

Abstract
Genetic variation plays a significant role in predisposing individuals to thoracic aortic aneurysms and dissections. Advances in genomic research have led to the discovery of 11 genes validated to cause heritable thoracic aortic disease (HTAD). Identifying the pathogenic variants responsible for aortic disease in affected patients confers substantial clinical utility by establishing a definitive diagnosis to inform tailored treatment and management, and enables identification of at-risk relatives to prevent downstream morbidity and mortality. The availability and access to clinical genetic testing has improved dramatically such that genetic testing is considered an integral part of the clinical evaluation for patients with thoracic aortic disease. This review provides an update on our current understanding of the genetic basis of thoracic aortic disease, practical recommendations for genetic testing, and clinical implications.
Keywords: Genetic therapy, Aneurysm, dissecting, Aortic diseases, Genetic testing
The major diseases affecting the thoracic aorta are aneurysms and acute dissections (TAAD).1 The natural history of thoracic aortic aneurysms, involving the aortic root and ascending aorta, is to asymptomatically enlarge over time until an acute tear in the intimal layer leads to an ascending aortic dissection (Stanford classification type A dissection). Type A dissections cause sudden death in up to 40% of afflicted individuals, with an increase in mortality of 1% per hour after dissection and 5% to 20% dying during or shortly after emergent surgical repair of the aorta.1 Less-deadly aortic dissections originating in the descending thoracic aorta just distal to the branching of the subclavian artery (type B dissections) are also part of the thoracic aortic disease spectrum. Although medical treatments can slow the rate of aneurysm growth, the mainstay of treatment to prevent type A aortic dissection-related deaths is prophylactic surgical repair of the ascending aneurysm. surgical repair is typically recommended when the aneurysm’s diameter reaches 5.0–5.5 cm; however, studies on patients presenting with acute type A dissections indicate that up to 60% present with aortic diameters smaller than 5.5 cm.2 Thus, success of this treatment approach relies on identifying at-risk individuals early in the disease process, prior to dissection to establish appropriate timing for repair of the aorta. Therefore, biomarkers are needed to identify individuals at risk for thoracic aortic disease and inform the timing of surgery to prevent dissections.
Along with genetic variants, the major risk factors for TAAD are increased biomechanical forces on the aorta, primarily due to hypertension and activities or drug use that increase force on the aorta (e.g., cocaine or methamphetamine abuse, weightlifting). Additional risk factors include bicuspid aortic valve (BAV), male sex, increasing age, inflammatory and infectious conditions, and pregnancy.1, 3 The genetic risk for TAAD extends from rare and highly penetrant genetic variants that trigger disease in all individuals who inherit the alteration (e.g., Marfan Syndrome due to FBN1 mutations) to common variants found in the general population that confer only a minimal risk for disease (variants identified in genome wide association studies [GWAS]). The frequency of a genetic change in the population (e.g., allele frequency) and effect size (e.g., risk for thoracic aortic disease) are generally inversely related, with rare variants having large effects e.g., mutations also termed pathogenic variants) and common variants conferring small effects. However, common variants with large effects are rare and rare variants with small effects are difficult to identify. This review provides an update on our current understanding of the genetic basis of TAAD and how this information impacts clinical care.
Heritable thoracic aortic disease (HTAD): rare variants in genes that confer a high risk for thoracic aortic disease
Our studies, along with others, established that up to 20% of all TAAD patients have similarly affected first-degree relatives, indicating a significant genetic predisposition to TAAD.3, 4 TAAD in these families is typically inherited in an autosomal dominant manner with decreased penetrance, particularly in women, indicating that heterozygous pathogenic variants, in a particular gene are responsible for the heritable thoracic aortic disease (HTAD).5 families with HTAD demonstrate variable expression of TAAD, including age of disease onset, risk for dissection at a small aortic diameter, risk for type A versus type B dissections, and the formation of aortic aneurysms of the aortic root, the ascending aorta, or both. Additionally, clinical heterogeneity is evident based on additional features co-segregating with TAAD in these families, which can include intracranial aneurysms, aneurysms, dissections/ruptures in other arteries, occlusive vascular diseases leading to early-onset strokes and coronary artery disease, BAV, and patent ductus arteriosus (PDA).
