

Abstract
A number of genes are reportedly responsible for hereditary hearing loss, which accounts for over 50% of all congenital hearing loss cases. Recent advances in genetic testing have enabled the identification of pathogenic variants in many cases, and systems have been developed to provide personalized treatment based on etiology. Gene therapy is expected to become an unprecedented curative treatment. Several reports have demonstrated the successful use of cochlear gene therapy to restore auditory function in mouse models of genetic deafness; however, many hurdles remain to its clinical application in humans. Herein, we focus on the frequency of deafness genes in patients with congenital and late‐onset progressive hearing loss and discuss the following points regarding which genes need to be targeted to efficiently proceed with clinical application: (a) which cells' genes are expressed within the cochlea, (b) whether gene transfer to the targeted cells is possible using vectors such as adeno‐associated virus, (c) what phenotype of hearing loss in patients is exhibited, and (d) whether mouse models exist to verify the effectiveness of treatment. Moreover, at the start of clinical application, gene therapy in combination with cochlear implantation may be useful for cases of progressive hearing loss.
Keywords: adeno‐associated virus, cochlear implant, electric‐acoustic stimulation, gene therapy, genetic deafness, hereditary hearing loss
1. INTRODUCTION
Congenital hearing loss is a relatively common disorder, occurring in 1 to 2 per 1000 newborns, among which 60%–70% of cases are hereditary. 1 Due to widespread newborn hearing screening, hearing loss has been increasingly detected in the early stages after birth. 1 Identifying the etiology of hearing loss has also improved due to genetic testing, imaging with CT and MRI, and testing for congenital cytomegalovirus infection following auditory testing. 2 Meanwhile, hearing aids (HAs) and cochlear implants (CIs) are indicated depending on the severity of hearing loss, with CIs currently being the standard treatment for severe‐to‐profound hearing loss patients. However, these devices are not biological treatments. Gene therapy is expected to be developed as a treatment targeting causal genes for genetic deafness. 3 Here, we report a summary of the current state of genetic testing in humans, gene therapy research in mouse models, and the areas requiring further study, as well as milestones toward the clinical application of gene therapy.
2. USEFULNESS OF GENETIC TESTING IN PATIENTS WITH HEREDITARY HEARING LOSS
Approximately 120 deafness genes have been identified as responsible for non‐syndromic hearing loss (NSHL) (https://hereditaryhearingloss.org/), which refers to hearing loss without other signs or symptoms. Among a number of deafness genes, assumptions regarding the causative gene of NSHL from the severity of hearing loss and mode of inheritance are difficult due to genetic heterogeneity 4 ; thus, an accurate diagnosis of NSHL requires genetic testing. Over 400 different syndromes have been reported for syndromic hearing loss (SHL), which is accompanied by various symptoms besides hearing loss. 5 Although most syndromes can be clinically diagnosed from the accompanying symptoms, many are associated with multiple genes, making accurate diagnosis difficult without genetic testing, similar to NSHL. For example, patients with Usher syndrome types 1 and 2, in which congenital hearing loss is accompanied by late‐onset retinitis pigmentosa, exhibit hearing loss alone in childhood, thus appearing similar to NSHL. 6 , 7 Patients later show vision symptoms such as night blindness, and Usher syndrome can be clinically diagnosed. Diagnosis before the appearance of vision symptoms can only be made by genetic testing; thus, in cases of SHL, genetic testing may allow early diagnosis and intervention. Identification of causal genes may enable not only accurate diagnosis, but also prediction of the accompanying symptoms, effectiveness of existing therapy, and rate of recurrence. 8 If gene therapy based on the genetic diagnosis of deafness becomes available in the future, genetic testing will become even more clinically important.
3. FREQUENCY OF GENES RESPONSIBLE FOR HEREDITARY HEARING LOSS
In recent years, due to the emergence of targeted genomic enrichment and massively parallel DNA sequencing, genetic testing has enabled comprehensive analysis of a number of genes responsible for hearing loss, which has led to a dramatic improvement in the diagnostic rate and turnaround time for diagnosis. 4 , 9 , 10 Sloan‐Heggen et al and our research team previously reported that pathogenic variants in deafness genes are identified in approximately 40% of patients with hearing loss. 9 , 10 As stated above, over 120 deafness genes have been identified, while their frequency differs and is known to vary by ethnicity. In Japan, GJB2, SLC26A4, and CDH23 are reported to be the prevalent deafness genes (Figure 1). 9 Sloan‐Heggen et al reported that GJB2 is the most frequently identified deafness gene (21.6%), followed by STRC (16.1%), SLC26A4 (6.6%), TECTA (5.2%), MYO15A (4.8%), MYO7A (4.5%), USH2A (4.3%), CDH23 (4.1%), ADGRV1 (2.7%), TMC1 (2.3%), PCDH15 (2.0%), OTOF (2.4% in autosomal recessive NHSL patients), and TMPRSS3 (2.4% in autosomal recessive NHSL patients) in 1119 deafness patients of Caucasian, Hispanic, African American, Asian, Middle Eastern, Ashkenazi Jewish, and mixed ethnicity. 10 When considering which gene should be targeted for gene therapy, estimating the number of patients is important while keeping in mind the frequency of the deafness gene in patients with hearing loss.
FIGURE 1.
