

ABSTRACT
Redondoviridae is a newly established family of circular Rep-encoding single-stranded (CRESS) DNA viruses found in the human ororespiratory tract. Redondoviruses were previously found in ∼15% of respiratory specimens from U.S. urban subjects; levels were elevated in individuals with periodontitis or critical illness. Here, we report higher redondovirus prevalence in saliva samples: four rural African populations showed 61 to 82% prevalence, and an urban U.S. population showed 32% prevalence. Longitudinal, limiting-dilution single-genome sequencing revealed diverse strains of both redondovirus species (Brisavirus and Vientovirus) in single individuals, persistence over time, and evidence of intergenomic recombination. Computational analysis of viral genomes identified a recombination hot spot associated with a conserved potential DNA stem-loop structure. To assess the possible role of this site in recombination, we carried out in vitro studies which showed that this potential stem-loop was cleaved by the virus-encoded Rep protein. In addition, in reconstructed reactions, a Rep-DNA covalent intermediate was shown to mediate DNA strand transfer at this site. Thus, redondoviruses are highly prevalent in humans, found in individuals on multiple continents, heterogeneous even within individuals and encode a Rep protein implicated in facilitating recombination.
IMPORTANCE Redondoviridae is a recently established family of DNA viruses predominantly found in the human respiratory tract and associated with multiple clinical conditions. In this study, we found high redondovirus prevalence in saliva from urban North American individuals and nonindustrialized African populations in Botswana, Cameroon, Ethiopia, and Tanzania. Individuals on both continents harbored both known redondovirus species. Global prevalence of both species suggests that redondoviruses have long been associated with humans but have remained undetected until recently due to their divergent genomes. By sequencing single redondovirus genomes in longitudinally sampled humans, we found that redondoviruses persisted over time within subjects and likely evolve by recombination. The Rep protein encoded by redondoviruses catalyzes multiple reactions in vitro, consistent with a role in mediating DNA replication and recombination. In summary, we identify high redondovirus prevalence in humans across multiple continents, longitudinal heterogeneity and persistence, and potential mechanisms of redondovirus evolution by recombination.
KEYWORDS: Redondoviridae, brisavirus, evolution, genetic recombination, redondovirus, rep protein, vientovirus, CRESS viruses
INTRODUCTION
Redondoviridae is a recently identified family of small DNA viruses predominantly found in the human ororespiratory tract (1–3). Redondoviruses were the second most prevalent DNA virus family identified through metagenomic sequence analysis in human airway samples (2). Elevated levels of redondovirus were observed in periodontitis, critical illness, and severe respiratory disease in humans (2, 4). The family Redondoviridae falls in the newly established phylum Cressdnaviricota, which contains CRESS (circular, Rep-encoding single-stranded) DNA viruses (5–7), and is the sole family in the new order Recrevirales (3, 8).
Redondoviruses have not yet been grown in pure culture, so direct evidence for their host is lacking. However, redondoviruses do not encode prokaryotic-type ribosome binding sites (2), and high-quality matches to redondovirus sequences have not been detected in databases of CRISPR spacer sequences. Based on this evidence, we infer that redondoviruses replicate either in human cells or in a human-associated eukaryotic organism.
The redondovirus genome is a covalently closed DNA circle of 3 kb encoding putative capsid (Cp) and replication-associated (Rep) proteins. Redondovirus Rep proteins contain helicase and nuclease domains that likely mediate viral DNA replication (10–12). Currently known redondoviruses can be classified into two species, Brisavirus and Vientovirus, whose members are defined by 50% or greater identity in the Rep amino acid sequence. All redondoviruses also encode a third open reading frame (ORF) of unknown function, ORF3, which overlaps the Cp ORF. Redondoviral genomes also contain a conserved stem-loop secondary structure proximal to the start codon of the Rep ORF (2), which, by analogy to other CRESS viruses, may be the initiation site of DNA replication (10, 11, 13, 14). Although this sequence differs between redondovirus species, the structure is conserved in all redondoviruses and could be a target for Rep cleavage to initiate DNA synthesis.
Redondovirus prevalence has been reported to be in the range of 2 to 15% in different populations, locales, and ororespiratory sample types as assayed by qPCR (2, 4, 15). These studies focused on industrialized populations from the United States and Europe and tested a limited number of sample types, so the global prevalence of redondovirus remains unclear. Redondovirus has been shown to persist in humans at least over a period of weeks (2), but nothing is known about their long-term persistence, genomic diversification, and evolution in humans over time.
Here, we investigated redondoviruses in U.S. urban and African rural populations and found much higher prevalence, 32 to 82%, when larger volumes of saliva were used as the analyte. We characterized redondovirus sequence diversity in different locales and within subjects over time using whole genomic sequencing and so began to define the global extent of redondovirus diversity. We identified signatures of redondoviral evolution involving recombination at a prominent hot spot, which overlaps the conserved DNA stem-loop structure. We demonstrated in vitro that purified Rep is capable of DNA cleavage, covalent intermediate formation, and DNA strand transfer localized at the stem-loop hot spot.
RESULTS
Redondovirus species are globally distributed and highly prevalent in four African populations.
Previous qPCR estimates of redondovirus prevalence in human ororespiratory samples range from 2% to 15% (2, 4, 15). However, these samples represented populations in industrialized countries (United States and Europe) and were collected from various sites along the ororespiratory tract, complicating assessment of global prevalence. Here, we performed qPCR screens for redondovirus DNA on saliva samples from subjects presenting as healthy and living in rural regions of four African countries (Botswana, Cameroon, Ethiopia, and Tanzania) and compared the results with those from healthy urban U.S. subjects residing in the Philadelphia area. Saliva was chosen as an analyte because of convenience of collection and because initial studies revealed relatively high viral levels.
We found that 32% of saliva samples from U.S. individuals (n = 50) were positive for redondovirus DNA. In contrast, the prevalence rates in African samples were 70% in Botswana (n = 96), 82% in Cameroon (n = 93), 69% in Ethiopia (n = 87), and 61% in Tanzania (n = 92) (Fig. 1A and Table 1). These samples were from hunter-gatherer (n = 179) and agriculturist/agropastoralist (n = 189) groups; no difference in prevalence was found between these two groups (70% and 71%, respectively). Overall, 70% of saliva samples from the four African populations were positive for redondovirus DNA, which was significantly greater than in the U.S. cohort (P < 0.00001 by chi-squared test; U.S. versus all African samples).
FIG 1.

Redondoviruses are globally distributed and highly prevalent in multiple African countries. (A) DNA extracted from saliva samples collected from subjects in different countries was subjected to redondovirus-selective whole-genome amplification, followed by qPCR. The proportion of samples positive by qPCR for each country is shown on the y axis. (B) Phylogenetic tree of single-genome-derived redondovirus Rep protein amino acid sequences along with sequences from databases. Rep proteins from SGS genomes are marked as circles, with colors representing the country of origin (purple for Cameroon and yellow for Ethiopia), while database redondovirus Reps are shown as black squares. Colored circles at each node show branch likelihood as determined by the approximate-likelihood ratio test. Clades representing the two redondovirus species are marked by colors: Vientovirus in cyan and Brisavirus in pink. (C) Global distribution of redondovirus species. The fraction of sequenced genomes in each of the redondovirus species in a particular country is shown in the pie charts. The number in parentheses after the country name represents the total number of redondovirus genomes sequenced from subjects in that country.
TABLE 1.
Global redondovirus prevalence determined by qPCR
| Reference(s) | Country | Site | No. (%) of samples |
Condition | |
|---|---|---|---|---|---|
| Total | qPCR positive | ||||
| 2, 20 | USA | Lung, oropharynx | 69 | 6 (9) | Critical illness |
| 2, 20 | USA | Oropharynx | 60 | 9 (15) | Healthy |
| 15 | Spain | Respiratory, multiple | 100 | 2 (2) | Ill (hospital patients) |
| 4 | Italy | Respiratory, multiple | 209 | 22 (11) | Ill (hospital patients) |
| 4 | Italy | Stool | 105 | 1 (0.9) | Ill (hospital patients) |
| This study | USA | Saliva | 50 | 16 (32) | Healthy |
| This study | Botswana | Saliva | 96 | 67 (70) | Healthy |
| This study | Ethiopia | Saliva | 87 | 60 (69) | Healthy |
| This study | Tanzania | Saliva | 92 | 56 (61) | Healthy |
| This study | Cameroon | Saliva | 93 | 76 (82) | Healthy |
To begin to define redondovirus genome sequence diversity in African subjects, we performed limiting-dilution single-genome sequencing (SGS) on a subset of subjects. Limiting-dilution analysis prevents recombination during PCRs, which can yield artefactual composite sequences (16, 17). We sequenced 18 complete redondovirus genomes from six individuals from Cameroon and Ethiopia (Fig. 1B; other samples failed to yield amplification products suitable for sequencing). These genomes did not cluster separately from previously sequenced genomes. Rep protein sequences are used to differentiate the two species, and the inferred Rep protein sequences met the criteria for membership in the species Vientovirus and Brisavirus (3).
