

ABSTRACT
Vaccination against the betaherpesvirus, human cytomegalovirus (HCMV) is a public health goal. However, HCMV has proved difficult to vaccinate against. Vaccination against single HCMV determinants has not worked, suggesting that immunity to a wider antigenic profile may be required. Live attenuated vaccines provide the best prospects for protection, but the question remains as to how to balance vaccine virulence with safety. Animal models of HCMV infection provide insights into identifying targets for virus attenuation and understanding how host immunity blocks natural, mucosal infection. Here, we evaluated the vaccine potential of a mouse cytomegalovirus (MCMV) vaccine deleted of a viral G protein-coupled receptor (GPCR), designated M33, that renders it attenuated for systemic spread. A single noninvasive olfactory ΔM33 MCMV vaccine replicated locally, but as a result of the loss of the M33 GPCR, it failed to spread systemically and was attenuated for latent infection. Vaccination did not prevent host entry of a superinfecting MCMV but spread from the mucosa was blocked. This approach to vaccine design may provide a viable alternative for a safe and effective betaherpesvirus vaccine.
IMPORTANCE Human cytomegalovirus (HCMV) is the most common cause of congenital infection for which a vaccine is not yet available. Subunit vaccine candidates have failed to achieve licensure. A live HCMV vaccine may prove more efficacious, but it faces safety hurdles which include its propensity to persist and to establish latency. Understanding how pathogens infect guide rational vaccine design. However, HCMV infections are asymptomatic and thus difficult to capture. Animal models of experimental infection provide insight. Here, we investigated the vaccine potential of a mouse cytomegalovirus (MCMV) attenuated for systemic spread and latency. We used olfactory vaccination and virus challenge to mimic its natural acquisition. We provide proof of concept that a single olfactory MCMV that is deficient in systemic spread can protect against wild-type MCMV superinfection and dissemination. This approach of deleting functional counterpart genes in HCMV may provide safe and effective vaccination against congenital HCMV disease.
KEYWORDS: cytomegalovirus, vaccination, animal model, cytomegalovirus, immunization
INTRODUCTION
Human cytomegalovirus (HCMV) is the most common cause of congenital infection and is diagnosed in approximately 1% of total live births (1, 2). Antivirals have limited efficacy in ameliorating sequelae in neonates such as sensorineural hearing loss and are toxic in pregnancy (3). Such harm is more likely to arise from a primary maternal infection and thus HCMV-seronegative women of childbearing age would benefit from an effective vaccine. Despite recognition of the public health and economic benefits for a HCMV vaccine over 20 years ago, successful licensure of a vaccine remains elusive (4). Since HCMV transmission to the neonate can still occur in the presence of natural immunity, the prospect of a vaccine achieving sterile immunity is unlikely. Nevertheless, the ability of a vaccine to prevent congenital disease would be a significant advance.
History has shown us that live attenuated viruses make excellent vaccines (5), and many were developed empirically several decades ago (6). Live herpesvirus vaccines against Marek’s disease virus, pseudorabies virus, and varicella-zoster virus have proved effective in preventing acute disease, even where transmission is not prevented (7–9), HCMV infection elicits an immune response to a broad antigenic profile that reveals what a vaccine to an immune-evasive virus like HCMV should mimic (10). The low efficacy of recombinant gp350 to reduce Epstein-Barr virus (EBV) infection (11), recombinant glycoprotein D (gD) to reduce herpes simplex virus infection (12) or glycoprotein B (gB) to reduce HCMV acquisition (13) argue that single component vaccines generally struggle to protect against herpesviruses. These results also brought into question the capacity of in vitro herpesvirus neutralization readouts to predict in vivo protection (14).
The hazards of congenital infection make difficult empirical testing of live HCMV vaccines. The challenge for the development of live HCMV vaccines is to achieve a balance between virulence and safety. Rational vaccine design is underpinned by knowledge of how a pathogen infects and an understanding of the immune correlates of protection (6). This approach was successful for PRV vaccination, as there was a clear molecular basis for virus attenuation (9). For HCMV, acquiring this knowledge has been problematic due to the silent nature of its acquisition and spread.
Studies in animal models of HCMV infection can provide insight in identifying virulence factors that are key to virus spread that are thus targets exploitable by antiviral therapies. It is crucial that animal models provide a realistic picture of virus-host interactions in vivo. Immune evasion is a key component of herpesvirus infections and is closely tied to its evolutionary context of normal host colonization. CMV infections long predate human speciation (15), and those of other mammals provide an opportunity to develop and test CMV vaccine strategies in a realistic context.
Mouse cytomegalovirus (MCMV) is among the best studied models of HCMV infection. Common to all herpesviruses is their ability to bind heparan sulfate (HS) (16). The unusual expression of HS on the apical, rather than basolateral surface of olfactory epithelium has provided a key factor for olfactory targeting and herpesvirus host entry (17). MCMV enters new hosts via the olfactory epithelium and then spreads systemically and almost exclusively via recirculating DC, driven by the constitutive signaling of the G protein-coupled receptor (GPCR) homolog, M33 (18). MCMV M33 drives the escape of infected dendritic cells (DC) back to the blood, allowing widespread dissemination and extravasation to peripheral tissues, including amplification in the salivary gland, where the MCMV CC chemokine homolog MCK-2 transfers infection from DC to acinar cells. In the absence of M33, infected DC are retained in the draining lymph nodes (LN); of the minority that do escape the LN, they are unable to extravasate to peripheral tissues. Notably, HCMV also binds HS and its pentamer complex has an olfactory receptor (19). Moreover, the constitutive signaling of the HCMV US28 GPCR appears to have a similar in vivo function as MCMV M33 in aiding infected DC recirculation (20). This reflects how related viruses typically conserved the key events of host colonization, even when their molecular details differ, and suggests that MCMV can provide a general model for HCMV vaccines.
Vaccination aims to protect against natural infection. HCMV causes disease by placental infection, but this is downstream of normal primary systemic spread that transmits to a vulnerable foetus. The key vaccine goal is to limit systemic spread of a superinfecting wild-type virus to undetectable levels. Vaccinations against MCMV have generally tried to protect against an acute liver and spleen infections after intraperitoneal (i.p.) challenge (21, 22). However, these infections result from a cell-free viremia that is specific to invasive, parenteral inoculation. It is not a feature of normal systemic spread after mucosal entry (23). Thus, evaluation of MCMV virulence factors using i.p. inoculation is not necessarily a useful guarantee of vaccine safety, and protection against hepatitis after i.p. challenge is not necessarily a useful indicator of vaccine efficacy. Olfactory infection provides an authentic basis. M33 is essential for MCMV to spread and has clear signaling and functional homology with HCMV (20, 24–26). We tested here the safety of an M33-deficient virus vaccine, particularly its systemic spread, and its capacity to prevent the spread of a subsequent wild-type mucosal challenge.
RESULTS
Acute olfactory infection with live MCMV is confined to the nose.
Our previous studies identified neuroepithelia as the natural entry site for MCMV. Given the small capacity of the upper respiratory airways, efficient infection requires a small inoculum, to allow optimal exposure. For adult mice, we have determined a 2-μl inoculum per nare of alert, adult mice provides detectable infection with acceptable reproducibility. Submicroliter amounts are required for neonates. We give olfactory infections to alert mice as loss of the “gag” reflex upon anesthesia results in inoculum being aspirated to the lung. As a preview to using olfactory vaccinations, we first determined kinetics of MCMV colonization spread from this site. We used luciferase-tagged wild type (WT) and ΔM33 MCMV to deliver olfactory infection (5 × 105 PFU; 4 μl delivered as 2 × 2 μl droplets to the nares to alert mice). This was compared to lung infection, using an equivalent PFU dose but in a larger volume (2 × 15 μl to the nares of anaesthetized mice), as described previously (18). We traced infection by live imaging. At day 3 postinfection (p.i.), light emission signals were confined to the nose in olfactory-inoculated mice; lung-infected mice exhibited higher signals from the thorax indicative of virus amplification (note different scale) (Fig. 1A). As strong signals from one anatomical site can mask dissemination to another, we quantified the light emission in dissected organs (Fig. 1B) and measured infectious virus by a plaque assay (Fig. 1C); both readouts confirmed live imaging data.
FIG 1.