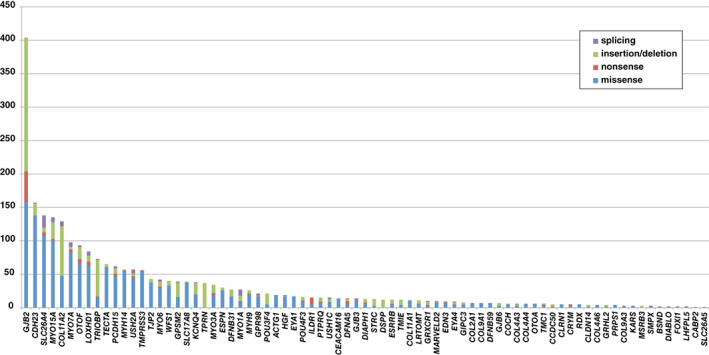
The frequency of genetic mutations found in 1120 Japanese patients with hearing loss (Nishio et al., 2015). 9 Relatively prevalent deafness genes, such as GJB2, CDH23, and SLC26A4, are described
4. SELECTION OF TARGET GENES FOR GENE THERAPY
As mentioned above, a preclinical study of the prevalent deafness genes in humans is needed prior to clinical application. Not only the genetic frequency needs to be determined, but also the type of cells in which each gene is expressed and the role it plays in cellular function. The cochlea, which has a highly complex structure, consists of various types of cells that possess functionally distinct roles owing to their distinct gene expression patterns (Figure 2). 11 For example, of the prevalent genes responsible for hearing loss shown in Table 1, those that are mainly expressed in the inner/outer hair cells include MYO15A, 26 MYO7A, 29 USH2A, 35 CDH23, 38 ADGRV1, 44 TMC1, 46 and PCDH15, 38 while STRC 17 is mainly expressed in the outer hair cells and OTOF 52 is mainly expressed in the inner hair cells. Moreover, GJB2 12 is reportedly expressed in the supporting cells, spiral limbus, and lateral wall, while SLC26A4 19 is expressed in the outer sulcus and the epithelial cells of the spiral prominence, TECTA 23 is expressed from the inter dental cells and encoding protein is localized in tectorial membrane, and TMPRSS3 54 is expressed in the hair cells and spiral ganglion. In the development of gene therapy, the target cells of the gene transfer will depend on where the target gene is expressed in the inner ear.
FIGURE 2.
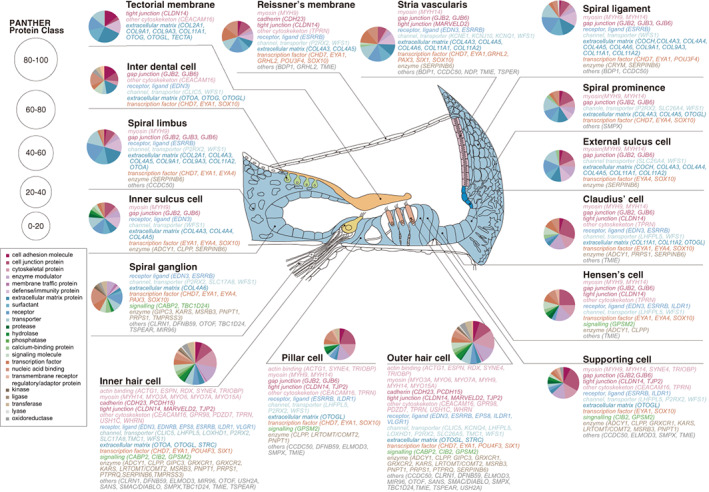
The expression sites of causal genes of hearing loss in the cochlea (Nishio et al., 2015). 11 The cochlea contains various types of cells that have distinct functions. The genes responsible for genetic deafness expressed in each cell type are shown
TABLE 1.
Summary of hearing loss phenotype and gene expression profiles of the prevalent deafness genes
| Length (bp) | |||||||
|---|---|---|---|---|---|---|---|
| Gene (OMIM) | [NM No.] | Locus (OMIM) | Hearing loss phenotype in human | Expression profiles in the cochlea | Mouse model | Hearing loss phenotype in mice | Reference |
| GJB2 | 2290 | DFNB1A | Congenital mild to profound | SCs | Conditional Gjb2 KO mouse | Moderate | 12, 13, 14, 15, 16 |
| [NM_004004.6] | DFNA3A | Late onset mild to moderate | Tg Cx26 R75W | Profound | |||
| STRC | 5515 | DFNB16 | Congenital/childhood onset mild to moderate, nonprogressive | Stereocilia of the OHC | Strc −/− | Progressive | 17, 18 |
| [NM_153700.2] | |||||||
| SLC26A4 | 4737 | DFNB4 | Congenital, high‐frequency, fluctuating | Outer sulcus | Pds −/− | Profound | 19, 20, 21, 22 |
| [NM_000441.2] | Spiral prominence epithelial cells | Slc26a4 insufficient | Fluctuating | ||||
| TECTA | 7353 | DFNA12 | Mild to moderate, mid‐ or high‐ frequency | Tectorial membrane | (None) | 23, 24, 25 | |
| [NM_005422.4] | |||||||
| MYO15A | 11 811 | DFNB3 | Congenital profound or late onset moderate to severe, progressive | HCs | Shaker‐2 | Profound | 26, 27, 28 |
| [NM_016239.4] | |||||||
| MYO7A | 7483 | DFNA11 | Late onset mild to severe, progressive | HCs, cuticular plate, synaptic region | Headbanger | Low frequency | 29, 30, 31, 32, 33, 34 |
| [NM_000260.4] | DFNB2, USH1B | Congenital profound | Shaker‐1 | Progressive | |||
| USH2A | 6372 | USH2A | Congenital moderate to severe | Ankle link of the HCs | Ush2a −/− | Moderate, nonprogressive | 35, 36, 37 |
| [NM_007123.6] | |||||||
| CDH23 | 11 138 | DFNB12 | Moderate to profound, high‐frequency progressive | Tip link of the HCs | salsa, C57BL/6J | Progressive | 38, 39, 40, 41, 42, 43 |
| [NM_022124.6] | USH1D | Congenital profound | Waltzer | Early‐onset profound | |||
| ADGRV1 | 19 557 | USH2C | High‐frequency | Ankle link of the HCs | CBA‐2ush/ush | Severe | 6, 44, 45 |
| [NM_032119.4] | |||||||
| TMC1 | 5340 | DFNA36 | Postlingual progressive | HCs | Beethoven | Progressive | 46, 47, 48, 49 |
| [NM_138691.3] | DFNB7/11 | Prelingual, severe to profound | Baringo | Early‐onset profound | |||
| PCDH15 | 6962 | DFNB23 | Prelingual profound | Tip link of the HCs | Ames waltzer | Profound | 38, 50, 51 |
| [NM_033056.4] | USH1F | Prelingual profound | |||||
| OTOF | 7214 | DFNB9 | Congenital or prelingual severe to profound | Synaptic vesicles of the IHC | Otof −/− | Profound | 52, 53 |
| [NM_194248.3] | |||||||
| TMPRSS3 | 2399 | DFNB8/10 | Congenital severe or postlingual progressive, high frequency | HCs, SGNs | Tmprss3 Y260X/Y260X | Profound | 54, 55, 56 |
| [NM_024022.4] |
Abbreviations: HC, hair cell; IHC, inner hair cell; KO, knockout; OHC, outer hair cell; SC, supporting cell; SGN, spiral ganglion neuron.
