

ABSTRACT
Rotaviruses are the causative agents of severe and dehydrating gastroenteritis in children, piglets, and many other young animals. They replicate their genomes and assemble double-layered particles in cytoplasmic electron-dense inclusion bodies called “viroplasms.” The formation of viroplasms is reportedly associated with the stability of microtubules. Although material transport is an important function of microtubules, whether and how microtubule-based transport influences the formation of viroplasms are still unclear. Here, we demonstrate that small viroplasms move and fuse in living cells. We show that microtubule-based dynein transport affects rotavirus infection, viroplasm formation, and the assembly of transient enveloped particles (TEPs) and triple-layered particles (TLPs). The dynein intermediate chain (DIC) is shown to localize in the viroplasm and to interact directly with nonstructural protein 2 (NSP2), indicating that the DIC is responsible for connecting the viroplasm to dynein. The WD40 repeat domain of the DIC regulates the interaction between the DIC and NSP2, and the knockdown of the DIC inhibited rotaviral infection, viroplasm formation, and the assembly of TEPs and TLPs. Our findings show that rotavirus viroplasms hijack dynein transport for fusion events, required for maximal assembly of infectious viral progeny. This study provides novel insights into the intracellular transport of viroplasms, which is involved in their biogenesis.
IMPORTANCE Because the viroplasm is the viral factory for rotavirus replication, viroplasm formation undoubtedly determines the effective production of progeny rotavirus. Therefore, an understanding of the virus-host interactions involved in the biogenesis of the viroplasm is critical for the future development of prophylactic and therapeutic strategies. Previous studies have reported that the formation of viroplasms is associated with the stability of microtubules, whereas little is known about its specific mechanism. Here, we demonstrate that rotavirus viroplasm formation takes advantage of microtubule-based dynein transport mediated by an interaction between NSP2 and the DIC. These findings provide new insight into the intracellular transport of viroplasms.
KEYWORDS: dynein intermediate chain (DIC), dynein transport, fusion, formation, microtubule based, NSP2, rotavirus, viroplasm
INTRODUCTION
Rotavirus (RV) infections are major causes of severe and dehydrating gastroenteritis in children, piglets, and many other young animals (1). RVs are divided into 10 groups, ranging from the group A rotaviruses (RVAs) to the group J rotaviruses, based on VP6 serology (2) and the website of the Rotavirus Classification Working Group (https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/rcwg). RVA is the RV group that most commonly infects humans and swine (3), killing more than 120,000 children annually throughout the world (4) and causing huge economic losses in the swine industry. RVs are nonenveloped viruses with three capsids and a genome consisting of 11 segments of double-stranded RNA, which encodes six structural proteins (VP1 to −4, VP6, and VP7) and five or six nonstructural proteins (NSP1 to −5/6) (5). In the replication stage of RVs, the viral proteins and viral RNAs assemble into cytoplasmic electron-dense inclusion bodies called “viroplasms,” in which the RV genome is replicated and new double-layered particles (DLPs) are assembled (6, 7). DLPs bud across the membrane of the endoplasmic reticulum (ER). During this process, the DLPs acquire a transient lipid envelope to become transient enveloped particles (TEPs). The transient lipid envelope is subsequently lost to yield the mature infectious triple-layered particles (TLPs) (8, 9). Viroplasms surrounded by TEPs and TLPs are detectable by electron microscopy.
The mature viroplasm contains viral proteins VP1, VP2, VP3, VP6, NSP2, and NSP5. Among these proteins, NSP2 and NSP5 are called the “scaffold proteins” (10). Studies have shown that the coexpression of NSP2 and NSP5 leads to the formation of viroplasm-like structures in uninfected cells (11), and mutation of NSP2 or knockdown of NSP5 impairs viroplasm formation in cells infected with RV (12–17). Therefore, NSP2 and NSP5 play a nucleation role in the formation of viroplasms. As well as viral proteins, several cellular proteins are also involved in viroplasms, including endoplasmic reticulum chaperones (18), cellular lipid droplet components (19), and heterogeneous nuclear ribonucleoproteins (20). Viroplasms have been observed embedded in the microtubule network, and their growth requires a functional microtubule network (21–23). In the course of an ongoing infection, smaller viroplasms fuse to form larger ones, thus increasing considerably in size and decreasing in number (7, 22). Viroplasm fusion is an undoubtedly dynamic process that is important for the production of progeny viruses. However, the mechanism of viroplasm movement is still unclear.
Microtubules are a common and highly efficient motorway for the transport of organelles and intracellular macromolecular substances. As members of the cytoskeletal filaments, microtubules are composed of heterodimers of α-tubulin and β-tubulin, an arrangement that appears to be ubiquitous in eukaryotes (24). Microtubule-based transport is based on two kinds of molecular motors, dyneins and kinesins (25). Most kinesins transport their cargo molecules to the plus ends, whereas a fraction of kinesins migrate to the minus ends of microtubules (26). Dyneins can be differentiated into cytoplasmic dynein 1 and cytoplasmic dynein 2. The function of cytoplasmic dynein 2 is restricted to the flagella and cilia. Dynein 1 exists in eukaryotic cells and drives its cargo molecules to the minus ends of microtubules (27). Dynein 1 (referred to here as “dynein”) is a large polysubunit complex containing double-dynein heavy chains (DHCs), double-dynein intermediate chains (DICs) (also known as DYNC1I1 and DYNC1I2), double-dynein light intermediate chains, and three couples of light chains (28–30). DHCs form the motor domain of dynein, with a hexameric AAA ring (31, 32). All subunits of dynein, except the DHCs, are nonmotor domains and have been shown to interact with dynein’s cargo molecules (33). There are several inhibitors of microtubule-based dynein transport, including ciliobrevins (34) and dynapyrazole-A. Dynapyrazole-A, in particular, specifically inhibits the ATPase activity of microtubule-stimulated dynein but has no effect on the microtubule-independent basal activity of dynein (35).
The dynamics of RV viroplasms and the association of RV viroplasm functions with microtubules prompted us to speculate that viroplasms require microtubular transport to complete their fusion, and microtubule-based transport cannot be separated from the motor proteins dynein and kinesins. Eichwald et al. have shown that kinesin participates in the fusion and localization of RV viroplasms (22). However, the role of dynein in RV infection, and especially in viroplasm formation, remains poorly understood. In this study, we clarify the mechanism by which RV viroplasms hijack microtubule-based dynein transport to complete their fusion via a direct interaction between NSP2 and the DIC, extending our understanding of the biological characteristics of RV viroplasms.
RESULTS
Rotavirus infection requires microtubule-based dynein transport.
To investigate whether microtubule-based dynein transport is involved in RVA strain pig/China/NMTL/2008/G9P[23] (RVA-NMTL) infection, a specific inhibitor of microtubule-based dynein transport, dynapyrazole-A (35), was used. First, to investigate the possible toxicity of dynapyrazole-A on MA-104 cells, the viability of drug-treated cells was determined with a CCK-8 (Cell Counting Kit-8) assay. The results showed no significant difference between the viabilities of the untreated cells and dynapyrazole-A-treated cells (Fig. 1A), indicating that treatment with <3 μg/ml dynapyrazole-A had no effect on MA-104 cell proliferation. Moreover, the 50% cytotoxic concentration (CC50) of dynapyrazole-A in MA-104 cells was 10.90 μg/ml (data not shown). Second, the influence of dynapyrazole-A on RVA-NMTL was examined by 50% tissue culture infective dose (TCID50) assays, Western blotting (WB), and immunofluorescence assays (IFAs). The timeline of the experimental procedure is shown in Fig. 1B. Compared with dimethyl sulfoxide (DMSO)-treated cells and untreated cells, the viral titers in cells treated with 1 to 3 μg/ml dynapyrazole-A decreased 5- to 1,000-fold, in a dose-dependent manner (P < 0.05, P < 0.01, or P < 0.0001) (Fig. 1C). The 50% effective concentration (EC50) of dynapyrazole-A was 0.73 μg/ml (data not shown). The expression of VP6, NSP2, and NSP5 was significantly reduced in the dynapyrazole-A-treated cells (Fig. 1D). We used the number of cells positive for NSP2 to indicate the infection rate of RV. The rate of RVA-NMTL infection was lower in the dynapyrazole-A-treated cells than in the DMSO-treated cells (Fig. 1E). These results suggest that microtubule-based dynein transport is involved in RV replication.
FIG 1.

An inhibitor of microtubule-based dynein transport, dynapyrazole-A, suppresses RVA-NMTL infection. (A) MA-104 cells were treated with 1 μg/ml, 2 μg/ml, and 3 μg/ml dynapyrazole-A for 14 h, and cell viability was analyzed using a CCK-8 assay. ns, not significant. (B to E) MA-104 cells were treated with 1 μg/ml, 2 μg/ml, and 3 μg/ml dynapyrazole-A or DMSO or remained untreated 1 h before infection and were then infected with RVA-NMTL at a multiplicity of infection (MOI) of 0.1. The concentrations of dynapyrazole-A used in panels C and D are 1 μg/ml, 2 μg/ml, and 3 μg/ml. The concentration of dynapyrazole-A used in panel E is 3 μg/ml. (B) Timeline of the experimental procedure. (C) Viral titers determined as TCID50s. Error bars represent the standard errors of the means (n = 3). *, P < 0.05; **, P < 0.01; ****, P < 0.0001. (D) VP6, NSP2, NSP5, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression in the cell lysates was analyzed by Western blotting (WB) with mouse anti-VP6 monoclonal antibody (1F4), rabbit anti-NSP2 polyclonal antibody, mouse anti-NSP5 monoclonal antibody (5E11), and rabbit anti-GAPDH polyclonal antibody, respectively. GAPDH was used as the loading control. (E) The infection rate of RVA-NMTL was determined by an immunofluorescence assay (IFA). NSP2 was stained with a rabbit anti-NSP2 polyclonal antibody (red), and the cell nuclei were stained with DAPI (blue).