HTAD was initially identified and characterized in individuals with Marfan syndrome (MfS) and relatively recently in individuals with Loeys-Dietz syndrome (LDS). MFS results from pathogenic variants in FBN1, which encodes fibrillin-1, a component of microfibrils that link the elastic lamellae to cell surface receptors on the SMCs.6, 7 LDS is diagnosed when an individual carries a heterozygous pathogenic variant in one of five genes encoding proteins in the TGF-β signaling pathway (TGFBR1, TGFBR2, SMAD3, TGFB2, or TGFB3) and clinically manifests with MFS-like skeletal features, thin skin with bruising and aberrant scar formation, craniofacial abnormalities, arterial tortuosity, and aneurysms and dissections of other arteries.8–13 Vascular Ehlers-Danlos syndrome (VEDS), resulting from mutations in COL3A1, which encodes the subunits of type III collagen, also confers a risk for TAAD but a greater risk for aneurysms and dissections of other arteries. These patients lack Marfan-like skeletal features but may present with thin skin with bruising and aberrant scar formation, spontaneous rupture of hollow organs, pneumothorax, an aged look to the hands, and a characteristic facial appearance.
Most families with HTAD do not have features suggestive of a genetic syndrome. Yet, approximately 10% of these families without syndromic features suggestive of MFS or LDS, have a pathogenic variant in the genes responsible for MFS and LDS.14–16 We and others have identified additional HTAD predisposition genes that disrupt proteins involved in SMC contraction, including the most frequently altered HTAD gene not associated with syndromic features of MFS or LDS, ACTA2, which encodes the SMC-specific isoform of α-actin, and MYH11, encoding the SMC-specific myosin heavy chain.17 We also determined that loss-of-function variants in the kinase that controls SMC contraction, myosin light chain kinase (MYLK), and activating variants in a kinase that drives SMC relaxation, PRKG1, cause HTAD.18, 19 Additional genes with evidence of HTAD predisposition include LOX, MFAP5, BGN, FOXE3, MAT2A, and ARIH1.20 Although greater than 20 genes have been described as associated with HTAD, after application of a systematic gene-disease curation process, only 11 were found to have sufficient data to validate the relationship between the gene and thoracic aortic disease; other genes await additional supporting data (Table I).21
TABLE I.—
Genes validated to cause heritable thoracic aortic disease (HTAD) and genes with putative HTAD association awaiting additional evidence to confirm disease association.
| Gene | Protein name | Function or molecular pathway | Primary phenotype or syndrome | Other major vascular findings | ||
|---|---|---|---|---|---|---|
| Validated genes | ACTA2 | Smooth muscle α-actin | SMC contraction | Smooth muscle dysfunction syndrome Nonsyndromic HTAD | Moyamoya disease Early-onset coronary artery disease |
|
| COL3A1 | Procollagen type III α1 | Extracellular matrix | Vascular Ehlers-Danlos syndrome | Aneurysms, dissections/ruptures of other arteries | ||
| FBN1 | Fibrillin-1 | Extracellular matrix | Marfan syndrome Nonsyndromic HTAD |
Consistent with syndrome | ||
| LOX | Lysyl oxidase | Extracellular matrix | Nonsyndromic HTAD | -- | ||
| MYH11 | Smooth muscle myosin heavy chain | SMC contraction | Nonsyndromic HTAD | |||
| MYLK | Myosin light chain kinase | SMC contraction | Nonsyndromic HTAD | -- | ||
| PRKG1 | Protein kinase cGMP-dependent type 1 | SMC contraction | Nonsyndromic HTAD | Coronary artery anomalies | ||
| SMAD3 | Mothers against decapentaplegic drosophila homolog 3 | TGF-β signaling | Loeys-Dietz syndrome 3 Aneurysms-osteoarthritis syndrome Nonsyndromic HTAD |
Aneurysms, dissections/ruptures of other arteries | ||
| TGFB2 | TGF-β2 | TGF-β signaling | Loeys-Dietz syndrome 4 Nonsyndromic HTAD |
Aneurysms, dissections/ruptures of other arteries | ||
| TGFBR1 | TGF-β receptor type I | TGF-β signaling | Loeys-Dietz syndrome 1 Nonsyndromic HTAD |