In addition, effective gene therapy for hearing loss requires an understanding of the audioprofile of deafness genes. In hereditary hearing loss, the time of onset, severity, and presence/absence of progression vary depending on the gene responsible. For example, GJB2‐related hearing loss, which is inherited in an autosomal recessive manner, is congenital with mild‐to‐profound hearing loss that rarely progresses. 13 Similarly, STRC‐, 18 TECTA‐, 24 , 25 USH2A‐, 36 and ADGRV1‐related 6 hearing loss are congenital with no progression, but the severity of hearing loss is relatively mild. SLC26A4 causes congenital and fluctuating progressive hearing loss. 20 In contrast, MYO15A, 27 MYO7A, 30 , 31 , 32 CDH23, 39 , 40 and TMPRSS3 55 are responsible for congenital profound or progressive hearing loss. CDH23 and PCDH15 are responsible for both NSHL and Usher syndrome. For these two genes, a genotype‐phenotype correlation has been reported, and Usher syndrome is known to result from a truncating mutation. 50 , 57 In CDH23‐related NSHL, hearing loss is progressive, starting with high‐frequency hearing loss that gradually affects the low frequencies and, finally, hearing loss occurs across all frequencies. 39 Moreover, for several deafness genes, the course of disease reportedly varies according to ethnicity. 58 Understanding the audioprofile for each deafness gene enables prediction of the appropriate therapeutic time window for gene therapy intervention.
5. LIMITATIONS OF MOUSE MODELS FOR HEREDITARY HEARING LOSS FOR PRECLINICAL STUDY
When considering the application of gene therapy to humans, preclinical studies using mouse models of hereditary hearing loss are essential (Table 1). However, mouse models of deafness that possess similar phenotypes and mutations as in humans with hearing loss are extremely rare. For example, truncating mutations (c.235delC in Asian countries or c.35delG in the non‐Asian population) in GJB2 are commonly reported in humans, 59 whereas GJB2 knockout is known to be lethal in embryonic mice. 60 At present, only conditional GJB2 knockout has been reported in a mouse model for the most common, autosomal recessive, GJB2‐related hearing loss. 14 A mouse model for TMC1‐related hearing loss, named Beethoven mice, demonstrates a similar genetic mutation, hearing loss severity, and presence/absence of progression as in humans. 47 Beethoven mice, generated by ENU mutagenesis, have a c.1235T>A mutation, which is orthologous to the TMC1 mutation found in patients with hearing loss. 61 , 62 As a phenotype of hearing loss, both are known to exhibit progressive hearing loss; thus, this is one of ideal mouse models for the study of human hereditary hearing loss. Unfortunately, it remains difficult to generate mice with mutations orthologous to those in each gene responsible for human hearing loss, and that exhibit similar hearing loss to that in humans. In preclinical studies, mice with a similar hearing loss phenotype to that in humans are considered useful. However, regenerating the lost hair cells and a denatured cochlear structure is considered extremely difficult; thus, using a mouse model of hereditary hearing loss exhibiting progressive hearing loss instead of congenital profound hearing loss is considered a straightforward strategy. 63 In addition, the therapeutic effectiveness of a model with progressive hearing loss is expected to be poor unless intervention is performed before irreversible changes occur. 64 Mouse models for CDH23‐related hearing loss, salsa 41 and C57BL/6J 42 mice, are attractive as they exhibit similar progressive hearing loss to that in humans. For congenital hearing loss, a mouse model of hearing loss caused by OTOF, associated with neurotransmission and not cochlear architecture, reportedly provides a good target to determine therapeutic effectiveness. 65 Furthermore, attention needs to be paid to the differences in inner ear development between mice and humans. The inner ear matures by 26 weeks' gestation in humans, whereas in mice, the inner ear does not mature until 15 days after birth. Therefore, the results of studies using neonatal mice are difficult to apply directly to humans, and experiments using adult mice are needed for clinical application in humans. 64
6. GENE THERAPY STRATEGIES FOR HEREDITARY HEARING LOSS
HAs and CIs are effective therapeutic devices for sensorineural hearing loss. However, gene therapy is expected to be a therapeutic method that can provide curative treatment. 3 Moreover, gene therapy may potentially suppress hearing loss progression or restore hearing function, which cannot be achieved with HAs or CIs. Gene therapy primarily includes three approaches. The first approach, “gene replacement,” is the most common method by which a functional protein is supplied by delivering a normal gene. The appropriate candidates for this strategy are inherited disorders caused by loss‐of‐function mutations, such as recessive diseases. The second approach, “gene silencing,” is a method to treat diseases with gain‐of‐function mutations by suppressing the expression of the mutated gene. The third approach, “gene editing,” enables correction of pathogenic variants by genome editing, such as the CRISPR/Cas9 system.
7. SELECTION OF TARGETED GENES, VECTORS, AND DELIVERY ROUTES
In the selection of any gene therapy strategy, genetic material needs to be delivered to the inner ear by use of a vector. Some papers have already summarized delivery routes and viral vectors for cochlear gene therapy. 3 , 66 Established routes for direct local administration have included (a) round window membrane (RWM) injection, (b) RWM injection with semi‐circular canal fenestration, (c) cochleostomy, and (d) canalostomy (Figure 3). 3 , 66 , 67 Among them, the two injection routes via the RWM enable the vector to be delivered into the perilymph, which is clinically feasible. While a number of viral or non‐viral vectors have been reported, adeno‐associated virus vectors (AAVs) have been used in most hereditary hearing loss studies due to their non‐pathogenicity or minimal immunogenicity. 66 There are several AAV serotypes, each of which exhibits distinct tropism. AAV cell tropism, as dictated by virus capsid proteins, is an important factor affecting transduction efficiency and specificity across cell types. The gene transfer efficiency to the murine inner ear by AAV vectors depends on the titer and promoter of the AAV, mouse age, delivery route, and presence/absence of an enhancer, in addition to tropism. 66 As approximately one‐third of deafness genes are expressed in the hair cells, 68 AAV serotypes that enable efficient gene transfer to the hair cells are important. We previously reported that AAV2/2 exhibited the highest total transduction rate for both the inner and outer hair cells when AAV serotypes 2/1, 2/2, 2/8, 2/9, and 2/Anc80L65 were introduced into the adult murine cochlea under the same titer. 69 Omichi et al 66 summarized the tropism of AAV serotypes in the adult mouse cochlea. To date, no AAV reportedly enables robust transduction in the supporting cells, in which the most common deafness gene (GJB2) is expressed, and in the outer sulcus and the spiral prominence cells, in which SLC26A4 is expressed; thus, further investigation of appropriate vectors (eg, synthetic AAV capsid) enabling efficient gene transfer to these types of cells is warranted.