The prevalence of redondovirus is substantially higher in the African individuals than the U.S. individuals surveyed, but the redondovirus genomes sequenced do not represent undescribed genera or species. Together with redondovirus genomes sequenced from additional countries (1, 2) (Fig. 1C and Table 1), these data indicate that the two redondovirus species are distributed in humans over multiple continents.
Multiple redondovirus genotypes can simultaneously colonize human individuals, and some persist over time.
We previously reported that hospitalized, acutely ill individuals can be persistently positive for redondovirus DNA over several weeks at multiple ororespiratory sites (2), but persistence and sequence diversity over longer times remain unstudied. Humans are known to be stably colonized by another family of small, circular DNA viruses, the anelloviruses (18–21). We therefore performed SGS on longitudinal samples taken from two individuals from our previously described cohort of critically ill patients (2), including a follow-up specimen obtained 2 years after initial sampling.
Subject 1 was positive for redondoviral DNA in endotracheal aspirate and oropharyngeal swab samples at two time points separated by 2 years; subject 2 was positive for redondoviral DNA in multiple endotracheal aspirate and oropharyngeal swab samples over 20 days. Using SGS, we sequenced 43 genomes in total, 31 from subject 1 and 12 from subject 2. Sequencing single redondovirus genomes showed marked heterogeneity of virus populations within each of the two subjects (Fig. 2A to C). We confirmed that our limiting dilution captured authentic single genomes by comparing the results of SGS, which yielded sequences aligning to only one redondovirus Rep sequence for each sample, to bulk sequencing without limiting dilution, where sequences could be found that aligned to multiple Rep types (data not shown).
FIG 2.

Longitudinal analysis of within-subject redondovirus diversity. (A) Highlighter plot of redondovirus genome nucleotide sequences from each subject analyzed after limiting-dilution SGS. The x axis represents the genomic position in a multiple-sequence alignment. Each gray horizontal line represents a sequenced redondoviral genome. Sequences are grouped by subject and then by time point, shown to the left of the plot. Nucleotide positions in each isolate that do not match the global consensus sequence are indicated by a red tick. Approximate positions of redondovirus ORFs are displayed below the plot. N, amino-terminal coding region; C, carboxyl-terminal coding region. The shapes to the right of the plot indicate the sample type: endotracheal aspirate (ET) or oropharyngeal (OP). The colored boxes indicate, from left to right, the redondovirus species to which each genome belongs, the genotype (cluster at 99% genomic nucleotide sequence identity) to which each sequence belongs, and the cluster (at 99% amino acid sequence identity) to which the Rep and Cp proteins belong. (B) Phylogenetic tree of Rep amino acid sequences from SGS redondovirus genomes sequenced from longitudinally sampled subjects, along with database sequences. Sequences from subject 1 are shown in purple, sequences from subject 3 are in green, and database sequences are in black. Colored circles at each node indicate branch likelihood as indicated by the approximate likelihood ratio test. Colored boxes delineate the two redondovirus species, Vientovirus (cyan) and Brisavirus (pink). (C) Schematic showing pairings between unique redondovirus Rep and Cp clusters. Each cluster is indicated by a different color. The table to the right of the schematic indicates the number of genomes sequenced at each time point belonging to each Cp-Rep pair.
We then analyzed redondovirus diversity over time within subjects. To query persistence of individual genotypes, we defined a cutoff between genotypes by performing pairwise alignments between all genomes identified from each subject and between all complete redondovirus genomes from the NCBI nucleotide database. Multiple groups of genomes within each subject exhibited extremely high (>99%) nucleotide sequence identity. In contrast, no two database genomes were as much as 99% identical. Thus, we used a cutoff of 99% identity to group redondovirus genome sequences within subjects into genotypes.
Subjects were positive for multiple genotypes of redondovirus both at single time points and over time (Fig. 2A to C). In subject 1, the same redondovirus genotype was present at time points separated by 2 years. Additionally, both redondovirus species could be simultaneously detected in both subjects. These data demonstrate that humans can be colonized by multiple redondovirus species and genotypes of Brisavirus and Vientovirus and that redondovirus genotypes can persist over time.
Redondovirus sequences are not found integrated in human cancers.
Some Rep-encoding viruses and transposable elements can become integrated into host genomic DNA (22–24), potentially contributing to transformation and cancer. We investigated whether redondovirus integration might contribute to human cancer by surveying whole-genome shotgun sequence data from The Cancer Genome Atlas (TCGA), an archive of genomic sequencing data from healthy and cancerous human cells and tissues (25). This strategy has been employed to identify novel DNA viruses integrated into the human genome (26). We queried 10,955 samples from 33 studies corresponding to a variety of cancer types and aligned the reads against all sequenced redondovirus genomes. As a positive control, we also aligned reads to human papillomavirus type 16 (HPV-16), which integrates and causes cancer in multiple body sites (27, 28). We found hits to HPV-16 in head-and-neck and cervical sites, but we did not identify reads aligning to redondovirus genomes in any normal or cancerous sample type tested (data not shown). These data do not support the idea that redondovirus integration contributes to human cancer.
Redondovirus recombination as a contributor to genomic diversity.
Within subjects, highly similar Cp proteins were often paired with divergent Rep proteins (Fig. 2A and C). To investigate this further, we clustered Rep and Cp amino acid sequences from SGS genomes at 99% amino acid sequence identity and plotted the pairings between Cp and Rep proteins, including SGS from all time points within each subject (Fig. 2C). This showed that the same Cp cluster was often paired with divergent Rep proteins. In subject 1, two distinct Reps were paired with the same Cp, while in subject 2, four distinct Reps were paired with the same Cp protein (Fig. 2C). Taken together with previous observations of discordance between redondovirus Cp and Rep protein phylogenies (2, 3), these data suggest that recombination may be a major mechanism contributing to redondoviral diversification. Recombination is known to be common in some families of CRESS DNA viruses (29–32).
To investigate recombination within Redondoviridae, we performed a recombination breakpoint analysis of redondovirus genome sequences using RDP4 (33) (Fig. 3A to C). We identified significant recombination hot spots in intergenic regions between Cp and Rep (Fig. 3A and C). Additionally, testing for imbalanced coinheritance of nucleotide pairs (34) showed that the Cp coding region was frequently separated from the Rep coding region by recombination breakpoints (Fig. 3C). Both of these observations are consistent with the pattern of divergent Cp-Rep pairings observed within individuals. A lack of phylogenetic link between Cp and Rep was found whether or not genomes were sequenced using single genome sequencing (SGS), indicating that recombination during the sample amplification and sequencing procedures does not explain the observed phylogenetic discordance between Cp and Rep (Fig. 3B). The lack of recombination breakpoints within Rep also parallels observations from some other CRESS virus families (32).
FIG 3.

Recombination favors redondovirus intergenic regions and occurs most frequently near the predicted origin of replication. (A) Plot showing the number of inferred recombination breakpoints in a 200-nucleotide (nt) sliding window across the redondovirus genome. The black ticks above the plot indicate observed recombination breakpoints. The positions of the redondovirus open reading frames and the stem-loop sequence are shown above the plot. N, amino-terminal coding region; C, carboxyl-terminal coding region. The black line indicates the count of breakpoints per 200-nt sliding window. The dark and light gray shaded areas represent local 95% and 99% confidence thresholds, respectively, for recombination hot and cold spots. Recombination hot spots (areas with more recombinants than expected by chance) are shaded in red; recombination cold spots (areas with fewer recombination breakpoints than expected by chance) are shown in blue. (B) Tanglegram showing relationships between Rep and Cp phylogenies. A Rep amino acid phylogeny is shown on the left; Cp amino acid phylogeny is shown to the right. Rep and Cp proteins originating from the same virus are connected by colored lines. The color code indicates whether each viral sequence was from a database (blue) or our SGS (orange). (C) Cp and Rep coding sequences are frequently separated by recombination events. Recombination region count matrix showing the frequency (top) and P value (bottom) of a recombination event separating any two nucleotide positions. In the top matrix, the color intensity represents the number of times each pair of nucleotides was separated. The bottom matrix shows nucleotide pairs that are most (red) and least (blue) frequently separated. The positions of redondovirus ORFs are displayed between the matrices.
The predicted recombination breakpoints are highly concentrated near a predicted stem-loop secondary structure (Fig. 3A) that is conserved in redondovirus genomes (2). Because stem-loop structures are often cleaved by Rep in other CRESS DNA viruses (10, 11) and free DNA ends can promote recombination (35–37), we hypothesized that redondovirus Rep might have a role in recombination at the stem-loop hot spot identified in the genomic sequence data and so assessed potential mechanisms with reactions using purified components in vitro.
Rep catalyzes DNA breaking and joining reactions potentially involved in recombination.