Acute olfactory vaccination with live MCMV is confined to the nose and spreads via SCLN. Luciferase-tagged wild-type (WT) and ΔM33 (5 × 105 PFU/mouse) MCMV was given to alert mice (delivered as 2 × 2 μl droplets to the nares) or to the lungs in anesthetized mice (2 × 15 μl to the nares). (A) Live imaging of infected mice at day 3 p.i., representative of five mice. The scale bars refer to the average radiance (p/s/cm2/sr). Colonization following olfactory infection was confined to the nose. Lung infection showed virus amplification in the thorax (note the different scale). (B and C) Quantification of light emission (B) and infectivity (C) in dissected organs (n = 5/group) confirmed site-specific virus replication. By day 6 p.i., live imaging detected cervical signals in mice nose vaccinated by WT and ΔM33 MCMV (indicated by open arrow in panel D) and in mice lung vaccinated with WT MCMV (G; each image representative of five mice). Quantification of light emission in tissues postmortem (E and H) and infectious virus recovered from homogenized organs (F and I) shows virus spread to the draining LN (SCLN, superficial cervical lymph nodes; MLN, mediastinal lymph nodes). Higher cervical luciferase signals exhibited in WT-infected mice were attributed to salivary gland colonization, which was absent in ΔM33-infected counterparts. (J) Persistence of the infectious inoculum was determined in BALB/c mice nose or lung infected with 5 × 105 PFU of the “single cycle” replication mutant ΔgL MCMV. Noses and lungs were harvested at the times p.i. indicated and assayed for levels of residual inoculum by infection of gL+ MEFs. Comparisons between virus groups were performed using Student t test (ns, not significant; ****, P < 0.0001; **, P < 0.01; *, P < 0.05). Symbols show the values for individual organs; the mean of each group is indicated by the solid bar. Horizontal dashed lines in each graph correspond the assay sensitivity limit.
By day 6 p.i., we detected cervical signals in WT MCMV-infected mice inoculated by either route (open arrows, Fig. 1D and G). Imaging of dissected organs revealed spread to the LN draining the nose or lung—the superficial cervical lymph nodes or mediastinal lymph nodes, respectively (Fig. 1E and H)—and was confirmed by plaque assay (Fig. 1F and I). Higher cervical luciferase signals exhibited in WT-infected mice were attributed to salivary gland colonization, which was absent in ΔM33 MCMV-infected counterparts (Fig. 1E, F, H, and I). In summary, ΔM33 MCMV infections at the mucosal surface were equivalent via olfactory or lung routes. However, consistent with previous studies, ΔM33 MCMV failed to colonize the salivary gland.
To exclude the possibility that infectious virus recovered at day 3 p.i. was due to residual inoculum, we infected mice with MCMV ΔgL (5 × 105 PFU/mouse) and harvested noses/lungs at 30 min, 1 day, and 3 days p.i. In vitro, ΔgL is pseudotyped gL+ when propagated in cells that deliver gL in trans. In vivo, ΔgL colonizes olfactory neurons but is incapable of further cell-cell spread at the inoculation site (23). By day 1 p.i., only 1% of olfactory and lung ΔgL inocula were detected in homogenized noses and lungs, respectively, and by day 3, no infectious inoculum was detected (Fig. 1J). Thus, infectious virus detected at day 3 p.i. was the result of amplification in situ and was much lower in olfactory compared to lung infection.
ΔM33 MCMV olfactory vaccine does not reactivate from latency.
M33 and US28 GPCRs drive extravasation of infected DC from the blood to tissues (18, 20). Olfactory ΔM33 MCMV failed to spread beyond the superficial cervical lymph nodes (SCLN), and thus we suspected that it would also be attenuated in its ability to establish a latent infection in peripheral tissues compared to WT MCMV. At 3 months after vaccination with either WT or ΔM33 vaccines (5 × 105 PFU), all spleen, lung, and bone marrow (BM) samples were negative for infectious virus as determined by plaque assay. We prepared genomic DNA from tissue samples to quantify levels of viral DNA by using virus-specific primers as detailed in Materials and Methods. Preliminary experiments using dilutions of target sequence in a 5-kb plasmid DNA estimated our sensitivity of detection at 100 copies of DNA (Fig. 2A). Analysis of up to 100 ng of DNA from tissues failed to detect latent MCMV DNA from the spleen (Fig. 2B and C) or from lung and BM (not shown). Thus, olfactory challenge resulted in levels of latent MCMV in peripheral tissues that were below the limit of detection.
FIG 2.
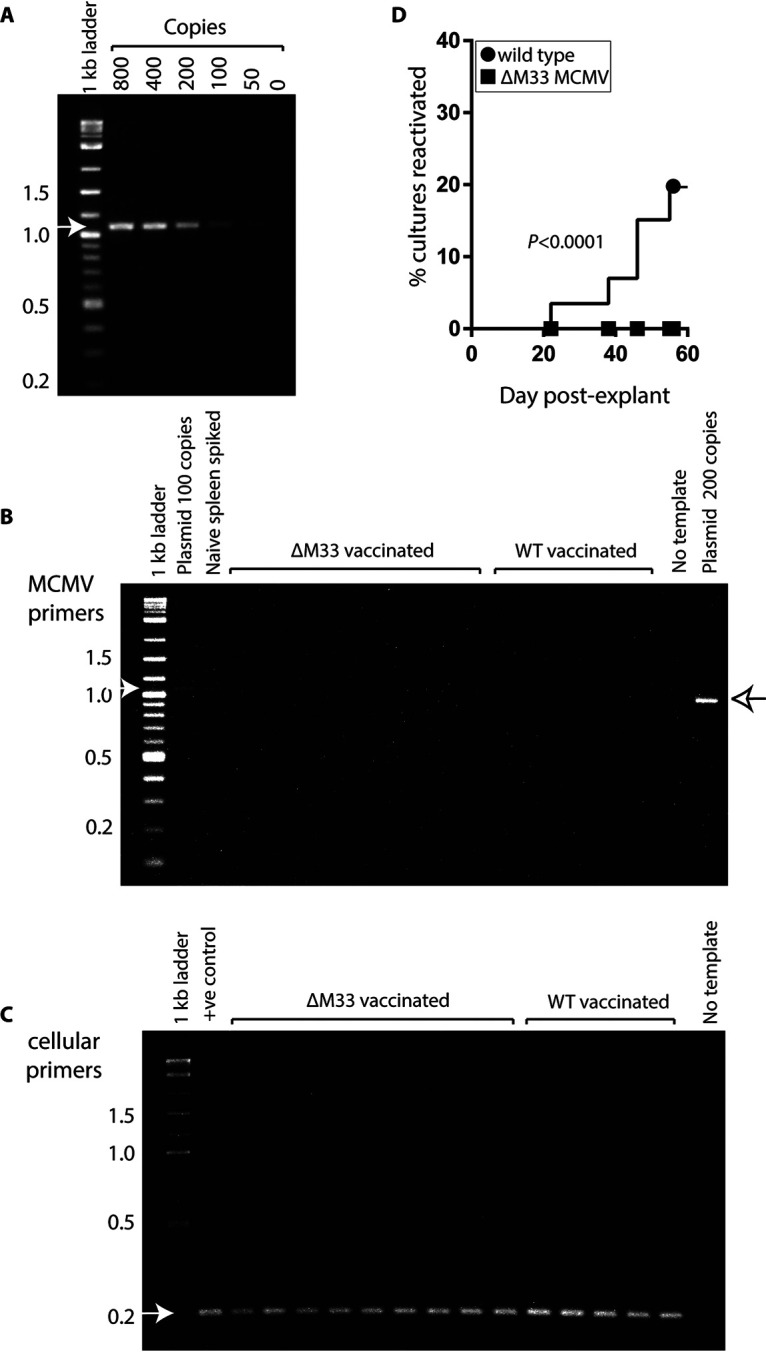
ΔM33 olfactory vaccine fails to reactivate. BALB/c were nose-vaccinated with either WT or ΔM33 MCMV (5 × 105 PFU/mouse; 5 mice/group). Spleen tissues from either WT- or ΔM33-vaccinated mice were analyzed by PCR for the presence of MCMV DNA 3 months later. (A) The sensitivity of PCR was determined to be 100 copies using a MCMV plasmid of known copy number. Spleen tissues (100 ng) were tested for MCMV DNA (B) and for cellular DNA (C). Positive controls included WT plasmid at known copy number and naive spleen tissue spiked with 100 copies of WT plasmid DNA. The primers used and cycling conditions are annotated in the methods. Arrows depict expected sizes of amplified products. After 3 months, the mice were euthanized, and spleen explants were established (n = 16 to 20 independent cultures/spleen) in 0.5-ml cultures in 48-well trays. Supernatants (100 μl) from cultures were tested twice weekly for 9 weeks for the presence of infectious virus on MEFs. MEFs were infected by centrifugal enhancement and examined for cytopathic effect (CPE) 3 to 4 days later. Reactivations was recorded on a contingency plot. (D) Differences in reactivation rates were significant at P < 0.0001 (Fisher two-tailed exact test).