FIGURE 3.
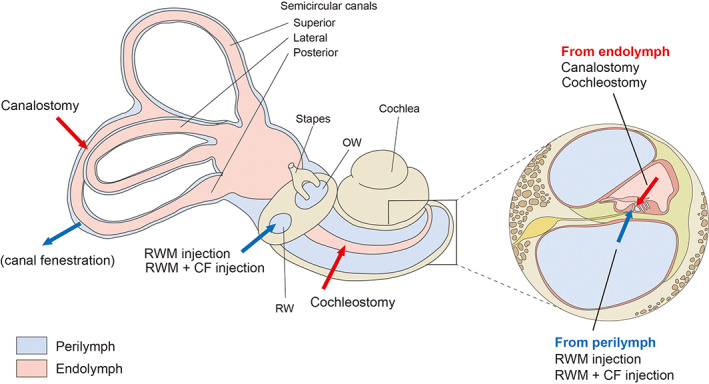
Inner ear schematic showing established delivery routes. Vector delivery into the perilymph via a round window membrane (RWM) or a RWM combined with a semicircular canal fenestration (CF); vector delivery into the endolymph via cochleostomy or canalostomy. OW indicates oval window
8. TRENDS IN PAST GENE THERAPY RESEARCH
In 2012, Akil et al 70 first reported the effectiveness of gene therapy in a mouse model of hereditary hearing loss using Vglut3 knockout mice. These researchers performed gene transfer by administering AAV1 containing Vgult3 cDNA to the inner ear of mouse pups via the round window and demonstrated improved hearing loss. Similarly, several successful studies in neonatal mice were reported using a gene replacement approach. 65 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 As described above, hereditary hearing loss is largely classified by mode of inheritance, including cases resulting from loss‐of‐function mutations (autosomal recessive inheritance) and from gain‐of‐function mutations (autosomal dominant inheritance). 3 The therapeutic target of gene replacement is hearing loss due to loss‐of‐function mutations. Lentz et al 84 and Shibata et al 63 reported the possibility of using gene therapy for hearing loss due to gain‐of‐function mutations by gene silencing via RNA interference. In addition, Gao et al 85 reported a study using gene editing in 2018. However, all the above studies were conducted with neonatal mice without mature inner ears (ie, mice younger than 2 weeks of age). Therefore, we performed gene therapy via gene silencing in a mouse model of human hereditary hearing loss in 2‐ to 8‐week‐old mice, demonstrating suppression of hearing loss progression, protection of the hair cells, and suppression of their degeneration. 64 This was the first study reporting the effectiveness of gene therapy in a mouse model of genetic deafness with fully developed inner ears. The targeted allele suppression by miRNA utilized in that study is a mutation‐specific therapy that requires miRNAs designed for each targeted variant. Thus, the target patients in any clinical application include only those who have a mutation orthologous to that in mice, which is an extremely limited population. It is impossible to generate a mouse model of genetic deafness to verify the therapeutic effectiveness of manipulating a number of deafness genes as well as genetic mutations. To address this limitation, the effectiveness of gene therapy may be verified in the future using disease‐specific induced pluripotent stem cells, 86 such as the outer sulcus cells in which SLC26A4 is expressed, as this technique enables induction of differentiation to various cell types in the cochlea.
9. PRECLINICAL STUDY REQUIRED FOR THE FUTURE DEVELOPMENT OF GENE THERAPY
Phase I/II gene therapy trials for patients with severe‐to‐profound hearing loss were started in the United States in 2014 (https://clinicaltrials.gov/ct2/show/NCT02132130); however, at present, gene therapy for hereditary hearing loss in humans has not been approved. The advantage of gene replacement is that it enables treatment regardless of the mutation position. However, the vector may need to be administered multiple times depending on the period of transduction to the target cells, in which immune responses to AAV in the inner ear need to be verified. In addition, the size of the cDNA that can be packaged using an AAV is approximately 4.7 kb, which may require splitting the transgene into two or three parts. Akil et al 87 summarized the deafness genes that need to be split to avoid exceeding the packaging capacity of AAV. We previously reported the feasibility of a dual AAV vector approach, 69 but no triple AAV vector approach has been reported and this warrants further study. In contrast, the disadvantage of gene silencing and gene editing is that miRNA and gRNA need to be designed for each targeted mutation. In gene editing using the CRISPR/Cas9 system, off‐target mutations have had serious adverse effects, although gene editing using a base editor that can minimize off‐target effects, in which double‐stranded DNA breaks are not included, has been reported in recent years 88 and future advancements are expected. Regardless of the method selected, achievements using prevalent deafness genes as targets in patients with hearing loss are desirable.