Aneurysms, dissections/ruptures of other arteries | ||
| TGFBR2 | TGF-β receptor type II | TGF-β signaling | Loeys-Dietz syndrome 2 Nonsyndromic HTAD |
Aneurysms, dissections/ruptures of other arteries | ||
|
| ||||||
| Putative genes | ARIH1 | Ariadne drosophila homolog 1 | LINC complex | Nonsyndromic HTAD | -- | |
| BGN | Biglycan | Extracellular matrix | Meester-Loeys syndrome | Consistent with syndrome | ||
| EFEMP2 | EGF containing fibulin-like extracellular matrix protein 2 (fibulin-4) | Extracellular matrix | Cutis laxa type 1B syndrome | Consistent with syndrome | ||
| ELN | Elastin | Extracellular matrix | Cutis laxa | Consistent with syndrome | ||
| FBN2 | Fibrillin 2 | Extracellular matrix | Congenital contractural arachnodactyly | Consistent with syndrome | ||
| FLNA | Filamin A | SMC contraction | Periventricular nodular heterotopia Cardiac valvular dysplasia | Consistent with syndrome | ||
| FOXE3 | Forkhead box E3 | SMC survival | Nonsyndromic HTAD | -- | ||
| HCN4 | Hyperpolarization-activated cyclic nucleotide-gated potassium channel 4 | Ion channel | Arrhythmias, sinus bradycardia, left ventricular non-compaction (cardiomyopathy) Nonsyndromic HTAD |
|||
| LTBP3 | Latent TGF-β3 binding protein 3 | Extracellular matrix and TGF-β signaling | Dental anomalies and short stature syndrome Geleophysic dysplasia 3 |
-- | ||
| MAT2A | Methionine adenosyltransferase II α | SMC metabolism | Nonsyndromic HTAD | -- | ||
| MFAP5 | Microfibrillar-associated protein 5 | Extracellular matrix | Nonsyndromic HTAD Musculoskeletal MFS manifestations |
-- | ||
| NOTCH1 | Notch 1 | Bicuspid aortic valve | ||||
| SKI | SKI protooncogene | TGF-β signaling | Shprintzen-Goldberg syndrome | Consistent with syndrome | ||
| SLC2A10 | Solute carrier family 2 member 10 | Glucose transporter | Arterial tortuosity syndrome | Consistent with syndrome | ||
| SMAD2 | Mothers against decapentaplegic drosophila homolog 2 | TGF-β signaling | -- | -- | ||
| SMAD4 | Mothers against decapentaplegic drosophila homolog 4 | TGF-β signaling | Juvenile polyposis Hereditary hemorrhagic telangiectasia Nonsyndromic HTAD |
Consistent with syndrome | ||
| TGFB3 | TGF-β3 | TGF-β signaling | Loeys-Dietz syndrome 5 Rienhoff syndrome Arrythmogenic right ventricular dysplasia 1 |
Aneurysms, dissections/ruptures of other arteries | ||
PDA: patent ductus arteriosus; cGMP: cyclic guanosine monophosphate; HTAD: heritable thoracic aortic disease; LINC: linker of nuclear skeleton and cytoskeleton; SMC: smooth muscle cell; TGF: transforming growth factor.
Despite the number of causative genes identified for HTAD to date, others remain unidentified. In HTAD families with systemic features of MFS and LDS, the majority will have an identifiable pathogenic variant. This is not true of HTAD families without syndromic features, as pathogenic variants in the 11 validated HTAD genes explain only approximately 30% of disease.
Identification of heritable thoracic aortic disease and the causative genes: impact of genetic testing and counseling on patients with TAAD
Genetic testing can establish, rule out, and differentiate genetic diagnoses. This information is used to identify family members at risk and enables appropriate management of patients with respect to medical therapy, surveillance strategy, timing of prophylactic aortic repair and surgical approach, and adoption of lifestyle modifications (Figure 1). Substantial clinical overlap of TAAD and systemic features among HTAD syndromes, such as MFS and LDS, often requires genetic testing to confirm the correct diagnosis; in fact, the only clinical characteristic that reliably distinguishes MfS from LDS is ocular lens dislocation. Correct classification of MFS and LDS is critical as management recommendations differ based on the underlying gene involved. Importantly, the majority of HTAD genes are not associated with systemic features, making genetic testing necessary to diagnose affected individuals and identify family members at risk for TAAD.
