

Abstract
Messenger RNA (mRNA) has emerged as a critical global technology that requires global joint efforts from different entities to develop a COVID-19 vaccine. However, the chemical properties of RNA pose a challenge in utilizing mRNA as a vaccine candidate. For instance, the molecules are prone to degradation, which has a negative impact on the distribution of mRNA among patients. In addition, little is known of the degradation properties of individual RNA bases in a molecule. Therefore, this study aims to investigate whether a hybrid deep learning can predict RNA degradation from RNA sequences. Two deep hybrid neural network models were proposed, namely GCN_GRU and GCN_CNN. The first model is based on graph convolutional neural networks (GCNs) and gated recurrent unit (GRU). The second model is based on GCN and convolutional neural networks (CNNs). Both models were computed over the structural graph of the mRNA molecule. The experimental results showed that GCN_GRU hybrid model outperform GCN_CNN model by a large margin during the test time. Validation of proposed hybrid models is performed by well-known evaluation measures. Among different deep neural networks, GCN_GRU based model achieved best scores on both public and private MCRMSE test scores with 0.22614 and 0.34152, respectively. Finally, GCN_GRU pre-trained model has achieved the highest AuC score of 0.938. Such proven outperformance of GCNs indicates that modeling RNA molecules using graphs is critical in understanding molecule degradation mechanisms, which helps in minimizing the aforementioned issues. To show the importance of the proposed GCN_GRU hybrid model, in silico experiments has been contacted. The in-silico results showed that our model pays local attention when predicting a given position’s reactivity and exhibits interesting behavior on neighboring bases in the sequence.
Keywords: COVID-19, Vaccine, mRNA degradation, Convolutional neural networks, Graph convolutional neural networks
1. Introduction
The first announced case of a novel coronavirus disease (COVID-19) was reported in December 2019 in Wuhan, China (Jin et al., 2020), which preceded the COVID 19 outbreak. Subsequently, the COVID-19 pandemic has had a continued immense impact on people’s lives around the world (Zhang and Ma, 2020, Arba et al., 2020, Bong et al., 2020). At the time of writing this paper, the global death toll of COVID-19 is at a staggering 1.36 million deaths and more, not including excess morbidity from the pandemic itself. Furthermore, the number of new cases each day is increasing by 620,000 globally (Esteban Ortiz-Ospina Max Roser, H2020). The global catastrophe has brought about an unprecedented effort to develop, approve, and distribute a vaccine against the novel virus in record time (Chung et al., 2020, Lazarus et al., 2020). Notably, the vaccination preparation process typically takes between 10 and 15 years (International Federation of Pharmaceutical Manufacturers, 2020). However, in the case of COVID 19, it is being accelerated to a timeframe of a year (Jeyanathan et al., 2020). Great appreciation has been given for the extraordinary support and collaboration across industry, academia, and governments across the globe. Such accelerated efforts motivate researchers to participate in vaccine production that serves the entire world.
Out of such initiatives to produce an effective vaccine, messenger RNA (mRNA) vaccines have taken the lead as the fastest vaccine candidates for COVID-19; currently however, it faces key potential limitations (Wang et al., 2020). One major challenge is in designing stable mRNA molecules under appropriate conditions. Conventional vaccines (seasonal flu shots) are packed in disposable syringes and shipped under refrigeration worldwide, but this is quite impossible for mRNA vaccines (Pardi et al., 2018). For instance, researchers have observed that RNA molecules tend to degrade spontaneously. For instance, a single cut can render the mRNA vaccine unusable, which is considered to be a severe limitation. Currently, little is known of the details of where the RNA backbone is most likely to be affected. Consequently, the current mRNA vaccines against COVID-19 must be prepared and shipped under intense refrigeration (Table 1 ) and are unlikely to reach more than a tiny fraction of human beings on the planet unless they can be stabilized. Particularly, the available knowledge on the stability profiles of the mRNA COVID-19 vaccine candidates in development is being updated regularly (Crommelin et al., 2021). Table 1 shows the latest shelf-life and temperature storage conditions released by three vaccine manufacturers (Modera, Pfizer-BioNTech, and CureVac). At the time of writing this paper (March 22, 2021), such information has been given solely by vaccine manufacturers, with no confirmation from regulatory authorities. However, the storage requirements during manufacturing, shipping, and at the end-user site are clearly essential characteristics of the mRNA vaccine drug product because they provide a competitive (dis)advantage.
Table 1.
mRNA COVID-19 Vaccine Candidates in Development: Current Stability Profile, Dose, and Dosing Schedule (Status March 22, 2021).
Such candidates have been reported to be effective with a percentage of ≥ 90% (Loftus et al., 2020). In contrast to traditional vaccines, which are composed of inactivated or attenuated components of the pathogen itself, the mRNA vaccine provides a template for the cellular synthesis of a viral component. The mRNA molecules are structures, as illustrated in Fig. 1 , wherein bases loop back on one another to form bonding interactions with linearly distant bases.
Fig. 1.
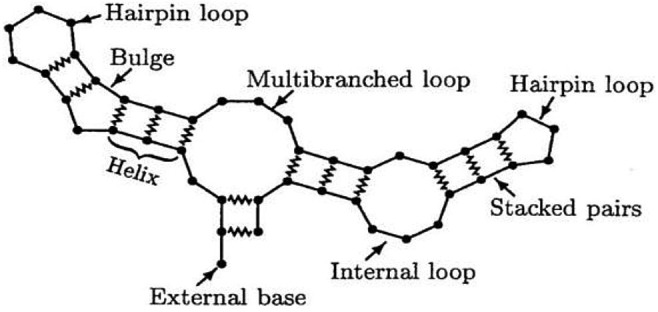
Diagram of different RNA loop structures. Covalent bonds are indicated by straight line segments, while jagged lines indicate H-bond base pairing (Lyngsø and Pedersen, 2000).
The development of these RNA sequences is comparatively less costly and time-consuming because it avoids the challenging purification processes of proteins (Jackson et al., 2020). However, mRNA vaccines are a novel technology and encounter unique challenges because of their chemical structure. In particular, mRNA molecules are known to degrade spontaneously over time (van Hoof and Parker, 2002). In a laboratory setting, RNA is generally stored in specialized freezers kept at −80 °C (Fabre et al., 2014). Thus, from a logistical perspective, this temperature preference poses a substantial hurdle in the successful administration of such an RNA-based vaccine. For instance, such freezers are not readily available, and the degradation of a single base could render the vaccine useless. Since Pfizer/BioNTech and Moderna's latest effectiveness announcements, the logistical challenges of distributing their vaccines at these requisite temperatures have been of great interest and concern (Ducharme, 2020). Relatively little is known of the tendencies for specific bases to degrade (OpenVaccine. Openvaccine: Covid-19 mrna vaccine degradation prediction. Stanford University, Eterna, Sept, 2020). Motivated by this problem, this study aims to improve the stability of mRNA vaccines during the shipping and transportation process by predicting the mRNA base-resolution degradation of RNA at each base of an RNA molecule through local characteristics using hybrid deep neural network (DNN) algorithms. Two types of deep hybrid neural network algorithms will be investigated and evaluated to achieve this aim. However, maintaining a continuous supply of high-quality, reliable, safe, and affordable medications is a critical component of a good health system (Juvin, 2019). The main contributions of this work are summarized as follows.