10. MILESTONES TOWARD THE CLINICAL APPLICATION OF GENE THERAPY IN HUMANS
As described above, there are many hurdles to overcome for the clinical application of gene therapy for hereditary hearing loss. However, in humans, it is important to consider how gene therapy will be combined with present therapies. At present, the therapeutic options for sensorineural hearing loss are HAs and CIs, which are not biological treatments, but are highly useful. Even in cases of congenital severe‐to‐profound hearing loss, favorable language development is frequently reported with bilateral CI at an early age. Similarly, the effectiveness of HAs for mild‐to‐moderate hearing loss has been demonstrated. Thus, even if gene therapy potentially becomes a curative treatment, it is unlikely to immediately become an alternative therapy to CIs and HAs. In addition, even when the gene transfer method to the inner ear is via round window injection, canalostomy, or a combination, 66 , 67 the method is invasive and patients with mild‐to‐moderate hearing loss are expected to prefer non‐invasive therapy with HAs. Therefore, starting with a hybrid therapy based on CIs combined with gene therapy for severe‐to‐profound hearing loss is considered clinically feasible. Drug delivery through CIs has been studied, and administration of neurotrophins and glucocorticosteroids through CIs reportedly reduces insertion trauma, immune reaction, degeneration of spiral ganglion cells, and fibrosis and ossification in cochlear implantation. 89 Gene transfer to the inner ear through CIs in patients with profound hearing loss is reasonable, and is considered a useful step to confirm the safety of gene therapy delivery. Further, as the achievement successful gene therapy for patients with profound hearing loss may be extremely challenging, patients with genetic progressive hearing loss, for which CIs are applied, are considered good targets. For example, in most patients with severe‐to‐profound high‐frequency hearing loss with only mild‐to‐moderate hearing loss at low frequencies, sufficient hearing could not be obtained with HAs. For such ski‐slope hearing loss, electric‐acoustic stimulation (EAS) was developed to perform acoustic stimulation that amplifies low‐frequency residual hearing and deliver electrical stimulation (ES) through a CI so as to improves high‐frequency hearing loss with a single device. 90 Greater improvements in speech recognition in noise, music appreciation, and sound localization by EAS than by ES alone have demonstrated the significance of preserving residual hearing. 91 , 92 Hearing at low frequencies is reportedly preserved after cochlear implantation, while most cases of low‐frequency hearing loss are progressive; consequently, most patients experience hearing loss across all frequencies as part of the natural course. 93 , 94 , 95 Therefore, preserving low‐frequency residual hearing by gene therapy via CIs is clinically significant (Figure 4). This strategy is considered applicable to hereditary hearing loss cases associated with the CDH23, TMPRSS3, and ACTG1 genes. In trials of drug delivery through CIs, coating and incorporating drugs into the CI itself is common at present, while a pump will conceivably need to be developed and installed in CIs to deliver multiple drug administration.
FIGURE 4.
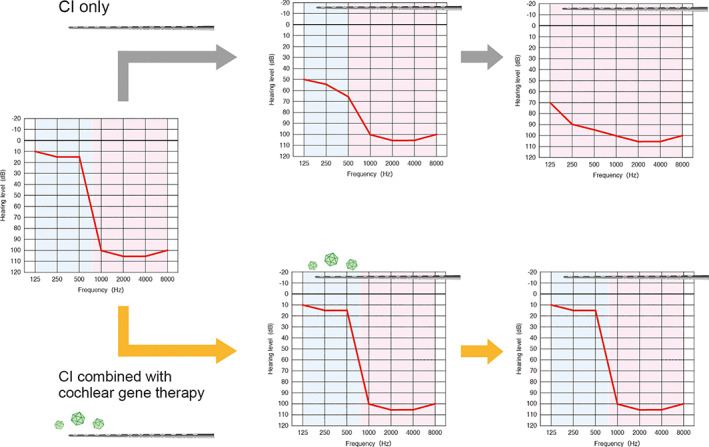
The schema of hybrid gene therapy based on a combination with cochlear implants. In patients with high‐frequency hearing loss, cochlear implants (CIs) improve hearing ability, while the natural course of low‐frequency hearing loss is observed (upper). Residual hearing can be preserved by performing cochlear gene therapy through CIs (lower)
11. CONCLUSION
Herein, we discussed gene therapy research for hereditary hearing loss, focusing on the frequency of deafness genes, hearing loss phenotypes in patients, and mouse models of genetic deafness. If gene therapy can be provided as a precise treatment for hereditary hearing loss in the future, restored hearing function and prevention of hearing loss progression, which cannot be achieved by conventional medicine, can be realized. The number of treatable deafness genes is expected to be expanded by further study, in which research based on data from patients with hearing loss is desirable. In order to actualize gene transfer to the human inner ear, gene therapy in combination with CIs is considered a reasonable strategy for clinical application in humans.
CONFLICT OF INTEREST
Authors declare no potential conflicts of interest.
ACKNOWLEDGMENTS
This research was funded by a Health and Labor Sciences Research Grant for Research on Rare and Intractable Diseases and Comprehensive Research on Disability Health and Welfare from the Ministry of Health, Labor and Welfare of Japan (S.U. 20FC1048), a Grant‐in‐Aid from Japan Agency for Medical Research and Development (AMED) (S.U.: 17kk0205010h0002, 18ek0109363h0001, H.Y.: 21ek0109432h0002), JSPS KAKENHI Grant Number JP 19K18727 (H.Y.), a grant provided by the Ichiro Kanehara Foundation (H.Y.) and grants from the Cell Science Foundation (H.Y.).