Small viroplasms move and fuse.
MA-104 cells were transfected with pEGFP-N1-NSP5 to visualize the viroplasms in living cells, according to the study by Eichwald et al. (7). The cells were simultaneously stained with SiR (Si—Rhodamines)-tubulin (a dye specific for microtubules in living cells) to visualize the microtubules. Under Airyscan microscopy, two viroplasms were observed to attach to microtubules and move. The two viroplasms moved increasingly closer as infection progressed and finally fused at 3 min 29 s. After 3 min 29 s, there was only one viroplasm in view (Fig. 2; see also Movie S1 in the supplemental material). These results indicate that small viroplasms move and fuse.
FIG 2.

Small viroplasms move and fuse. MA-104 cells were transfected with pEGFP-N1-NSP5 for 24 h and then infected with RVA-NMTL (MOI = 1), followed by incubation with 1 μM SiR-tubulin and 10 μM verapamil after 7 hpi to stain the microtubules (red). The inclusion bodies containing enhanced green fluorescent protein (EGFP)-NSP5 are viroplasms (green dots). Snapshots were taken with an Airyscan microscope at 9 hpi at intervals of 40 s. The gray boxes show the magnified images.
Microtubule-based dynein transport is involved in both the early and late stages of viroplasm formation.
We found that RVA-NMTL started to be released at 5 h postinfection (hpi) and was released in large quantities at 6 hpi (data not shown), so most viruses had completed their first replication cycle at 6 hpi. Therefore, we divided the stages of viroplasm formation into the early stage (before 6 hpi) and the late stage (after 6 hpi). Because dynein is the motor of microtubule-based transport, we investigated whether it is involved in viroplasm formation in the early or late stage of viral replication. To study viroplasm formation in the early stage, the experiment shown in Fig. 3A was performed. RVA-NMTL-neutralizing hyperimmune serum was added to block infection by the progeny viruses. When dynapyrazole-A and RVA-NMTL-neutralizing hyperimmune serum were added starting at 1.5 hpi, the viroplasms in the dynapyrazole-A-treated cells appeared smaller and fewer than those in the DMSO-treated control cells at 6 hpi (Fig. 3B). Statistical analysis showed that the viroplasms in the dynapyrazole-A-treated cells were about half the size of those in the DMSO-treated cells (Fig. 3C). Most of the dynapyrazole-A-treated cells contained <3 viroplasms per cell, whereas most of the DMSO-treated cells contained >4 viroplasms per cell (Fig. 3D). There was no difference in the expression of VP6, NSP2, or NSP5 between dynapyrazole-A-treated cells and untreated cells (Fig. 3E). These results demonstrate that dynapyrazole-A disrupts viroplasm formation as early as the first replication cycle of RV, indicating that microtubule-based dynein transport participates in viroplasm formation in the early stage of RV replication.
FIG 3.

Dynapyrazole-A inhibits the early stage of viroplasm formation. MA-104 cells were infected with RVA-NMTL (MOI = 0.1) and then treated with 3 μg/ml dynapyrazole-A and RVA-NMTL-neutralizing hyperimmune serum starting at 1.5 hpi. (A) Timeline of the experimental procedure. (B) LSCM was used to examine viroplasms (green) stained with a rabbit anti-NSP2 polyclonal antibody. Nuclei were stained with DAPI (blue). (C) Sizes of viroplasms were measured in 40 cells with Zen 2.3 software and analyzed statistically. (D) The numbers of viroplasms were analyzed statistically in three parallel experiments with 40 cells each time. Cells containing similar numbers of viroplasms were grouped, and the proportion of each group of cells of the total number of cells (40 cells) is the ordinate. (E) VP6, NSP2, NSP5, and GAPDH expression in the cell lysates was analyzed by WB with mouse anti-VP6 monoclonal antibody (1F4), rabbit anti-NSP2 polyclonal antibody, mouse anti-NSP5 monoclonal antibody (5E11), and rabbit anti-GAPDH polyclonal antibody, respectively. GAPDH was used as the loading control. The error bars represent the standard errors of the means (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
According to studies by Eichwald et al., the fusion of viroplasms mainly occurs in the late stage of viroplasm formation, and the viroplasms increase in size and decrease in number as they fuse during RV infection (7, 22). Therefore, we examined the effect of dynapyrazole-A on viroplasm fusion in the late stage of replication. The experiment was performed as shown in Fig. 4A. The viroplasms in the dynapyrazole-A-treated cells looked dramatically smaller but were more numerous than those in DMSO-treated cells (Fig. 4B). Statistical analysis of the sizes of the viroplasms indicated that those in the dynapyrazole-A-treated cells were about one-quarter of the size of those in the DMSO-treated cells (P < 0.0001) (Fig. 4C). In contrast, statistical analysis of the number of viroplasms per cell indicated that the dynapyrazole-A-treated cells contained more small viroplasms per cell than the DMSO-treated cells (Fig. 4D). There was no significant difference in the expression of VP6, NSP2, or NSP5 between dynapyrazole-A-treated cells and untreated cells (Fig. 4E). These results establish that viroplasm fusion is disrupted by dynapyrazole-A in the late stage of viroplasm formation, which was unrelated to the expression of RV proteins. Moreover, washing out dynapyrazole-A rescued the formation of larger viroplasms (data not shown). In summary, microtubule-based dynein transport is involved in viroplasm formation in both the early and late stages of RV replication.
FIG 4.

Dynapyrazole-A inhibits viroplasm fusion in the late stage of viroplasm formation. MA-104 cells were infected with RVA-NMTL (MOI = 0.1) and then treated with 3 μg/ml dynapyrazole-A and RVA-NMTL-neutralizing hyperimmune serum at 5 to 9 hpi. (A) Timeline of the experimental procedure. (B) Confocal microscopy was used to examine viroplasms (green) stained with a rabbit anti-NSP2 polyclonal antibody. Nuclei were stained with DAPI (blue). (C) The sizes of viroplasms were measured in 40 cells with Zen 2.3 software and analyzed statistically. (D) The numbers of viroplasms were analyzed statistically in three parallel experiments with 40 cells each time. Cells containing similar numbers of viroplasms were grouped, and the proportion of each group of cells of the total number of cells (40 cells) is the ordinate. (E) VP6, NSP2, NSP5, and GAPDH expression in the cell lysates was analyzed by WB with mouse anti-VP6 monoclonal antibody (1F4), rabbit anti-NSP2 polyclonal antibody, mouse anti-NSP5 monoclonal antibody (5E11), and rabbit anti-GAPDH polyclonal antibody, respectively. GAPDH was used as the loading control. Error bars represent the standard errors of the means (n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Participation of microtubule-based dynein transport in viroplasm formation is not associated with the expression of viroplasm proteins.
The expression of viroplasm proteins inevitably affects viroplasm formation. NSP2, NSP5, and VP6 are important viral proteins and the main components of viroplasms. In particular, NSP2 and NSP5 are essential elements in the assembly of functional viroplasms (7, 11, 12, 14, 36). Therefore, to understand the mechanism by which dynein transport participates in viroplasm formation, we determined whether the expression of these viroplasm proteins was inhibited by dynapyrazole-A in MA-104 cells infected with RVA-NMTL, with or without treatment with RVA-NMTL-neutralizing hyperimmune serum. Surprisingly, without treatment with RVA-NMTL-neutralizing hyperimmune serum, the expression levels of NSP2, NSP5, and VP6 were lower in dynapyrazole-A-treated cells than in cells of the control group (Fig. 5B). However, after treatment with RVA-NMTL-neutralizing hyperimmune serum (blocking the entry of the progeny viruses), the expression of NSP2, NSP5, and VP6 did not differ between the dynapyrazole-A-treated and control cells (Fig. 5C). This indicates that microtubule-based dynein transport does not participate directly in the expression of viroplasm proteins after blocking the entry of the progeny viruses. It is noteworthy that although the cells were treated with RVA-NMTL-neutralizing hyperimmune serum, viroplasm formation was inhibited by dynapyrazole-A (as described above), indicating that the involvement of microtubule-based dynein transport in viroplasm formation is not associated with the expression of viroplasm proteins.
FIG 5.