Figure 1.—
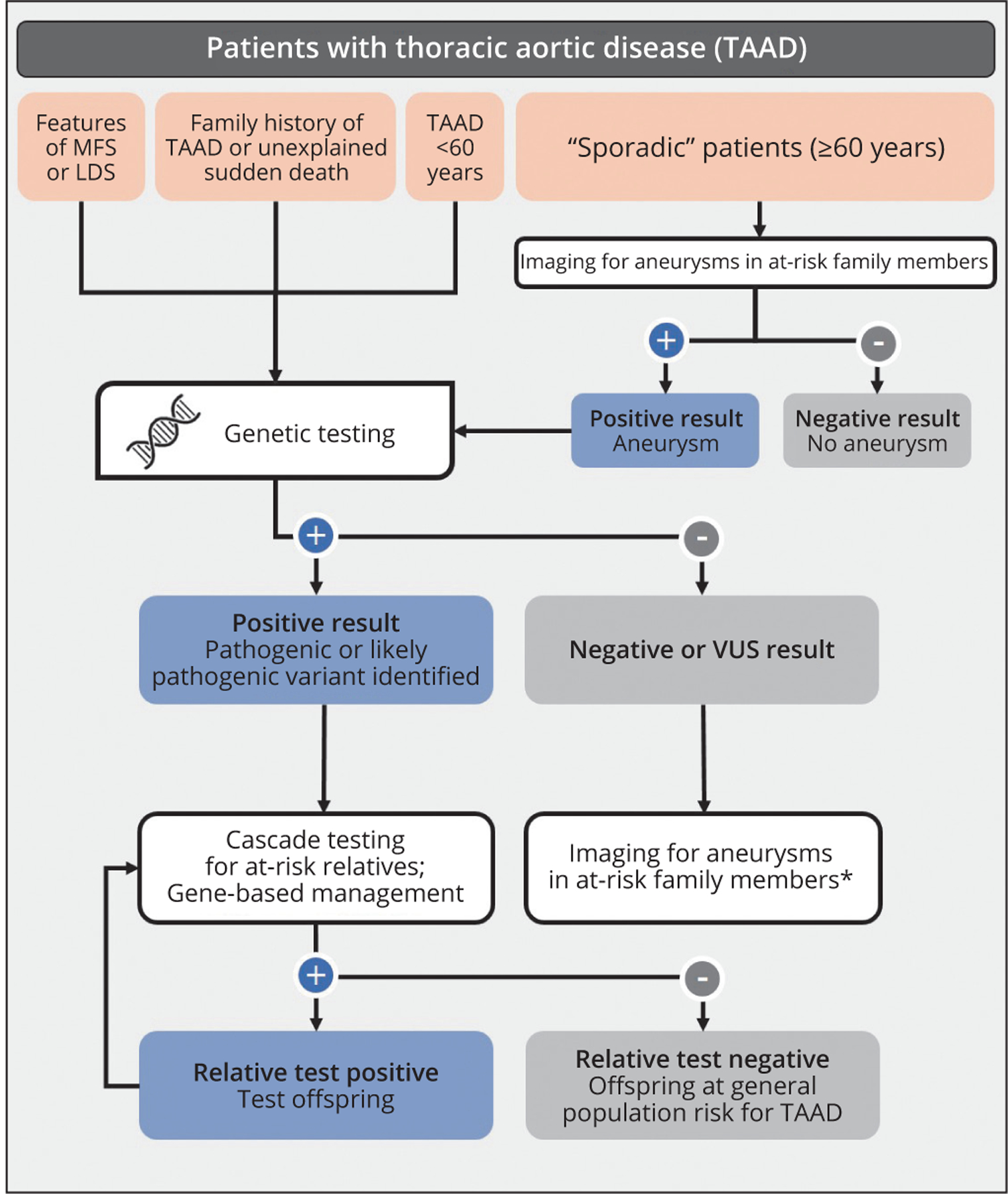
Evaluation and genetic testing process for patients with thoracic aortic disease. Genetic testing is recommended for individuals with syndromic features, family history of TAAD and/or early age of disease onset. Thoracic aortic imaging is recommended for first-degree relatives of all individuals with TAAD regardless of age of onset to detect asymptomatic aneurysms. Genetic testing can lead to a positive result, triggering gene-based management and cascade testing of at-risk relatives. Individuals with negative or VUS results should be managed according to standards of care, which includes the recommendation for imaging in first-degree relatives.
LDS: Loeys-Dietz syndrome; MFS: Marfan syndrome; TAAD: thoracic aneurysms and acute dissections; VUS: variant of unknown significance.
*May be asymptomatic aneurysm.