To investigate further the existence of low-level latent infection, we repeated the experiment to determine whether it we could detect reactivated MCMV in spleen explants ex vivo. We chose to evaluate reactivation from the spleen since this site is an established reservoir of latent MCMV that has demonstrated efficient rates of reactivation ex vivo (27). Furthermore, spleen tissue is amenable to long-term explant culture. Explant cultures were sampled twice weekly for the presence of lytic MCMV. We detected reactivation in 17% of splenic cultures from WT-vaccinated animals (Fig. 2D). Reactivations were first detected 3 weeks after explantation. In contrast, we did not detect reactivation in any splenic explants over an 8-week time course for ΔM33-vaccinated animals (n = 90 to 96 cultures from 5 mice/vaccine). In summary, data from olfactory vaccination demonstrate that M33 is attenuated for latency.
Olfactory vaccination induces local and systemic MCMV-specific antibody responses.
Next, we sought to determine whether an olfactory vaccine was immunogenic. We compared the kinetics of MCMV-specific antibodies elicited by olfactory WT and ΔM33 MCMV vaccinations. An enzyme-linked immunospot (ELISPOT) assay was used to enumerate MCMV-specific antibody-secreting cells (ASCs) either local or distal to the vaccination site. Tissue ASCs from unvaccinated mice were included to provide background numbers.
Mice were nose-vaccinated with WT or ΔM33 MCMV (5 × 105 PFU). At 7, 14, and 28 days postvaccination, MCMV-specific ASCs were determined in the nasal-associated lymphoid tissue (NALT), SCLN, spleen, and BM. Early ASC responses in the NALT were low across all antibody isotypes, but elevated levels were detected subsequently (Fig. 3A). In the SCLN, ASC numbers following ΔM33 MCMV vaccination emerged more rapidly compared with WT (Fig. 3B). While IgM and IgA ASCs contracted for both vaccination groups in the SCLN by day 28 p.i., IgG ASCs remained elevated. Spleens and BM exhibited early IgM and delayed IgG/IgA consistent with a classical primary immune response and while responses in the spleen contracted for both vaccines by day 28, they remained elevated in the BM, including an IgA response (Fig. 3C and D).
FIG 3.

Olfactory vaccination stimulates local and systemic immunity. BALB/c mice were nose vaccinated with 5 × 105 PFU of either WT K181 or ΔM33 MCMV. At days 7, 14, and 28 p.i., NALT (A), SCLN (B), spleens (C), and bone marrow (D) were assessed for MCMV-specific ASCs. Tissues from unvaccinated mice were used to determine baseline numbers. Closed symbols refer to mean ASCs from WT-vaccinated animals; open symbols are from ΔM33 MCMV-infected animals (n = 5 mice per virus group). Symbols for tissue harvests on particular days p.i. are indicated and represent the mean ASCs ± the standard deviations (SD; n = 5 mice/group). Comparisons between vaccination groups were performed by a two-tailed Student t test (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001). Sera from these mice were tested for IgM antibodies (E) or IgG antibodies (F) to MCMV by ELISA on the days p.i. indicated. All sera were assayed independently, and the results show the average absorbance at each dilution ± the SEM. The background absorbance of sera from unvaccinated mice was subtracted from vaccinated sera. The dashed horizontal line indicates the assay detection limit.
In summary, despite absence of systemic spread by olfactory ΔM33 MCMV, its immunogenicity was similar to that of WT MCMV. Both vaccinations induced virus-specific ASCs in all lymphoid tissues examined, consistent with widespread dispersion of these cells. Enhanced, early ASC responses in the SCLN of ΔM33 MCMV-vaccinated mice may be the result of infected DC accumulation at this site (18).
We also compared MCMV-specific serum isotype responses from the same vaccinated mice. Consistent with serum antibody responses to olfactory infections with the mouse gammaherpesvirus MuHV-4 (28), IgM and IgG responses to MCMV were consistently low (Fig. 3E and F); for IgA they were not detectable across the time course (data not shown). In WT-vaccinated mice the IgM levels were modestly higher than ΔM33 MCMV counterparts at 14 days p.i. (P < 0.05) and matched the kinetics of the ASC response in the spleen. Nevertheless, this had no impact on subsequent IgG serum titers. Thus, while substantive regional and systemic B cell development was detected following olfactory vaccination, this was not reflected in serum antibody titers.
Olfactory vaccination protects against superinfection.
BALB/c mice were vaccinated with either luciferase-tagged WT or ΔM33 MCMV (5 × 105 PFU/mouse in 4 μl; n = 10 per group). We traced acute infection to confirm nose colonization as in Fig. 1. One month later, when luciferase expression was absent from all vaccinated mice, we superinfected with a genotypically distinct K181 MCMV, Δm157 pARK, using the same route and dose. Δm157 pARK is also derived from the K181 (Perth) MCMV and possesses a small deletion in m157 that blocks ligation with the NK cell Ly49H activating receptor in C57BL/6 mice; in BALB/c mice Δm157 is phenotypically equivalent to WT K181 (29). Unvaccinated mice (n = 5) were infected as controls. Serum IgG and IgM antibody levels were quantified by enzyme-linked immunosorbent assay (ELISA) 1 day prior to challenge and 14 days postchallenge. Overall, the results demonstrate weak, but detectable MCMV-specific antibody responses within the first month postvaccination (Fig. 4A), consistent with our previous studies (Fig. 3E and F). After challenge with Δm157 pARK, the IgG but not the IgM titers of vaccinated mice were significantly enhanced compared to unvaccinated controls, a finding indicative of an effective recall response. There was no difference between vaccinated groups in titers of either IgG or IgM antibodies elicited (P > 0.05).
FIG 4.

Olfactory vaccination with spread-deficient ΔM33 MCMV protects against superinfection with Δm157 pARK MCMV. Adult BALB/c mice were given olfactory vaccinations with either WT MCMV or ΔM33 MCMV (n = 10 per virus group; 5 × 105 PFU in 4 μl). Controls were unvaccinated mice (n = 5). After 1 month, all mice were challenged with 5 × 105 PFU Δm157 pARK MCMV via the same route. (A) Serum IgM and IgG levels determined pre- and postchallenge by ELISA. Each point represents the means ± the SEM from 10 individual serum samples per group. Differences in antibody titers were compared between vaccinated and unvaccinated groups (**, P < 0.01; ***, P < 0.001). (B) Infectious MCMV titers in salivary glands of vaccinated and unvaccinated mice from panel A were determined at day 14 postchallenge with Δm157 pARK MCMV. One of ten mice in the WT MCMV-vaccinated group showed a reduced level of infection. No virus was detected ΔM33 MCMV-vaccinated counterparts. Symbols show the values for individual mice; bars depict means. The dashed line corresponds to the assay sensitivity limit. The data are representative of three separate experiments. (C) DNA was extracted from randomly selected salivary gland homogenates from panel B and assessed for MCMV gDNA by qPCR. Unvaccinated, Δm157pARK-challenged mice exhibited high levels of gDNA; levels for ΔM33MCMV salivary glands were below the limit of detection. (D) Adult BALB/c mice were given olfactory vaccinations with either ΔM33 MCMV or ΔgL (n = 10 per virus group; 5 × 105 PFU/mouse). Controls were unvaccinated mice. After 1 month, all mice were challenged with 5 × 105 PFU Δm157pARK MCMV. IgM and IgG antibodies were quantified by ELISA; each point represents the means ± the SEM of 10 mice. Differences in antibody titers were compared between vaccinated and unvaccinated groups (**, P < 0.01; ***, P < 0.001). ΔgL-vaccinated mice did not exhibit antibody titers significantly different from unvaccinated controls. (E) MCMV titers in the salivary glands of vaccinated and unvaccinated mice in panel D were determined 14 days postchallenge and showed modest reduction by ΔgL vaccination; no infectious virus was detected in ΔM33 MCMV-vaccinated mice.