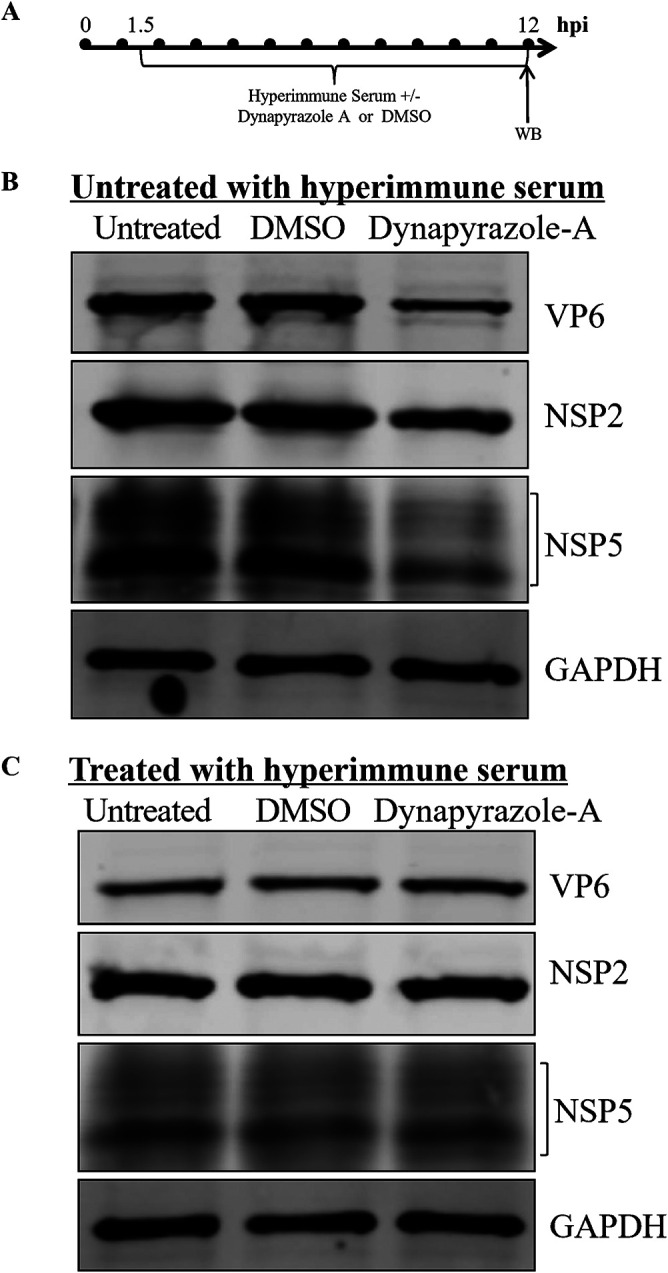
Expression of viroplasm proteins is inhibited by dynapyrazole-A, but this inhibition is eliminated by RVA-NMTL-neutralizing hyperimmune serum. (A) Timeline of the experimental procedure. (B) MA-104 cells were infected with RVA-NMTL (MOI = 0.1) and then treated with 3 μg/ml dynapyrazole-A or DMSO or left untreated starting at 1.5 hpi. (C) MA-104 cells were infected with RVA-NMTL (MOI = 0.1) and then treated with 3 μg/ml dynapyrazole-A or DMSO or left untreated and at the same time with RVA-NMTL-neutralizing hyperimmune serum starting at 1.5 hpi. In panels B and C, viroplasm proteins NSP2, NSP5, and VP6 were detected by WB with rabbit anti-NSP2 polyclonal antibody, mouse anti-NSP5 monoclonal antibody (5E11), and mouse anti-VP6 monoclonal antibody (1F4), respectively. As the loading control, GAPDH was immunoblotted with a rabbit anti-GAPDH polyclonal antibody.
The DIC anchors within viroplasms and interacts with NSP2.
To investigate whether dynein is the motor allowing viroplasms to move along microtubules, the subcellular localization of dynein and viroplasms in MA-104 cells was observed by Airyscan microscopy. The DIC was used to mark dynein, and NSP2 was used to mark the viroplasms. The DIC was always observed to colocalize with NSP2 and, thus, with viroplasms (Fig. 6A) and even in two approaching viroplasms (Fig. 6B), indicating that dynein anchors in the viroplasms. The Pearson, Manders, k1, k2, m1, and m2 colocalization coefficients of the two approaching viroplasms were high, at 0.9137, 0.9780, 0.9047, 0.9222, 0.9734, and 0.9618, respectively (Fig. 6C1 to C3), supporting their colocalization. We tested all the antibodies used in this study against purified RVA-NMTL using WB. DIC-, Myc-, glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-, and hemagglutinin (HA)-directed antibodies did not react with purified RVA-NMTL, whereas the anti-VP6 antibody reacted with purified RVA-NMTL (data not shown). It demonstrates that there was free of rotavirus antibodies in these antibodies. These results provide the first evidence that dynein is the motor for viroplasm transport.
FIG 6.

The DIC colocalizes with viroplasms. MA-104 cells were infected with RVA-NMTL (MOI = 0.1) and fixed at 9 hpi. Airyscan microscopy was used to examine viroplasms (red) stained with a rabbit anti-NSP2 polyclonal antibody, dynein stained with a mouse anti-DIC monoclonal antibody (green), and nuclei stained with DAPI (blue). Panel C1 is a magnification of the gray box in panel B. In panels C2 and C3, scatterplot analysis of the fluorescence intensity and analysis of the colocalization coefficients from panel C1 were performed using Zen 2.3 software. The closer the colocalization coefficient is to 1, the more reliable the colocalization relationship is.
To investigate the colocalization mechanism of the DIC and viroplasms, a coimmunoprecipitation assay on the lysates of MA-104 cells infected with RVA-NMTL was performed. The results of protein mass spectrometry analysis showed that NSP2 was the only viral protein in the precipitates (data not shown). This result was confirmed by WB, which showed that the DIC interacted with NSP2 but not NSP5 (Fig. 7A). These results indicate that the DIC interacts with NSP2 in MA-104 cells infected with RVA-NMTL. Moreover, in a coimmunoprecipitation assay of the lysate of MA-104 cells infected with RVA-NMTL, the amount of NSP2 that coprecipitated with NSP5 was not affected by small interfering RNA (siRNA) targeting DIC mRNA (siDIC) (data not shown), indicating that siDIC did not affect the interaction between NSP2 and NSP5.
FIG 7.

The DIC interacts directly with NSP2. (A) A coimmunoprecipitation assay was performed on lysates of RVA-NMTL-infected or uninfected cells. The lysates were precipitated with a mouse anti-DIC monoclonal antibody. The precipitates were analyzed by WB with a rabbit anti-DIC monoclonal antibody, mouse anti-NSP5 monoclonal antibody 5E11, a rabbit anti-NSP2 polyclonal antibody, and a rabbit anti-GAPDH polyclonal antibody. IB, immunoblotting. (B) HEK-293T cells were cotransfected with pCMV-Myc-DIC and pCAGGS-HA-NSP2, pCMV-Myc-DIC and pCAGGS-HA-NSP5, pCMV-Myc-DIC and pCAGGS-HA, pCMV-Myc and pCAGGS-HA-NSP2, or pCMV-Myc-DIC and pCAGGS-HA-NSP5. The cell lysates were coimmunoprecipitated with a mouse anti-Myc monoclonal antibody. The precipitates were analyzed by WB with a mouse anti-Myc monoclonal antibody, a rabbit anti-HA polyclonal antibody, and a rabbit anti-GAPDH polyclonal antibody.
The interaction between NSP2 and the DIC was confirmed by WB of HEK-293T cells cotransfected with pCMV-Myc-DIC and pCAGGS-HA-NSP2 (Fig. 7B), indicating that NSP2 directly interacts with the DIC. These results provide a second line of evidence that dynein is the motor of viroplasm transport.
The region of the DIC that interacts with NSP2 was investigated. The DIC was divided into two segments: the region spanning amino acids (aa) 1 to 280 and the region spanning aa 281 to 584 (Fig. 8A). The latter region is the WD40 repeat domain. In a coimmunoprecipitation assay, NSP2 coimmunoprecipitated with the region from aa 281 to 584 of the DIC but not with the region from aa 1 to 280 (Fig. 8B), indicating that NSP2 interacts directly with the region from aa 281 to 584, which is the WD40 repeat domain.
FIG 8.
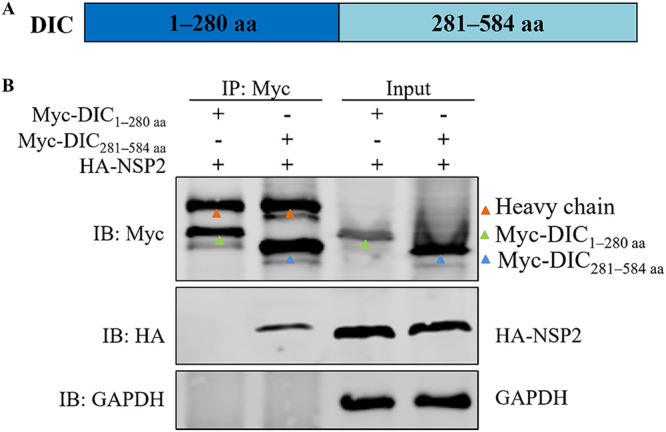
The region spanning aa 281 to 584 of the DIC interacts with NSP2. HEK-293T cells were cotransfected with pCMV-Myc-DIC1–280aa and pCAGGS-HA-NSP2 or with pCMV-Myc-DIC281–584aa and pCAGGS-HA-NSP2. (A) Timeline of the experimental procedure. (B) Cell lysates were coimmunoprecipitated with a mouse anti-Myc monoclonal antibody. The precipitates were analyzed by WB with a mouse anti-Myc monoclonal antibody, a rabbit anti-HA polyclonal antibody, and a rabbit anti-GAPDH polyclonal antibody.
Knockdown of the DIC inhibits RVA-NMTL infection and disrupts viroplasm formation.
The direct interaction between NSP2 and the DIC prompted us to study the role of the DIC in the life cycle of RVA-NMTL infection. Therefore, we constructed siDIC and a negative-control siRNA (siCON). To assess the possible toxic effects of transfecting MA-104 cells with siRNA, the viability of the transfected cells was determined with a CCK-8 assay. The viability of the siDIC-transfected cells did not differ significantly from that of the siCON-transfected or untreated cells (Fig. 9A). WB analysis confirmed that DIC expression was significantly reduced in MA-104 cells transfected with siDIC for 36 h (Fig. 9B). The effect of knocking down the DIC on the expression of VP6, NSP5, and NSP2 in MA-104 cells infected with RVA-NMTL was also determined by WB. The results showed a significant reduction in viral protein expression in the DIC-silenced cells compared with that in the siCON-transfected or untreated cells (Fig. 9C). Similarly, the viral titer was about 10-fold lower (P < 0.01) in the DIC-silenced cells than in the siCON-transfected or untreated cells according to the results of a TCID50 analysis (Fig. 9D). Together, these results indicate that the DIC directly and markedly affects RVA-NMTL infection.