1.1. Contribution
In this study, the authors explored the hybridization of GCN and GRU models in the mRNA degradation field to predict the stability/reactivity and degradation risk of mRNA sequences. As per the authors’ knowledge, there is no such works that use a hybrid GCN-GRU model in this field. Second, this study emphasizes on the efficiency and effectiveness of the hybrid GCN-GRU model by comparing the proposed model with GCN_CNN. An intensive experiment was conducted in this work based on the Stanford COVID-19 mRNA vaccine dataset. Third, we validated the proposed DL-based hybrid models of COVID-19 mRNA vaccine degradation through well-known evaluation metrics such as mean columnwise root mean squared error (MCRMSE) and Area Under the Curve (AUC).
The rest of the paper is organized as follows. Section 2 gives an overview of the methodology and the dataset description. Results are discussed in Section 3, while model analysis and validation are given in section 4. The conclusion and the future work are provided in Section 5.
2. Related Works
Artificial intelligence and machine learning, particularly deep learning, have resulted in significant advances in a wide range of contexts of science and engineering due to their capacity to thoroughly understand features. The most profound impact has been on vaccine discovery (Keshavarzi Arshadi et al., 2020). Recent advances in deep learning techniques such as GCN, GRU and CNN have enabled the modelling of DNA and RNA sequences. AI can be used to combat the COVID-19 pandemic and generate solutions in a variety of fields, including drug research, vaccine development, public communication, and integrative medicine (Ahuja et al., 2020).
Recurrent neural networks (RNNs) were utilized in the early days of machine learning (or deep learning) to deal with data representations in directed acyclic graphs (Frasconi et al., 1998). Later, as a generalization of RNNs, Graph Neural Networks (GNNs) (Gori et al., 2005) are developed to process general directed and undirected graphs. Convolutional neural networks (CNNs) are then designed to handle data representations from a spatial domain to a graph domain. Graph convolutional networks (GCNs) are the methods created in this regard, and they are divided into two categories: spectral approaches and non-spectral approaches. GCNs have shown cutting-edge performance in a variety of complex mining tasks (for example, semi-supervised node classification and sequence prediction) (Hamilton et al., 2017, Kipf and Welling, 2016).
Based on the extensive research conducted, only two similar studies have been done so far. Authors in (Singhal, 2020) have proposed three single-DL methods (LSTM, GRU, and GCN) to predict mRNA vaccine degradation. The authors claimed that among the three methods developed GRU performed the best with an accuracy of 76%. The critical drawback of (Singhal, 2020) work is that a single algorithm has limited accuracy and cannot capture the mRNA degradation features. Additionally, authors in (Singhal, 2020) did not consider the stability of the vaccine, unlike our proposed study that suggested two-hybrid DNN models and conducted extensive experimentation to predict the RNA sequence degradation. Besides, the experiments that show a general reactivity score, degradation likelihood prediction after incubation at 10pH with magnesium, and degradation likelihood prediction at a temperature of 50 °C with magnesium were not provided.
Similarly, authors in (Qaid et al., 2021) proposed a bidirectional GRU integrating with the LSTM model. This model has been tested and evaluated with the same benchmark dataset provided by Stanford University scientists. However, only the MCRMSE score was reported, and no further experimentation was conducted on temperature storage conditions which is a vital element to evaluate the effectiveness of their model.
3. The proposed approach
This section provides the dataset description and the pre-processing techniques used. It also illustrates the proposed methodology and the deep learning algorithms utilized in this study.
3.1. Dataset collection and pre-processing
3.1.1. Dataset description (Sequence, loop type, and base pairing)
In September 2020, Das Lab at Stanford Biochemistry and Eterna partnered to sponsor a Kaggle competition focused on RNA degradation problems (OpenVaccine. Openvaccine: Covid-19 mrna vaccine degradation prediction. Stanford University, Eterna, Sept, 2020). To achieve the goals of the current study, the authors used their published dataset of 3029 RNA sequences, which are annotated with base-wise information relevant to degradation. Each sequence in the training set comprises 107 bases. The data include base identities (A, G, U, C) and secondary structure information indicating which bases are paired with each other. This pairing is denoted by a string of opening and closing parentheses, where matching pairs indicate paired bases at those indices. Additionally, the data provide a prediction of the RNA loop structure type in a base resolution according to the local characteristics of the sequence structure, including bulge, hairpin loop, paired stem, etc. following the results of the bpRNA prediction algorithm in (Lorenz et al., 2011). Fig. 1 shows prototypical examples of these loop structures (Watters and Lucks, 2016), and Table 2 shows the three-input data used in this study.
Table 2.
Three-input data used in this study and their examples.
| Input Label | Example |
|---|---|
| sequence | GGAAAAGCUCUAAUAACAGGAGA |
| structure | ….….((((((.......)))).)).. |
| predicted_loop_type | EEEEESSSSSSHHHHHHHSSSSBSSX |
The labels of the dataset are a set of reactivity, and the degradation values are measured experimentally in different conditions at each base. Reactivity is measured using SHAPE-Seq and features the structural flexibility of the nucleotide (Seetin et al., 2014). The degradation rates are measured using MAP-Seq under four conditions and feature the likelihood of degradation in each condition (Yan et al., 2020). The dataset includes five metrics of reactivity and degradation, as listed in Table 3 . However, this study focuses on evaluating the first three metrics, namely, reactivity, deg_Mg_ph10, and deg_Mg_50C, as ruled by the Das Lab competition. Motivated by this we focused on these three matrices to conduct a fair comparative analysis with the related works. Thus, this study will develop a multi-task network that takes the RNA sequence information as input and produces three predictions at each base: reactivity, deg_Mg_pH10, and deg_Mg_50C. The performance of the proposed model will be evaluated on two test sets, including a public test set and a private test set (defined by the original Kaggle competition). Moreover, the proposed approach performance will be evaluated using Mean Column-wise Root Mean Squared Error (MCRMSE), which has been used in “OpenVaccine: COVID-19 mRNA Vaccine Degradation Prediction” competition. The MCRMSE evaluation matrix is described in Equation (1).
| (1) |
where Nt is the number of columns/tasks (i.e., reactivity/stability, deg_Mg_pH10, and deg_Mg_50C) and (yij, ŷij) ground-truth and predicted values for reactivity type and RNA sequence at a specific base, respectively.