Reps carry out an ordered series of reactions to facilitate DNA replication (11, 12, 23, 38–40). Reps first bind to specific DNA stem-loops in the virus or element genome and then nick the substrate to form a covalent Rep-DNA 5′ linkage, which provides a free 3′ end for DNA polymerization. Reps also commonly contain an ATP-dependent helicase that facilitates polymerization and possibly viral DNA packaging (10, 41, 42). In some CRESS viruses, recombination hot spots have been located near the Rep cleavage site (30).
To investigate whether redondovirus Rep plays a role in redondovirus evolution by recombination, we first expressed and purified a representative redondovirus Rep protein from Vientovirus FB in Escherichia coli and investigated its activities in vitro with model substrates (Fig. 4). We reacted the purified Rep protein with a 5′ fluorescently labeled oligonucleotide stem-loop matching that of Vientovirus FB (Fig. 4A and B). Vientovirus FB Rep catalyzed strand-specific, magnesium-dependent cleavage of the oligonucleotide stem-loop (Fig. 4C). Coelectrophoresis of cleavage products with synthetic standards mapped the cleavage site to a single location in the conserved loop sequence (Fig. 4D). No cleavage activity was detected in assays containing an oligonucleotide matching the complementary strand of the stem-loop (Fig. 4C).
FIG 4.
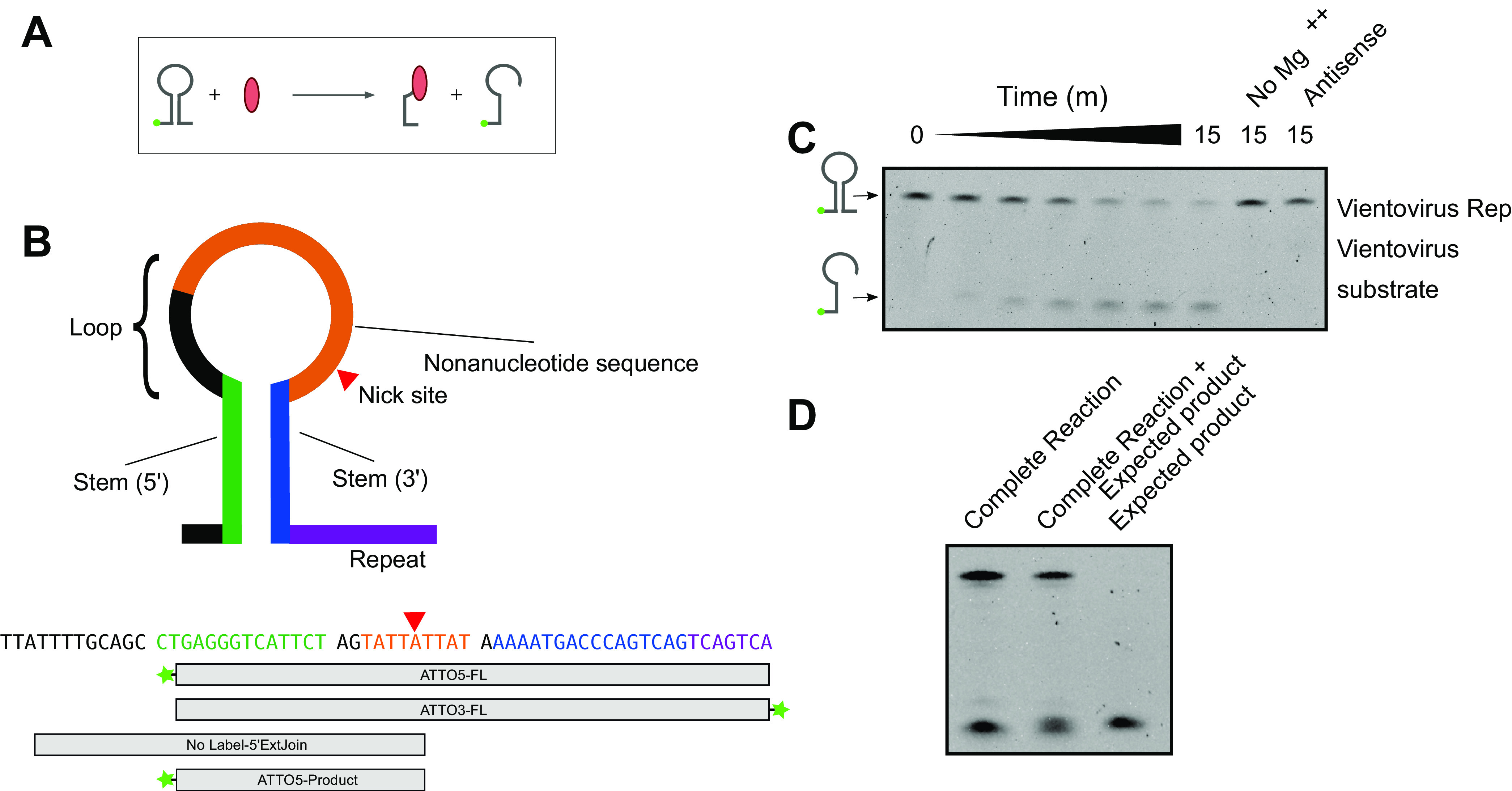
Redondovirus Rep cleaves at the DNA stem-loop recombination hot spot. (A) Reaction schematic. Rep is shown in red. Rep nicks a DNA stem-loop, which is shown fluorescently labeled on the DNA 5′ end (green ball). The cleavage reaction produces a shorter labeled oligonucleotide and a covalent Rep-DNA intermediate. (B) (Top) Diagram of the DNA stem-loop sequences and reaction substrates. The Rep nick site is indicated by a red triangle. (Bottom) Sequence of the Vientovirus FB stem-loop with boxes showing the regions included in each oligonucleotide. Fluorescent ATTO488 labels (Integrated DNA Technologies) are indicated by green stars. All sequences are shown from 5′ to 3′ (left to right). (C) Results of electrophoresis on a TBE-urea acrylamide gel showing fluorescent products after reacting Vientovirus FB Rep with the labeled stem-loop from Vientovirus FB. The expected products are diagrammed to the left of the gel. “No Mg++” indicates a complete reaction with magnesium omitted; “antisense” indicates a substrate that is antisense in sequence compared to the expected correct orientation of the stem-loop. (D) Specificity of cleavage by the Vientovirus Rep protein. The product of the complete reaction (left) was mixed with the hypothetical product (middle) to demonstrate coelectrophoresis. The hypothetical product alone is shown in the right lane.
To assess the specificity of the cleavage reaction, Vientovirus FB Rep was reacted with a labeled Brisavirus stem-loop substrate (Fig. 4B). Little cleavage activity was detected (quantified in Fig. 4C), indicating species-specific cleavage activity.
To probe specificity further, we purified a Brisavirus Rep and compared cleavage of the Brisavirus and Vientovirus substrates (Fig. 5A and B). Vientovirus Rep showed robust cleavage of the Vientovirus substrate, but only slight cleavage of the Brisavirus substrate (quantified in Fig. 5A). Brisavirus Rep showed robust cleavage activity on Brisavirus stem-loop substrate but undetectable activity on the Vientovirus substrate (Fig. 5B). Thus, redondovirus Rep proteins catalyze sequence-specific DNA nicking more efficiently on stem-loop oligonucleotides derived from their cognate species.
FIG 5.

Specificity of cleavage by Vientovirus and Brisavirus Rep proteins. Quantification of cleavage products produced in the presence of Vientovirus Rep (A) and Brisavirus Rep (B). Results with the two substrates are color coded (Vientovirus substrate in blue, Brisavirus substrate in red). Time is on the x axis, and the amount of cleavage product is on the y axis.
Several Rep proteins have been reported to form a covalent intermediate with viral DNA, which then breaks down by a transesterification reaction with a 3′ DNA hydroxyl group late in replication to form circular molecules (13, 14, 43). To test for covalent intermediate formation, we reacted Vientovirus FB Rep with a 3′-end-labeled fluorescent oligonucleotide and analyzed the products by SDS-PAGE (Fig. 6A). Reactions yielded a fluorescent protein band consistent with covalent complex formation. The putative covalent complex migrated at a molecular weight ∼10 kDa greater that the size expected for Rep alone (50 kDa versus 40 kDa) (Fig. 6A).
FIG 6.

Redondovirus Rep forms a covalent DNA-protein intermediate and catalyzes re-joining to de novo-introduced DNA strands. (A) (Top) Schematic of Rep-DNA covalent intermediate formation. Rep nicks a 3′-end-labeled oligonucleotide, forming a fluorescently labeled protein-DNA covalent complex. (Bottom) Reaction products electrophoresed on an SDS-PAGE gel and visualized by fluorescence detection (left); purified Rep visualized by Coomassie staining (right). Locations of Rep and Rep-DNA are indicated on the far left. (B) (Top) Reaction schematic. Rep is mixed with 3′-end-labeled oligonucleotide and, after 2 min, chased with a 100× molar excess of longer, unlabeled substrate. Rep joining activity in trans results in the production of a longer, fluorescently labeled oligonucleotide species. (Bottom) The products of this reaction were electrophoresed on a TBE-urea acrylamide gel and then visualized by fluorescence detection.