FIG 9.

DIC knockdown inhibits RVA-NMTL infection. (A) MA-104 cells were transfected with siDIC or siCON for 48 h, and cell viability was analyzed by a CCK-8 assay. (B) Western blotting was performed on the cells with a rabbit anti-DIC monoclonal antibody and a rabbit anti-GAPDH polyclonal antibody (loading control). (C and D) MA-104 cells transfected with siDIC or siCON for 36 h were infected with RVA-NMTL for 12 h (MOI = 0.1). (C) The expression of VP6, NSP5, NSP2, and GAPDH was analyzed by WB with mouse anti-VP6 monoclonal antibody 1F4, a rabbit anti-NSP2 polyclonal antibody, mouse anti-NSP5 monoclonal antibody 5E11, and a rabbit anti-GAPDH polyclonal antibody, respectively. GAPDH was the loading control. (D) Viral titers determined as TCID50s.
To further study the effects of the DIC on viroplasm formation, viroplasms were observed at 9 hpi by laser scanning confocal microscopy (LSCM) in MA-104 cells that had been transfected with siDIC or siCON before infection with RVA-NMTL. The viroplasms in the DIC-silenced cells were smaller than those in the siCON-transfected cells (Fig. 10A). Although there was no difference in the viroplasm numbers in the DIC-silenced cells and siCON-transfected cells (Fig. 10C), statistical analysis showed that the viroplasms in the DIC-silenced cells were about one-third of the size of those in the siCON-transfected cells (Fig. 10B). The reduction in the viroplasm size was sufficient to confirm that the knockdown of the DIC disrupted the formation of large viroplasms, providing a third line of evidence that dynein is the motor of viroplasm transport.
FIG 10.

DIC knockdown disrupts viroplasm formation. MA-104 cells were transfected with siDIC or siCON for 36 h and then infected with RVA-NMTL for 9 h (MOI = 0.1). (A) Viroplasms were immunofluorescently stained with a rabbit anti-NSP2 polyclonal antibody and observed by LSCM at 9 hpi. (B) The sizes of the viroplasms in 40 cells were measured with Zen 2.3 software and analyzed statistically. (C) The numbers of viroplasms were analyzed statistically in three parallel experiments with 40 cells each time. Cells containing similar numbers of viroplasms were grouped, and the proportion of each group of cells of the total number of cells (40 cells) is the ordinate. Error bars represent the standard errors of the means (n = 3). ****, P < 0.0001.
Dynapyrazole-A or knockdown of the DIC induces the production of small viroplasms, which are surrounded by fewer TEPs and TLPs.
Transmission electron microscopy (TEM) was used to observe TEPs and TLPs around the viroplasms in MA-104 cells infected with RVA-NMTL. The viroplasms in dynapyrazole-A-treated cells were distinctly smaller than those in DMSO-treated cells. Remarkably, far fewer TEPs and TLPs assembled from the viroplasms in the dynapyrazole-A-treated cells than from those in the DMSO-treated cells (Fig. 11A). Similar results were observed in DIC-silenced MA-104 cells. Both the size of the viroplasms and the numbers of TEPs and TLPs assembled from the viroplasms were reduced by the knockdown of the DIC compared to those in the siCON-transfected cells (Fig. 11B). These results indicate that the smaller viroplasms induced by dynapyrazole-A or siDIC lacked the ability to assemble TEPs and TLPs (which determines the production of progeny viral particles).
FIG 11.
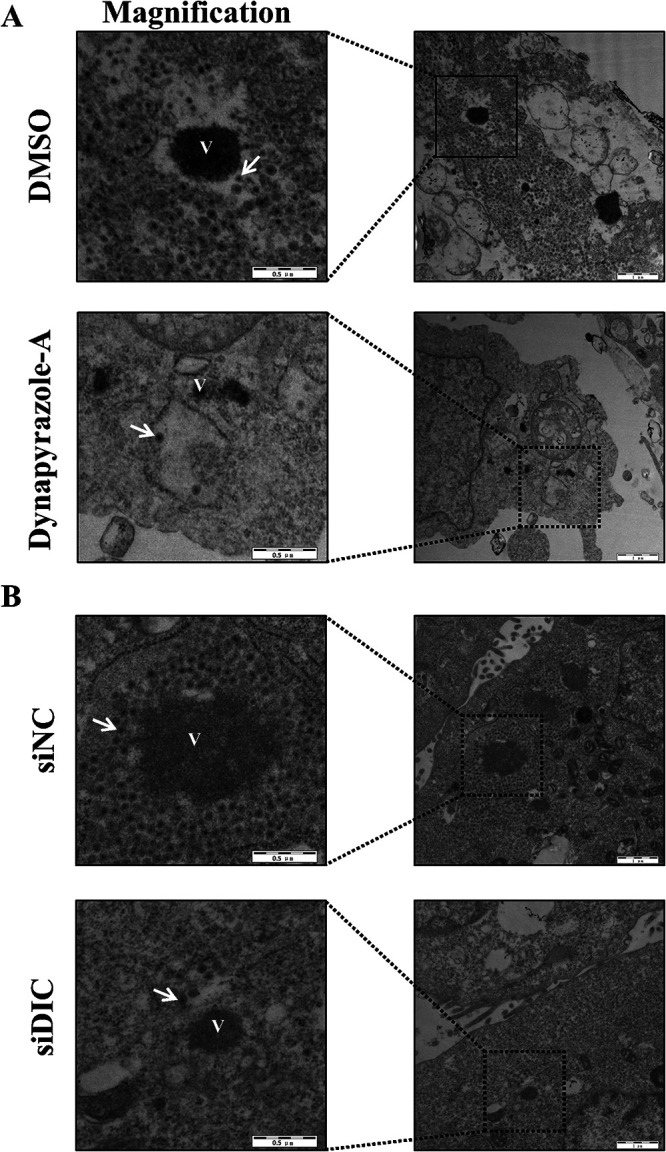
Dynapyrazole-A or DIC knockdown prevents the assembly of DLPs from viroplasms. (A) MA-104 cells were infected with RVA-NMTL (MOI = 0.1) and then treated with 3 μg/ml dynapyrazole-A or DMSO and RVA-NMTL-neutralizing hyperimmune serum starting at 1.5 hpi. (B) MA-104 cells were transfected with siDIC or siCON for 36 h and then infected with RVA-NMTL (MOI = 0.1). In panels A and B, TEM was used to analyze the cells at 12 hpi. “V” represents viroplasms. Arrows show DLPs assembled from viroplasms. Black boxes show magnified images.
DISCUSSION
Unlike other viruses, RV replicates its genome and assembles its DLPs in virus-induced cytoplasmic inclusion bodies called viroplasms. It is known that RV viroplasms are observed from 2 hpi and increase in number before 6 hpi. Next, after 6 hpi, RV viroplasms grow in size and decrease in number through fusion. Viroplasm fusion is clearly a dynamic process (7, 22). Therefore, we hypothesized that the fusion of viroplasms requires their intracellular transport. In recent studies, the mature viroplasms of RVs have been considered to be organelles, with five concentric layers composed of multiple viral proteins (10, 37). Of these proteins, NSP2 and NSP5 play key roles in determining viroplasm formation (11, 12, 14), and even the phosphorylation of NSP2 and NSP5 determines viroplasm assembly (15, 36). However, the effect of intracellular transport on viroplasm biogenesis was still unknown. In this study, we have demonstrated that RVA-NMTL viroplasms utilize dynein, the motor of microtubules to drive retrograde transport, to move along microtubules through a direct interaction between NSP2 and the DIC. The movement of viroplasms causes two viroplasms to fuse into a single viroplasm. Microtubule-based dynein transport promotes the formation of large viroplasms surrounded by more TEPs and TLPs. In other words, it enhances the reproduction of progeny virions (Fig. 12). Our results clarify the significant function of NSP2 in viroplasm formation and even in the efficient replication of RVs. However, the effect of posttranslational modification of NSP2, such as its phosphorylation (which can affect viroplasm formation), on dynein transport remains to be investigated.
FIG 12.

Schematic model depicting the formation of RV viroplasms via microtubule-based dynein transport. The dynein intermediate chain (DIC) is located in the RV viroplasms, where the DIC interacts with NSP2. RV viroplasms utilize the interaction to move along microtubules (MTs). The movement of viroplasms not only causes two viroplasms to fuse into a single viroplasm but also increases the ability of viroplasms to assemble DLPs. DLC, dynein light chain; DLIC, dynein light intermediate chain; DHC, dynein heavy chain.
Several previous studies support our results. For example, RV viroplasms were shown to be associated with the microtubule network in space and to require stable microtubules to grow (23). In fact, it is common among members of the family Reoviridae that the fusion of viroplasms (also known as “viral factories”) depends on stabilized microtubules, such as the mammalian orthoreoviruses (38). The fusion of viral factories in cells infected with infectious bursal disease viruses (members of the family Birnaviridae) also involves stabilized microtubules (39). Therefore, the fusion of viroplasms or viral factories in these viruses may also be driven by microtubule-based transport. Microtubule-based dynein transport may also be used by other viruses to complete the fusion of their viroplasms or viral factories. This inference is supported by research on measles virus (MeV), which showed that dynein promotes the formation of large inclusion bodies (considered the viral factories of MeV) (40).
Microtubule-based dynein transport has been shown to be involved in viroplasm formation (Fig. 3 and 4), and the viroplasms move and fuse in cells infected with RV (Fig. 2). However, the effect of microtubule-based dynein transport on the movement of viroplasms should be investigated further to better understand why microtubule-based transport is involved in viroplasm formation. Unfortunately, we failed to determine the movement velocity of the viroplasms because the viroplasm moved too fast and did not move in straight lines, which resulted in the trajectory being difficult capture. Although the current experimental conditions did not allow statistical analysis of viroplasm movement along microtubules, the effect of microtubule-based dynein transport on the movement of viroplasms warrants further research in the future.