Table 3.
Reactivity labels and their descriptions. The first three metrics are the predicted outputs of our proposed models, while the last two are not evaluated.
| No | Output Label | Description |
|---|---|---|
| 1 | reactivity | General reactivity score. |
| 2 | deg_Mg_pH10 | Likelihood of degradation after incubation at high pH with magnesium. |
| 3 | deg_Mg_50C | Likelihood of degradation at high temperature with magnesium. |
| 4 | deg_pH10 | Likelihood of degradation after incubation at high pH (pH 10). |
| 5 | deg_50C | Likelihood of degradation at high temperature (50 deg C) |
3.1.2. Features engineering
Both proposed hybrid model (GCN_GRU and GCN_CNN) model begins by extracting features using feature engineering, then uses a sequence input to predict the mRNA sequences responsible for degradation by predicting three reactivity values for each location in the sequence. Categorical features (sequence, structure, and predicted loop type) are the types of features extracted by features engineering. First, categorical features are stored, and an embedding layer is used to capture relationships in sequences that would otherwise be challenging to captured. Then, as shown in Fig. 2 , feature extraction is used to extract categorical features utilizing statistical and mathematical calculations.
Fig. 2.

Steps in the base technique for the encoding of sequence, structure, and expected loop type data.
3.1.3. Data augmentation with pseudo labeling
Given the limited training data, the authors in (Lee, 2013) tried to use pseudo labeling to leverage additional, non-labeled data for training. Pseudo-labeling is a data augmentation technique that uses unlabeled and labeled data during the training; it is a semi-supervised learning algorithm. The process is shown in Fig. 3 . We first train the network on the labeled data for 20 epochs. Then we introduced the unlabeled data by mixing training on unlabeled and labeled batches. The authors used two types of unlabeled data for pseudo labeling. First, we used the end of the sequences (bases 68 to 107) of the training set. After training the network on labeled data for 20 epochs, we started using unlabeled data. For each epoch, we compute the loss on the labeled and unlabeled parts using the pseudo labels. The pseudo labels were computed by a forward pass of the unlabeled data in the network at the state obtained from the previous epoch. This procedure is repeated for every epoch after the 20th. The exception is that for every 1 out of 10 epochs, we go back to training only on labeled data. Second, we used the private test set sequences as unlabeled data. After training the network on labeled data for 20 epochs, we trained the model for 2 epochs on unlabeled data alone, followed by 5 epochs on labeled data. Therefore, we used a “save and load” method to prevent the pseudo labeling from deteriorating the model performances too much. At the end of each pseudo labeling section, we compared the current model with the model right before the pseudo labeling. If the current loss is higher than the loss before pseudo labeling by 0.04, the authors did not accept the current model and reloaded the model's weights before the pseudo labeling section.
Fig. 3.

Data augmentation technique using Pseudo-labeling.
3.1.4. Aggregator functions selection approach
Because there are various forms of aggregator functions in GraphSAGE, the need to evaluate such functions is urgent. Thus, an experiment was conducted to examine three aggregator functions (mean, convolution, and LSTM (Cho et al., 2014) whereby their performance was compared in RNA degradation prediction. The mean aggregator takes the average of neighboring nodes and concatenates it with the original node embedding. On the other side, the LSTM aggregator feeds the embeddings of the neighboring nodes sequentially to the LSTM and concatenates the final output with the original node embedding. For LSTM and mean aggregators, the node embeddings after concatenation are fed into a linear layer to reduce it to the original dimension. Lastly, the convolution operation aggregates over the neighboring nodes (including the node itself) and passes the result to a linear layer for the final embedding. Based on the comparison of different aggregate functions, the authors extracted the node embeddings from the GCN and then passed it to the Gated Recurrent Units (GRU) (He et al., 2016), which is a variant of the Recurrent Neural Network (RNN). The output from the GRU is fed into a fully connected layer to make the final prediction.
3.2. Candidate Deep Model Training and Optimization
3.2.1. Baseline convolutional neural network model
At the beginning, the authors implemented a simple baseline model that would give a lower performance bound. It consisted of a simple convolutional neural network (CNN) algorithm with two 1D convolution layers followed by two fully connected layers (Naseer et al., 2021). This model encoded small window size features (21) around each base in the sequence and predicted the three outputs: reactivity, deg_Mg_pH10, and deg_Mg_50C. Based on the observation, the authors noted that the performance of the first baseline model is sensitive to the selected window size. The model’s prediction is limited to a local window of neighboring bases along the primary sequence. Thus, the authors designed a second baseline model, whereby the sequence and structure of the entire RNA molecule were encoded as input to the CNN algorithm. Thus, the model could leverage global sequencing information during the prediction process by considering the sequence, loop type, and base pairing of each base in the RNA sequence as one-hot encoded and concatenated final input. The inputs are passed to three convolution layers, which apply average pooling in windows of all three. All the layers apply batch normalization, rectified linear units, and dropout. The CNN's final output is passed to a linear layer to predict the RNA degradation rates at each base.
3.2.2. Graph Convolutional Network (GCNs)
As illustrated above, the proposed 1D convolution-based baseline only aggregates information from neighboring bases along the primary sequence of the molecule. However, the mRNA molecule structure holds bases that loop back on one another to form bonding interactions with linearly distant bases, as depicted in Fig. 4 . To reflect a more realistic 3D structure, the RNA molecules can be represented as graphs, where the nodes represent the information of each base, and the edges represent bases adjacent or paired by bonding interactions (Duvenaud et al., 2015). Traditional CNNs cannot operate directly on graphs because of their irregular structure. Thus, a generalized form of the CNN called graph convolutional network (GCN) was developed for this specific purpose (Hamilton et al., 2017, Naseer et al., 2021). According to (Duvenaud et al., 2015), GCN is an attractive architecture to infer RNA structures, and has been used several times in the literature.
Fig. 4.

Architecture of hybrid GCN_GRU model, from input sequence to reactivity prediction.
In the implementation stage, the sequence, loop type, and base pairing information are used to generate the embedding for each node after passing the integer encoded input to an embedding layer, as shown in Fig. 4. The edges are represented using adjacency matrices calculated from the secondary structure. We used a type of GCN architecture called GraphSAGE. GraphSAGE (Hochreiter and Schmidhuber, 1997) is an instance of GCNs developed for representational learning. Instead of learning node embeddings directly, GraphSAGE learns the aggregator function and computes the node embeddings by applying the aggregator function to the neighboring nodes. In our application, we trained GraphSAGE in a supervised fashion. The node embeddings were extracted from the GCN and fed to another neural network to make the final prediction.