This led us to hypothesize that Rep could join a covalently bound genome in trans to another genome within the same cell, potentially accounting in part for the observed high level of recombination at the stem-loop target. To test this hypothesis, we queried whether the Rep covalent complex was capable of joining the linked DNA to a newly introduced DNA strand in vitro. We incubated Vientovirus FB Rep with a 3′-end fluorescently labeled oligonucleotide to form a covalent intermediate, then chased with an excess of a longer, unlabeled DNA strand (Fig. 6B). Electrophoresis of reaction products showed accumulation of labeled DNA of a larger size than the input stem-loop. Thus, we infer that Rep re-joined the covalently bound stem-loop DNA to the newly introduced substrate, consistent with a role for Rep in intermolecular recombination at this site.
These data support a potential model for redondoviral DNA replication and intragenomic recombination. Host DNA polymerase likely synthesizes the complementary strand of the redondoviral ssDNA genome. Nicking of this dsDNA replication intermediate by Rep forms a covalent protein-DNA intermediate and exposes a 3′-OH, allowing continued DNA extension by host DNA polymerase. After the viral genome has been fully replicated, Rep performs a joining reaction. Joining in cis results in the release of a circular, single-stranded-DNA (ssDNA) viral genome, which can then be packaged or undergo further rounds of replication. Joining in trans to a different redondovirus genome could link the edge of the Rep-encoding DNA to new sequences, accounting for the observed recombination hot spot. In this model, another DNA break would then be required to complete strand transfer.
DISCUSSION
In this study, we investigated redondovirus diversity and dynamics at scales from global to molecular. We demonstrated that redondoviruses are globally distributed and highly prevalent in oral samples from four nonindustrialized African populations. Use of saliva as an analyte revealed higher prevalence than was seen in previous studies, averaging 70% in rural African populations and 32% in a U.S. urban population. Using limiting-dilution single-genome sequencing, we found that humans can be positive for multiple species and genotypes of redondovirus at a single time point. We also identified redondovirus genotypes that persisted over time, including up to 2 years, and identified signatures of recombination. Recombination break points were concentrated in or near intergenic regions, providing an explanation for the apparent phylogenetic independence of redondovirus Cp and Rep proteins. The highest frequency of predicted recombination was near a DNA stem-loop, so we investigated possible Rep cleavage and strand transfer at this site. We found that purified redondovirus Rep catalyzed DNA nicking, covalent Rep-DNA intermediate formation and joining to a de novo introduced DNA strand. These data support a role for Rep in redondovirus evolution by facilitating recombination through stem-loop cleavage and DNA strand transfer.
Redondoviruses have been identified by us and others in samples from four continents: North America, Asia, Europe, and Africa (1, 2, 4, 15). The redondovirus prevalence of 70% found in saliva samples from the four rural African populations was higher than in our urban U.S. saliva samples (32%). The reason for this difference is unclear. Previously redondoviruses were reported to be elevated in samples from individuals with periodontitis (2), but we lack data on oral health for the subjects sampled here, precluding further investigation. The African individuals sampled come from hunter/gatherer and small-scale agriculturist/agropastoralist subsistence groups and live in rural areas; the higher prevalence of redondoviruses may be linked to factors such as diet, lifestyle, or other conditions. The finding of both redondovirus species in samples from multiple continents indicates that both have likely colonized humans long term.
In our longitudinally sampled individuals, we detected multiple species and genotypes of redondovirus at a single time point, reminiscent of human colonization by diverse anellovirus swarms (18–21). Furthermore, single redondovirus genotypes were detected at multiple time points, including over 2 years, the longest interval investigated. Although these were not healthy subjects, these data demonstrate that multiple redondovirus lineages may persist in some individuals. Further longitudinal study of healthy volunteers is necessary to determine whether redondoviruses establish persistent infections in healthy humans.
Anelloviruses, the most common known human circular ssDNA viruses, share some attributes with redondoviruses. Anelloviruses persistently colonize humans, with some estimates of prevalence above 90% in healthy adults (44). Anelloviruses are found in blood and multiple human tissues (20, 45–48). Redondoviruses also persistently colonize humans but are predominantly present in the ororespiratory tract and have not been found in blood (2, 4). Redondovirus prevalence based on quantitative PCR (qPCR) detection varies from 2 to 82% in different locales and sample types. Multiple strains of anellovirus can be present within individuals at a single time point, as shown here for redondoviruses (19, 20, 49). However, a unique feature of redondovirus is the apparent modular nature of genomes, with apparent frequent swapping of Cp and Rep regions, suggesting that recombination plays a major role in diversification and evolution.
Our biochemical data provide evidence consistent with a role for redondovirus Rep in intergenomic recombination. We demonstrate that Rep nicks the conserved stem-loop, which is likely the viral replication origin, at the recombination hot spot. After nicking, Rep forms a covalent protein-DNA intermediate and is capable of re-joining the covalently bound DNA fragment to a newly introduced DNA 3′ end. These functions are required activities for initiation and termination of DNA replication (12, 38, 50) and are also consistent with a role for Rep in recombination. Further DNA breaking-and-joining reactions would then be required to complete the recombination reaction.
In summary, we report that redondovirus prevalence in saliva is approximately 70% in four African countries, significantly higher than in a U.S. cohort (32%) and much higher than in previous reports for other sample types in the United States and two European countries (2 to 15%). We found that both species of redondovirus are present globally, that diverse redondovirus genotypes colonize humans, and that genotypes can persist over at least 2 years. Comparing viral genome sequences sampled globally, we found that recombination in intergenic regions, and especially at a hot spot near a conserved stem-loop structure, commonly contributes to redondovirus genomic diversity. Using in vitro assays with purified Rep protein, we demonstrate that redondovirus Rep performs nicking and joining reactions consistent with a role for Rep in mediating replication initiation and recombination at the observed hot spot. These data thus specify redondoviruses as widespread human colonists that encode an enzyme which drives their diversification.
MATERIALS AND METHODS
Ethics statement.
Ororespiratory samples from U.S. subjects were collected after written informed consent was obtained under protocols approved by the University of Pennsylvania Institutional Review Board (protocols 842613 and 823392).
For all African study participants, written, informed consent was obtained and research/ethics approvals were obtained from the following institutions prior to the start of sample collection: Institutional Review Board of the University of Pennsylvania (protocol 807981), the Cameroonian National Ethics Committee, the Cameroonian Ministry of Public Health, the Tanzanian Commission for Science and Technology and National Institute for Medical Research in Dar es Salaam, the Ministry of Health in the Republic of Botswana, the Federal Democratic Republic of Ethiopia Ministry of Science, and the Technology National Health Research Ethics Review Committee of Ethiopia. All samples were coded with an alphanumeric identifier to protect participant confidentiality.
A compilation of new genome sequences determined is in Table 2.
TABLE 2.