Dynapyrazole-A inhibited RVA-NMTL entry (data not shown), but it was added at 1.5 hpi and 5 hpi in Fig. 3 and 4, respectively, after RV had completed its entry (in general, RV completes its entry within 10 min [41]). Therefore, dynapyrazole-A did not affect RVA-NMTL entry in Fig. 3 and 4. Multiple rounds of RV infection were avoided by the addition of RVA-NMTL-neutralizing hyperimmune serum, which neutralized the progeny virus shown in Fig. 3 and 4. In summary, when RVA-NMTL-neutralizing hyperimmune serum was added and the time of drug administration was controlled, dynapyrazole-A acted only on the replication stage of RVA-NMTL. siDIC also inhibited RVA-NMTL entry (data not shown). There are two possible explanations for the inhibition of viroplasm size by siDIC, as shown in Fig. 10. One is that siDIC inhibited viroplasm formation by disrupting dynein transport. The other is that siDIC reduced the number of virions that infected the cells by inhibiting RV entry.
NSP2 is reported to interact with NSP5 and is located in viroplasms (7). Moreover, NSP2 and NSP5 are considered to be the proteins that determine viroplasm formation (12–14). In this study, the DIC is shown to interact with NSP2 and localize in viroplasms, and dynein transport is shown to be involved in viroplasm formation. However, siDIC did not affect the interaction between NSP2 and NSP5 (data not shown). Therefore, the involvement of dynein transport in viroplasm formation does not result from the disruption of the interaction between NSP2 and NSP5 by the interaction between NSP2 and the DIC.
Viroplasms are mainly responsible for the assembly of DLPs, from which both TEPs and TLPs assemble (6, 7), and microtubule-based dynein transport demonstrably affects the assembly of TEPs and TLPs (Fig. 11A) and viroplasm formation (Fig. 3B and Fig. 4B). We infer that microtubule-based dynein transport promotes the assembly of DLPs by participating in the formation of large viroplasms. However, this inference must be tested with further experiments. Why viroplasm formation affects the assembly of DLPs by viroplasms is still unknown.
Our results also show that dynapyrazole-A inhibited the expression of viral proteins during multiple rounds of infection (Fig. 5B). However, this inhibition was eliminated by blocking the invasion of progeny viruses with RVA-NMTL-neutralizing hyperimmune serum (Fig. 5C). It has been demonstrated that secondary transcription is the main source of rotaviral proteins in infected cells (42). It is clear that in the absence of RVA-NMTL-neutralizing hyperimmune serum, the inhibition of viral protein expression by dynapyrazole-A resulted from its effect on the production of progeny viruses because it disrupted secondary or subsequent viral infection. The expression of NSP2, NSP5, and VP6 was not influenced by dynapyrazole-A when dynapyrazole-A disrupted the formation of viroplasms (Fig. 3 and 4). Therefore, the inhibition of viral protein expression by dynapyrazole-A during normal infection is an indirect result of the involvement of microtubule-based dynein transport in viroplasm formation and enlargement.
The RV genome has been detected in the cerebrospinal fluid of RV-infected children (43–45), suggesting that RV can spread from the site of initial infection to the central nervous system (CNS). In general, RV infection occurs through the fecal-oral route, but how it spreads to the CNS is unknown. In this study, microtubule-based dynein transport has been shown to be involved in the formation of viroplasms. It is noteworthy that several viruses can use microtubule-based transport to spread along axons and invade the CNS, including poliovirus and rabies virus (46–48). Whether RV viroplasms can spread along axons by microtubule-based transport to invade the CNS warrants investigation.
Rotavirus gastroenteritis is a serious diarrheal disease with high infection and mortality rates in children under 5 years old in middle- and low-income countries (4, 49, 50). Moreover, the economic losses in livestock-breeding industries caused by RV should not be underestimated. At present, inhibiting RV viroplasm formation has become a novel and effective antiviral strategy in the development of new drugs, such as thiazolides, a new class of anti-RV agents (51). In the present study, an inhibitor of dynein transport, dynapyrazole-A, was shown to effectively inhibit viroplasm formation (Fig. 3B to D). Therefore, dynapyrazole-A and other inhibitors of dynein transport potentially offer new anti-RV drugs. Small-molecule drugs that directly break protein-protein interactions have also successfully reached clinical development (52), and NSP2 has been shown to interact directly with the WD40 repeat domain of the DIC (Fig. 7A and B). The WD40 repeat domain can form a propeller-like donut structure, and the central cavity of the propeller often mediates its interactions with other proteins. It also has the appropriate size and physical properties to allow it to bind to drug-like small molecules with high affinity (53). Therefore, drugs targeting the WD40 repeat domain of the DIC, which interacts with NSP2, offer a new direction in anti-RV drug development.
In summary, it is known that microtubule-based dynein transport participates in various stages of viral replication (54–56), and RV viroplasms are associated with microtubules (21, 22). In this study, we have shown that microtubule-based dynein transport facilitates RV replication by promoting the formation of viroplasms. Our findings provide insights into the development of therapeutic anti-RV strategies.
MATERIALS AND METHODS
Cells and hyperimmune serum.
MA-104 cells (Cercopithecus aethiops embryo kidney epithelial cells) were cultured in RPMI 1640 basic medium (catalog number C11875500BT; Gibco) with 10% fetal bovine serum (catalog number 10099-141C; Gibco) at 37°C under 5% CO2–air. Human embryonic kidney cells expressing simian virus 40 (SV40) T antigen (HEK-293T) were cultured in Dulbecco’s modified Eagle’s medium (catalog number RNBJ2913; Gibco) containing 10% fetal bovine serum (catalog number 10099-141C; Gibco) at 37°C under 5% CO2–air. The RVA-NMTL-neutralizing hyperimmune serum was prepared with purified RVA-NMTL (see below) in guinea pigs, and the neutralization titer was 1:1,200.
Virus and infection.
Rotavirus A strain pig/China/NMTL/2008/G9P[23] (abbreviated RVA-NMTL) of genotype G9P[23] was isolated from a pig farm in China and propagated in MA-104 cells, as previously described (57). Before infection, RVA-NMTL was treated with 8 μl/ml 0.25% trypsin (catalog number 15050-065; Gibco) for 1.5 h at 37°C, and MA-104 cells were washed three times with phosphate-buffered saline (PBS). The MA-104 cells were then incubated with RVA-NMTL for 1 h at 37°C under 5% CO2–air. The cells were then cultured in RPMI 1640 basic medium containing 0.5 μl/ml 0.25% trypsin.
Rotavirus purification.
The rotavirus-containing culture (500 ml) was centrifuged at 10,000 × g at 4°C for 10 min. The supernatant was centrifuged at 30,000 × g at 4°C for 15 h. The pellet was suspended in 15 ml of PBS (virus suspension). Next, 9 ml of a 70% sucrose solution was placed at the base of a 38.5-ml ultracentrifuge tube and overlaid with 13 ml of a 30% sucrose solution. The virus suspension (15 ml) was placed on top of the 30% sucrose solution. The ultracentrifuge tube containing the virus suspension was ultracentrifuged with an SW 32 Ti swinging-bucket rotor at 175,000 × g at 4°C for 2 h. The white band above the 70% sucrose solution (crude viral fraction) was extracted.
Western blotting.
For Western blotting (WB), the cell samples were lysed with radioimmunoprecipitation assay (RIPA) buffer (catalog number R0278; Sigma) and boiled with 5× SDS-PAGE protein loading buffer (catalog number P0015L; Beyotime). The samples were subjected to 12.5% SDS-PAGE and then transferred to nitrocellulose membranes (catalog number 66485; Pall). The nitrocellulose membranes were blocked with 5% skim milk (catalog number 232100; BD) after protein transfer for 2 h at room temperature and then washed three times with PBS containing 0.05% Tween (PBS-T). The membranes were incubated with the appropriate primary antibodies (Table 1) for 2 h at room temperature, washed three times with PBS-T, incubated with the corresponding secondary antibodies (Table 1) for 2 h at room temperature, and washed three times with PBS-T. The membranes were scanned and analyzed with a near-infrared bicolor fluorescence imaging system (Odyssey CLX; Licor).
TABLE 1.
Antibodies for Western blotting
| Proteins | Primary antibody (dilution, catalog no.; source) | Secondary antibody (dilution, catalog no.; source) |
|---|---|---|
| NSP5 | Mouse anti-NSP5 monoclonal antibody 5E11 (1:1,000; prepared in our laboratory) | Goat anti-mouse IgG (1:20,000, 926-32210; Licor) |
| VP6 | Mouse anti-VP6 monoclonal antibody 1F4 (1:1,000; prepared in our laboratory) | |
| Myc | Mouse anti-Myc monoclonal antibody (1:1,000, M4439; Sigma) | |
| GAPDH | Rabbit anti-GAPDH polyclonal antibody (1:1,000, G9545; Sigma) | Goat anti-rabbit IgG (1:20,000, 926-32211; Licor) |
| DIC | Rabbit anti-DIC monoclonal antibody (1:1,000, ab171964; Abcam) | |
| NSP2 | Rabbit anti-NSP2 polyclonal antibody (1:1,000; prepared in our laboratory) | |
| HA | Rabbit anti-HA polyclonal antibody (1:5,000, ab9110; Abcam) | |
Indirect immunofluorescence assay.