The proposed hybrid model architecture is shown in Fig. 4. The graph embeddings generated by GCNs can be fed into a GRU, and we refer to this architecture as the GCN_GRU architecture. As an alternative, we also experimented by using CNNs on top of the graph embeddings and passed the CNN's output to a fully connected layer to make the prediction. This model is referred to as the GCN_CNN architecture, and we tried CNN architectures with and without residual connections (Kingma and Ba, 2017) between layers. In addition to varying the model components, we also experimented with the number of GCN layers (K). The number of GCN layers, K, affects how the node embedding is generated. For instance, when K = 2, the node embedding is generated by aggregating all neighbors located at most two edges apart. However, this is issue considered a multi-task learning problem. We also experimented with weight loss by assigning higher weights for explicitly evaluated tasks (this will be further explained in section 3). The aggregator functions are also trained jointly with the RNA degradation prediction. Therefore, the node embeddings are a representation of the sequence and structure of each base. A GCN is used to compute a graph embedding for each node. The node embeddings are then passed through a GRU (or CNN) and a fully connected layer to make the final reactivity prediction.
3.3. GCN_GRU Model Enhancement Using Pretrain Node Embedding Technique.
The size of the training set is relatively small. Only 2096 examples are used in training after filtering measurements with a low signal-to-noise ratio. Therefore, we also experimented on pretraining the node embedding in an auto-encoder (Lee, 2013) fashion using the sequences in the training set and the 3000 sequences in the test set. The node embeddings of the GCN layer were fed into a fully connected network to reconstruct the original sequences and structures (i.e., base pairing and loop types). The Mean Squared Error (MSE) between the input and the reconstruction was used as the auto-encoder loss function. After the auto-encoder converged, the node embeddings were extracted to initialize the node embeddings in the GCN_GRU architecture.
Synthesizing the proposed model performance in terms of public and private test scores helps us quantify whether the proposed model will accurately assess the actual degradation rates of mRNA sequences in practice. However, as with many black-box methods, we are also interested in understanding the proposed model's behavior and interpretability. With relatively small data, we can implement statistical in silico mutagenesis (ICM) to probe our model's behavior. Given the computational costs of running a forward pass over many mutated inputs, we restricted our analysis to a sample of the sequences and perturbed the sequences at intervals of 5 rather than at every base. In implementing ICM, we perturbed the input data at every five positions in the sequence and measured the output predictions for each of the five different reactivity and degradation measures. Fig. 5 portrays an example of this process for a perturbation on the first nucleotide of a given sequence. In Step 1, we select a sample of our original sequence and structure data to work with. We sampled 250 examples from the 2000 examples in the original public training dataset for the following analyses. In Step 2, we perturb the bases in the sequences: at every five base positions (0, 5, 10, etc.), we change the base value to each of the other three nitrogenous bases that can be used in an RNA sequence. In our example above, because base position 0 is “A”, we change this base to each of “C”, “G”, and “U”. With the perturbed sequence, the original secondary structure is no longer valid: perturbing even a single base can have ripple effects on the molecule's overall structure (Danaee et al., 2018).
Fig. 5.

Inference Workflow. Note that µB,N is a vector of reactivity outputs of length 107 where B is the base value position perturbed (1 in the example above) and N is the base value it was perturbed.
Thus, we must compute the secondary structure of the new perturbed sequence. The RNA.fold function from the Vienna RNA 2.0 package (OpenVaccine. Openvaccine: Covid-19 mrna vaccine degradation prediction. Stanford University, Eterna, Sept, 2020) is used to extract the predicted base pairing for the perturbed sequence. Then, we fed the perturbed sequence and the predicted base-pair data to bpRNA to predict the loop type of each base (van der Maaten and Hinton, 2008). We repeated step 2 for each of the 250 sequences in the sample to have complete input data for each of the perturbed sequences. In step 3, we take these modified sequences and their secondary structure annotations and feed them into the proposed GCN_GRU model. The model outputs all five predicted reactivity and degradation values for each modified sequence (although, recall that the eventual evaluation is computed only using the first three reactivity and degradation values). Hence, these predicted values can be analyzed to understand the behavior of the proposed model better.
4. Experimental results and discussion
4.1. The proposed model performance evaluation
We evaluated the proposed models based on two test sets: a public test set and a private test set (as defined by the Kaggle competition). The length of the RNA sequence in the public test is 107 while the length of the RNA sequence in the private test is 130. The measurement of RNA degradation rates does not cover the last 39 bases in the sequence because of technological challenges. Thus, the prediction length is 68 and 91 for the public, and private mean column-wise root mean squared error (MCRMSE) test, respectively. Because the test scores are MCRMSE between predictions and labels, a lower score represents better model performance.
The results of the two baseline models are shown in Table 4 . Compared to the first baseline model that focuses only on the local structure, the second baseline model outperforms it by a large margin. This finding is within our expectation because the second model contains the structure and sequence information over the entire RNA molecule.
Table 4.
Public and private MCRMSE test scores for the baseline model.
| Model | Public MCRMSE test score | Private MCRMSE test score |
|---|---|---|
| Baseline1 | 0.3798 | 0.4727 |
| Baseline2 (CNN over the entire sequence) | 0.30424 | 0.41348 |
The test results for GCNs with different aggregator functions are shown in Table 5 . The ability of GCNs to perform convolution on data with irregular structures helps to capture better and leverage the structure information of RNA molecules: neighboring bases in secondary structure (not just primary structure) can have an immediate effect on predicted reactivity. As expected, all GCN models outperformed our CNN-based baseline. The mean aggregator function achieved the best performance during test time. Thus, the mean aggregator was chosen for comparing different model architectures.
Table 5.
Public and private MCRMSE test scores for GCNs with different aggregate functions.
| Aggregate Function | Public test score | Private test score |
|---|---|---|
| Mean | 0.22614 | 0.34571 |
| Conv | 0.22173 | 0.34989 |
| LSTM | 0.23126 | 0.35904 |
The test results for GCNs with different model architectures are summarized in Table 6 . Replacing GRU only with a CNN does not boost our model performance. After adding residue connections between convolutional layers, the model’s performance is comparable but still slightly worse than the original GCN_GRU architectures. One of the possible explanations for the performance of the GCN_CNN model is that the GCN is a generalized form of CNNs. Thus, GCN and CNN will have similar operations. On the contrary, combining GCNs with GRUs, which operate recurrently, will add more diversity to the model architecture and better capture sequential data.
Table 6.