Clinical genetic testing is integral to the diagnostic evaluation of TAAD patients who have clinical indicators or “red flags” suggestive of HTAD. TAAD patients with the following should be considered for genetic testing: a family history of TAAD or sudden premature death; systemic or vascular features suggestive of an HTAD genetic syndrome; TAAD diagnosed at a relatively young age (<60 years).1, 2 It is important to initiate testing in an individual diagnosed with thoracic aortic disease. A multigene panel comprised of all genes established or suspected to cause HTAD is currently the most cost effective and clinically useful approach to testing (Table I). These gene panels include the 11 HTAD genes validated to cause disease along with genes that have not been validated. These multigene “aortopathy” panels are available from DNA diagnostic laboratories worldwide and are diagnostically superior to targeted single-gene sequencing due to the phenotypic overlap between genes. Importantly, the same panel for aortopathies can be used to test for causative mutations in individuals with aneurysms, dissection or ruptures of other arteries at young ages and older individuals with multiple aneurysms/dissections/ruptures of other arteries with or without thoracic aortic disease. A more comprehensive approach to genetic testing, exome sequencing, may be used to evaluate protein-coding regions (exons) throughout the genome; however, this is typically only ordered for patients with thoracic aortic disease associated with complex multisystem disease that does not fit a pattern of a known syndrome.
The genetic testing laboratory provides classification of rare variants in HTAD genes identified in a patient into the following classes: pathogenic, likely pathogenic, variant of uncertain/unknown significance (VUS), benign and likely benign (not issued on test report). If a pathogenic or likely pathogenic variant is identified, this establishes a definitive diagnosis, enabling adoption of gene-based management and diagnosis of at-risk relatives through cascade testing of family members, (e.g., testing all relatives who are at risk for inheriting the disease-causing variant). VUSs are commonly identified through multi-gene panel testing and refer to an alteration in a gene with insufficient evidence to determine whether it is responsible for TAAD. Therefore, VUSs are not clinically actionable and should not be used for cascade testing of family members or to inform management. However, these classifications are based on available data at the time and thus, are subject to change and may be modified over time.
While a negative test result signifies that no pathogenic variant or VUS was identified in the genes tested, it does not imply absence of heritable aortic disease. Rather, a negative result suggests that the genetic etiology remains unidentified. When a pathogenic variant is not identified in an individual with TAAD and/or genetic testing was not pursued due to lack of family history or older age of disease onset, imaging of the aortic root and ascending aorta is still recommended for that patient’s first-degree relatives to identify family members with asymptomatic aneurysms in accordance with guidelines.3 If other vascular diseases, such as intracranial aneurysms, segregate with TAAD in a family, imaging should be expanded to include other relevant arteries. When imaging reveals an asymptomatic aneurysm in a relative, this signifies a hereditary form of disease and genetic testing of an affected family member is recommended if no other affected relatives have undergone testing. If no relative is found to have thoracic aortic disease through familial imaging, the affected patient is considered to have sporadic thoracic aortic disease.
Importantly, the family history of some HTAD families suggest that only men are affected, but most of the genes responsible thoracic aortic disease are inherited in a sex-independent autosomal dominant manner. The predominance of disease in males evident in some HTAD families is likely due to the decreased penetrance of the disease in women rather than X-linked inheritance. Therefore, the sex of the family member should not influence testing or imaging recommendations.
Through clinical characterization of HTAD families with pathogenic variants in novel genes, data have emerged that the underlying gene predicts not only who in the family is at risk for TAAD, but also: 1) the aortic disease presentation; 2) risk for dissection at a given range of aortic diameters as described above; and 3) risk for additional vascular diseases. for example, TGFBR2 mutations predispose to TAAD, but also intracranial aneurysms and aneurysms and dissections of other arteries, whereas ACTA2 mutations lead to TAAD and occlusive vascular disease, including early onset stroke and coronary artery disease.9, 22 Based on the strength of these clinical correlates, the ACCf/AHA Treatment Guidelines for Thoracic Aortic Diseases emphasize gene-based management of aortic disease once the defective gene is identified.1 To define the clinical phenotype and spectrum of pathogenic variants more rapidly and effectively in novel HTAD genes, we established the first thoracic aortic disease international consortium, the Montalcino Aortic Consortium (MAC). The Montalcino Aortic Consortium (MAC) was founded with the mission to collect vascular disease data on individuals with pathogenic variants in HTAD genes worldwide to improve the management and outcomes of these patients through generation of evidence-based treatment guidelines. In the largest cohort of cases with TGFBR1 and TGFBR2 pathogenic variants (N.