We used salivary gland colonization as a marker of systemic spread of superinfecting MCMV, quantified 14 days postchallenge, corresponding to titers in unvaccinated animals ranging from 104 to 105 PFU/organ. Despite weak MCMV-specific antibody levels at the time of challenge, we did not detect salivary gland titers above the assay detection limit for ΔM33 MCMV-vaccinated mice. For WT-vaccinated mice, we detected one breakthrough of vaccine protection (Fig. 4B). These results are representative of three experiments, with a cumulative total of n = 30 vaccinees for each vaccine. Salivary gland replication was not detected in 30/30 ΔM33 MCMV and in 25/30 WT MCMV vaccine cohorts.
To exclude the possibility that MCMV-specific antibodies in the salivary glands of vaccinees obscured virus detection, we screened homogenates for genomic MCMV DNA by qPCR. We tested salivary gland homogenates from the ΔM33 MCMV-vaccinated/Δm157 pARK challenged cohort, since the absence of M33 renders the vaccine unable to colonize the salivary glands (30). These were compared to salivary glands from unvaccinated/WT-challenged mice. In contrast to the unvaccinated/challenged cohort that exhibited high levels of MCMV gDNA, all qPCR signals from ΔM33 MCMV-vaccinated/challenged cohort were below the limit of detection (Fig. 4C). Thus, olfactory vaccination prevented salivary gland colonization by a superinfecting MCMV.
Given that ΔM33 MCMV exhibited robust protection against superinfection, despite low in vivo replication and spread, we next investigated whether replication-deficient ΔgL MCMV could protect similarly. We nose-vaccinated mice with either ΔM33-MCMV or a ΔgL. After 4 weeks, we challenged vaccinated mice with Δm157 pARK MCMV via the same route. The IgG and IgM levels from ΔgL-vaccinated mice were equivalent to those of unvaccinated controls, both 4 weeks after vaccination (not shown) and 14 days after postchallenge with Δm157 pARK (Fig. 4D). Although ΔgL vaccination resulted in a 5-fold decrease in virus titers in the salivary gland compared to unvaccinated controls, its efficacy was significantly weaker than ΔM33 MCMV (P < 0.0001). Thus, local replication of ΔM33 MCMV contributed to its ability to protect.
Vaccination does not prevent virus entry but protects against spread.
To determine where vaccination arrests MCMV, we vaccinated mice with ΔM33 MCMV (5 × 105 PFU). We used MCMV-GFP challenge virus and compared colonization and spread between ΔM33 MCMV-vaccinated mice and unvaccinated controls. At 3 days p.i., sporadic EGFP+ cells were detected in both vaccinated and naive nasal epithelia (indicated by open arrows) demonstrating that vaccine protection was not afforded by establishing sterile immunity (Fig. 5A). Quantification of infectious virus from homogenized noses showed a reduced titer in vaccinated mice, but this reduction was not significant (Fig. 5B).
FIG 5.

Olfactory vaccination does not prevent initial colonization but blunts systemic spread. BALB/c mice were given olfactory vaccination with ΔM33 MCMV (5 × 105 PFU) or not (n = 5 per virus group). After 1 month, mice were challenged with 5 × 105 PFU MCMV-GFP by the same route. (A) Noses were harvested 3 days p.i. and examined histologically for evidence of GFP+ infected cells. Open arrows indicate GFP+ infected cells; the solid white bar denotes 50 μm. Infected GFP+ cells were observed sporadically in both vaccinated and naive mice in the nasal epithelium; at least one GFP+ section per 20 independent sections was detected per mouse. (B) BALB/c mice were infected or not as in panel A, and virus infection in the noses from MCMV-GFP challenged mice was quantified at day 3 p.i. by a plaque assay (n = 4 mice per virus group). Differences between titers were not significant. BALB/c mice were infected with ΔM33 MCMV or not as in panel A and challenged 1 month later with MCMV-GFP via lung infection of anaesthetized animals. Infectious virus loads in vaccinated and naive mice in lungs (C), MLN (D), and salivary glands (F) were quantified by plaque assay on the days p.i. indicated. Viremia was quantified by qPCR at day 6 p.i. (E) Significance levels refer to a comparison of vaccinates with their respective naive controls. The dashed bar indicates the limit of detection for each tissue in each assay.
We next sought to determine whether olfactory ΔM33 MCMV vaccination could provide protection against challenge at a distal mucosal site. Lung MCMV infection also spreads to the draining LN (the mediastinal LN) exclusively via CD11c+ DC and thus uses an equivalent pathway to olfactory infection, but with more predictable kinetics (18). In lungs from ΔM33 MCMV-vaccinated mice MCMV-GFP superinfection was significantly reduced compared to unvaccinated controls (Fig. 5C). However, levels of GFP+ MCMV in vaccinates at sites downstream of the lung—the mediastinal LN (MLN), blood, and salivary glands (Fig. 5D to F)—were at the limit of detection for each assay, suggesting that vaccination blunts spread of infectious virus prior to, or within, the draining LN.
Olfactory vaccination augments mucosal innate and adaptive effector cell populations.
Since quantitative histology in the lung is considerably easier than in the nasopharynx, we used lung superinfection to compare the nature of the inflammatory infiltrates in vaccinates versus naive mice. As expected, EGFP+ infected cells were detected in the lungs of vaccinated mice at day 3 p.i. (open arrowheads), albeit with the EGFP signal markedly diminished and with significantly reduced numbers compared to naive controls (Fig. 6A). Significantly higher numbers of CD68+, CD4+, CD8+, and NK+ cells were detected in the lungs of vaccinated mice compared to naive controls, consistent with a host recall immune response (Fig. 6A and B). In summary, these studies demonstrate that superinfecting virus was not neutralized prior to host entry in vaccinated mice. Rather, vaccination induces the recruitment of diverse immune cells that likely contribute to preventing virus spread.
FIG 6.

Vaccinated mice exhibit amplified levels of local innate and adaptive lymphocytes. Adult BALB/c mice were given olfactory vaccinations with either WT MCMV or ΔM33 MCMV (n = 4 mice per virus group; 5 × 105 PFU in 4 μl). BALB/c mice vaccinated as per Fig. 4 were lung challenged with 5 × 105 PFU of MCMV MCMV-GFP (30 μl to sedated animals) 1 month after vaccination. (A) At 3 days p.i. lungs were examined to identify superinfection (GFP+), and the cell surface phenotypes of local lymphocytes, including CD4, CD8, NK, and CD68 cells, was determined using specific antibodies. The scale bar denotes 25 μm. (B) Quantification of EGFP+ cells and lymphocyte markers per field from three to five mice. Each symbol shows an independent value per field of view (×20 magnification), and the bar indicates the mean value per group. A two-tailed Student t test assessed differences between vaccinated and unvaccinated groups (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
In vitro serum neutralization titers do not correlate with in vivo protection.
We compared the ability of sera from WT- and ΔM33-vaccinated mice to neutralize cell-free MCMV in vitro, using NIH 3T3 fibroblasts, SVEC-4 endothelial cells, NMuMG epithelial cells, or RAW 264 macrophages as readouts of residual infection. Half-log dilutions of sera were incubated with WT MCMV-GFP for 1 h at 37°C, and the residual infectivity was quantified on NIH 3T3, NMuMG, or RAW cells by enumerating GFP+ cells at 20 h p.i. by flow cytometry.
The potency of antisera in preventing infection varied across the cell lines, with RAW macrophages providing the most sensitive readout and SVEC-4 endothelial cells the weakest (Table 1). Indeed, the serum dilution from WT-vaccinated mice required for 50% virus neutralization was 15- to 60-fold higher using SVEC-4 cells as a readout compared to RAW cells. Serum neutralization efficacy from WT- versus ΔM33-vaccinated mice was markedly disparate within cell lines, with up to a 10-fold difference in serum dilution required for 50% neutralization for RAW macrophages and up to a 160-fold difference for NIH 3T3 cells. Irrespective of differences in potency and efficacy, neutralization of cell-free virus using fibroblast, epithelial, and endothelial cells as readouts did not correlate with in vivo protection. In contrast, the sensitivity of RAW cells as a readout for neutralization, coupled with similar titers between vaccine sera, was more predictive of in vivo protection (31).
TABLE 1.
