correct level of knowledge and information, as well as biological subsystems to solve the problem of model interpretability. Meanwhile, sort out a small number of high-quality mathematical representations based on known knowledge and combine them with clinical knowledge and professional knowledge to produce a more focused output. (ii) The low-level exhaustiveness docking algorithm used in most studies encounters difficulties in finding receptor-ligand interactions with a local minimal, resulting in a high degree of affinity variability. In addition, the high computational burden of docking algorithm limits its application to large compound libraries. Also, the docking algorithm generates a divergent candidate set using diverse evaluation scoring criteria in many cases. Therefore, the selection of docking algorithm, the design of algorithm structure and the setting of evaluation criteria all need to be improved systematically.
Clinical trials
The translational gap between computational efforts for drug or vaccine development and clinical application is a major and widely recognized bottleneck in the fields of computational biology and medicine. Many predicted drugs and vaccines have not advanced to clinical trials. Even though, there is a lack of optimization of clinical benefits due to the difficulty in both determining clinical endpoints and recruiting patient cohorts.
In conclusion, we expect future advanced AI/ML approaches for drugs and vaccines development to improve in terms of output generation, multi-dimensional data integration, algorithm structure deployment and working mechanism interpretation.
Limitations
In this review, we have not delved into the criteria or rationale of different researches when selecting AI/ML algorithms. In addition, there is a lack of systematic screening of candidate drugs predicted by different studies to further evaluate the reliability of experimental results.
Key Points
We screened out methods for drug discovery and vaccine design using AI/ML techniques from the blowout of COVID-19-related studies.
We classify the selected studies and systematically analyze the solutions provided by different AI/ML architectures in response to the SARS-CoV-2 pandemic.
We identified the current challenges of existing approaches and future directions to encourage researchers to make efforts in developing innovative solutions to fight against COVID-19.
Acknowledgments
We would like to thank the anonymous reviewers for valuable suggestions.
Hao Lv is a PhD candidate of Center for Informational Biology at University of Electronic Science and Technology of China. His research interests include bioinformatics.
Lei Shi is an attending surgeon and medical doctor at Department of Spine Surgery, Changzheng Hospital, Naval Medical University, Shanghai, China. His research interests include spinal injury and degenarative disease, bioinformatics and molecular biology.
Joshua William Berkenpas is an undergraduate student at Pacific Lutheran University, his research interest includes Bioinformatics.
Fu-Ying Dao is a PhD candidate of Center for Informational Biology at University of Electronic Science and Technology of China. Her research interests include bioinformatics.
Hasan Zulfiqar is a PhD candidate of Center for Informational Biology at University of Electronic Science and Technology of China. His research interests include computational biology.
Hui Ding is an associate professor of Center for Informational Biology at University of Electronic Science and Technology of China. Her research is in the areas of computational biology and system biology.
Yang Zhang is an associate professor of Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine. His interests are in the areas of bioinformatics, machine learning and biological data mining.
Liming Yang is a professor at Department of Pathophysiology, Harbin Medical University-Daqing, Harbin, China. His research interests include cardiovascular disease, biomedical engineering and bioinformatics.
Renzhi Cao is an Assistant Professor at Pacific Lutheran University, USA. His research interest mainly focuses on developing and applying machine learning and data mining techniques to solve biomedical problems, such as protein structure and function predictions.
Contributor Information
Hao Lv, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China.
Lei Shi, Department of Spine Surgery, Changzheng Hospital, Naval Medical University, Shanghai 200433, China.
Joshua William Berkenpas, Department of Computer Science, Pacific Lutheran University, Tacoma 98447, USA.
Fu-Ying Dao, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China.
Hasan Zulfiqar, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China.
Hui Ding, Center for Informational Biology, University of Electronic Science and Technology of China, Chengdu 610054, China.
Yang Zhang, Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu 611137, China.
Liming Yang, Department of Pathophysiology, Harbin Medical University-Daqing, Daqing, 163319, China.
Renzhi Cao, Department of Computer Science, Pacific Lutheran University, Tacoma 98447, USA.
Author contributions
Conceptualization: H.D., Y.Z., L.-M.Y., R.-Z.C.; Investigation: H.L., L.S., J.W.B., F.-Y.D., H.Z.; Writing (original draft): H.L., F.-Y.D.; Writing (review and editing): J.W.B., H.D., Y.Z., R.-Z.C.; Funding acquisition: R.-Z.C.
Funding
This work is supported by the Natural Sciences Undergraduate Research Program at Pacific Lutheran University.