Genomes sequenced in this study
| GenBank no. | Country | Subject ID | Internal ID |
|---|---|---|---|
| MZ405022 | Ethiopia | ET203 | A203-9_polished |
| MZ405023 | Ethiopia | ET239 | A239-2_polished |
| MZ405028 | Ethiopia | ET239 | A239-11_polished |
| MZ405029 | Ethiopia | ET239 | A239-12_polished |
| MZ405030 | Ethiopia | ET724 | A724-2_polished |
| MZ405031 | Ethiopia | ET724 | A724-6_polished |
| MZ405032 | Ethiopia | ET724 | A724-7_polished |
| MZ405033 | Ethiopia | ET724 | A724-8_polished |
| MZ405034 | Ethiopia | ET738 | A738-6_polished |
| MZ405035 | Ethiopia | ET738 | A738-12_polished |
| MZ405024 | Ethiopia | ET895 | D895-8_polished |
| MZ405025 | Ethiopia | ET895 | D895-9_polished |
| MZ405026 | Ethiopia | ET895 | D895-10_polished |
| MZ405027 | Ethiopia | ET895 | D895-11_polished |
| MZ405018 | Cameroon | CM207 | E2_polished |
| MZ405019 | Cameroon | CM239 | E6_polished |
| MZ405020 | Cameroon | CM239 | E7_polished |
| MZ405021 | Cameroon | CM239 | E8_polished |
| MZ405067 | USA | CORE0067 | p67_d0_ET_B2 |
| MZ405068 | USA | CORE0067 | p67_d0_ET_C7 |
| MZ405069 | USA | CORE0067 | p67_d0_OP_A5 |
| MZ405070 | USA | CORE0067 | p67_d0_OP_D2 |
| MZ405071 | USA | CORE0067 | p67_d0_OP_D5 |
| MZ405072 | USA | CORE0067 | p67_d0_OP_D6 |
| MZ405073 | USA | CORE0067 | p67_d0_OP_D7 |
| MZ405074 | USA | CORE0067 | p67_d0_OP_D9 |
| MZ405075 | USA | CORE0067 | p67_d8_ET_B10 |
| MZ405076 | USA | CORE0067 | p67_d8_ET_C1 |
| MZ405077 | USA | CORE0067 | p67_d15_ET_A1 |
| MZ405078 | USA | CORE0067 | p67_d20_ET_WGA_Rep1 |
| MZ405079 | USA | CORE0067 | p67_d20_ET_WGA_Rep2 |
| MZ405036 | USA | CORE0048 | p48_v1_ET_w2_c4 |
| MZ405037 | USA | CORE0048 | p48_v1_ET_w1_c2 |
| MZ405038 | USA | CORE0048 | p48_v1_ET_w1_c3 |
| MZ405039 | USA | CORE0048 | p48_v1_ET_w1_c4 |
| MZ405040 | USA | CORE0048 | p48_v1_ET_w1_c5 |
| MZ405041 | USA | CORE0048 | p48_v1_ET_w2_c1 |
| MZ405042 | USA | CORE0048 | p48_v1_ET_w2_c2 |
| MZ405043 | USA | CORE0048 | p48_v1_ET_w2_c3 |
| MZ405044 | USA | CORE0048 | p48_v1_ET_w4_c1 |
| MZ405045 | USA | CORE0048 | p48_v1_ET_w4_c2 |
| MZ405046 | USA | CORE0048 | p48_v1_ET_w4_c3 |
| MZ405047 | USA | CORE0048 | p48_v1_ET_w4_c5 |
| MZ405048 | USA | CORE0048 | p48_v1_ET_w5_c1 |
| MZ405049 | USA | CORE0048 | p48_v1_ET_w5_c2 |
| MZ405050 | USA | CORE0048 | p48_v1_ET_w5_c4 |
| MZ405051 | USA | CORE0048 | p48_v1_ET_w5_c5 |
| MZ405052 | USA | CORE0048 | p48_v2_ET_w6_c1 |
| MZ405053 | USA | CORE0048 | p48_v2_ET_w6_c3 |
| MZ405054 | USA | CORE0048 | p48_v2_ET_w6_c4 |
| MZ405055 | USA | CORE0048 | p48_v2_ET_w6_c5 |
| MZ405056 | USA | CORE0048 | p48_v2_ET_w8_c2 |
| MZ405057 | USA | CORE0048 | p48_v2_ET_w8_c3 |
| MZ405058 | USA | CORE0048 | p48_v2_ET_w8_c4 |
| MZ405059 | USA | CORE0048 | p48_v2_ET_w8_c5 |
| MZ405060 | USA | CORE0048 | p48_v2_ET_w9_c1 |
| MZ405061 | USA | CORE0048 | p48_v2_ET_w9_c2 |
| MZ405062 | USA | CORE0048 | p48_v2_ET_w9_c4 |
| MZ405063 | USA | CORE0048 | p48_v2_ET_w10_c1 |
| MZ405064 | USA | CORE0048 | p48_v2_ET_w10_c4 |
| MZ405065 | USA | CORE0048 | p48_v2_ET_w10_c5 |
| MZ405066 | USA | CORE0048 | p48_v2_ET_w11_c2 |
Sample collection and DNA isolation.
Saliva samples were collected from participants across multiple seasons of fieldwork in four sub-Saharan African countries: Cameroon (collection in 2015), Tanzania (2011/2012), Ethiopia (2010), and Botswana (2012 to 2013). Within each country, participants were sampled from hunting and gathering and agropastoralist subsistence groups. We sampled Baka hunter-gatherers (n = 46) and Tikari agriculturists (n = 47) in Cameroon, Hadza hunter-gatherers (n = 46) and Burunge agriculturists (n = 46) in Tanzania, Chabu hunter-gatherers (n = 39) and Amhara agriculturists (n = 48) from Ethiopia, and Ju'Hoan hunter-gatherers (n = 48) and Tswana agropastoralists (n = 48) in Botswana, for a total of 368 African saliva samples. Saliva samples from healthy Philadelphians (n = 50) were collected in 2020. Negative controls consistently showed a cycle threshold (CT) of >40, so positive samples were called as anything below this value. Median qPCR CT values for positive samples from each country are as follows: Botswana, 30.1; Cameroon, 29.9; Ethiopia, 26.3; Tanzania, 30.3; United States, 21.9.
For Ethiopian populations, 2 ml saliva was collected using the Oragene kit. All other African populations had 2 ml of saliva collected and stored in 2 ml lysis buffer. Following collection, samples were kept at room temperature until extraction.
DNA was extracted using the Qiagen DNeasy blood and tissue kit (Qiagen Ltd., West Sussex, United Kingdom) with a user-developed protocol for saliva (https://www.qiagen.com/us/resources/download.aspx?id=22471a48-832e-488d-8be6-2b308133b88a) with the following modifications: 250 μl saliva was used as the starting input instead of 1 ml in step 1, 250 μl of ethanol was used instead of 200 μl in step 5, and a repeat elution step was done using the initial eluate to maximize DNA yield. DNA purity was determined using a Nanodrop 2000/200C spectrophotometer (Thermo Fisher Scientific, USA) and DNA yield was measured using PicoGreen (Affymetrix) quantification.
DNA oligonucleotides used in this study.
All synthetic DNA oligonucleotides used in this study are in Table 3.
TABLE 3.
Oligonucleotides used in this study
| Identifier | Sequence (5′–3′) | Description |
|---|---|---|
| LJT-011 | CCTTTGGTCTCGAAATCTTCCTATACTGG | Redondovirus whole-genome amplification F, set A (3-bp overlap with LJT-035) |
| LJT-035 | AGGCCTCTCTCCCTTCCATTTGG | Redondovirus whole-genome amplification R, set A (3-bp overlap with LJT-011) |
| LJT-036 | GGTTATCGTTCATTTGATCATGCATTAGTACC | Redondovirus whole-genome amplification F, set B (3-bp overlap with LJT-037) |
| LJT-037 | ACCAAGATGTTTAAGCCCTTTAGTTAATGTTTC | Redondovirus whole-genome amplification R, set B (3-bp overlap with LJT-036) |
| HCRVswga1 | TACGAATATTA | Redondovirus SWGA primer |
| HCRVswga2 | TATCGTAATAT | Redondovirus SWGA primer |
| HCRVswga3 | GTAATAATCTAT | Redondovirus SWGA primer |
| HCRVswga4 | ATTATAATACG | Redondovirus SWGA primer |
| HCRVswga5 | TATTACGATAA | Redondovirus SWGA primer |
| HCRVswga6 | TAATAATACTAG | Redondovirus SWGA primer |
| HCRVswga7 | TAGTATAACTC | Redondovirus SWGA primer |
| HCRVswga8 | TTATCGTAATA | Redondovirus SWGA primer |
| HCRVswga9 | ATATTACGATA | Redondovirus SWGA primer |
| HCRVswga10 | GAGTTATACTA | Redondovirus SWGA primer |
| HCRVswga11 | CAATATTACG | Redondovirus SWGA primer |
| HCRVswga12 | CGTAATATTG | Redondovirus SWGA primer |
| HCRVswga13 | ATTAGTATTATG | Redondovirus SWGA primer |
| HCRVswga14 | ATATTATTGTAG | Redondovirus SWGA primer |
| HCRVswga15 | CTACAATAATAT | Redondovirus SWGA primer |
| HCRVswga16 | CTAGTATTATTA | Redondovirus SWGA primer |
| HCRVswga17 | GTATTATTAGAA | Redondovirus SWGA primer |
| HCRVswga18 | CATAATACTAAT | Redondovirus SWGA primer |
| HCRVswga19 | TAATATTCGTA | Redondovirus SWGA primer |
| HCRVswga20 | GTTATTATATTG | Redondovirus SWGA primer |
| Pan-HCRV-AA-Fwd | GCAGAGTTGTCAGCACATTT | Redondovirus qPCR forward primer |
| Pan-HCRV-AA-Rev | ATACCAGTATAGGAAGATTTCGAG | Redondovirus qPCR reverse primer |
Redondovirus SWGA and qPCR.
Redondovirus selective whole-genome amplification (SWGA) and qPCR were performed as previously described (2, 51). SWGA was carried out using the Phi29 DNA polymerase kit (New England Biolabs [NEB]) and a 100 μM primer pool consisting of 20 primers (Integrated DNA Technologies [IDT]; Table 3) that encompass conserved segments of the redondovirus genome. DNA extracted from samples was subject to SWGA using the following PCR conditions on a Veriti 96-well thermocycler (Thermo Fisher Scientific, USA): 35°C for 5 min, 34°C for 10 min, 33°C for 15 min, 32°C for 20 min, 31°C for 30 min, 30°C for 16 h, and a final extension at 65°C for 15 min.
To detect redondovirus, SWGA-amplified samples were run in duplicate in a real-time qPCR using TaqMan Fast universal PCR (2×) MasterMix (Applied Biosystems) and a combination primer/probe mix (IDT) based on conserved segments of the redondovirus genome (F, 5′-GGATGCCATGAAACTTTGATAC-3′; R, 5′-TCTTCCTCCTTATTTGTATGGC-3′; probe, 5′-CCCATACTTACGCCGGTTACCTGC-3′). Primers and probe had final concentrations of 18 μM and 5 μM, respectively. To quantify positive samples, a standard curve made from serial dilutions of a plasmid containing the cloned genome of Brisavirus AA was run in triplicate on every qPCR. qPCRs were run on a QuantStudio5 (Thermo Fisher) machine using the “fast” mode and 45 cycles.