Sera from vaccinated mice show potency in preventing infection of myeloid cells in vitroa
| Vaccine | Cell line neutralization readoutb |
|||
|---|---|---|---|---|
| RAW 264.7 macrophage | NIH 3T3 fibroblast | NMuMG (epithelial) | SVEC4-10 (endothelial) | |
| WT-MCMV | 1:1,428–1:3,333 | 1:400–1:2,000 | 1:111–1:167 | 1:20–1:100 |
| ΔM33-MCMV | 1:333–1:1,000 | 1:14–1:25 | 1:9–1:14 | <1:10 |
Half-log dilutions of sera taken from mice vaccinated or not with either WT-MCMV or ΔM33 MCMV were mixed with MCMV-GFP for 90 min at 37°C. Antibody-virus mixtures (in triplicate) were added to monolayers of either RAW macrophages, NIH 3T3 fibroblasts, NMuMG epithelial cells, or SVEC4-10 endothelial cells. After 20 h, the proportions of GFP+ cells in each culture were quantified by flow cytometry. The percentages of cells infected in the absence of sera were equivalent across all cell lines. Pooled sera from unvaccinated (i.e., naive) animals failed to exhibit 50% neutralization on any cell line, even at the highest concentration (10% [vol/vol]) used.
Values are expressed as the serum dilutions required to neutralize 50% of the virus inoculum.
Vaccine protection in olfactory vaccinated mice is independent of T cell effectors.
Previous studies have established the role of T cells in control of MCMV infection, particularly in the salivary glands (32, 33). Given that vaccination did not prevent olfactory superinfection but arrested its cell-associated spread to the salivary glands, we tested the requirement of T cells in restricting systemic MCMV spread in vaccinated mice. BALB/c mice were nose vaccinated or not with ΔM33 MCMV (5 × 105 PFU/mouse) and T cell depleted or not 1 month later. All mice were then nose challenged with Δm157 pARK MCMV. Collectively, the %CD4+ T cells derived from undepleted mouse spleens was 15.4 ± 2.2 but 1.5 ± 0.1 from depleted mice. The %CD8+ splenic T cells for undepleted mice was 8.6 ± 1.5 but 0.7 ± 0.1 for depleted mice (Fig. 7A).
FIG 7.

Olfactory vaccination protects against systemic MCMV spread via a T cell-independent mechanism. BALB/mice were vaccinated or not with 5 × 105 PFU ΔM33 MCMV (n = 10 per group). 1 month later, half the mice from each group were depleted of Thy 1+ cells beginning day −1 relative to Δm157 pARK MCMV olfactory challenge (5 × 105 PFU). Control mice were challenged in the absence of T cell depletion. (A) Mice were euthanized 14 days postchallenge, and CD4+ and CD8+ T cell depletion was confirmed by flow cytometry of spleens from three mice per group. (B) Serum IgM/IgG levels were quantified by ELISA. Each point represents the means ± the SD from five individual serum samples per group. (C) Virus titers in the salivary glands were quantified by plaque assay. Virus loads in unvaccinated T cell-depleted mice were significantly higher than the nondepleted cohort (P < 0.021). Virus loads of vaccinated mice and T cell-depleted mice were below the limit of detection. The dashed horizontal line indicates the assay detection limit.
In naive mice, T cell depletion ablated the host IgG, but not IgM responses consistent with T cell-directed antibody switching (Fig. 7B). In vaccinated mice, IgM levels accumulated following T cell-depletion, while IgG titers were unaffected. We quantified virus load in the salivary glands 14 days p.i., shown in Fig. 7C. As expected, significantly higher MCMV titers were sustained in the salivary glands of unvaccinated, T cell-depleted mice compared with untreated controls (P < 0.021), consistent with their importance for recovery from primary MCMV infection. However, T cell depletion did not impair the efficacy of vaccination against salivary gland infection. Thus, vaccine protection against MCMV spread to the salivary glands was independent of T cell effectors.
DISCUSSION
Results from these studies provide proof of concept that a single olfactory MCMV vaccine deficient in systemic spread can protect against WT MCMV superinfection and dissemination. In situ imaging and virus quantification in tissues confirmed that olfactory ΔM33 MCMV replication was confined to the nose and draining SCLN. Moreover, by 3 months postvaccination, we did not detect reactivation of latent ΔM33 MCMV from the spleen, suggesting that spread deficiency resulted in attenuated latency or reactivation from latency. The protection afforded by ΔM33 MCMV vaccination was markedly superior to a replication-disabled ΔgL vaccine counterpart, demonstrating that the limited, local replication by ΔM33 MCMV was necessary and sufficient to prime an immune response to limit MCMV systemic spread and colonization of the salivary glands. Efficient protection by olfactory vaccination suggests that mucosal immunization “educates” host immunity to provide responses relevant to limiting natural acquisition of superinfecting virus.
Here, we demonstrated that the protection afforded by olfactory ΔM33 MCMV vaccination was not attributed to blocking host entry. Olfactory vaccination elicited weak antibody responses, similar to previous studies of alpha- and gammaherpesvirus infections and is consistent with the highly cell-associated nature of herpesvirus dissemination (30, 34). While intranasal vaccination blunts mucosal entry by acute respiratory infections (35, 36), our results support previous observations that herpesviruses are still able to colonize the mucosa in vaccinated hosts (37, 38). Antibodies are generally regarded to be important in limiting MCMV disease and preventing reactivation from latency (39). As with most myeloid cells, MCMV replication in DC is not highly productive (40), and their cell-associated spread likely renders them resistant to neutralization by antibody. Nevertheless, DC are potential targets for antibody-dependent cellular cytotoxicity (ADCC). Mucosal vaccination against persistent viruses elicits higher levels of ADCC than parenteral routes (41). Notably, vaccination with MuHV-4 is dependent on FcγR engagement consistent with ADCC-dependent mechanisms of virus clearance (34, 42). Although the ADCC contribution to vaccine protection was not investigated here, phagocytes and NK cells, whose numbers increased markedly in the SCLN of vaccinated mice, are candidate effectors.
Vaccinated mice exhibited a marked lymphocytic infiltrate consistent with a primed immune response. Tracking of challenge virus in in situ revealed that infection in vaccinated mice was blunted at, or upstream of, the draining LN. Data from our previous studies have demonstrated the role for M33/US28 GPCR signaling in facilitating the rapid transit of MCMV+ DC from the draining LN to the blood (18). Enhanced B cell responses in the SCLN in ΔM33 MCMV-vaccinated mice may be attributable to the accumulation of ΔM33 MCMV in the LN (18).
We used salivary gland infection as the readout for systemic spread since MCMV infection amplifies to high titers in naive mice 2 weeks postinfection. Once MCMV reaches the salivary gland acinar cells, infection is shedding from the salivary glands is resistant to antibodies (43). We were unable to detect MCMV colonization of the salivary glands 2 weeks postchallenge. Surprisingly, protection against salivary gland superinfection was achieved in T cell-depleted, vaccinated mice. A role for T cells in protection is not excluded but, given that antibody responses provide the basis to most antiviral vaccines, these data support the idea that antibody is important for preventing virus spread. In addition, it is also possible that naive rather than primed T cells in vaccinated mice were preferentially eliminated by antibody (44). Further studies quantifying the roles of antibody and T cell responses on vaccine efficacy are in progress to discriminate between these interpretations.
MCMV spreads from the mucosa to peripheral tissues via DC; extravasation of infected DC from the blood is dependent on M33 (18). We reasoned that the failure of ΔM33 MCMV+ DC to colonize peripheral tissues may account for previous reports of a latency defect for ΔM33 MCMV in lung- challenged mice (27). Olfactory WT MCMV infection results in latent MCMV loads below the level of detection in peripheral tissues and thus similar to the clinical picture of HCMV-seropositive immunocompetent subjects. Nevertheless, long-term explant cultures demonstrated that the spleen was a reservoir of latent MCMV that was capable of reactivation. Notably, we were unable to reactivate ΔM33 MCMV, providing an indication of vaccine safety. Studies are in progress to determine whether ΔM33 MCMV vaccination protects against the establishment of a latent infection following superinfection.