=441), the MAC found that type A dissections at diameters <4.5 cm in patients with LDS were rare,23 which led to changes in previous consensus-based recommendations.24, 25 We also identified differences in aortic disease presentation in patients with TGFBR1 versus TGFBR2 variants, including dramatic sex differences in the age of onset of aortic events in patients with TGFBR1 mutations, but not TGFBR2.23 Subsequently, MAC sites provided data on patients with pathogenic variants and VuSs in ACTA2, SMAD3 and MYLK, along with assessment of the contribution of SMAD4 variants to HTAD.26–29 Thus, the data support that once the causative gene is identified, gene-based recommendations should be used to inform treatment and management. Notably, this includes the genes for LDS as the vascular disease management and systemic complications vary between the LDS genes.4, 5
Genetic predisposition in sporadic TAAD patients
Most patients with thoracic aortic disease, approximately 80%, do not carry a mutation in a single gene predisposing them to disease. At the same, data emerging from research indicates that these individuals, termed sporadic cases, have variants in their genome that predispose them to the disease. We were the first to do a (GWAS) on patients of European American descent with sporadic TAAD. Surprisingly, there was only one major region of the human genome associated with sporadic TAAD in this study, and this region fell on top of the gene for the MfS, FBN1, suggesting that the molecular pathogenesis leading to TAAD in MfS patients, also drives thoracic aortic disease in the general population.30, 31 However, a mutation in FBN1 that causes MfS, increases an individual’s risk for TAAD to nearly 100%, whereas the increased risk associated with the GWAS signaling, only increased risk 1.6-fold and therefore, does not help identify individuals at high risk for aortic disease. Our GWAS also identified a chromosomal duplication at 16p13.1, encompassing the HTAD gene (MYH11). This duplication was present at low levels in the general population and led to a 10-fold increased risk for sporadic thoracic aortic disease, much higher than the GWAS peak, but much lower than a pathogenic variant in MYH11.32 A separate GWAS on patients of European American descent with sporadic aortic dissections confirmed the association of single nucleotide polymorphisms (SNPs) at FBN1 with disease, and also identified significant associations of SNPs at ULK4 and LRP1 loci with thoracic aortic dissection.33
We pursued exome sequencing on a cohort of 355 individuals with aortic dissections at ≤56 years of age, and identified pathogenic variants in the 11 known HTAD genes in 9% of the cohort.34 other exome sequencing studies on patients with sporadic TAAD have confirmed that pathogenic variants in known HTAD genes were present in 9–20% of patients.35–37 In our study, burden analysis showed the patients of European American descent have a significantly higher burden of variants with minor allele frequency <0.005 in the Exome Aggregation Consortium Database compared to ethnicity-matched controls and an increased burden of VUSs in these same genes in the cases compared to controls.34, 38 Assays on variants identified in LOX and MYLK found that pathogenic variants completely disrupted protein function, whereas a subset of the VUSs disrupted protein function to a lesser extent than the mutations.39 These results suggest that there are variants in the validated HTAD genes that increase the risk for an acute aortic dissection but are not sufficiently penetrant to be classified as pathogenic variants.
Two recent GWAS were completed on over 30,000 individuals from the United Kingdom Biobank whose aortic images were available to investigate the association of SNPs with aortic diameters. Most of the individuals studied were of European descent and did not have TAAD. Common variants or SNPs in FBN1, the gene for MFS, showed significant association with increased aortic diameters. In addition, SNPs at the ULK4 locus also showed significant association with diameters of aortas.40, 41 A meta-analysis of GWAS data including patients with TAAD, abdominal aortic aneurysms, and intracranial aneurysms, indicated that there was no evidence for polygenic overlap among these diseases.42
Conclusions
In conclusion, the expanding list of HTAD genes has significantly impacted the care and management of patients with TAAD and their families. At the same time, genetic studies on sporadic TAAD and thoracic aortic diameters in the general population have identified variants in novel genes. Most of these genetic variants confer either a low risk for TAAD or no risk for progression to TAAD. Identifying individuals in the general population who are at risk for acute aortic dissection will require integrating genetic data with environmental risk factors (e.g., hypertension, drugs, and activity prior to dissection) from a large cohort of dissection cases to establish a dissection risk profile. Only then can we move towards the goal of preventing all acute aortic dissections and the associated mortality and morbidity.
Footnotes
Conflicts of interest.—Dianna M. Milewicz, Alana C. Cecchi, Ellen Hostetler, Isabella Marin, Amelie C. Pinard and Dongchuan Guo received research grants from NIH, AHA and GADA and received donations from patients and foundations. Dianna M. Milewicz received employment funds.