References
- 1.Morens DM, Fauci AS. Emerging pandemic diseases: how we got to COVID-19. Cell 2020;183(3):837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng L, Han X, Zhu Z, et al. . Functional alterations caused by mutations reflect evolutionary trends of SARS-CoV-2. Brief Bioinform 2021;22(2):1442–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li JW, Wang XY, Li N, et al. . Feasibility of mesenchymal stem cell therapy for COVID-19: a mini review. Curr Gene Ther 2020;20(4):285–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ren X, Wen W, Fan X, et al. . COVID-19 immune features revealed by a large-scale single-cell transcriptome atlas. Cell 2021;184(7):1895–1913.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nasir MA, Nawaz S, Huang J. A mini-review of computational approaches to predict functions and findings of novel micro peptides. Curr Bioinform 2020;15(9):1027–35. [Google Scholar]
- 6.Mokhtari T, Hassani F, Ghaffari N, et al. . COVID-19 and multiorgan failure: a narrative review on potential mechanisms. J Mol Histol 2020;51(6):613–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng L, Zhu Z, Wang C, et al. . COVID-19 induces lower levels of IL-8, IL-10, and MCP-1 than other acute CRS-inducing diseases. Proc Natl Acad Sci U S A 2021;118(21):e2102960118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 2019;17(3):181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paules CI, Marston HD, Fauci AS. Coronavirus infections-more than just the common cold. JAMA 2020;323(8):707–8. [DOI] [PubMed] [Google Scholar]
- 10.Gordon DE, Jang GM, Bouhaddou M, et al. . A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020;583(7816):459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu F, Zhao S, Yu B, et al. . Author correction: a new coronavirus associated with human respiratory disease in China. Nature 2020;580(7803):E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomson EC, Rosen LE, Shepherd JG, et al. . Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021;184(5):1171–1187.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li F, Luo M, Zhou W, et al. . Single cell RNA and immune repertoire profiling of COVID-19 patients reveal novel neutralizing antibody. Protein Cell 2020. doi: 10.1007/s13238-020-00807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Zhang T, Lei H, et al. . Research on gastric Cancer's drug-resistant gene regulatory network model. Curr Bioinform 2020;15(3):225–34. [Google Scholar]
- 15.Chenthamarakshan V, Das P, Padhi I, et al. . Mojsilovic AJapa: target-specific and selective drug design for covid-19 using deep generative models. arXiv 2020. https://doi.org/2004.01215. [Google Scholar]
- 16.Zhao T, Hu Y, Peng J, et al. . DeepLGP: a novel deep learning method for prioritizing lncRNA target genes. Bioinformatics 2020;36(16):4466–4472. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Lian X, Liu F, et al. . Identification of novel key targets and candidate drugs in oral squamous cell carcinoma. Curr Bioinform 2020;15(4):328–37. [Google Scholar]
- 18.Wang H, Liang P, Zheng L, et al. . eHSCPr discriminating the cell identity involved in endothelial to hematopoietic transition. Bioinformatics (Oxford, England) 2021. doi: 10.1093/bioinformatics/btab071. [DOI] [PubMed] [Google Scholar]
- 19.Liang P, Yang W, Chen X, et al. . Machine learning of single-cell transcriptome highly identifies mRNA signature by comparing F-score selection with DGE analysis. Mol Ther Nucleic Acids 2020;20:155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y. Artificial intelligence for bioinformatics and biomedicine. Curr Bioinform 2020;15(8):801–2. [Google Scholar]
- 21.Ma X, Xi B, Zhang Y, et al. . A machine learning-based diagnosis of thyroid cancer using thyroid nodules ultrasound images. Curr Bioinform 2020;15(4):349–58. [Google Scholar]
- 22.Chen L, Li J, Chang M. Cancer diagnosis and disease gene identification via statistical machine learning. Curr Bioinform 2020;15(9):956–62. [Google Scholar]
- 23.Liu L, Zhang LR, Dao FY, et al. . A computational framework for identifying the transcription factors involved in enhancer-promoter loop formation. Molecular therapy Nucleic acids 2021;23:347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, Li QZ, Jin W, et al. . Revealing gene function and transcription relationship by reconstructing gene-level chromatin interaction. Comput Struct Biotechnol J 2019;17:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Law JN, Akers K, Tasnina N, et al. . Murali TJapa: identifying human interactors of SARS-CoV-2 proteins and drug targets for COVID-19 using network-based label propagation. arXiv 2020. https://doi.org/2006.01968. [Google Scholar]