Public and private MCRMSE test scores for GCNs with different model architectures.
| Model Architecture | Public test score | Private test score |
|---|---|---|
| GCN_GRU | 0.22614 | 0.34571 |
| GCN_CNN | 0.23275 | 0.35280 |
| GCN_CNN (with residue connection) | 0.22729 | 0.34822 |
| GCN_GRU (weighted loss) | 0.22514 | 0.34494 |
| GCN_GRU (pre-trained embedding) | 0.22614 | 0.34152 |
The test results for models with different GCN layers (K) are plotted in Fig. 6 b. As the number of GCN layers increases, the test loss also increases. We hypothesized that the small size of the graph causes this deterioration in performance. The graph representation of RNA molecules only contains roughly 100 nodes. When the number of GCN layers increases, the node embedding begins to capture more global information in favor of local information. As a result, the model’s performance in predicting the degradation rate at the base resolution deteriorates.
Fig. 6.

Performance evaluation: (a) GCN_GRU model using mean squared error (MSE) during training and validation; (b) Public and private test losses for the model with a different number of GCN layers.
The test results for using weighted loss and pre-trained node embeddings are shown in Table 6. As expected, assigning higher weights for tasks evaluated during test time improved the model performance. Pretraining, which refers to node embedding in an auto-encoder fashion, also improved the model on the public and private test sets. However, the improvement of the performance on the private test set is marginal. The test results for pseudo labeling on the GCN_GRU architecture are presented in Table 7 . We used the GCN_GRU model with the weighted loss but without pre-trained node embeddings as the pseudo labeling architecture. The end of the sequences and the private test set improved the model performances. However, pseudo labeling improvement using the private test set is smaller than that of pseudo labeling using the training set. This effect can be explained by the training set sequences following a distinct distribution compared to the private test set sequences. We discuss this observed effect in detail in Section 4.2. This difference in the distribution could make training the model using pseudo labels on the private test set less stable, making it more challenging for the model to converge to an optimal solution.
Table 7.
Public and private MCRMSE test scores for GCN_GRU architecture using pseudo labeling.
| Approach | Public test score | Private test score |
|---|---|---|
| GCN_GRU (before pseudo labeling) | 0.22514 | 0.34494 |
| Train set - bases 68 to 107 | 0.22345 | 0.34113 |
| Private test set - bases 1 to 91 | 0.22434 | 0.34368 |
The related works (Metzker, 2010) have observed that measurement error increased for bases at the end of the RNA sequence because of technological challenges, and thus, only the measurements of the first 68 bases are reported. The predictions are truncated to the first 68 bases for all the models to calculate the loss, which is the mean squared error (MSE) between the predictions and ground truth. All models were trained using batch gradient descent with a learning rate adapted via Adam optimizer (Hinton and Salakhutdinov, 2006). The proposed deep models were also trained with five-fold cross-validation, and the prediction was the average of all folds. The MSE performance evaluation of the GCN_GRU model during training and validation is shown in Fig. 6a. The gap between training and validation loss does not indicate over-fitting because we stopped the model when the validation loss did not decrease for a certain number of epochs. Stopping the model earlier led to worse performance during test time.
Additionally, it is often beneficial to summarize the ROC curve insights of a model to a single scalar value that indicates the model's output. One of these common techniques is the region under the ROC curve, known as the AUC. The AUC reduces the effects of the ROC curve to a single value and illuminates mathematical insights into the success of the model. AUC is equal to the probability that a randomly chosen positive sample will be classified higher than a randomly chosen negative instance by the classifier. The AUC values for the models built in this study are presented in Table 8 . The highest AUC value of 0.938 was achieved by the GCN_GRU pre-trained model, while the model developed with GCN_CNN with residue connection obtained the lowest ratings at 0.838. For three more DNN-based models, the scores obtained differed from the two performance values. The three models achieved an AUC rate of 0.928, 0.925, and 0.844 for GCN_GRU weighted loss, GCN_GRU, and GCN_CNN, respectively.
Table 8.
Receiver operating characteristic (AUC) score for the proposed models’ architectures.
| DNN Model Architecture | ROC-AuC |
|---|---|
| GCN_GRU | 0.925 |
| GCN_CNN | 0.844 |
| GCN_CNN (with residue connection) | 0.838 |
| GCN_GRU (weighted loss) | 0.928 |
| GCN_GRU (pre-trained embedding) | 0.938 |
4.2. Model performance benchmarking
The proposed model has been tested according to benchmark the different proposed model based on the conditions given in Table 1. The proposed GCN_GRU model outperformed the GCN_CNN model by a large margin on both private and public tests. To better understand the model performance, we calculated the MSE and plotted the labels versus predictions for each task. The MSE for the reactivity, the degradation rate with Mg at pH = 10, and the degradation rate with Mg at 50 °C are 0.087, 0.255, and 0.125, respectively. From the MSE scores, we observed that the proposed model performed much better in predicting reactivity than predicting degradation rate with Mg at pH = 10. Thus, performing task-specific optimizations, such as stopping some tasks early or training a separate model to only predict degradation rates at pH = 10, may be beneficial. As shown in Fig. 7 , the GCN model generally underestimated the degradation rate and rarely predicted any degradation rate of more than 5. In the training example, bases with a degradation rate of more than 5 appeared with a low frequency (less than 0.2 %), which explains why the proposed model barely predicted any high degradation rate.Fig. 8.
Fig. 7.

General reactivity score (Left), degradation likelihood prediction after incubation at 10pH with magnesium (middle), and degradation likelihood prediction at a temperature of 50 °C with magnesium.
Fig. 8.

Predictions results of private test score; (a) The first 68 bases in the private test set and (b) Bases 69–91 in the private test set. The green represents the general reactivity score, the orange represents the likelihood of degradation after incubation at high pH with magnesium, and the blue denotes the likelihood of degradation at high temperature with magnesium.
During our experiment, a consistent performance gap between the public test set and the private test set was observed, similar to what was observed in the Kaggle competition models. The MCRMSE on the private test set is 0.12 higher than that on the public test set. The measurement of degradation rates is up to 91 bases in the private test set and is only up to 68 bases in the public and training sets. Thus, the authors hypothesized that bases 69–91 in the private test set would have a higher loss, contributing to the private test set's worse performance. We calculated the MCRMSE scores and plotted labels versus predictions for the first 68 bases and bases 69–91 in the private test set, respectively. Surprisingly, as shown in Fig. 6, our model performed better on bases 69–91 than in the first 68 bases. The MCRMSE for the first 68 bases is 0.405, and the MCRMSE for the bases 69–91 is only 0.317. Thus, the later bases do not contribute to the worse performance on the private test set.