Cells were fixed with 4% paraformaldehyde at 4°C for 30 min and then washed three times with PBS. The cell membranes were permeabilized with 0.25% Triton X-100 (catalog number 2235C022; Amresco) diluted in PBS for 15 min and washed three times with PBS. They were then blocked with 5% skim milk diluted in PBS for 2 h at 37°C and washed three times with PBS. The cells were incubated with the appropriate primary antibodies (Table 2) overnight at 4°C and washed three times with PBS. They were then incubated with the corresponding secondary antibodies (Table 2) for 1 h at 37°C and washed three times with PBS. The nuclei were stain with 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (catalog number C1006; Beyotime) for 15 min. The cells were washed three times with PBS and covered with Fluoroshield mounting medium (catalog number ab104135; Abcam). Images were taken with an inverted fluorescence microscope (Evos M5000; Life), by laser scanning confocal microscopy (LSCM) (LSM800; Zeiss), or with an Airyscan microscope (LSM800; Zeiss). The Alexa Fluor 594-labeled goat anti-rabbit immunoglobulin G (IgG), Alexa Fluor 488-labeled goat anti-rabbit IgG, and Alexa Fluor 488-labeled goat anti-mouse IgG antibodies and DAPI emitted signals at 617 nm, 517 nm, 517 nm, and 461 nm, respectively. LSCM and Airyscan microscopic images were taken at a ×63 magnification and processed with Zen 2.3 software (Zeiss, Germany).
TABLE 2.
Antibodies for IFA
| Protein(s) | Primary antibody (dilution, catalog no.; source) | Secondary antibody (dilution, catalog no.; source) |
|---|---|---|
| NSP2 | Rabbit anti-NSP2 polyclonal antibody (1:500; prepared in our laboratory) | Fig. 1E, Alexa Fluor 594-labeled goat anti-rabbit IgG (1:500, A11012; Thermo Fisher Scientific) |
| Fig. 3B, Fig. 4B, and Fig. 10A, Alexa Fluor 488-labeled goat anti-rabbit IgG (1:500, A11034; Thermo Fisher Scientific) | ||
| NSP2 and DIC | Rabbit anti-NSP2 polyclonal antibody (1:500; prepared in our laboratory) | Fig. 6A–C, Alexa Fluor 594-labeled goat anti-rabbit IgG (1:500, A11012; Thermo Fisher Scientific) |
| Mouse anti-DIC monoclonal antibody (1:20, ab23905; Abcam) | Alexa Fluor 488-labeled goat anti-mouse IgG (1:500, A10680; Thermo Fisher Scientific) | |
Live-cell imaging microscopy.
MA-104 cells were seeded in a 35-mm dish (catalog number D35-10-1.5-N; Cellvis). To analyze the movement of the viroplasms, the cells were first transfected with pEGFP-N1-NSP5 (see below) for 24 h and then infected with RVA-NMTL at a multiplicity of infection (MOI) of 1, as described above. At 7 h postinfection (hpi), the cell microtubules were stained with 1 μM SiR-tubulin (catalog number CY-SC002; Cytoskeleton) and 10 μM verapamil (catalog number CY-SC002; Cytoskeleton). The movement of the viroplasms was recorded starting at 9 hpi at 40-s intervals by Airyscan microscopy (LSM800; Zeiss).
Viral titer quantification.
MA-104 cells were seeded in 96-well plates. The viral samples to be tested were treated with 8 μl/ml 0.25% trypsin (catalog number 15050-065; Gibco) for 1.5 h at 37°C. The MA-104 cells were then inoculated with serially 10-fold-diluted viral samples. Each dilution was tested eight times. The median tissue culture infective dose (TCID50) was calculated using the Karber method (58, 59), and the cytopathic effect was observed at 72 hpi with an optical microscope. Statistical analysis of the TCID50 and EC50 values was performed with GraphPad Prism 6.
Plasmid construction and transfection.
To construct plasmids pCMV-Myc-DIC, pCMV-Myc-DIC1–280aa, and pCMV-Myc-DIC281–584aa, the total RNA of MA-104 cells was extracted with the RNeasy minikit (catalog number 74106; Qiagen). The cDNA of MA-104 cells was prepared by reverse transcription (RT) using total RNA with the PrimeScript II 1st-strand cDNA synthesis kit (catalog number 6210A; TaKaRa). The DIC gene, the DIC1–280 gene, and the DIC281–584 gene were amplified from the cDNA of MA-104 cells by PCR using the corresponding primers listed in Table 3 and PrimeSTAR Max DNA polymerase (catalog number R045Q; TaKaRa). The plasmids pCMV-Myc-DIC, pCMV-Myc-DIC1–280aa, and pCMV-Myc-DIC281–584aa were constructed from the DIC gene, the DIC1–280 gene, and the DIC281–584 gene, respectively, and the plasmid pCMV-Myc (linearized at the EcoRI restriction site) was constructed by homologous recombination with the In-Fusion HD cloning kit (catalog number 639649; TaKaRa).
TABLE 3.
Primers
| Gene | Primer | Sequence (5′→3′) | Product size (bp) |
|---|---|---|---|
| DIC | DIC-F | ATGGAGGCCCGAATTATGTCAGACAAAAGTGAATTAAAGG | 1,869 |
| DIC-R | TCGGTCGACCGAATTCTAAGCAGGTATTCGGGTAGC | ||
| DIC1–280 | DIC1–280aa-F | ATGGAGGCCCGAATTATGTCAGACAAAAGTGAATTA | 873 |
| DIC1–280aa-R | TCGGTCGACCGAATTTTAAGGGGCATCTTCATTGTTATT | ||
| DIC281–584 | DIC281–584aa-F | ATGGAGGCCCGAATTCATGAGCCTGATGGTGTGGCC | 945 |
| DIC281–584aa-R | TCGGTCGACCGAATTTTATTCATCATTGCGGGGAACAGC | ||
| NSP2 | NSP2-F | AGATTACGCTGAATTCGCTGAGCTAGCTTGCTT | 985 |
| NSP2-R | CGATGAGCTCGAATTTTAAATTCCGACATGTGAAACTTCA | ||
| NSP5 | HA-NSP5-F | AGATTACGCTGAATTCATGTCTCTCAGCATTGACGTA | 621 |
| HA-NSP5-R | CGATGAGCTCGAATTTTACAAATCTTCGATCAATTG | ||
| NSP5 | NSP5-F | CGCGGGCCCGGGATCCATGTCTCTCAGCATTGACGTA | 621 |
| NSP5-R | GCGACCGGTGGATCCAAATCTTCGATCAATTGCAT | ||
| VP6 | VP6-qPCR-F | CCACAATCTGAAGCACTAAGGA | 187 |
| VP6-qPCR-R | GCGCTGGCTGTGATCTATTC | ||
To construct plasmids pCAGGS-HA-NSP2 and pCAGGS-HA-NSP5, RNA of RVA-NMTL was extracted with the QIAamp viral RNA minikit (catalog number 53706; Qiagen). The NSP2 and NSP5 genes were amplified from RNA of RVA-NMTL by PCR using the corresponding primers (the primers HA-NSP5-F and HA-NSP5-R were used for the NSP5 gene) in Table 3 and the PrimeScript one-step RT-PCR kit (catalog number RR055A; TaKaRa). The plasmids pCAGGS-HA-NSP2 and pCAGGS-HA-NSP5 were constructed from the NSP2 gene and the HA-NSP5 gene, respectively, and the plasmid pCAGGS-HA (linearized at the EcoRI restriction site) was constructed by homologous recombination with the In-Fusion HD cloning kit (catalog number 639649; TaKaRa). Plasmid pEGFP-N1-NSP5 was constructed using a method similar to that used to construct pCAGGS-HA-NSP2. After amplification with primers NSP5-F and NSP5-R (Table 3), the RVA-NMTL NSP5 gene was inserted into plasmid pEGFP-N1 at the BamHI restriction site. HEK-293T or MA-104 cells were transfected with these plasmids using X-tremeGENE HP DNA transfection reagent (catalog number 06366546001; Roche), according to the manufacturer’s instructions.
Real-time absolute fluorescence quantitative PCR.
Viral RNA was extracted with the QIAamp viral RNA minikit (catalog number 52906; Qiagen). The copies of VP6 mRNA were detected with primers VP6-qPCR-F and VP6-qPCR-R (Table 3) and probe 5′-TTGGAATTTACAAAACAGACGACAGCG-3′. Real-time absolute fluorescence quantitative PCR was performed with the one-step PrimeScript RT-PCR kit (catalog number RR064A; TaKaRa) on the QuantStudio 5 real-time PCR system (Applied Biosystems), according to the manufacturers’ instructions.
Coimmunoprecipitation assay.
For the coimmunoprecipitation assay of MA-104 cells infected with RVA-NMTL, the cells were seeded in 75-cm2 bottles (catalog number 430720; Corning) and then infected with RVA-NMTL at an MOI of 0.1 for 18 h. The control cells were not infected. The cells were then lysed with 1 ml of Pierce immunoprecipitation (IP) lysis buffer (catalog number 87788; Thermo Scientific) containing phenylmethylsulfonyl fluoride (catalog number ST506; Beyotime) and a protease inhibitor (catalog number 4693132001; Roche) for 30 min at 4°C and centrifuged at 12,000 × g for 5 min at 4°C. An aliquot (80 μl) of the supernatant was boiled with 20 μl of 5× SDS-PAGE protein loading buffer. To avoid nonspecific reactions, the remaining supernatant was shaken with protein A/G magnetic beads (catalog number UI287210A; Thermo) and negative mouse IgG (catalog number A7028; Beyotime) for 2 h at 4°C. The beads were removed, and the supernatant was shaken overnight with a mouse anti-DIC monoclonal antibody (catalog number ab23905; Abcam) at 4°C. Protein A/G magnetic beads were added, and the samples were shaken again at 4°C for 4 h. The beads were washed five times with Pierce IP lysis buffer and boiled in 5× SDS-PAGE protein loading buffer. All the samples were analyzed by Western blotting, as described above. For the coimmunoprecipitation assay in HEK-293T cells, the cells were transfected with plasmids pCMV-Myc-DIC and pCAGGS-HA-NSP2 for 48 h, as described above. The control group of cells was cotransfected with pCMV-Myc-DIC and pCAGGS-HA, pCMV-Myc-DIC and pCAGGS-HA-NSP5, pCMV-Myc and pCAGGS-HA-NSP2, or pCMV-Myc and pCAGGS-HA-NSP5. The remaining procedures were the same as those described above for the MA-104 cells but with a mouse anti-Myc monoclonal antibody (catalog number M4439; Sigma) instead of the mouse anti-DIC monoclonal antibody.