To be considered positive, samples had to show qPCR amplification in both technical replicates. Nontemplate controls and extraction controls were included in qPCR assays; no negative controls showed amplification.
Limiting-dilution single-genome amplification and sequencing.
Positive sample DNA was diluted from stock DNA concentrate across 96-well plates to achieve 1:5, 1:50, 1:100, 1:200, 1:400, 1:800, 1:1,600, and 1:3,200 dilution technical replicates that were then subjected to SWGA and qPCR as described above. Positive wells from rows with four or fewer positive wells were subjected to redondovirus whole-genome PCR using primers landing back-to-back on the redondoviral genome. At that level of positivity, each well had a >90% chance to contain only one redondoviral genome by the binomial distribution (17). To recover complete redondovirus genomes, two whole-genome PCRs were performed with nonoverlapping primer sets (set A F, 5′-CCTTTGGTCTCGAAATCTTCCTATACTGG -3′; set A R, 5′-AGGCCTCTCTCCCTTCCATTTGG -3′; set B F, 5′-GGTTATCGTTCATTTGATCATGCATTAGTACC-3′; set B R, 5-ACCAAGATGTTTAAGCCCTTTAGTTAATGTTTC-3′) using the Phusion enzyme kit (New England Biolabs) and the following PCR settings: 98°C for 30 s, then 35 cycles of 98°C for 10 s, 55°C for 15 s, and 72°C for 1 min 30 s, followed by a final extension of 10m at 72°C. The ∼3-kb PCR products were visualized on a 1% agarose gel and then excised and purified from gels using a Monarch gel elution kit (New England Biolabs) and the manufacturer’s protocols with one modification of a 15-μl water final elution. After gel extraction, libraries were prepared from PCR products using the Nextera XT library preparation kit (FC-131-1002; Illumina). Libraries were sequenced using the Illumina MiSeq platform (Illumina). We were able to amplify genomes from Cameroon and Ethiopia (Table 2) but unable to amplify whole genomes from Botswana and Tanzania, possibly due to issues of sample integrity after long-term storage.
Read processing and genome assembly.
Genomes (Table 2) were built from FASTQ-formatted reads processed using Sunbeam version 2.1 (52), a Snakemake-based pipeline (53), as follows.
(i) Quality control and host decontamination.
Where applicable, reads were downloaded using grabseqs and sra-tools (54, 55). Quality control was performed as previously described (2, 52). Adapter trimming was performed by Trimmomatic (56); quality control was performed using FastQC (57). Low-complexity reads were filtered using complexity (52). Host reads were mapped to human and PhiX genomes using bwa (58) and removed.
(ii) Genome assembly and polishing.
Contigs were assembled from quality-controlled reads using MEGAHIT (59) and annotated using BLAST against a database of 20 published redondovirus genome sequences (2, 60). Using the sbx_select_contigs Sunbeam extension (https://github.com/ArwaAbbas/sbx_select_contigs), contigs with homology to redondoviruses (as annotated by BLAST) were extracted and overlap-assembled using CAP3 (61). The resulting contigs were manually inspected to identify draft genomes, circularized based on the overlaps identified by sbx_select_contigs, and polished by aligning quality-controlled reads to the draft genomes. Rare assembly errors were manually corrected by visualization of alignments using Integrated Genomics Viewer (IGV) (62).
DNA and amino acid sequence analysis.
Phylogenetic analyses were performed as previously described (2). DNA and protein sequence alignments were performed using MUSCLE (version 3.8.31) (63). Phylogenetic trees were constructed from sequence alignments using PHYML (64); branch support was quantified by the approximate likelihood ratio test (65). Phylogenetic trees were visualized using FigTree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree). For genotype classification and protein clustering, full-length redondovirus genomes or redondovirus amino acid sequences were clustered using VSEARCH (66). Analysis of recombination patterns in redondovirus genomes were carried out using RDP4 (33). as described by Lefeuvre et al. (34). All available genome sequences were used for this analysis, but to avoid bias introduced from repeated isolation and sequencing of the same or highly similar genomes in our limiting-dilution SGS, we included only one sequence per redondovirus genotype in the recombination pattern analysis. The entangled Rep and Cp phylogeny was constructed using baltic (https://github.com/evogytis/baltic) (67) as previously described (3), using one genome from each subject sampled.
Rep protein purification and biochemical assays.
Codon-optimized Vientovirus FB and Brisavirus AA Rep proteins with an N-terminal His-FLAG-SUMO tag (68) were expressed in HI-Control BL21(DE3) cells (Lucigen). One-liter cultures in LB broth were grown to an optical density at 600 nm (OD600) of 0.6 to 0.8 at 37°C and then induced by adding 250 μl of 1 M IPTG (isopropyl-β-d-thiogalactopyranoside) and incubating for 3 h at 37°C. After induction, bacterial pellets were resuspended in 30 ml chilled Ni-binding buffer (20 mM HEPES [pH 7.5], 1 M NaCl, 20 mM imidazole) with cOmplete Mini protease inhibitor (Roche), followed by addition of 100 μl of 100 mg/ml lysozyme and incubation on ice for 30 min. All subsequent steps were performed at 4°C. Then, lysates were sonicated six times for 30 s each, followed by centrifugation at 15,000 × g for 30 min. Cleared lysates were applied to 5 ml nickel-nitrilotriacetic acid (Ni-NTA) columns (Qiagen) and then washed with 10 column volumes (CV) of Ni-binding buffer. Protein was then eluted with Ni elution buffer (20 mM HEPES [pH 7.5], 1 M NaCl, 200 mM imidazole). After elution, 50 μg His-SUMO protease (kindly provided by G. D. Van Duyne) was added to the eluate; then the eluate was dialyzed into 20 mM HEPES (pH 7.5), 1 M NaCl, 10 mM β-mercaptoethanol (BME) overnight at 4°C during tag cleavage. After dialysis, the sample was applied to a 5 ml Ni-NTA column to remove the cleaved tag and SUMO protease, sized on a SuperDex column, and then concentrated to 1 to 2 mg/ml using 30,000-molecular-weight-cutoff (MWCO) Amicon spin columns (Millipore). Analytical SDS-PAGE followed by Coomassie staining was performed to confirm protein purity, and gels were visualized with a GelDoc XR system (Bio-Rad).
Oligonucleotides were ordered from Integrated DNA Technologies. For Rep nicking assays, oligonucleotides labeled on the 5′ end with an ATTO488 fluorophore (IDT) were used; for covalent intermediate formation and joining assays, 3′-end-labeled oligonucleotides were used. Reaction mixtures were incubated at 37°C for the indicated times, in 20-μl amounts. The following buffer was used: 50 mM potassium acetate, 20 mM Tris-acetate, 100 μg/ml bovine serum albumin (BSA), at pH 7.9, together with 1.5 μg of Rep and 5 pmol oligonucleotide per reaction. Reactions were performed with either 10 mM magnesium acetate or 10 mM EDTA added as a negative control. Reactions were stopped with loading dye containing an excess of EDTA. For cleavage and joining assays, 1 μg proteinase K was added, and reaction mixtures were incubated at 50°C for 30 min prior to electrophoresis. Cleavage and joining assay products were electrophoresed on Tris-borate-EDTA (TBE)–urea acrylamide gels and then visualized using a GelDoc XR system (Bio-Rad). Covalent intermediate formation assays were electrophoresed on SDS-PAGE gels and then visualized using a GelDoc XR system (Bio-Rad). Quantification and image analysis was performed using ImageJ (69).
Data availability.
Newly determined genome sequences described in this paper have been deposited in GenBank under the accession numbers MZ405018 to MZ405079.
ACKNOWLEDGMENTS
We are extremely grateful to the subjects who donated samples, data, and time to this study. We thank the State Institutions of the different countries where sampling was carried out for authorizing these studies. We thank Anya Bauer, Katie Bar, and members of the Bushman and Collman labs for helpful discussions and suggestions. We thank Greg Van Duyne for kindly providing the pHFS plasmid, SUMO protease enzyme, and the use of laboratory equipment.
This work was supported by NIH grant R33HL137063 (R.G.C. and F.D.B.), the PennCHOP Microbiome Program, and NIH grant R35 GM134957-01 and American Diabetes Association Pathway to Stop Diabetes grant 1-19-VSN-02 (S.A.T.). L.J.T. was supported by T32AI007324, M.I.D. by R25GM071745 and R33HL137063-S1, M.A.R. was supported in part by the Lewis and Clark Fund, the University of Pennsylvania, the Leakey Foundation, the Wenner-Gren Foundation (9299), an NIH training grant in Parasitology (5T32AI007532-18), and the National Science Foundation (BCS-1540432). We acknowledge assistance from multiple Cores of the Penn Center for AIDS Research (P30-AI45008).