References
- 1.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE Jr, et al. ; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010;121:e266–369. [DOI] [PubMed] [Google Scholar]
- 2.Pape LA, Tsai TT, Isselbacher EM, Oh JK, O’gara PT, Evangelista A, et al. ; International Registry of Acute Aortic Dissection (IRAD) Investigators. Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation 2007;116:1120–7. [DOI] [PubMed] [Google Scholar]
- 3.Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg 1997;25:506–11. [DOI] [PubMed] [Google Scholar]
- 4.Albornoz G, Coady MA, Roberts M, Davies RR, Tranquilli M, Riz-zo JA, et al. Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006;82:1400–5. [DOI] [PubMed] [Google Scholar]
- 5.Milewicz DM, Chen H, Park ES, Petty EM, Zaghi H, Shashidhar G, et al. Reduced penetrance and variable expressivity of familial thoracic aortic aneurysms/dissections. Am J Cardiol 1998;82:474–9. [DOI] [PubMed] [Google Scholar]
- 6.Pyeritz RE, McKusick VA. The Marfan syndrome: diagnosis and management. N Engl J Med 1979;300:772–7. [DOI] [PubMed] [Google Scholar]
- 7.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet 1995;4:1799–809. [DOI] [PubMed] [Google Scholar]
- 8.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005;37:275–81. [DOI] [PubMed] [Google Scholar]
- 9.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006;355:788–98. [DOI] [PubMed] [Google Scholar]
- 10.Regalado ES, Guo DC, Villamizar C, Avidan N, Gilchrist D, McGillivray B, et al. ; NHLBI GO Exome Sequencing Project. Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circ Res 2011;109:680–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet 2011;43:121–6. [DOI] [PubMed] [Google Scholar]
- 12.Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, et al. ; National Heart, Lung, and Blood Institute (NHLBI) Go Exome Sequencing Project. TGfB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet 2012;44:916–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, fert-Bober J, et al. Loss-of-function mutations in TGfB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet 2012;44:922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pannu H, fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, et al. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation 2005;112:513–20. [DOI] [PubMed] [Google Scholar]
- 15.Tran-fadulu V, Pannu H, Kim DH, Vick GW 3rd, Lonsford CM, Lafont AL, et al. Analysis of multigenerational families with thoracic aortic aneurysms and dissections due to TGfBR1 or TGfBR2 mutations. J Med Genet 2009;46:607–13. [DOI] [PubMed] [Google Scholar]
- 16.LeMaire SA, Pannu H, Tran-fadulu V, Carter SA, Coselli JS, Mile-wicz DM. Severe aortic and arterial aneurysms associated with a TGfBR2 mutation. Nat Clin Pract Cardiovasc Med 2007;4:167–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo DC, Pannu H, Tran-fadulu V, Papke CL, Yu RK, Avidan N, et al. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 2007;39:1488–93. [DOI] [PubMed] [Google Scholar]
- 18.Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, et al. Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 2010;87:701–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo DC, Regalado E, Casteel DE, Santos-Cortez RL, Gong L, Kim JJ, et al. ; GenTAC Registry Consortium; National Heart, Lung, and Blood Institute Grand Opportunity Exome Sequencing Project. Recurrent gain-of-function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. Am J Hum Genet 2013;93:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo DC, Gong L, Regalado ES, Santos-Cortez RL, Zhao R, Cai B, et al. ; GenTAC Investigators, National Heart, Lung, and Blood Institute Go Exome Sequencing Project; Montalcino Aortic Consortium. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am J Hum Genet 2015;96:170–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renard M, francis C, Ghosh R, Scott Af, Witmer PD, Adès LC, et al. Clinical Validity of Genes for Heritable Thoracic Aortic Aneurysm and Dissection. J Am Coll Cardiol 2018;72:605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo DC, Papke CL, Tran-fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet 2009;84:617–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jondeau G, Ropers J, Regalado E, Braverman A, Evangelista A, Teixedo G, et al. ; Montalcino Aortic Consortium. International Registry of Patients Carrying TGfBR1 or TGfBR2 Mutations: Results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet 2016;9:548–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiratzka Lf, Bakris GL, Beckman JA, Bersin RM, Carr Vf, Casey DE Jr, et al. ; American College of Cardiology foundation/American Heart Association Task force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. 2010 ACCf/AHA/AATS/ACR/ASA/SCA/SCAI/ SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology foundation/American Heart Association Task force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology,American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons,and Society for Vascular Medicine. J Am Coll Cardiol 2010;55:e27–129. [DOI] [PubMed] [Google Scholar]