Knockdown by small interfering RNA.
Based on the coding sequence of the DIC gene in MA-104 cells, a small interfering RNA (siRNA) targeting DIC mRNA (siDIC) (targeting sequence, 5′-GCAUGGAGUUGGUUCAUAA-3′) and a negative-control siRNA (siCON) (targeting sequence, 5′-UUCUCCGAACGUGUCACGU-3′) were designed and synthesized by Shanghai GenePharma Co., Ltd. MA-104 cells were transfected with these siRNAs using Lipofectamine RNAiMAX reagent (catalog number 13778-150; Invitrogen), according to the manufacturer’s instructions.
Cell viability assay.
After treatment with various concentrations of dynapyrazole-A for 12 h or transfection with siRNA for 48 h, MA-104 cells were incubated with CCK-8 (catalog number NN699; Dojindo) for 2 h at 37°C. The optical density at a wavelength of 450 nm (OD450) was then determined. Statistical analysis of cell viability and calculation of CC50 values were performed with GraphPad Prism 6.
Transmission electron microscopy.
MA-104 cells were fixed with 2.5% glutaraldehyde prepared in 0.1 M phosphate buffer (pH 7.2), postfixed with osmium tetroxide, dehydrated with acetone, and embedded in Epon 812 resin. Sections were cut and stained with uranyl acetate and lead citrate. Images were taken on a Hitachi H-7650 electron microscope (Hitachi, Tokyo, Japan).
Statistical analysis.
All statistical differences identified in the comparisons of experimental data sets with the controls were assessed with Student’s t test, and the results are presented as means ± standard deviations (SD) from at least three independent experiments. The results were plotted with GraphPad Prism 6 software. Asterisks indicate the significance of differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
ACKNOWLEDGMENTS
This study was supported by the Heilongjiang Province Natural Science Foundation of China (TD2020C002), the National Key Research and Development Program of China (2017YFD0501603), the Agricultural Science and Technology Innovation Program (CAAS-ZDRW202008), and the Heilongjiang Science Foundation Project (C2018066).
Footnotes
Supplemental material is available online only.
Contributor Information
Xin Zhang, Email: zhangxin01@caas.cn.
Li Feng, Email: fengli@caas.cn.
Susana López, Instituto de Biotecnologia/UNAM.
REFERENCES
- 1.Crawford SE, Ramani S, Tate JE, Parashar UD, Svensson L, Hagbom M, Franco MA, Greenberg HB, O’Ryan M, Kang G, Desselberger U, Estes MK. 2017. Rotavirus infection. Nat Rev Dis Primers 3:17083. 10.1038/nrdp.2017.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matthijnssens J, Otto PH, Ciarlet M, Desselberger U, Van Ranst M, Johne R. 2012. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch Virol 157:1177–1182. 10.1007/s00705-012-1273-3. [DOI] [PubMed] [Google Scholar]
- 3.Vlasova AN, Amimo JO, Saif LJ. 2017. Porcine rotaviruses: epidemiology, immune responses and control strategies. Viruses 9:48. 10.3390/v9030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Troeger C, Khalil IA, Rao PC, Cao S, Blacker BF, Ahmed T, Armah G, Bines JE, Brewer TG, Colombara DV, Kang G, Kirkpatrick BD, Kirkwood CD, Mwenda JM, Parashar UD, Petri WA, Jr, Riddle MS, Steele AD, Thompson RL, Walson JL, Sanders JW, Mokdad AH, Murray CJL, Hay SI, Reiner RC, Jr.. 2018. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 years. JAMA Pediatr 172:958–965. 10.1001/jamapediatrics.2018.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramig RF. 1997. Genetics of the rotaviruses. Annu Rev Microbiol 51:225–255. 10.1146/annurev.micro.51.1.225. [DOI] [PubMed] [Google Scholar]
- 6.Patton JT, Silvestri LS, Tortorici MA, Vasquez-Del Carpio R, Taraporewala ZF. 2006. Rotavirus genome replication and morphogenesis: role of the viroplasm. Curr Top Microbiol Immunol 309:169–187. 10.1007/3-540-30773-7_6. [DOI] [PubMed] [Google Scholar]
- 7.Eichwald C, Rodriguez JF, Burrone OR. 2004. Characterization of rotavirus NSP2/NSP5 interactions and the dynamics of viroplasm formation. J Gen Virol 85:625–634. 10.1099/vir.0.19611-0. [DOI] [PubMed] [Google Scholar]
- 8.Lopez T, Camacho M, Zayas M, Najera R, Sanchez R, Arias CF, Lopez S. 2005. Silencing the morphogenesis of rotavirus. J Virol 79:184–192. 10.1128/JVI.79.1.184-192.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki H, Konno T, Numazaki Y. 1993. Electron microscopic evidence for budding process-independent assembly of double-shelled rotavirus particles during passage through endoplasmic reticulum membranes. J Gen Virol 74(Part 9):2015–2018. 10.1099/0022-1317-74-9-2015. [DOI] [PubMed] [Google Scholar]
- 10.Garces Suarez Y, Martinez JL, Torres Hernandez D, Hernandez HO, Perez-Delgado A, Mendez M, Wood CD, Rendon-Mancha JM, Silva-Ayala D, Lopez S, Guerrero A, Arias CF. 2019. Nanoscale organization of rotavirus replication machineries. Elife 8:e42906. 10.7554/eLife.42906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabbretti E, Afrikanova I, Vascotto F, Burrone OR. 1999. Two non-structural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. J Gen Virol 80(Part 2):333–339. 10.1099/0022-1317-80-2-333. [DOI] [PubMed] [Google Scholar]
- 12.Silvestri LS, Taraporewala ZF, Patton JT. 2004. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J Virol 78:7763–7774. 10.1128/JVI.78.14.7763-7774.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vascotto F, Campagna M, Visintin M, Cattaneo A, Burrone OR. 2004. Effects of intrabodies specific for rotavirus NSP5 during the virus replicative cycle. J Gen Virol 85:3285–3290. 10.1099/vir.0.80075-0. [DOI] [PubMed] [Google Scholar]
- 14.Campagna M, Eichwald C, Vascotto F, Burrone OR. 2005. RNA interference of rotavirus segment 11 mRNA reveals the essential role of NSP5 in the virus replicative cycle. J Gen Virol 86:1481–1487. 10.1099/vir.0.80598-0. [DOI] [PubMed] [Google Scholar]
- 15.Papa G, Venditti L, Arnoldi F, Schraner EM, Potgieter C, Borodavka A, Eichwald C, Burrone OR. 2020. Recombinant rotaviruses rescued by reverse genetics reveal the role of NSP5 hyperphosphorylation in the assembly of viral factories. J Virol 94:e01110-19. 10.1128/JVI.01110-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Criglar JM, Crawford SE, Zhao B, Smith HG, Stossi F, Estes MK. 2020. A genetically engineered rotavirus NSP2 phosphorylation mutant impaired in viroplasm formation and replication shows an early interaction between vNSP2 and cellular lipid droplets. J Virol 94:e00972-20. 10.1128/JVI.00972-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez T, Rojas M, Ayala-Breton C, Lopez S, Arias CF. 2005. Reduced expression of the rotavirus NSP5 gene has a pleiotropic effect on virus replication. J Gen Virol 86:1609–1617. 10.1099/vir.0.80827-0. [DOI] [PubMed] [Google Scholar]
- 18.Maruri-Avidal L, Lopez S, Arias CF. 2008. Endoplasmic reticulum chaperones are involved in the morphogenesis of rotavirus infectious particles. J Virol 82:5368–5380. 10.1128/JVI.02751-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung W, Gill M, Esposito A, Kaminski CF, Courousse N, Chwetzoff S, Trugnan G, Keshavan N, Lever A, Desselberger U. 2010. Rotaviruses associate with cellular lipid droplet components to replicate in viroplasms, and compounds disrupting or blocking lipid droplets inhibit viroplasm formation and viral replication. J Virol 84:6782–6798. 10.1128/JVI.01757-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhillon P, Tandra VN, Chorghade SG, Namsa ND, Sahoo L, Rao CD. 2018. Cytoplasmic relocalization and colocalization with viroplasms of host cell proteins, and their role in rotavirus infection. J Virol 92:e00612-18. 10.1128/JVI.00612-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin D, Duarte M, Lepault J, Poncet D. 2010. Sequestration of free tubulin molecules by the viral protein NSP2 induces microtubule depolymerization during rotavirus infection. J Virol 84:2522–2532. 10.1128/JVI.01883-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eichwald C, Arnoldi F, Laimbacher AS, Schraner EM, Fraefel C, Wild P, Burrone OR, Ackermann M. 2012. Rotavirus viroplasm fusion and perinuclear localization are dynamic processes requiring stabilized microtubules. PLoS One 7:e47947. 10.1371/journal.pone.0047947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cabral-Romero C, Padilla-Noriega L. 2006. Association of rotavirus viroplasms with microtubules through NSP2 and NSP5. Mem Inst Oswaldo Cruz 101:603–611. 10.1590/s0074-02762006000600006. [DOI] [PubMed] [Google Scholar]