- 26.Zhang H, Saravanan KM, Yang Y, et al. . Deep learning based drug screening for novel coronavirus 2019-nCov. Interdiscip Sci 2020;12(3):368–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beck BR, Shin B, Choi Y, et al. . Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput Struct Biotechnol J 2020;18:784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Majumdar S, Nandi SK, Ghosal S, et al. . Deep learning-based potential ligand prediction framework for COVID-19 with drug-target interaction model. Cognit Comput 2021. doi: 10.1007/s12559-021-09840-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Unterthiner T, Mayr A, Klambauer G, Steijaert M, Wegner JK, Ceulemans H, Hochreiter S: Deep learning as an opportunity in virtual screening. In: Proceedings of the Deep Learning Workshop at NIPS : 2014; 2014: 1–9. [Google Scholar]
- 30.Zeng X, Song X, Ma T, et al. . Repurpose open data to discover therapeutics for COVID-19 using deep learning. J Proteome Res 2020;19(11):4624–36. [DOI] [PubMed] [Google Scholar]
- 31.Hsieh K, Wang Y, Chen L, et al. . Drug repurposing for COVID-19 using graph neural network with genetic, mechanistic, and epidemiological validation. arXiv 2020. https://doi.org/2009.10931. [Google Scholar]
- 32.Che M, Yao K, Che C, et al. . Knowledge-graph-based drug repositioning against COVID-19 by graph convolutional network with attention mechanism. Future Internet 2021;13(1):13. [Google Scholar]
- 33.Ge Y, Tian T, Huang S, et al. . An integrative drug repositioning framework discovered a potential therapeutic agent targeting COVID-19. Signal Transduct Target Ther 2021;6(1):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morselli Gysi D, Valle I, Zitnik M, et al. . Network medicine framework for identifying drug-repurposing opportunities for COVID-19. Proc Natl Acad Sci U S A 2021;118(19):e2025581118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Killick R, Ballard C, Doherty P, et al. . Transcription-based drug repurposing for COVID-19. Virus Res 2020;290:198176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu J, Deng Y-Q, Wang X, et al. . An artificial intelligence system reveals liquiritin inhibits SARS-CoV-2 by mimicking type I interferon. bioRxiv 2020. 10.1101/2020.05.02.074021. [DOI] [Google Scholar]
- 37.Belyaeva A, Cammarata L, Radhakrishnan A, et al. . Causal network models of SARS-CoV-2 expression and aging to identify candidates for drug repurposing. Nat Commun 2021;12(1):1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pham TH, Qiu Y, Zeng J, et al. . A deep learning framework for high-throughput mechanism-driven phenotype compound screening and its application to COVID-19 drug repurposing. Nat Mach Intell 2021;3(3):247–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zulfiqar H, Masoud MS, Yang H, et al. . Screening of prospective plant compounds as H1R and CL1R inhibitors and its antiallergic efficacy through molecular docking approach. Comput Math Methods Med 2021;2021:6683407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen D, Gao K, Chen J, et al. . Potentially highly potent drugs for 2019-nCoV. bioRxiv 2020. 10.1101/2020.02.05.936013. [DOI] [Google Scholar]
- 41.Batra R, Chan H, Kamath G, et al. . Sankaranarayanan SJapa: screening of therapeutic agents for covid-19 using machine learning and ensemble docking simulations. arXiv 2020. https://doi.org/2004.03766. [DOI] [PubMed] [Google Scholar]
- 42.Karki N, Verma N, Trozzi F, et al. . Predicting potential SARS-COV-2 drugs-in depth drug database screening using deep neural network framework SSnet, classical virtual screening and docking. Int J Mol Sci 2021;22(4):1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mohapatra S, Nath P, Chatterjee M, et al. . Repurposing therapeutics for COVID-19: rapid prediction of commercially available drugs through machine learning and docking. PLoS One 2020;15(11):e0241543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hasan MM, Alam MA, Shoombuatong W, et al. . NeuroPred-FRL: an interpretable prediction model for identifying neuropeptide using feature representation learning. Brief Bioinform 2021. doi: 10.1093/bib/bbab167. [DOI] [PubMed] [Google Scholar]
- 45.Charoenkwan P, Nantasenamat C, Hasan MM, et al. . BERT4Bitter: a bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides. Bioinformatics 2021. doi: 10.1093/bioinformatics/btab133. [DOI] [PubMed] [Google Scholar]
- 46.Charoenkwan P, Chiangjong W, Nantasenamat C, et al. . StackIL6: a stacking ensemble model for improving the prediction of IL-6 inducing peptides. Brief Bioinform 2021. doi: 10.1093/bib/bbab172. [DOI] [PubMed] [Google Scholar]
- 47.Hasan MM, Schaduangrat N, Basith S, et al. . HLPpred-fuse: improved and robust prediction of hemolytic peptide and its activity by fusing multiple feature representation. Bioinformatics 2020;36(11):3350–6. [DOI] [PubMed] [Google Scholar]
- 48.Basith S, Manavalan B, Hwan Shin T, et al. . Machine intelligence in peptide therapeutics: a next-generation tool for rapid disease screening. Med Res Rev 2020;40(4):1276–314. [DOI] [PubMed] [Google Scholar]