Yoshimura H, Nishio S‐Y, Usami S‐I. Milestones toward cochlear gene therapy for patients with hereditary hearing loss. Laryngoscope Investigative Otolaryngology. 2021;6(5):958‐967. 10.1002/lio2.633
Funding information Cell Science Foundation (H.Y.); Ichiro Kanehara Foundation for the Promotion of Medical Sciences and Medical Care (H.Y.); Japan Agency for Medical Research and Development, Grant/Award Numbers: S.U.: 17kk0205010h0002, 18ek0109363h0001, H.Y.: 21ek0109432h0002; JSPS KAKENHI, Grant/Award Number: JP 19K18727 (H.Y.); Ministry of Health, Labor and Welfare of Japan, Grant/Award Number: S.U. 20FC1048
REFERENCES
- 1. Morton CC, Nance WE. Newborn hearing screening—a silent revolution. N Engl J Med. 2006;354:2151‐2164. [DOI] [PubMed] [Google Scholar]
- 2. Korver AM, Smith RJ, Van Camp G, et al. Congenital hearing loss. Nat Rev Dis Primers. 2017;3:16094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ahmed H, Shubina‐Oleinik O, Holt JR. Emerging gene therapies for genetic hearing loss. J Assoc Res Otolaryngol. 2017;18:649‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sloan‐Heggen CM, Smith RJ. Navigating genetic diagnostics in patients with hearing loss. Curr Opin Pediatr. 2016;28:705‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shearer AE, Hildebrand MS, Smith RJH. Hereditary hearing loss and deafness overview. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. Seattle (WA); University of Washington; 2017. [Google Scholar]
- 6. Moteki H, Yoshimura H, Azaiez H, et al. USH2 caused by GPR98 mutation diagnosed by massively parallel sequencing in advance of the occurrence of visual symptoms. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):123S‐128S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoshimura H, Iwasaki S, Nishio SY, et al. Massively parallel DNA sequencing facilitates diagnosis of patients with Usher syndrome type 1. PLoS One. 2014;9:e90688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abe S, Yamaguchi T, Usami S. Application of deafness diagnostic screening panel based on deafness mutation/gene database using invader assay. Genet Test. 2007;11:333‐340. [DOI] [PubMed] [Google Scholar]
- 9. Nishio SY, Usami S. Deafness gene variations in a 1120 nonsyndromic hearing loss cohort: molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):49S‐60S. [DOI] [PubMed] [Google Scholar]
- 10. Sloan‐Heggen CM, Bierer AO, Shearer AE, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441‐450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishio SY, Hattori M, Moteki H, et al. Gene expression profiles of the cochlea and vestibular endorgans: localization and function of genes causing deafness. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):6S‐48S. [DOI] [PubMed] [Google Scholar]
- 12. Rabionet R, Gasparini P, Estivill X. Molecular genetics of hearing impairment due to mutations in gap junction genes encoding beta connexins. Hum Mutat. 2000;16:190‐202. [DOI] [PubMed] [Google Scholar]
- 13. Tsukada K, Nishio S, Usami S, Deafness Gene Study C. A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clin Genet. 2010;78:464‐470. [DOI] [PubMed] [Google Scholar]
- 14. Cohen‐Salmon M, Ott T, Michel V, et al. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol. 2002;12:1106‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kudo T, Kure S, Ikeda K, et al. Transgenic expression of a dominant‐negative connexin26 causes degeneration of the organ of Corti and non‐syndromic deafness. Hum Mol Genet. 2003;12:995‐1004. [DOI] [PubMed] [Google Scholar]
- 16. Morle L, Bozon M, Alloisio N, et al. A novel C202F mutation in the connexin26 gene (GJB2) associated with autosomal dominant isolated hearing loss. J Med Genet. 2000;37:368‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verpy E, Weil D, Leibovici M, et al. Stereocilin‐deficient mice reveal the origin of cochlear waveform distortions. Nature. 2008;456:255‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yokota Y, Moteki H, Nishio SY, et al. Frequency and clinical features of hearing loss caused by STRC deletions. Sci Rep. 2019;9:4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Everett LA, Morsli H, Wu DK, Green ED. Expression pattern of the mouse ortholog of the Pendred's syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci U S A. 1999;96:9727‐9732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miyagawa M, Nishio SY, Usami S, Deafness Gene Study C. Mutation spectrum and genotype‐phenotype correlation of hearing loss patients caused by SLC26A4 mutations in the Japanese: a large cohort study. J Hum Genet. 2014;59:262‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Everett LA, Belyantseva IA, Noben‐Trauth K, et al. Targeted disruption of mouse Pds provides insight about the inner‐ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10:153‐161. [DOI] [PubMed] [Google Scholar]
- 22. Choi BY, Kim HM, Ito T, et al. Mouse model of enlarged vestibular aqueducts defines temporal requirement of Slc26a4 expression for hearing acquisition. J Clin Invest. 2011;121:4516‐4525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Verhoeven K, Van Laer L, Kirschhofer K, et al. Mutations in the human alpha‐tectorin gene cause autosomal dominant non‐syndromic hearing impairment. Nat Genet. 1998;19:60‐62. [DOI] [PubMed] [Google Scholar]
- 24. Moteki H, Nishio SY, Hashimoto S, et al. TECTA mutations in Japanese with mid‐frequency hearing loss affected by zona pellucida domain protein secretion. J Hum Genet. 2012;57:587‐592. [DOI] [PubMed] [Google Scholar]
- 25. Yasukawa R, Moteki H, Nishio SY, et al. The prevalence and clinical characteristics of TECTA‐associated autosomal dominant hearing loss. Genes (Basel). 2019;24(10):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Belyantseva IA, Boger ET, Naz S, et al. Myosin‐XVa is required for tip localization of whirlin and differential elongation of hair‐cell stereocilia. Nat Cell Biol. 2005;7:148‐156. [DOI] [PubMed] [Google Scholar]
- 27. Miyagawa M, Nishio SY, Hattori M, et al. Mutations in the MYO15A gene are a significant cause of nonsyndromic hearing loss: massively parallel DNA sequencing‐based analysis. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):158S‐168S. [DOI] [PubMed] [Google Scholar]
- 28. Liang Y, Wang A, Probst FJ, et al. Genetic mapping refines DFNB3 to 17p11.2, suggests multiple alleles of DFNB3, and supports homology to the mouse model shaker‐2. Am J Hum Genet. 1998;62:904‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hasson T, Heintzelman MB, Santos‐Sacchi J, Corey DP, Mooseker MS. Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc Natl Acad Sci U S A. 1995;92:9815‐9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guilford P, Ayadi H, Blanchard S, et al. A human gene responsible for neurosensory, non‐syndromic recessive deafness is a candidate homologue of the mouse sh‐1 gene. Hum Mol Genet. 1994;3:989‐993. [DOI] [PubMed] [Google Scholar]