- 25.MacCarrick G, Black JH 3rd, Bowdin S, El-Hamamsy I, frischmeyer-Guerrerio PA, Guerrerio AL, et al. Loeys-Dietz syndrome: a primer for diagnosis and management. Genet Med 2014;16:576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Regalado ES, Guo DC, Prakash S, Bensend TA, flynn K, Estrera A, et al. ; Montalcino Aortic Consortium. Aortic Disease Presentation and Outcome Associated With ACTA2 Mutations. Circ Cardiovasc Genet 2015;8:457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace SE, Regalado ES, Gong L, Janda AL, Guo DC, Russo Cf, et al. MYLK pathogenic variants aortic disease presentation, pregnancy risk, and characterization of pathogenic missense variants. Genet Med 2019;21:144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hostetler EM, Regalado ES, Guo DC, Hanna N, Arnaud P, Muiño-Mosquera L, et al. SMAD3 pathogenic variants: risk for thoracic aortic disease and associated complications from the Montalcino Aortic Consortium. J Med Genet 2019;56:252–60. [DOI] [PubMed] [Google Scholar]
- 29.Duan XY, Guo DC, Regalado ES, Shen H, Coselli JS, Estrera AL, et al. ; University of Washington Center for Mendelian Genomics. SMAD4 rare variants in individuals and families with thoracic aortic aneurysms and dissections. Eur J Hum Genet 2019;27:1054–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.LeMaire SA, McDonald ML, Guo DC, Russell L, Miller CC 3rd, Johnson RJ, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning fBN1 at 15q21.1. Nat Genet 2011;43:996–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolford BN, Hornsby WE, Guo D, Zhou W, Lin M, farhat L, et al. Clinical Implications of Identifying Pathogenic Variants in Individuals With Thoracic Aortic Dissection. Circ Genom Precis Med 2019;12:e002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuang SQ, Guo DC, Prakash SK, McDonald ML, Johnson RJ, Wang M, et al. ; GenTAC Investigators. Recurrent chromosome 16p13.1 duplications are a risk factor for aortic dissections. PLoS Genet 2011;7:e1002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo DC, Grove ML, Prakash SK, Eriksson P, Hostetler EM, LeMaire SA, et al. ; GenTAC Investigators; BAVCon Investigators. Genetic Variants in LRP1 and ULK4 Are Associated with Acute Aortic Dissections. Am J Hum Genet 2016;99:762–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo DC, Hostetler EM, fan Y, Kulmacz RJ, Zhang D, Nickerson DA, et al. ; GenTAC Investigators. Heritable Thoracic Aortic Disease Genes in Sporadic Aortic Dissection. J Am Coll Cardiol 2017;70:2728–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Overwater E, Marsili L, Baars MJ, Baas Af, van de Beek I, Dulfer E, et al. Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Hum Mutat 2018;39:1173–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pan M, Chen S, Wang H, Wu S, Ding Z, Wang Y, et al. Exploring the genetic pathogenicity of aortic dissection from 72 Han Chinese individuals using next-generation sequencing. Clin Genet 2020;97:704–11. [DOI] [PubMed] [Google Scholar]
- 37.Zhao H, Yang Y, Pan X, Li W, Sun L, Guo J. Identification of clinically relevant variants by whole exome sequencing in Chinese patients with sporadic non-syndromic type A aortic dissection. Clin Chim Acta 2020;506:160–5. [DOI] [PubMed] [Google Scholar]
- 38.Iakoubova OA, Tong CH, Rowland CM, Luke MM, Garcia VE, Catanese JJ, et al. Genetic variants in fBN-1 and risk for thoracic aortic aneurysm and dissection. PLoS One 2014;9:e91437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kwartler CS, Gong L, Chen J, Wang S, Kulmacz R, Duan XY, et al. Variants of Unknown Significance in Genes Associated with Heritable Thoracic Aortic Disease Can Be Low Penetrant “Risk Variants”. Am J Hum Genet 2018;103:138–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pirruccello JP, Chaffin MD, Fleming SJ, Arduini A, Lin H, Khurshid S, et al. Deep learning enables genetic analysis of the human thoracic aorta. bioRxiv 2020:091934. [DOI] [PMC free article] [PubMed]
- 41.Tcheandjieu C, Xiao K, Tejeda H, Lynch JA, Ruotsalainen S, Bellomo T, et al. High heritability of ascending aortic diameter and multi-ethnic prediction of thoracic aortic disease. medRxiv 2020:20102335. [DOI] [PubMed]
- 42.van ‘t Hof FN, Ruigrok YM, Lee CH, Ripke S, Anderson G, de Andrade M, et al. ; Aneurysm Consortium; Vascular Research Consortium of New Zealand. Shared Genetic Risk Factors of Intracranial, Abdominal, and Thoracic Aneurysms. J Am Heart Assoc 2016;5:e002603. [DOI] [PMC free article] [PubMed] [Google Scholar]