Therefore, the authors hypothesized that the sequences of the two test sets are distinct, which contributes to the gap in performance. We analyzed the training sequences, public test set, and private test set using dimension reduction techniques following the suggestions from the Kaggle posts. We encoded the sequences as an array of integers and performed t-SNE dimensionality reduction on them. The sequences of the private test set are truncated to 107 bases to ensure they have the same dimension as that in the training and public test sets. The result of the t-SNE reduction on sequences is plotted in Fig. 9 . We found that the private test sequences were sequences that perform a completely different distribution compared to the training and public test sets. Although we truncated the sequences in the private test set, all sequences, including the training, public, and private test sets, are not complete RNA molecules and were truncated before they were released. Thus, we believe the distribution difference contributes to the worse performance on the private test sets. We believe that data augmentation techniques are necessary to further improve the model and help it generalize better on the private test sets.
Fig. 9.

t-SNE reduction on the sequences in the training set, public test set, and private test set.
Additionally, a key important argument is that reducing the length of the poly(A) tail can have beneficial and significant roles in cell biology and vaccine production as observed in this study. Short poly(A) tails are needed in embryos to repress translation until the appropriate stage of development is reached (Subtelny et al., 2014). Furthermore, short poly(A) tails that tend to denote mRNAs are essential for early development and may be used to control translation in a dose and time-dependent manner (Gohin et al., 2014). Thus, the optimal length of poly(A) was considered in this study and is reported to be 250.
5. Model analysis and validation
5.1. Inferential analysis
We can prove the model from several different angles to capture the model’s behavior and understand where the model sees necessary signals for its prediction task and how bases at different positions can affect the model’s attention. We break down our inference tasks into three buckets: point to point analysis, point to sequence analysis, and sequence to point analysis. We restricted our focus to a single reactivity metric (reactivity) from the overall five available in each analysis. Each of these analyses could be extended to the other reactivity metrics as well.
5.1.1. Point to point analysis
In point-to-point analysis, we aim to understand the effect the original base at position B has on its reactivity. The effect is normalized by the predicted reactivity values that position B could have in all its perturbations. Once the proposed ICM has been run, we have a L × 4 × S × 5 × L multi-dimensional array, where L is the sequence length (107) and S is the number of sampled sequences (250). To understand the effects of base position B on its reactivity, we must compute the original normalized reactivity, which is the original reactivity normalized by the reactivities of all perturbed reactivities at position B. To put it more concretely, we compute the following mean:
| (2) |
where µ is the vector of reactivity values, the B in superscript is the index of µ, the B in the subscript is the base position we have perturbed, and N is the modified base value perturb to. The original reactivity at position B is normalized as follows:
| (3) |
where O is the original reactivity value at position B. We display a sampling of the sequence reactivity profiles produced using this method. The other sequences displayed similar behavior to the sequences displayed here.
During the experiments, each sequence expressed noticeably different normalized effects across their bases. The effects for sequence 135, for example, oscillate between slightly below 0 to 0.4 for most of its positions, but position 75 expressed a large effect size, at around 1. Meanwhile, several base positions in sequence 20 have a noticeable effect size relative to 0, with position 20 and position 75 producing the most pronounced effects.
Interestingly, the profiles of both sequences revealed a large effect size at 75. To investigate this behavior, we averaged the absolute normalized effect of each sequence, and the result is displayed in the “Average Absolute Normalized Effect” plot (bottom of Fig. 10 ). The average absolute effect confirmed this pattern: position 75 appears to have an outsized effect on its reactivity value relative to other bases, whose averaged effect mellows below 0.4. Another noticeable pattern is the behavior between the 85th and 100th positions. In most of the sequences, position 80 has a low effect, position 85 has a slightly higher effect than does position 80, position 90 has more significant effect than does position 85, position 95′s effect drops compared to position 90, position 100′s effect drops to nearly 0, and finally position 105 has a very slightly larger effect size than position 100. Recall that in our training data, we do not have reactivity values for base positions higher than 68. Thus, the patterns that we see for position 75 and above may simply be artifacts of our model and our data construction. Upon further examination of position 75, we found that the original base pair was uracil (U) for all 2096 training sequences, which likely led to a more significant perturbation effect than that in other bases. In general, it appears that our model suggests that only a few positions have a large effect (in magnitude) on their reactivity values, while many of the positions in the sequence, especially near the middle, have minimal effect on their reactivity scores.
Fig. 10.

Absolute average base to sequence reactivity plot: (a) effects at position 25 base; (b) at effects position 85 base; and (c) effects at position 100 base.
5.1.2. Point to sequence analysis
In point to sequence analysis, we aim to understand the effect of changing the base value at position B on the rest of the sequence's reactivity values and how they vary by the base value we perturb B. To understand how perturbing base values at position B affects the whole sequence, we compute the average absolute base to sequence effect for every possible perturbation at position B. Specifically, for position B, we compute the 250 sequence reactivity profiles for all four possible perturbations. We average the absolute reactivity profile over the 250 sequences for each perturbation, and thus, we are essentially looking at the effect that perturbing position B to base value N has on average upon the reactivity values of the entire sequence. In concrete terms, the following for perturbation of position B to base value N was computed as follows:
| (4) |
where,
| (5) |
where i is the sequence number, B is the base position we are perturbing, N is the base value we perturb to, and j is the position in the sequence. Thus, ΛB, N is the vector of average absolute reactivity effect for perturbing base position B to base value N. We show a sampling of the results below. Each plot overlays ΛB, N for N = A, C, G, U for a select few positions B. Note that the positions for which we have selected plots show patterns similar to their relative neighbors’ average absolute base to sequence profiles.
Fig. 10 shows that positions and the sequence have some similar features and some markedly different features. All positions have tapering effects; that is, the influence of a given position B on reactivity values of positions far away from B is negligible. Changing the base value of position 25, in particular, has steep effects on very close neighbors, with the effect tapering off very quickly as we move away from position 25 in either direction. position 100 reflects a similar pattern. However, position 85 appears to sustain its effect more sharply because it results in spikes of reactivity for bases more than 15 positions away. Generally, these patterns show the model does not appear to influence changing one base located very far.
Interestingly, the effect size also differs. While positions 25 and 85 appear to have relatively similar effect sizes, around 0.3 to 0.4 absolute effect on reactivity, position 100 appears to much larger magnitude of effect size, reaching 0.8 when perturbed to U. For perturbations to other bases, however, its effect size is comparable to that of positions 25 and 85. This analysis suggests that the model has a “short-term” attention span; that is, the model does not tend to use information from positions far away from B when predicting the reactivity value of B.
5.1.3. Sequence to point analysis
In sequence to point analysis, we aim to understand the effect of perturbing the sequence located some distance (offset) away from a given position in the sequence. Precisely, we measure the reactivity at position B after perturbing bases +/− offset away from that position. The authors computed the effect of the perturbation by subtracting the non-perturbed (old) value from the perturbed (new) value. These relative effects were computed in a sliding window fashion across all base positions in a sequence. We group these effects by the offset and the base type (U, C, A, or G) that the base was perturbed. We can then compute the mean effect of perturbations at the offset for all base positions and sequences. The results are displayed in Fig. 11 .