- 24.Goodson HV, Jonasson EM. 2018. Microtubules and microtubule-associated proteins. Cold Spring Harb Perspect Biol 10:a022608. 10.1101/cshperspect.a022608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sweeney HL, Holzbaur ELF. 2018. Motor proteins. Cold Spring Harb Perspect Biol 10:a021931. 10.1101/cshperspect.a021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muller MJ, Klumpp S, Lipowsky R. 2008. Tug-of-war as a cooperative mechanism for bidirectional cargo transport by molecular motors. Proc Natl Acad Sci USA 105:4609–4614. 10.1073/pnas.0706825105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts AJ, Kon T, Knight PJ, Sutoh K, Burgess SA. 2013. Functions and mechanics of dynein motor proteins. Nat Rev Mol Cell Biol 14:713–726. 10.1038/nrm3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olenick MA, Holzbaur ELF. 2019. Dynein activators and adaptors at a glance. J Cell Sci 132:jcs227132. 10.1242/jcs.227132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfister KK, Fisher EM, Gibbons IR, Hays TS, Holzbaur EL, McIntosh JR, Porter ME, Schroer TA, Vaughan KT, Witman GB, King SM, Vallee RB. 2005. Cytoplasmic dynein nomenclature. J Cell Biol 171:411–413. 10.1083/jcb.200508078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kardon JR, Vale RD. 2009. Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol 10:854–865. 10.1038/nrm2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt H, Gleave ES, Carter AP. 2012. Insights into dynein motor domain function from a 3.3-Å crystal structure. Nat Struct Mol Biol 19:492–497, S1. 10.1038/nsmb.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhabha G, Cheng HC, Zhang N, Moeller A, Liao M, Speir JA, Cheng Y, Vale RD. 2014. Allosteric communication in the dynein motor domain. Cell 159:857–868. 10.1016/j.cell.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bremner KH, Scherer J, Yi J, Vershinin M, Gross SP, Vallee RB. 2009. Adenovirus transport via direct interaction of cytoplasmic dynein with the viral capsid hexon subunit. Cell Host Microbe 6:523–535. 10.1016/j.chom.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roossien DH, Miller KE, Gallo G. 2015. Ciliobrevins as tools for studying dynein motor function. Front Cell Neurosci 9:252. 10.3389/fncel.2015.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinman JB, Santarossa CC, Miller RM, Yu LS, Serpinskaya AS, Furukawa H, Morimoto S, Tanaka Y, Nishitani M, Asano M, Zalyte R, Ondrus AE, Johnson AG, Ye F, Nachury MV, Fukase Y, Aso K, Foley MA, Gelfand VI, Chen JK, Carter AP, Kapoor TM. 2017. Chemical structure-guided design of dynapyrazoles, cell-permeable dynein inhibitors with a unique mode of action. Elife 6:e25174. 10.7554/eLife.25174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Criglar JM, Anish R, Hu L, Crawford SE, Sankaran B, Prasad BVV, Estes MK. 2018. Phosphorylation cascade regulates the formation and maturation of rotaviral replication factories. Proc Natl Acad Sci USA 115:E12015–E12023. 10.1073/pnas.1717944115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boudreaux CE, Kelly DF, McDonald SM. 2015. Electron microscopic analysis of rotavirus assembly-replication intermediates. Virology 477:32–41. 10.1016/j.virol.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Bussiere LD, Choudhury P, Bellaire B, Miller CL. 2017. Characterization of a replicating mammalian orthoreovirus with tetracysteine-tagged muNS for live-cell visualization of viral factories. J Virol 91:e01371-17. 10.1128/JVI.01371-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campbell EA, Reddy V, Gray AG, Wells J, Simpson J, Skinner MA, Hawes PC, Broadbent AJ. 2020. Discrete virus factories form in the cytoplasm of cells coinfected with two replication-competent tagged reporter birnaviruses that subsequently coalesce over time. J Virol 94:e02107-19. 10.1128/JVI.02107-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou Y, Su JM, Samuel CE, Ma D. 2019. Measles virus forms inclusion bodies with properties of liquid organelles. J Virol 93:e00948-19. 10.1128/JVI.00948-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salgado EN, Upadhyayula S, Harrison SC. 2017. Single-particle detection of transcription following rotavirus entry. J Virol 91:e00651-17. 10.1128/JVI.00651-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papa G, Venditti L, Braga L, Schneider E, Giacca M, Petris G, Burrone OR. 2020. CRISPR-Csy4-mediated editing of rotavirus double-stranded RNA genome. Cell Rep 32:108205. 10.1016/j.celrep.2020.108205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Villiers FP, Steele AD, Driessen M. 2003. Central nervous system involvement in neonatal rotavirus infection. Ann Trop Paediatr 23:309–312. 10.1179/027249303225007789. [DOI] [PubMed] [Google Scholar]
- 44.Medici MC, Abelli LA, Guerra P, Dodi I, Dettori G, Chezzi C. 2011. Case report: detection of rotavirus RNA in the cerebrospinal fluid of a child with rotavirus gastroenteritis and meningism. J Med Virol 83:1637–1640. 10.1002/jmv.22156. [DOI] [PubMed] [Google Scholar]
- 45.Iturriza-Gomara M, Auchterlonie IA, Zaw W, Molyneaux P, Desselberger U, Gray J. 2002. Rotavirus gastroenteritis and central nervous system (CNS) infection: characterization of the VP7 and VP4 genes of rotavirus strains isolated from paired fecal and cerebrospinal fluid samples from a child with CNS disease. J Clin Microbiol 40:4797–4799. 10.1128/JCM.40.12.4797-4799.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohka S. 2006. Dissemination pathways for poliovirus cells to animals models. Uirusu 56:51–58. (In Japanese.) 10.2222/jsv.56.51. [DOI] [PubMed] [Google Scholar]
- 47.Bauer A, Nolden T, Schroter J, Romer-Oberdorfer A, Gluska S, Perlson E, Finke S. 2014. Anterograde glycoprotein-dependent transport of newly generated rabies virus in dorsal root ganglion neurons. J Virol 88:14172–14183. 10.1128/JVI.02254-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klingen Y, Conzelmann KK, Finke S. 2008. Double-labeled rabies virus: live tracking of enveloped virus transport. J Virol 82:237–245. 10.1128/JVI.01342-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tate JE, Burton AH, Boschi-Pinto C, Steele AD, Duque J, Parashar UD, WHO-Coordinated Global Rotavirus Surveillance Network. 2012. 2008 estimate of worldwide rotavirus-associated mortality in children younger than 5 years before the introduction of universal rotavirus vaccination programmes: a systematic review and meta-analysis. Lancet Infect Dis 12:136–141. 10.1016/S1473-3099(11)70253-5. [DOI] [PubMed] [Google Scholar]
- 50.Tate JE, Burton AH, Boschi-Pinto C, Parashar UD, World Health Organization-Coordinated Global Rotavirus Surveillance Network. 2016. Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000-2013. Clin Infect Dis 62(Suppl 2):S96–S105. 10.1093/cid/civ1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.La Frazia S, Ciucci A, Arnoldi F, Coira M, Gianferretti P, Angelini M, Belardo G, Burrone OR, Rossignol JF, Santoro MG. 2013. Thiazolides, a new class of antiviral agents effective against rotavirus infection, target viral morphogenesis, inhibiting viroplasm formation. J Virol 87:11096–11106. 10.1128/JVI.01213-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scott DE, Bayly AR, Abell C, Skidmore J. 2016. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov 15:533–550. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- 53.Schapira M, Tyers M, Torrent M, Arrowsmith CH. 2017. WD40 repeat domain proteins: a novel target class? Nat Rev Drug Discov 16:773–786. 10.1038/nrd.2017.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banerjee I, Miyake Y, Nobs SP, Schneider C, Horvath P, Kopf M, Matthias P, Helenius A, Yamauchi Y. 2014. Influenza A virus uses the aggresome processing machinery for host cell entry. Science 346:473–477. 10.1126/science.1257037. [DOI] [PubMed] [Google Scholar]
- 55.Boulant S, Douglas MW, Moody L, Budkowska A, Targett-Adams P, McLauchlan J. 2008. Hepatitis C virus core protein induces lipid droplet redistribution in a microtubule- and dynein-dependent manner. Traffic 9:1268–1282. 10.1111/j.1600-0854.2008.00767.x. [DOI] [PubMed] [Google Scholar]
- 56.Brault JB, Kudelko M, Vidalain PO, Tangy F, Despres P, Pardigon N. 2011. The interaction of flavivirus M protein with light chain Tctex-1 of human dynein plays a role in late stages of virus replication. Virology 417:369–378. 10.1016/j.virol.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 57.Shi H, Chen J, Li H, Sun D, Wang C, Feng L. 2012. Molecular characterization of a rare G9P[23] porcine rotavirus isolate from China. Arch Virol 157:1897–1903. 10.1007/s00705-012-1363-2. [DOI] [PubMed] [Google Scholar]
- 58.Spearman C. 1908. The method of ‘right and wrong cases’ (‘constant stimuli’) without Gauss’s formulae. Br J Psychol 2:227–242.
- 59.Kärber G. 1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Archiv Exper Pathol Pharmakol 162:480–483. 10.1007/BF01863914. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie Legend<br>. Download JVI.01246-21-s0001.pdf, PDF file, 0.08 MB (87.4KB, pdf)
Movie S1. Download JVI.01246-21-s0002.avi, AVI file, 14.1 MB (14.1MB, avi)