L.J.T., M.I.D., M.A.R., R.G.C., and F.D.B. conceived and designed the experiments. L.J.T. designed and performed biochemical Rep activity assays, performed bioinformatics analyses, assembled and polished redondovirus genomes, performed the recombination analysis, analyzed and visualized data, and wrote the initial manuscript draft. L.J.T., A.A.A., and Y.H. designed, optimized, and performed Rep protein purification. M.I.D. designed and optimized the single-genome isolation protocol, performed the wet-side portion of the longitudinal single-genome isolations, and assembled redondovirus genomes. A.M.R. performed MiSeq sequencing of redondovirus whole-genome PCR products. M.A.R. designed the sampling strategy and executed the wet-side portion of redondoviral single-genome isolation from the African samples. L.J.T., A.S.F., L.A.K. and J.G.-W. collected and processed samples from healthy Philadelphians. A.R., S.R.T., W.R.B., E.M., D.W., G.G.M., S.W.M., C.F., A.K.N., G.B., T.N., S.A.T., and M.C.C. collected and provided access to oral samples from African countries. R.G.C. and F.D.B. provided supervision, mentorship, and funding. L.J.T., R.G.C., and F.D.B. revised the manuscript. All authors approved the final manuscript.
Contributor Information
Ronald G. Collman, Email: collmanr@pennmedicine.upenn.edu.
Frederic D. Bushman, Email: bushman@pennmedicine.upenn.edu.
Colin R. Parrish, Cornell University
REFERENCES
- 1.Cui L, Wu B, Zhu X, Guo X, Ge Y, Zhao K, Qi X, Shi Z, Zhu F, Sun L, Zhou M. 2017. Identification and genetic characterization of a novel circular single-stranded DNA virus in a human upper respiratory tract sample. Arch Virol 162:3305–3312. 10.1007/s00705-017-3481-3. [DOI] [PubMed] [Google Scholar]
- 2.Abbas A, Taylor LJ, Dothard MI, Leiby JS, Fitzgerald AS, Khatib LA, Collman RG, Bushman FD. 2019. Redondoviridae, a family of small, circular DNA viruses of the human oro-respiratory tract associated with periodontitis and critical illness. Cell Host Microbe 25:719–729. 10.1016/j.chom.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abbas A, Taylor LJ, Collman RG, Bushman FD. 2020. ICTV virus taxonomy profile: Redondoviridae. J Gen Virol. 102:jgv001526. 10.1099/jgv.0.001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spezia PG, Macera L, Mazzetti P, Curcio M, Biagini C, Sciandra I, Turriziani O, Lai M, Antonelli G, Pistello M, Maggi F. 2020. Redondovirus DNA in human respiratory samples. J Clin Virol 131:104586. 10.1016/j.jcv.2020.104586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosario K, Duffy S, Breitbart M. 2012. A field guide to eukaryotic circular single-stranded DNA viruses: insights gained from metagenomics. Arch Virol 157:1851–1871. 10.1007/s00705-012-1391-y. [DOI] [PubMed] [Google Scholar]
- 6.Tisza MJ, Pastrana DV, Welch NL, Stewart B, Peretti A, Starrett GJ, Pang YYS, Krishnamurthy SR, Pesavento PA, McDermott DH, Murphy PM, Whited JL, Miller B, Brenchley J, Rosshart SP, Rehermann B, Doorbar J, Ta’ala BA, Pletnikova O, Troncoso JC, Resnick SM, Bolduc B, Sullivan MB, Varsani A, Segall AM, Buck CB. 2020. Discovery of several thousand highly diverse circular DNA viruses. Elife 9:e51971. 10.7554/eLife.51971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao L, Rosario K, Breitbart M, Duffy S. 2019. Eukaryotic circular Rep-encoding single-stranded DNA (CRESS DNA) viruses: ubiquitous viruses with small genomes and a diverse host range. Adv Virus Res 103:71–133. 10.1016/bs.aivir.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 8.Krupovic M, Varsani A, Kazlauskas D, Breitbart M, Delwart E, Rosario K, Yutin N, Wolf YI, Harrach B, Zerbini FM, Dolja VV, Kuhn JH, Koonin EV. 2020. Cressdnaviricota: a virus phylum unifying seven families of Rep-encoding viruses with single-stranded, circular DNA genomes. J Virol 94:e00582-20. 10.1128/JVI.00582-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reference deleted. [Google Scholar]
- 10.Laufs J, Traut W, Heyraud F, Matzeit V, Rogers SG, Schell J, Gronenborn B. 1995. In vitro cleavage and joining at the viral origin of replication by the replication initiator protein of tomato yellow leaf curl virus. Proc Natl Acad Sci USA 92:3879–3883. 10.1073/pnas.92.9.3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinfeldt T, Finsterbusch T, Mankertz A. 2006. Demonstration of nicking/joining activity at the origin of DNA replication associated with the rep and rep’ proteins of porcine circovirus type 1. J Virol 80:6225–6234. 10.1128/JVI.02506-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandler M, De La Cruz F, Dyda F, Hickman AB, Moncalian G, Ton-Hoang B. 2013. Breaking and joining single-stranded DNA: the HUH endonuclease superfamily. Nat Rev Microbiol 11:525–538. 10.1038/nrmicro3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hafner GJ, Stafford MR, Wolter LC, Harding RM, Dale JL. 1997. Nicking and joining activity of banana bunchy top virus replication protein in vitro. J Gen Virol 78(Pt 7):1795–1799. 10.1099/0022-1317-78-7-1795. [DOI] [PubMed] [Google Scholar]
- 14.Marsin S, Forterre P. 1998. A rolling circle replication initiator protein with a nucleotidyl- transferase activity encoded by the plasmid pGT5 from the hyperthermophilic archaeon Pyrococcus abyssi. Mol Microbiol 27:1183–1192. 10.1046/j.1365-2958.1998.00759.x. [DOI] [PubMed] [Google Scholar]
- 15.Lázaro-Perona F, Dahdouh E, Román-Soto S, Jiménez-Rodríguez S, Rodríguez-Antolín C, de la Calle F, Agrifoglio A, Membrillo FJ, García-Rodríguez J, Mingorance J. 2020. Metagenomic detection of two vientoviruses in a human sputum sample. Viruses 12:327. 10.3390/v12030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carmichael A, Jin X, Sissons P, Borysiewicz L. 1993. Quantitative analysis of the human immunodeficiency virus type 1 (HIV-1)-specific cytotoxic T lymphocyte (CTL) response at different stages of HIV-1 infection: differential CTL responses to HIV-1 and Epstein-Barr virus in late disease. J Exp Med 177:249–256. 10.1084/jem.177.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenbloom DIS, Elliott O, Hill AL, Henrich TJ., Siliciano JM, Siliciano RF. 2015. Designing and interpreting limiting dilution assays: general principles and applications to the latent reservoir for human immunodeficiency virus-1. Open Forum Infect Dis 2:ofv123. 10.1093/ofid/ofv123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spandole S, Cimponeriu D, Berca LM, Mihăescu G, Miha G. 2015. Human anelloviruses: an update of molecular, epidemiological and clinical aspects. Arch Virol 160:893–908. 10.1007/s00705-015-2363-9. [DOI] [PubMed] [Google Scholar]
- 19.Abbas A, Diamond JM, Chehoud C, Chang B, Kotzin JJ, Young JC, Imai I, Haas AR, Cantu E, Lederer DJ, Meyer KC, Milewski RK, Olthoff KM, Shaked A, Christie JD, Bushman FD, Collman RG. 2017. The perioperative lung transplant virome: torque teno viruses are elevated in donor lungs and show divergent dynamics in primary graft dysfunction. Am J Transplant 17:1313–1324. 10.1111/ajt.14076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abbas A, Young JC, Clarke EL, Diamond JM, Imai I, Haas AR, Cantu E, Lederer DJ, Meyer K, Milewski RK, Olthoff KM, Shaked A, Christie JD, Bushman FD, Collman RG. 2019. Bidirectional transfer of Anelloviridae lineages between graft and host during lung transplantation. Am J Transplant 19:1086–1097. 10.1111/ajt.15116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaczorowska J, van der Hoek L. 2020. Human anelloviruses: diverse, omnipresent and commensal members of the virome. FEMS Microbiol Rev 44:305–313. 10.1093/femsre/fuaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krupovic M, Forterre P. 2015. Single-stranded DNA viruses employ a variety of mechanisms for integration into host genomes. Ann N Y Acad Sci 1341:41–53. 10.1111/nyas.12675. [DOI] [PubMed] [Google Scholar]