Fig. 11.

Experimental results analysis for a sequence to base. Each figure describes the effect of perturbing a base, offset bases away from the reference. The left figure describes the mean absolute difference, whereas the right figure describes just the mean difference.
As depicted in Fig. 11 (left), the perturbations with small offsets have the most significant average absolute effect, with the maximum effect realized when perturbing the reference base itself (offset = 0). The effect dissipates as the offset increases in magnitude, meaning that moving the perturbation further and further away corresponds to a decrease in the average effect. This makes sense intuitively because the chemical properties of a base distant in linear sequence will, on average, have a smaller effect on the chemical properties of the reference base. This result serves primarily as a sanity check that the model behaves as we might expect it.
Fig. 11 (right) depicts the mean raw difference in the predicted perturbation. We notice that the mean raw effects of perturbing bases beyond about +/− 25 are near zero, which suggests that the absolute effect observed (left) at those offset values may simply be noise-amplified by the absolute value. However, within an offset of magnitude 10, we do see a noisy effect on the reactivity. Interestingly, we see a variable effect based on the perturbation type. On average, perturbations to U and A (red and purple traces) appear to have a local destabilizing effect (increase the base’s reactivity), while perturbations to C and G appear to have local stabilizing effects. This finding confers a biological understanding of RNA base pairing. Bases G and C form a base-pair bond with three hydrogen bonds, whereas bases A and U form a base-pair bond with two hydrogen bonds. Thus, a perturbation to A or U nearby has the likely effect of decreasing the number of base pair bonds by one, which has a destabilizing effect. Conversely, a perturbation to G or C nearby has the likely effect of increasing the number of base pair bonds by one, which has a stabilizing effect. Although these observations are preliminary, it is encouraging, because it indicates that our model appears to understand these differences in base pairing despite never being “taught” the underlying chemistry of the base pair bonding.
5.2. Model complexity
The recent success of neural networks has sparked renewed interest in sequence-based prediction and drug discovery research. Deep learning's success in a wide variety of fields is due to the rapid development of computational resources (e.g., GPU), the large training data availability, and the DL effectiveness in extracting latent representations from data (e.g., texts (Akbar et al., 2021), video, and image (Muneer et al., 2021).
Encouraged by the CNNs success in the computer vision field, a wide number of methods are being developed concurrently that redefine the concept of convolution for graph data. These techniques are referred to as convolutional graph neural networks (ConvGNNs). ConvGNNs and recurrent graph neural networks (RGNN) are closely related. Rather than iterating over node states using contractive constraints, ConvGNNs address cyclic mutual dependencies structurally by employing a set number of layers with varying weights in each layer. Since GCN (Singhal, 2020) overcame the barrier between spectral and spatial approaches, spatial-based methods have grown in popularity in recent years due to their remarkable efficiency, flexibility, and generality. To improve the ConvGNNs training efficiency, several methods have been proposed in the literature, such as
GCN (Singhal, 2020) is frequently essential to save the entire graph's data and intermediate transitions in memory. However, the full-batch training technique for ConvGNNs is greatly affected by the memory overflow problem, which is magnified when a graph has millions of nodes. GraphSage (Hamilton et al., 2017) presents a batch-training method for ConvGNNs in order to save memory. It samples a tree rooted at each node using a fixed sample size by recursively expanding the root node's vicinity by K steps. GraphSage calculates the root node's hidden representation for each sampled tree by hierarchically aggregating hidden node representations from bottom to top. A different work suggested by (Giulini and Potestio, 2019) uses CNN for structural analysis of proteins in molecule, and the network-based method takes less than 5 min to process the full training set and predict the appropriate eigenvalues on a single-core CPU.
As shown in Table 9 , GCN (Singhal, 2020) is the baseline that conducts full-batch training. GraphSage saves memory by sacrificing time efficiency as a cost. Meanwhile, when K and r increase, the time and memory complexity of GraphSage increases exponentially. However, in our suggested hybrid model, we used GraphSage, which requires significantly less computational memory than CNN. Complexity reduction is critical for deploying large CNNs models in mRNA sequence degradation with limited hardware and energy resources.
Table 9.
Comparison of the time and memory complexity of ConvGNN learning models.
| Complexity | GCN (Singhal, 2020) | GraphSage (Hamilton et al., 2017) | CNN (Giulini and Potestio, 2019) |
|---|---|---|---|
| Time | N/A | ||
| Memory | N/A |
Where n denotes the number of nodes in total. The total number of edges is denoted by m. The number of layers is denoted by K. The batch size is denoted by s. r is the number of neighbors that each node is sampled. For simplicity, the dimensions of the node's hidden features, represented by d, remain constant.
5.3. Comparison with the literature
To predict the COVID-19 mRNA vaccine degradation, we were unable to find any research contribution that has been evaluated, but we have compared our contribution with the two recently proposed models for mRNA reactivity prediction. However, authors in (Singhal, 2020) have suggested three single-DL methods (LSTM, GRU, and GCN) to predict mRNA vaccine degradation. The authors reported that among the three methods developed GRU performed the best with an accuracy of 76%. However, the critical drawback is that a single algorithm has limited accuracy and cannot capture the mRNA degradation features. Additionally, the authors did not consider the stability of the vaccine and only showed the reactivity prediction, unlike our proposed study that suggested two hybrid DNN models and conducted extensive experimentation to predict the RNA sequence degradation and the stability affect was considered by analyzing B cell epitopes present in the mRNA with a different position. This proves the significance of the model from several different angles to capture the model’s behavior and understand where the model sees necessary signals for its prediction task and how bases at different positions can affect the model’s attention.
Additionally, authors in (Qaid et al., 2021) proposed a bidirectional GRU integrating with the LSTM model. This model has been tested and evaluated with the same benchmark dataset provided by Stanford University scientists. However, only the MCRMSE score was reported, and no further experimentation was conducted on temperature storage conditions and vaccine stability, which is a vital element to evaluate the model behavior and its effectiveness. This key limitation makes the model suggested in (Qaid et al., 2021) not practical for accurately predicting the vaccine stability under real storage conditions and during temperature excursions. Therefore, a further comparison is conducted in the next section with the top state-of-the-art Stanford models.