- 31. Sun Y, Chen J, Sun H, et al. Novel missense mutations in MYO7A underlying postlingual high‐ or low‐frequency non‐syndromic hearing impairment in two large families from China. J Hum Genet. 2011;56:64‐70. [DOI] [PubMed] [Google Scholar]
- 32. Weil D, Blanchard S, Kaplan J, et al. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60‐61. [DOI] [PubMed] [Google Scholar]
- 33. Gibson F, Walsh J, Mburu P, et al. A type VII myosin encoded by the mouse deafness gene shaker‐1. Nature. 1995;374:62‐64. [DOI] [PubMed] [Google Scholar]
- 34. Rhodes CR, Hertzano R, Fuchs H, et al. A Myo7a mutation cosegregates with stereocilia defects and low‐frequency hearing impairment. Mamm Genome. 2004;15:686‐697. [DOI] [PubMed] [Google Scholar]
- 35. Adato A, Lefevre G, Delprat B, et al. Usherin, the defective protein in Usher syndrome type IIA, is likely to be a component of interstereocilia ankle links in the inner ear sensory cells. Hum Mol Genet. 2005;14:3921‐3932. [DOI] [PubMed] [Google Scholar]
- 36. Hmani‐Aifa M, Ben Arab S, Kharrat K, et al. Distinctive audiometric features between USH2A and USH2B subtypes of Usher syndrome. J Med Genet. 2002;39:281‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu X, Bulgakov OV, Darrow KN, et al. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci U S A. 2007;104:4413‐4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kazmierczak P, Sakaguchi H, Tokita J, et al. Cadherin 23 and protocadherin 15 interact to form tip‐link filaments in sensory hair cells. Nature. 2007;449:87‐91. [DOI] [PubMed] [Google Scholar]
- 39. Miyagawa M, Nishio SY, Usami S. Prevalence and clinical features of hearing loss patients with CDH23 mutations: a large cohort study. PLoS One. 2012;7:e40366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wayne S, Der Kaloustian VM, Schloss M, et al. Localization of the Usher syndrome type ID gene (Ush1D) to chromosome 10. Hum Mol Genet. 1996;5:1689‐1692. [DOI] [PubMed] [Google Scholar]
- 41. Schwander M, Xiong W, Tokita J, et al. A mouse model for nonsyndromic deafness (DFNB12) links hearing loss to defects in tip links of mechanosensory hair cells. Proc Natl Acad Sci U S A. 2009;106:5252‐5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kane KL, Longo‐Guess CM, Gagnon LH, Ding D, Salvi RJ, Johnson KR. Genetic background effects on age‐related hearing loss associated with Cdh23 variants in mice. Hear Res. 2012;283:80‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Di Palma F, Holme RH, Bryda EC, et al. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27:103‐107. [DOI] [PubMed] [Google Scholar]
- 44. McGee J, Goodyear RJ, McMillan DR, et al. The very large G‐protein‐coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J Neurosci. 2006;26:6543‐6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yan W, Long P, Chen T, et al. A natural occurring mouse model with Adgrv1 mutation of usher syndrome 2C and characterization of its recombinant inbred strains. Cell Physiol Biochem. 2018;47:1883‐1897. [DOI] [PubMed] [Google Scholar]
- 46. Pan B, Akyuz N, Liu XP, et al. TMC1 forms the pore of mechanosensory transduction channels in vertebrate inner ear hair cells. Neuron. 2018;99:736‐753. e736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vreugde S, Erven A, Kros CJ, et al. Beethoven, a mouse model for dominant, progressive hearing loss DFNA36. Nat Genet. 2002;30:257‐258. [DOI] [PubMed] [Google Scholar]
- 48. Kurima K, Peters LM, Yang Y, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair‐cell function. Nat Genet. 2002;30:277‐284. [DOI] [PubMed] [Google Scholar]
- 49. Manji SS, Miller KA, Williams LH, Dahl HH. Identification of three novel hearing loss mouse strains with mutations in the Tmc1 gene. Am J Pathol. 2012;180:1560‐1569. [DOI] [PubMed] [Google Scholar]
- 50. Ahmed ZM, Riazuddin S, Ahmad J, et al. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum Mol Genet. 2003;12:3215‐3223. [DOI] [PubMed] [Google Scholar]
- 51. Raphael Y, Kobayashi KN, Dootz GA, Beyer LA, Dolan DF, Burmeister M. Severe vestibular and auditory impairment in three alleles of Ames waltzer (av) mice. Hear Res. 2001;151:237‐249. [DOI] [PubMed] [Google Scholar]
- 52. Roux I, Safieddine S, Nouvian R, et al. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277‐289. [DOI] [PubMed] [Google Scholar]
- 53. Iwasa Y, Nishio SY, Yoshimura H, et al. OTOF mutation screening in Japanese severe to profound recessive hearing loss patients. BMC Med Genet. 2013;14:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guipponi M, Vuagniaux G, Wattenhofer M, et al. The transmembrane serine protease (TMPRSS3) mutated in deafness DFNB8/10 activates the epithelial sodium channel (ENaC) in vitro. Hum Mol Genet. 2002;11:2829‐2836. [DOI] [PubMed] [Google Scholar]
- 55. Miyagawa M, Nishio SY, Sakurai Y et al. The patients associated with TMPRSS3 mutations are good candidates for electric acoustic stimulation. Ann Otol Rhinol Laryngol 2015; 124 Suppl 1:193S‐204S. [DOI] [PubMed] [Google Scholar]
- 56. Fasquelle L, Scott HS, Lenoir M, et al. Tmprss3, a transmembrane serine protease deficient in human DFNB8/10 deafness, is critical for cochlear hair cell survival at the onset of hearing. J Biol Chem. 2011;286:17383‐17397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Astuto LM, Bork JM, Weston MD, et al. CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet. 2002;71:262‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walls WD, Moteki H, Thomas TR, et al. A comparative analysis of genetic hearing loss phenotypes in European/American and Japanese populations. Hum Genet. 2020;139:1315‐1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tsukada K, Nishio SY, Hattori M, Usami S. Ethnic‐specific spectrum of GJB2 and SLC26A4 mutations: their origin and a literature review. Ann Otol Rhinol Laryngol. 2015;124(Suppl 1):61S‐76S. [DOI] [PubMed] [Google Scholar]