- 23.Grabundzija I, Messing SA, Thomas J, Cosby RL, Bilic I, Miskey C, Gogol-Doring A, Kapitonov V, Diem T, Dalda A, Jurka J, Pritham EJ, Dyda F, Izsvak Z, Ivics Z. 2016. A Helitron transposon reconstructed from bats reveals a novel mechanism of genome shuffling in eukaryotes. Nat Commun 7:10716. 10.1038/ncomms10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Fu Y, Li B, Yu X, Xie J, Cheng J, Ghabrial SA, Li G, Yi X, Jiang D. 2011. Widespread horizontal gene transfer from circular single-stranded DNA viruses to eukaryotic genomes. BMC Evol Biol 11:276. 10.1186/1471-2148-11-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinstein JN, Collisson EA, Mills GB, Mills Shaw KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM, Cancer Genome Atlas Research Network. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45:1113–1120. 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cantalupo PG, Katz JP, Pipas JM. 2018. Viral sequences in human cancer. Virology 513:208–216. 10.1016/j.virol.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arias-Pulido H, Peyton CL, Joste NE, Vargas H, Wheeler CM. 2006. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J Clin Microbiol 44:1755–1762. 10.1128/JCM.44.5.1755-1762.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lace MJ, Anson JR, Klussmann JP, Wang DH, Smith EM, Haugen TH, Turek LP. 2011. Human papillomavirus type 16 (HPV-16) genomes integrated in head and neck cancers and in HPV-16-immortalized human keratinocyte clones express chimeric virus-cell mRNAs similar to those found in cervical cancers. J Virol 85:1645–1654. 10.1128/JVI.02093-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Walt E, Rybicki EP, Varsani A, Polston JE, Billharz R, Donaldson L, Monjane AL, Martin DP. 2009. Rapid host adaptation by extensive recombination. J Gen Virol 90:734–746. 10.1099/vir.0.007724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin DP, Biagini P, Lefeuvre P, Golden M, Roumagnac P, Varsani A. 2011. Recombination in eukaryotic single stranded DNA viruses. Viruses 3:1699–1738. 10.3390/v3091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stenzel T, Piasecki T, Chrząstek K, Julian L, Muhire BM, Golden M, Martin DP, Varsani A. 2014. Pigeon circoviruses display patterns of recombination, genomic secondary structure and selection similar to those of beak and feather disease viruses. J Gen Virol 95:1338–1351. 10.1099/vir.0.063917-0. [DOI] [PubMed] [Google Scholar]
- 32.Kazlauskas D, Varsani A, Krupovic M. 2018. Pervasive chimerism in the replication-associated proteins of uncultured single-stranded DNA viruses. Viruses 10:187. 10.3390/v10040187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. 2015. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:vev003. 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lefeuvre P, Lett J-M, Varsani A, Martin DP. 2009. Widely conserved recombination patterns among single-stranded DNA viruses. J Virol 83:2697–2707. 10.1128/JVI.02152-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth DB, Wilson JH. 1985. Relative rates of homologous and nonhomologous recombination in transfected DNA. Proc Natl Acad Sci USA 82:3355–3359. 10.1073/pnas.82.10.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang XB, Wilson JH. 1987. Modification of DNA ends can decrease end joining relative to homologous recombination in mammalian cells. Proc Natl Acad Sci USA 84:4959–4963. 10.1073/pnas.84.14.4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cromie GA, Connelly JC, Leach DRF. 2001. Recombination at double-strand breaks and DNA ends: conserved mechanisms from phage to humans. Mol Cell 8:1163–1174. 10.1016/S1097-2765(01)00419-1. [DOI] [PubMed] [Google Scholar]
- 38.Arai N, Arai K, Kornberg A. 1981. Complexes of Rep protein with ATP and DNA as a basis for helicase action. J Biol Chem 256:5287–5293. 10.1016/S0021-9258(19)69400-7. [DOI] [PubMed] [Google Scholar]
- 39.Jeske H. 2009. Geminiviruses. Curr Top Microbiol Immunol 331:185–226. 10.1007/978-3-540-70972-5_11. [DOI] [PubMed] [Google Scholar]
- 40.Jeske H, Lütgemeier M, Preiss W. 2001. DNA forms indicate rolling circle and recombination-dependent replication of Abutilon mosaic virus. EMBO J 20:6158–6167. 10.1093/emboj/20.21.6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mankertz A, Mankertz J, Wolf K, Buhk H-JJ. 1998. Identification of a protein essential for replication of porcine circovirus. J Gen Virol 79(Pt 2):381–384. 10.1099/0022-1317-79-2-381. [DOI] [PubMed] [Google Scholar]
- 42.Cheung AK. 2005. Mutational analysis of the direct tandem repeat sequences at the origin of DNA replication of porcine circovirus type 1. Virology 339:192–199. 10.1016/j.virol.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 43.Laufs J, Schumacher S, Geisler N, Jupin I, Gronenborn B. 1995. Identification of the nicking tyrosine of geminivirus Rep protein. FEBS Lett 377:258–262. 10.1016/0014-5793(95)01355-5. [DOI] [PubMed] [Google Scholar]
- 44.Lolomadze EA, Rebrikov DV. 2020. Constant companion: clinical and developmental aspects of torque teno virus infections. Arch Virol 165:2749–2757. 10.1007/s00705-020-04841-x. [DOI] [PubMed] [Google Scholar]
- 45.Vasilyev EV, Trofimov DY, Tonevitsky AG, Ilinsky VV, Korostin DO, Rebrikov DV. 2009. Torque Teno Virus (TTV) distribution in healthy Russian population. Virol J 6:134. 10.1186/1743-422X-6-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsiao KL, Wang LY, Lin CL, Liu HF. 2016. New phylogenetic groups of torque teno virus identified in eastern Taiwan indigenes. PLoS One 11:e0149901. 10.1371/journal.pone.0149901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Al-Qahtani AA, Alabsi ES, AbuOdeh R, Thalib L, El Zowalaty ME, Nasrallah GK. 2016. Prevalence of anelloviruses (TTV, TTMDV, and TTMV) in healthy blood donors and in patients infected with HBV or HCV in Qatar. Virol J 13:208. 10.1186/s12985-016-0664-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moustafa A, Xie C, Kirkness E, Biggs W, Wong E, Turpaz Y, Bloom K, Delwart E, Nelson KE, Venter JC, Telenti A. 2017. The blood DNA virome in 8,000 humans. PLoS Pathog 13:e1006292. 10.1371/journal.ppat.1006292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Young JC, Chehoud C, Bittinger K, Bailey A, Diamond JM, Cantu E, Haas AR, Abbas A, Frye L, Christie JD, Bushman FD, Collman RG. 2015. Viral metagenomics reveal blooms of anelloviruses in the respiratory tract of lung transplant recipients. Am J Transplant 15:200–209. 10.1111/ajt.13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.King JA, Dubielzig R, Grimm D, Kleinschmidt JA. 2001. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J 20:3282–3291. 10.1093/emboj/20.12.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Clarke EL, Sundararaman SA, Seifert SN, Bushman FD, Hahn BH, Brisson D. 2017. Swga: a primer design toolkit for selective whole genome amplification. Bioinformatics 33:2071–2077. 10.1093/bioinformatics/btx118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clarke EL, Taylor LJ, Zhao C, Connell J, Lee J-J, Fett B, Bushman FD, Bittinger K. 2019. Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome 7:46. 10.1186/s40168-019-0658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koster J, Rahmann S. 2012. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28:2520–2522. 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 54.Leinonen R, Sugawara H, Shumway M, International Nucleotide Sequence Database Collaboration. 2011. The sequence read archive. Nucleic Acids Res 39:D19–D21. 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Taylor LJ, Abbas A, Bushman FD. 2020. Grabseqs: simple downloading of reads and metadata from multiple next-generation sequencing data repositories. Bioinformatics 36:3607–3609. 10.1093/bioinformatics/btaa167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Babraham Bioinformatics. FastQC: a quality control tool for high throughput sequence data. v0.11.9. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 58.Li H, Li H, Durbin R, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li D, Luo R, Liu CM, Leung CM, Ting HF, Sadakane K, Yamashita H, Lam TW. 2016. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102:3–11. 10.1016/j.ymeth.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 60.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic Local Alignment Search Tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 61.Huang XQ, Madan A. 1999. CAP3: a DNA sequence assembly program. Genome Res 9:868–877. 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative Genomics Viewer. Nat Biotechnol 29:24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guindon S, Lethiec F, Duroux P, Gascuel O. 2005. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res 33:D557–D559. 10.1093/nar/gki352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 66.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. 2016. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4:e2584. 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dudas G, Bedford T, Lycett S, Rambaut A. 2015. Reassortment between influenza B lineages and the emergence of a coadapted PB1-PB2-HA gene complex. Mol Biol Evol 32:162–172. 10.1093/molbev/msu287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eilers G, Gupta K, Allen A, Zhou J, Hwang Y, Cory MB, Bushman FD, Van Duyne G. 2020. Influence of the amino-terminal sequence on the structure and function of HIV integrase. Retrovirology 17:28–16. 10.1186/s12977-020-00537-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. 2015. The ImageJ ecosystem: an open platform for biomedical image analysis. Mol Reprod Dev 82:518–529. 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Newly determined genome sequences described in this paper have been deposited in GenBank under the accession numbers MZ405018 to MZ405079.