Neutralizing antibody titers are the “gold standard” readout for predictions of HCMV vaccine efficacy (45). The HCMV gB+MF59 adjuvant subunit vaccine elicited robust nonneutralizing antibodies that have been attributed to its 50% efficacy, although the mechanism(s) of protection were not determined (14, 46). Dismissal of vaccine candidates on the basis that they elicit nonneutralizing serum antibodies may be thus predicated on assays irrelevant to in vivo outcome. Here, the varied neutralizing potency of serum antibodies from WT- and ΔM33-vaccinated animals to efficiently block MCMV infection in vitro suggested that direct neutralization of cell-free virus as the sole mechanism in preventing infection was unlikely. WT-vaccinated mice are exposed to a higher antigenic load compared to ΔM33-vaccinated counterparts that may account in part for increased neutralizing potency of antisera, yet disparities in potency between cell lines also suggest differences in antibody efficacy. Nevertheless, except for RAW cells, in vitro readouts did not provide a correlate of in vivo protection. RAW cells provided a sensitive readout of antibody function which may be attributable to antibody-dependent phagocytosis, and this suggests the compelling possibility that antibody-dependent cellular phagocytosis or other Fc receptor-dependent effector functions may contribute and enhance the protection afforded by antibodies.
Preclinical studies in animal models that mimic natural infection provide insights into what vaccination modalities may work in humans. Herpesviruses have coevolved with their hosts (15), and thus infectious pathways and protective host responses are likely conserved. Rodent models of natural infection provide the opportunity to trace when, where, and how vaccines block superinfection. We have demonstrated that vaccination does not induce sterile immunity at the point of virus entry, similar to humans. Nevertheless, we demonstrated that local immunity downstream of the mucosa blunts DC-dependent systemic spread. Animal models for HCMV congenital infection, such as rhesus and guinea pig cytomegaloviruses, will be invaluable to evaluate whether the deletion of GPCR counterparts also prevents DC-dependent spread and, if so, their vaccine potential against congenital disease.
MATERIALS AND METHODS
Mice.
Female 6-week-old BALB/c mice were derived from the Animal Resource Centre (Perth, Australia). They were infected either intranasally without anesthesia (5 × 105 PFU in 4 μl; here designated “nose” infection) or under isoflurane anesthesia (5 × 105 PFU in 30 μl, here designated “lung” infection). For luciferase imaging, mice were given 2 mg of luciferin i.p. and then scanned for light emission with a charge-coupled device camera (Xenogen, IVIS-2000). For vaccinations, groups of 5 to 10 mice were nose infected with either WT K181 strain MCMV (here referred to as “WT MCMV”) or ΔM33 MCMV (30). Blood samples were collected at 4 weeks p.i. to quantify virus-specific antibody levels. At 1 month postvaccination, mice were challenged by the same route and dose with Δm157 pARK MCMV (29). Tissues were recovered postmortem for virus quantification. For T cell depletions, vaccinated and age matched naive mice were i.p. administered 250 μg of anti-Thy1 (CD90; clone T24/31) on days −1, +1, +3, +5, +7, and +9 relative to nose challenge. T cell subset depletion, measured by flow cytometry of spleen cells with antibodies to a distinct CD4 epitope (MAb RM4-4) or CD8β (MAb H35-17.2), was 90 and 93% complete, respectively, in anti-Thy1-treated animals, compared to nondepleted counterparts. Experiments were approved by the University of Queensland Animal Ethics Committee in accordance with Australian National Health and Medical Research Council guidelines (project 207/18).
Cells and viruses.
MCMV strain K181 (WT) and ΔM33, used with or without the luciferase tag, have been described previously (18). Briefly, luciferase was expressed in tandem with the M78 lytic gene by autocatalytic release. EGFP-tagged WT and ΔM33 MCMV used for histological studies have also been described (18). M33 was disrupted by insertion of a premature translational stop codon (25). Δm157 pARK was used as the challenge virus; it was derived from WT strain K181 and, although it is genotypically distinct to WT by virtue of a small deletion in m157, it is phenotypically equivalent to WT K181 in BALB/c mice (29). These viruses were propagated on NIH 3T3 cells (ATCC CRL-1658). The preparation of MCMV deleted of the essential glycoprotein gL and the supporting gL+ NIH 3T3 cell line has been described (18). MCMV ΔgL is pseudotyped gL+ by growth on the gL cell line, but since it is genetically gL–, it is incapable of cell-cell spread. Cells were grown in Dulbecco modified Eagle medium supplemented with 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (here, DMEM-10). Infected cells were precleared of cell debris by low-speed centrifugation (500 × g, 10 min), and then virus was concentrated by ultracentrifugation (35,000 × g, 2 h) and resuspended in 2% of the original volume. Infectious virus in cultured cells or tissue homogenates was quantified by plaque assay on murine embryonic fibroblasts (MEFs). Mouse epithelial cells, NMuMG cells (ATCC CRL-1636), mouse endothelial SVEC4-10 cells (ATCC CRL-2181), mouse macrophages, and RAW 264.7 cells (ATCC TIB-71) were all propagated in DMEM-10.
Immunostaining.
Organs were fixed in 1% formaldehyde–10 mM sodium periodate–75 mM l-lysine (18 h, 4°C), equilibrated in 30% sucrose (18 h, 4°C), and then frozen in optimal cutting temperature compound (OCT). Sections (6 μm) were blocked with 0.1% Triton X-100–5% normal goat serum in phosphate-buffered saline (PBS) and then incubated (18 h, 4°C) with antibodies to CD45R/B220 (rat MAb RA3-6B2; Santa Cruz); CD68 (rat MAb; FA-11; Abcam); CD11c (hamster MAb HL3), CD4 (rat MAb GK1.5), and CD8 (rat Mab clone 2.43), all from BD Pharmingen; and NKp46 (rat MAb clone 29A1.4) and PNAd (rat MAb MECA-79), both from BioLegend. Virus-expressed GFP was visualized directly. After incubation with primary antibodies, sections were washed three times in PBS, incubated (1 h, 23°C) with combinations of Alexa 488-, Alexa 568-, or Alexa 647-conjugated goat polyclonal antibodies (Abcam) and then washed three times in PBS, stained with DAPI (4′,6′-diamidino-2-phenylindole), and mounted in ProLong Gold (Life Technologies). For quantification of marker+ and MCMV-infected cells, at least 20 independent sections were counted from three to five mice. Images were acquired with a Zeiss LSM510 microscope and analyzed with Zen software.
Flow cytometry.
For measurement of the efficacy of T cell depletion, spleen homogenates were prepared, and red blood cell were lysed. The cells were washed in FACS buffer (PBS containing 0.2% EDTA and 1 μg/ml bovine serum albumin [BSA]) and Fc receptors blocked with anti-CD16/32 (2.4G2) for 30 min at 4°C. Cells were then incubated with the rat monoclonal antibodies: PerCP-Cy5.5-conjugated anti-CD8 (clone 53-6.7), FITC-conjugated anti-CD4 (clone RM4-4), and Alexa Fluor 647-conjugated anti-CD19 (clone 1D3) (all from BD Biosciences) for 1 h at 4°C. Cells were washed three times in FACS buffer and analyzed with an Accuri flow cytometer (BD Biosciences).
Neutralization assay.
Immune sera from vaccinated mice, or sera from aged-matched naive controls, were harvested, and dilutions (in triplicate) were added to EGFP+ MCMV. After 90 min at 37°C, the virus-serum mixtures were added to either RAW, NIH 3T3, NMuMG, or SVEC cells, and 20 h later, the level of infection was determined by flow cytometry of GFP expression using an Accuri flow cytometer (BD Biosciences). Serum neutralization is expressed as the dilution of serum required to neutralize 50% of the virus infectivity.
ELISA.
MCMV virions concentrated by centrifugation were resuspended in carbonate-bicarbonate buffer (pH 9.6), and 100 μl per well was added to enzyme immunoassay plates (Corning; 4°C, 18 h). The plates were washed three times with PBS/0.05% Tween 20, blocked with 2% BSA in PBS/0.05% Tween 20 (18°C, 1 h), and incubated (3 h, 37°C) with 3-fold dilutions of individual serum samples from vaccinated (n = 10) and age-matched naive (n = 5) mice. The plates were washed three times with PBS–0.05% Tween 20, followed by incubation with alkaline phosphatase-conjugated isotype-specific antibodies (Southern Biotechnology; 1 h, 37°C). After four washes with PBS–0.05% Tween 20, the plates were developed with nitrophenylphosphate substrate (Sigma-Aldrich). The absorbance at 405 nm was read with a Gen5 microplate reader (BioTeK).
B cell ELISPOT assay.