- 49.Wei L, Chen H, Su R. M6APred-EL: a sequence-based predictor for identifying N6-methyladenosine sites using ensemble learning. Molecular Therapy-Nucleic Acids 2018;12:635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei L, Hu J, Li F, et al. . Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief Bioinform 2020;21(1):106–19. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Shi Y, Wang X, et al. . A drug target interaction prediction based on LINE-RF learning. Curr Bioinform 2020;15(7):750–7. [Google Scholar]
- 52.Wei L, Liao M, Gao Y, et al. . Improved and promising identification of human MicroRNAs by incorporating a high-quality negative set. IEEE/ACM Trans Comput Biol Bioinform 2014;11(1):192–201. [DOI] [PubMed] [Google Scholar]
- 53.Wei L, Zhou C, Chen H, et al. . ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 2018;34(23):4007–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeng X, Zhu S, Lu W, et al. . Target identification among known drugs by deep learning from heterogeneous networks. Chem Sci 2020;11(7):1775–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zeng X, Zhu S, Liu X, et al. . Cheng FJB: deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019;35(24):5191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Wang H, Wang X, et al. . Predicting drug-target interactions via FM-DNN learning. Curr Bioinform 2020;15(1):68–76. [Google Scholar]
- 57.Yang H, Luo Y, Ren X, et al. . Risk prediction of diabetes: big data mining with fusion of multifarious physical examination indicators. Information Fusion 2021;75:140–9. [Google Scholar]
- 58.Long J, Yang H, Yang Z, et al. . Integrated biomarker profiling of the metabolome associated with impaired fasting glucose and type 2 diabetes mellitus in large-scale Chinese patients. Clin Transl Med 2021;11(6):e432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu B, Liu D, Wang Z, et al. . Multi-substrate selectivity based on key loops and non-homologous domains: new insight into ALKBH family. Cell Mol Life Sci 2021;78(1):129–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Z, Liu D, Xu B, et al. . Modular arrangements of sequence motifs determine the functional diversity of KDM proteins. Brief Bioinform 2020. doi: 10.1093/bib/bbaa215. [DOI] [PubMed] [Google Scholar]
- 61.Hatzis Y, Thireou T, Viennas E, et al. . RGDtrip: a database for the investigation of proteins containing the RGD tripeptide. Curr Bioinform 2018;13(5):518–528. [Google Scholar]
- 62.Zhavoronkov A, Aladinskiy V, Zhebrak A, et al. . Orekhov PJIMHKLA: potential COVID-2019 3C-like protease inhibitors designed using generative deep learning approaches. Insilico Medicine Hong Kong Ltd A 2020;307:E1. [Google Scholar]
- 63.Tang B, He F, Liu D, et al. . AI-aided design of novel targeted covalent inhibitors against SARS-CoV-2. bioRxiv 2020. 10.1101/2020.03.03.972133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ton AT, Gentile F, Hsing M, et al. . Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol Inform 2020;39(8):e2000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chenthamarakshan V, Das P, Hoffman SC, et al. . Cogmol: target-specific and selective drug design for covid-19 using deep generative models. arXiv 2020. https://doi.org/2004.01215. [Google Scholar]
- 66.Bung N, Krishnan SR, Bulusu G, et al. . De novo design of new chemical entities for SARS-CoV-2 using artificial intelligence. Future Med Chem 2021;13(6):575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang S, Sun Q, Xu Y, et al. . A transferable deep learning approach to fast screen potential antiviral drugs against SARS-CoV-2. Brief Bioinform 2021. 10.1093/bib/bbab211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Srinivasan S, Batra R, Chan H, et al. . Artificial intelligence-guided De novo molecular design targeting COVID-19. ACS Omega 2021;6(19):12557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wong KKL. Optimization in the design of natural structures, biomaterials, bioinformatics and biometric techniques for solving physiological needs and ultimate performance of bio-devices. Curr Bioinform 2019;14(5):374–5. [Google Scholar]
- 70.Fast E, Chen B. Potential T-cell and B-cell epitopes of 2019-nCoV. bioRxiv 2020. 10.1101/2020.02.19.955484. [DOI] [Google Scholar]
- 71.Ong E, Wong MU, Huffman A, et al. . COVID-19 coronavirus vaccine design using reverse vaccinology and machine learning. Front Immunol 2020;11:1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Prachar M, Justesen S, Steen-Jensen DB, et al. . Identification and validation of 174 COVID-19 vaccine candidate epitopes reveals low performance of common epitope prediction tools. Sci Rep 2020;10(1):20465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang Z, Bogdan P, Nazarian S. An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci Rep 2021;11(1):3238. [DOI] [PMC free article] [PubMed] [Google Scholar]