- 60. Gabriel HD, Jung D, Butzler C, et al. Transplacental uptake of glucose is decreased in embryonic lethal connexin26‐deficient mice. J Cell Biol. 1998;140:1453‐1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang H, Wu K, Guan J, et al. Identification of four TMC1 variations in different Chinese families with hereditary hearing loss. Mol Genet Genomic Med. 2018;14:504‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao Y, Wang D, Zong L, et al. A novel DFNA36 mutation in TMC1 orthologous to the Beethoven (Bth) mouse associated with autosomal dominant hearing loss in a Chinese family. PLoS One. 2014;9:e97064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shibata SB, Ranum PT, Moteki H, et al. RNA interference prevents autosomal‐dominant hearing loss. Am J Hum Genet. 2016;98:1101‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yoshimura H, Shibata SB, Ranum PT, Moteki H, Smith RJH. Targeted allele suppression prevents progressive hearing loss in the mature murine model of human TMC1 deafness. Mol Ther. 2019;27:681‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Akil O, Dyka F, Calvet C, et al. Dual AAV‐mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci U S A. 2019;116:4496‐4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Omichi R, Shibata SB, Morton CC, Smith RJH. Gene therapy for hearing loss. Hum Mol Genet. 2019;28:R65‐R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yoshimura H, Shibata SB, Ranum PT, Smith RJH. Enhanced viral‐mediated cochlear gene delivery in adult mice by combining canal fenestration with round window membrane inoculation. Sci Rep. 2018;8:2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Scheffer DI, Shen J, Corey DP, Chen ZY. Gene expression by mouse inner ear hair cells during development. J Neurosci. 2015;35:6366‐6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Omichi R, Yoshimura H, Shibata SB, Vandenberghe LH, Smith RJH. Hair cell transduction efficiency of single‐ and dual‐AAV serotypes in adult murine cochleae. Mol Ther Methods Clin Dev. 2020;17:1167‐1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Akil O, Seal RP, Burke K, et al. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron. 2012;75:283‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Al‐Moyed H, Cepeda AP, Jung S, Moser T, Kugler S, Reisinger E. A dual‐AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock‐out mice. EMBO Mol Med. 2019;11:e9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Askew C, Rochat C, Pan B, et al. Tmc gene therapy restores auditory function in deaf mice. Sci Transl Med. 2015;7:295ra108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chang Q, Wang J, Li Q, et al. Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange‐Nielsen deafness syndrome. EMBO Mol Med. 2015;7:1077‐1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chien WW, Isgrig K, Roy S, et al. Gene therapy restores hair cell Stereocilia morphology in inner ears of deaf Whirler mice. Mol Ther. 2016;24:17‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dulon D, Papal S, Patni P, et al. Clarin‐1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J Clin Invest. 2018;128:3382‐3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Geng R, Omar A, Gopal SR, et al. Modeling and preventing progressive hearing loss in Usher syndrome III. Sci Rep. 2017;7:13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gyorgy B, Sage C, Indzhykulian AA, et al. Rescue of Hearing by gene delivery to inner‐ear hair cells using exosome‐associated AAV. Mol Ther. 2017;25:379‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Iizuka T, Kamiya K, Gotoh S, et al. Perinatal Gjb2 gene transfer rescues hearing in a mouse model of hereditary deafness. Hum Mol Genet. 2015;24:3651‐3661. [DOI] [PubMed] [Google Scholar]
- 79. Isgrig K, Shteamer JW, Belyantseva IA, et al. Gene therapy restores balance and auditory functions in a mouse model of usher syndrome. Mol Ther. 2017;25:780‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kim MA, Cho HJ, Bae SH, et al. Methionine sulfoxide reductase B3‐targeted in utero gene therapy rescues hearing function in a mouse model of congenital sensorineural hearing loss. Antioxid Redox Signal. 2016;24:590‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Nist‐Lund CA, Pan B, Patterson A, et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat Commun. 2019;10:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pan B, Askew C, Galvin A, et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat Biotechnol. 2017;35:264‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yu Q, Wang Y, Chang Q, et al. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 2014;21:71‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lentz JJ, Jodelka FM, Hinrich AJ, et al. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat Med. 2013;19:345‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gao X, Tao Y, Lamas V, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553:217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hosoya M, Fujioka M, Sone T, et al. Cochlear cell modeling using disease‐specific iPSCs unveils a degenerative phenotype and suggests treatments for congenital progressive hearing loss. Cell Rep. 2017;18:68‐81. [DOI] [PubMed] [Google Scholar]
- 87. Akil O. Dual and triple AAV delivery of large therapeutic gene sequences into the inner ear. Hear Res. 2020;394:107912. [DOI] [PubMed] [Google Scholar]
- 88. Yeh WH, Shubina‐Oleinik O, Levy JM, et al. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci Transl Med. 2020;3:eaay9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Plontke SK, Gotze G, Rahne T, Liebau A. Intracochlear drug delivery in combination with cochlear implants: current aspects. HNO. 2017;65:19‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. von Ilberg C, Kiefer J, Tillein J, et al. Electric‐acoustic stimulation of the auditory system. New technology for severe hearing loss. ORL J Otorhinolaryngol Relat Spec. 1999;61:334‐340. [DOI] [PubMed] [Google Scholar]
- 91. von Ilberg CA, Baumann U, Kiefer J, Tillein J, Adunka OF . Electric‐acoustic stimulation of the auditory system: a review of the first decade. Audiol Neurootol. 2011;16(Suppl 2):1‐30. [DOI] [PubMed] [Google Scholar]
- 92. Welch C, Dillon MT, Pillsbury HC. Electric and acoustic stimulation in cochlear implant recipients with hearing preservation. Semin Hear. 2018;39:414‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Moteki H, Nishio SY, Miyagawa M, Tsukada K, Iwasaki S, Usami SI. Long‐term results of hearing preservation cochlear implant surgery in patients with residual low frequency hearing. Acta Otolaryngol. 2017;137:516‐521. [DOI] [PubMed] [Google Scholar]
- 94. Usami S, Moteki H, Tsukada K, et al. Hearing preservation and clinical outcome of 32 consecutive electric acoustic stimulation (EAS) surgeries. Acta Otolaryngol. 2014;134:717‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yoshimura H, Moteki H, Nishio SY, Usami SI. Electric‐acoustic stimulation with longer electrodes for potential deterioration in low‐frequency hearing. Acta Otolaryngol. 2020;140:632‐638. [DOI] [PubMed] [Google Scholar]