Single-cell suspensions of SCLN and spleens were prepared by mashing each organ through a wire sieve. BM was harvested from dissected tibias and femurs by flushing them with RPMI 1640 medium, 25 mM HEPES (pH 7.4), 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 10% fetal calf serum (complete RPMI). NALT was recovered by dissecting out the palate and mashing it through a wire sieve. Red blood cells were removed by brief incubation in ammonium chloride solution. All cells were washed in complete RPMI before assay. Multiscreen polyvinylidene difluoride (polyvinylidene difluoride) plates (Millipore) were washed with 50% methanol in water for 2 min, washed twice with PBS, and then coated (18 h; 4°C) with MCMV virions resuspended in 0.05% Triton X-100/PBS. The plates were then washed three times with PBS and blocked with complete RPMI. Threefold cell dilutions of cell preparations in complete RPMI were added to duplicate wells, followed by incubation for 4 h at 37°C in 5% CO2. The plates were then washed three times in 0.1% Tween 20/PBS, incubated with alkaline phosphatase-conjugated isotype-specific secondary antibodies (Southern Biotechnology; 18 h; 4°C), washed four times in PBS/0.1% Tween 20, and developed with BCIP (5-bromo-4-chloro-3-indolylphosphate)–nitroblue tetrazolium substrate (Sigma-Aldrich). Spots were counted with a dissecting microscope. Statistical comparisons were done by using a Student unpaired two-tailed t test.
PCR of latently infected tissues.
Samples of lung, spleen, and bone marrow were harvested from mice nose vaccinated 3 months previously with either WT MCMV or ΔM33 LUC MCMV and genomic DNA isolated (Invitrogen Pure Link). MCMV was amplified using primers to amplify MCMV DNA genomic coordinates 41685 to 42755 (5′-GAGCGACTACCTGCACGTG and 5′-GACCTCTGCAGCTCCGAAC). Samples were also amplified for cellular DNA using in-house genotyping primers (5′-CAAATGTTGCTTGTCTGGTG and 5′-GTCAGTCGAGTGCACAGTTT). The limit of sensitivity was determined using WT MCMV plasmid at a known copy number.
Quantitative PCR to measure viremia.
DNA was prepared from blood samples (Wizard genomic DNA purification; Promega). The quality of DNA recovered, monitored by determining the OD260/OD280 ratio, was >1.8. MCMV genomic coordinates 111218 to 111461 (5′-CTAGGGAGCCTCATCCTCT and 5′-ATCGAGCGTGAGGTACAGGT) were amplified by qPCR (LightCycler 480 SYBR green; Roche Diagnostics) and converted to genome copies by comparison to plasmid standards amplified in parallel. Cellular DNA loads were quantified for each sample by amplifying a mouse titin gene fragment (5′-AAAACGAGCAGTGACGTGAGC and 5′-TTCAGTCATGCTGCTAGCGC). Viral DNA levels were then normalized by comparison to cellular DNA loads.
Reactivation of latent MCMV from spleen explants.
Spleens from vaccinated or unvaccinated mice were cut into 1-mm3 pieces. Individual pieces were placed into the wells of a 48-well tissue culture plate containing RPMI supplemented with 2 mM glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanal, and 10% heat-inactivated fetal calf serum. Medium was changed twice weekly for 8 weeks. Explant supernatants were sampled for infectious virus twice weekly by plating onto MEF monolayers. After 3 days, MEFs were fixed with 0.4% formaldehyde solution and stained with toluidine blue. Monolayers were inspected for evidence of cytopathic effect.
Statistical analyses.
Data were analyzed using GraphPad Prism 6.0. Comparisons were evaluated by using a two-tailed Student t test with or without Welch’s correction; multiple groups within experiments were compared by two-way analysis of variance (ANOVA) test with Tukey’s multiple comparison, and reactivation data were compared using a two-tailed Fisher exact test. Results with >95% confidence were considered significant.
ACKNOWLEDGMENTS
This study was supported by grants from the Australian Research Council, the National Health and Medical Research Council, and Queensland Health. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We declare there are no competing interests.
Contributor Information
Helen E. Farrell, Email: h.farrell1@uq.edu.au.
Rozanne M. Sandri-Goldin, University of California—Irvine
REFERENCES
- 1.Naing ZW, Scott GM, Shand A, Hamilton ST, van Zuylen WJ, Basha J, Hall B, Craig ME, Rawlinson WD. 2016. Congenital cytomegalovirus infection in pregnancy: a review of prevalence, clinical features, diagnosis, and prevention. Aust N Z J Obstet Gynaecol 56:9–18. 10.1111/ajo.12408. [DOI] [PubMed] [Google Scholar]
- 2.Fowler KB, Stagno S, Pass RF. 2003. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA 289:1008–1011. 10.1001/jama.289.8.1008. [DOI] [PubMed] [Google Scholar]
- 3.Andrei G, De Clercq E, Snoeck R. 2009. Drug targets in cytomegalovirus infection. Infect Disord Drug Targets 9:201–222. 10.2174/187152609787847758. [DOI] [PubMed] [Google Scholar]
- 4.Stratton KR, Durch JS, Lawrence RS (ed). 2000. Vaccines for the 21st century: a tool for decision-making. Institute of Medicine/National Academies Press, Washington, DC. [PubMed] [Google Scholar]
- 5.Minor PD. 2015. Live attenuated vaccines: historical successes and current challenges. Virology 479-480:379–392. 10.1016/j.virol.2015.03.032. [DOI] [PubMed] [Google Scholar]
- 6.Rueckert C, Guzmán CA. 2012. Vaccines: from empirical development to rational design. PLoS Pathog 8:e1003001. 10.1371/journal.ppat.1003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy SM, Izumiya Y, Lupiani B. 2017. Marek’s disease vaccines: current status, and strategies for improvement and development of vector vaccines. Vet Microbiol 206:113–120. 10.1016/j.vetmic.2016.11.024. [DOI] [PubMed] [Google Scholar]
- 8.Yoshikawa T, Kawamura Y, Ohashi M. 2016. Universal varicella vaccine immunization in Japan. Vaccine 34:1965–1970. 10.1016/j.vaccine.2016.02.058. [DOI] [PubMed] [Google Scholar]
- 9.Freuling CM, Müller TF, Mettenleiter TC. 2017. Vaccines against pseudorabies virus (PrV). Vet Microbiol 206:3–9. 10.1016/j.vetmic.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 10.Sylwester AW, Mitchell BL, Edgar JB, Taormina C, Pelte C, Ruchti F, Sleath PR, Grabstein KH, Hosken NA, Kern F, Nelson JA, Picker LJ. 2005. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med 202:673–685. 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sokal EM, Hoppenbrouwers K, Vandermeulen C, Moutschen M, Léonard P, Moreels A, Haumont M, Bollen A, Smets F, Denis M. 2007. Recombinant gp350 vaccine for infectious mononucleosis: a phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis 196:1749–1753. 10.1086/523813. [DOI] [PubMed] [Google Scholar]
- 12.Belshe RB, Leone PA, Bernstein DI, Wald A, Levin MJ, Stapleton JT, Gorfinkel I, Morrow RLA, Ewell MG, Stokes-Riner A, Dubin G, Heineman TC, Schulte JM, Deal CD, Herpevac Trial for Women. 2012. Efficacy results of a trial of a herpes simplex vaccine. N Engl J Med 366:34–43. 10.1056/NEJMoa1103151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pass RF, Zhang C, Evans A, Simpson T, Andrews W, Huang M-L, Corey L, Hill J, Davis E, Flanigan C, Cloud G. 2009. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med 360:1191–1199. 10.1056/NEJMoa0804749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson CS, Huffman T, Jenks JA, Cisneros de la Rosa E, Xie G, Vandergrift N, Pass RF, Pollara J, Permar SR. 2018. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc Natl Acad Sci USA 115:6267–6272. 10.1073/pnas.1800177115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGeoch DJ, Cook S, Dolan A, Jamieson FE, Telford EA. 1995. Molecular phylogeny and evolutionary timescale for the family of mammalian herpesviruses. J Mol Biol 247:443–458. 10.1006/jmbi.1995.0152. [DOI] [PubMed] [Google Scholar]
- 16.Shukla D, Spear PG. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest 108:503–510. 10.1172/JCI13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milho R, Frederico B, Efstathiou S, Stevenson PG. 2012. A heparan-dependent herpesvirus targets the olfactory neuroepithelium for host entry. PLoS Pathog 8:e1002986. 10.1371/journal.ppat.1002986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farrell HE, Bruce K, Lawler C, Oliveira M, Cardin R, Davis-Poynter N, Stevenson PG. 2017. Murine cytomegalovirus spreads by dendritic cell recirculation. mBio 8:e01264-17. 10.1128/mBio.01264-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.E X, Meraner P, Lu P, Perreira JM, Aker AM, McDougall WM, Zhuge R, Chan GC, Gerstein RM, Caposio P, Yurochko AD, Brass AL, Kowalik TF. 2019. OR14I1 is a receptor for the human cytomegalovirus pentameric complex and defines viral epithelial cell tropism. Proc Natl Acad Sci USA 116:7043–7052. 10.1073/pnas.1814850116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farrell HE, Bruce K, Ma J, Davis-Poynter N, Stevenson PG. 2018. Human cytomegalovirus US28 allows dendritic cell exit from lymph nodes. J Gen Virol 99:1509–1514. 10.1099/jgv.0.001154. [DOI] [PubMed] [Google Scholar]
- 21.Sandford GR, Burns WH. 1988. Use of temperature-sensitive mutants of mouse cytomegalovirus as vaccines. J Infect Dis 158:596–601. [PubMed] [Google Scholar]
- 22.Morello CS, Ye M, Hung S, Kelley LA, Spector DH. 2005. Systemic priming-boosting immunization with a trivalent plasmid DNA and inactivated murine cytomegalovirus (MCMV) vaccine provides long-term protection against viral replication following systemic or mucosal MCMV challenge. J Virol 79:159–175. 10.1128/JVI.79.1.159-175.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farrell HE, Lawler C, Tan CSE, MacDonald K, Bruce K, Mach M, Davis-Poynter N, Stevenson PG. 2016. Murine cytomegalovirus exploits olfaction to enter new hosts. mBio 7:e00251-16. 10.1128/mBio.00251-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waldhoer M, Kledal TN, Farrell H, Schwartz TW. 2002. Murine cytomegalovirus (CMV) M33 and human CMV US28 receptors exhibit similar constitutive signaling activities. J Virol 76:8161–8168. 10.1128/jvi.76.16.8161-8168.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farrell HE, Abraham AM, Cardin RD, Sparre-Ulrich AH, Rosenkilde MM, Spiess K, Jensen TH, Kledal TN, Davis-Poynter N. 2011. Partial functional complementation between human and mouse cytomegalovirus chemokine receptor homologues. J Virol 85:6091–6095. 10.1128/JVI.02113-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farrell HE, Abraham AM, Cardin RD, Mølleskov-Jensen AS, Rosenkilde MM, Davis-Poynter N. 2013. Identification of common mechanisms by which human and mouse cytomegalovirus seven-transmembrane receptor homologues contribute to in vivo phenotypes in a mouse model. J Virol 87:4112–4117. 10.1128/JVI.03406-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cardin RD, Schaefer GC, Allen JR, Davis-Poynter NJ, Farrell HE. 2009. The M33 chemokine receptor homolog of murine cytomegalovirus exhibits a differential tissue-specific role during in vivo replication and latency. J Virol 83:7590–7601. 10.1128/JVI.00386-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan CS, Stevenson PG. 2014. B cell response to herpesvirus infection of the olfactory neuroepithelium. J Virol 88:14030–14039. 10.1128/JVI.02345-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McWhorter AR, Smith LM, Masters LL, Chan B, Shellam GR, Redwood AJ. 2013. Natural killer cell dependent within-host competition arises during multiple MCMV infection: consequences for viral transmission and evolution. PLoS Pathog 9:e1003111. 10.1371/journal.ppat.1003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis-Poynter NJ, Lynch DM, Vally H, Shellam GR, Rawlinson WD, Barrell BG, Farrell HE. 1997. Identification and characterization of a G protein-coupled receptor homolog encoded by murine cytomegalovirus. J Virol 71:1521–1529. 10.1128/JVI.71.2.1521-1529.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tay MZ, Wiehe K, Pollara J. 2019. Antibody-dependent cellular phagocytosis in antiviral immune responses. Front Immunol 10:332. 10.3389/fimmu.2019.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jonjić S, Pavić I, Lucin P, Rukavina D, Koszinowski UH. 1990. Efficacious control of cytomegalovirus infection after long-term depletion of CD8+ T lymphocytes. J Virol 64:5457–5464. 10.1128/JVI.64.11.5457-5464.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jonjić S, Mutter W, Weiland F, Reddehase MJ, Koszinowski UH. 1989. Site-restricted persistent cytomegalovirus infection after selective long-term depletion of CD4+ T lymphocytes. J Exp Med 169:1199–1212. 10.1084/jem.169.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glauser DL, Milho R, Lawler C, Stevenson PG. 2019. Antibody arrests gammaherpesvirus olfactory superinfection independently of neutralization. J Gen Virol 100:246–258. 10.1099/jgv.0.001183. [DOI] [PubMed] [Google Scholar]
- 35.Sano K, Ainai A, Suzuki T, Hasegawa H. 2018. Intranasal inactivated influenza vaccines for the prevention of seasonal influenza epidemics. Expert Rev Vaccines 17:687–696. 10.1080/14760584.2018.1507743. [DOI] [PubMed] [Google Scholar]
- 36.Holmgren J, Czerkinsky C. 2005. Mucosal immunity and vaccines. Nat Med 11:S45–S53. 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 37.Sato A, Suwanto A, Okabe M, Sato S, Nochi T, Imai T, Koyanagi N, Kunisawa J, Kawaguchi Y, Kiyono H. 2014. Vaginal memory T cells induced by intranasal vaccination are critical for protective T cell recruitment and prevention of genital HSV-2 disease. J Virol 88:13699–13708. 10.1128/JVI.02279-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lawler C, Stevenson PG. 2020. Limited protection against gammaherpesvirus infection by replication-deficient virus particles. J Gen Virol 101:420–425. 10.1099/jgv.0.001391. [DOI] [PubMed] [Google Scholar]
- 39.Jonjić S, Pavić I, Polić B, Crnković I, Lucin P, Koszinowski UH. 1994. Antibodies are not essential for the resolution of primary cytomegalovirus infection but limit dissemination of recurrent virus. J Exp Med 179:1713–1717. 10.1084/jem.179.5.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farrell HE, Davis-Poynter N, Bruce K, Lawler C, Dolken L, Mach M, Stevenson PG. 2015. Lymph node macrophages restrict murine cytomegalovirus dissemination. J Virol 89:7147–7158. 10.1128/JVI.00480-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matchett WE, Anguiano-Zarate SS, Nehete PN, Shelton K, Nehete BP, Yang G, Dorta-Estremera S, Barnette P, Xiao P, Byrareddy SN, Villinger F, Hessell AJ, Haigwood NL, Sastry KJ, Barry MA. 2019. Divergent HIV-1-directed immune responses generated by systemic and mucosal immunization with replicating single-cycle adenoviruses in rhesus macaques. J Virol 93:e02016-18. 10.1128/JVI.02016-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wright DE, Colaco S, Colaco C, Stevenson PG. 2009. Antibody limits in vivo murid herpesvirus-4 replication by IgG Fc receptor-dependent functions. J Gen Virol 90:2592–2603. 10.1099/vir.0.014266-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. 2003. Pathogenesis of murine cytomegalovirus infection. Microbes Infect 5:1263–1277. 10.1016/j.micinf.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Rep MH, van Oosten BW, Roos MT, Adèr HJ, Polman CH, van Lier RA. 1997. Treatment with depleting CD4 monoclonal antibody results in a preferential loss of circulating naive T cells but does not affect IFN-gamma secreting TH1 cells in humans. J Clin Invest 99:2225–2231. 10.1172/JCI119396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabanova A, Perez L, Lilleri D, Marcandalli J, Agatic G, Becattini S, Preite S, Fuschillo D, Percivalle E, Sallusto F, Gerna G, Corti D, Lanzavecchia A. 2014. Antibody-driven design of a human cytomegalovirus gHgLpUL128L subunit vaccine that selectively elicits potent neutralizing antibodies. Proc Natl Acad Sci USA 111:17965–17970. 10.1073/pnas.1415310111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cui X, Meza BP, Adler SP, McVoy MA. 2008. Cytomegalovirus vaccines fail to induce epithelial entry neutralizing antibodies comparable to natural infection. Vaccine 26:5760–5766. 10.1016/j.vaccine.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]