5.4. Comparison with the Winning Models from Kaggle
The mRNA dataset used in this study is derived from a Kaggle competition, which was launched on 11 September 2020 and lasted 26 days. However, we have conveniently compared our model’s performance to others from the competition. Many of the top-performing competitors have released informal write-ups describing their approaches and general architectures. In Fig. 12 , we compare the proposed best hybrid model’s performance (the GCN with GRU model) against the top-three performing models based on MCRMSE measure to ensure fair comparative analysis since this measure was used in “OpenVaccine: COVID-19 mRNA Vaccine Degradation Prediction” competition. We observed that our best model outperformed the winning models from the Kaggle competition, although only by 0.04 MCRMSE on the private test set. Notably, many of the leading models achieved very similar loss scores (around 0.34 overall), and the difference from 1st to 10th place is marginal (0.00375). This observation suggests that the top-performing models maxed out their performance at around the same level. What might account for the gap in performance between our best model and the best models of the Kaggle competition would be interesting to explore. As a disclaimer, we observed that because we were not participating directly in the competition, we prioritized constructing and interpreting the proposed models rather than only improving the proposed scores on the Kaggle leaderboard.
Fig. 12.

Comparison of our top-performing model (far right) against the three top-performing models from the Kaggle competition.
Nevertheless, a comparison of the different approaches is also useful. Many of the top competitors have reported basing their final models on an architecture released about midway through the competition: “AE pretrain + GCN + Attention.” This architecture uses a graph convolutional network fit with attention modules. Its weights are pre-trained using an auto-encoder-like and unsupervised process wherein inputs are reconstructed.
After random dropout in the sequence, we observed that this approach is similar to what we performed when pretraining the node-embedding in the GCN. Indeed, pre-training the model to recover details of the RNA sequence appears to benefit training. We also note the addition of attention in this architecture. Future work could investigate the benefits of using attention with our model architecture. Another general theme of the top-performing models is extensive and creative data augmentation. As mentioned earlier, the overall dataset is relatively small, and thus, data augmentation strategies would be essential to avoid pitfalls like overfitting. Fig. 9 shows that the private test set is distributional distinct from the training and public test sets and thus, any methods that successfully augment training data to account for this difference would benefit model generalization to the private test set. Competitors have reported various data augmentation strategies such as pseudo labeling, inverting the sequence, and perturbing the RNA sequence at various base pairs. Future work could investigate robust and effective methods for data augmentation in RNA structure data.
6. Conclusions
We investigated the utility and interpretability of hybrid deep neural network architectures for the relevant problem of predicting mRNA sequence degradation. We find that GCNs consistently outperformed standard CNN architectures for the task of base-wise reactivity prediction. However, in silico vaccine prediction and design had a high efficacy value and emphasized vaccine stability considering B-cell and T-cell epitopes. Two-hybrid DNN algorithms were proposed in this research. These algorithms used the AI-based approach to rapidly predict mRNA base-resolution degradation of RNA at each base of an RNA molecule, thereby implementing a new method for achieving much higher speed and efficiency in silico vaccine design. The aim is to predict the possible vaccine mRNA degradation directly without having to perform a large number of different predictions. We can avoid at least 95 percent of unnecessary predictions by allowing the machine to evaluate and predict the reactivity using this AI-based approach. Additionally, increasing the availability of ground truth data, rational data augmentation strategies, and a better understanding of distributional shifts across different datasets would be most beneficial in developing and training better models. Despite existing data and modeling challenges, inference and preliminary interpretation of the proposed trained model still provided valuable insights. When predicting a given position B, we found that the model focuses on the identity of relevant bases near B’s vicinity. The proposed approach allows a researcher to predict base-wise reactivity, degradation at high temperature, and pH with magnesium for a new virus and verify its quality in less than an hour.
Additionally, the model can recover without prior knowledge, the effects of physical and chemical differences or properties in base pair bonding characteristics (i.e., the H-bond count in G-C versus A-U). These preliminary investigations suggest a well-trained model with access to a representative dataset could provide valuable clues to researchers working to understand essential mRNA degradation factors. In future, we aim to incorporate the improved therapeutic efficacy of mRNA COVID-19 vaccine. Finally, a well-trained model may guide engineering insights into the process of developing synthetic mRNA molecules.
7. Data Availability Statement
The proposed model source code is available on https://gitlab.com/hackshields/rna-paper, and the dataset used in this study also available on https://www.kaggle.com/c/stanford-covid-vaccine. We expect researchers to add new ideas to take this model in interesting directions.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This research received no external funding.
Footnotes
Peer review under responsibility of King Saud University.
References
- A dose-confirmation study to evaluate the safety, reactogenicity and immu- nogenicity of vaccine CVnCoV in healthy adults for COVID-19. https:// clinicaltrials.gov/ct2/show/NCT04515147. Accessed March 20, 2021.
- A phase 1/2/3, placebo-controlled, randomized, observer-blind, dose-finding study to evaluate the safety, tolerability, immunogenicity, and efficacy of SARS- CoV-2 RNA vaccine candidates against COVID-19 in healthy individuals. pfe- pfizer.com. https://pfe-pfizercom-d8-prod.s3.amazonaws.com/2020-11/C4591 001_Clinical_Protocol_Nov2020.pdf. Accessed March 20, 2021.
- A study to evaluate efficacy, safety, and immunogenicity of mRNA-1273 vac- cine in adults aged 18 Years and older to prevent COVID-19. https://clini caltrials.gov/ct2/show/NCT04470427. Accessed March 20, 2021.
- Ahuja A.S., Reddy V.P., Marques O. Artificial intelligence and COVID-19: A multidisciplinary approach. Integr. Med. Res. 2020;9(3) doi: 10.1016/j.imr.2020.100434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar N.A., Darmayanti I., Fati S.M., Muneer A. Deep Learning of a Pre-trained Language Model's Joke Classifier Using GPT-2. J. Hunan Univ. Natural Sci. 2021;48(9) [Google Scholar]
- Arba M., Nur-Hidayat A., Usman I., Yanuar A., Wahyudi S.T., Fleischer G., Brunt D.J., Wu C. Virtual screening of the Indonesian medicinal plant and zinc databases for potential inhibitors of the rna-dependent rna polymerase (Rdrp) of 2019 novel coronavirus. Indonesian J. Chem. 2020 [Google Scholar]
- Bong C.L., Brasher C., Chikumba E., McDougall R., Mellin-Olsen J., Enright A. The COVID-19 pandemic: effects on low-and middle-income countries. Anesthesia Analgesia. 2020 doi: 10.1213/ANE.0000000000004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyunghyun Cho, Bart Van Merriënboer, Dzmitry Bahdanau, and Yoshua Bengio. On the properties of neural machine translation: Encoder-decoder approaches. arXiv preprint arXiv:1409.1259, 2014.
- Chung Y.H., Beiss V., Fiering S.N., Steinmetz N.F. COVID-19 vaccine frontrunners and their nanotechnology design. ACS Nano. 2020;14(10):12522–12537. doi: 10.1021/acsnano.0c07197. [DOI] [PubMed] [Google Scholar]
- Crommelin Daan J.A., Anchordoquy Thomas J., Volkin David B., Jiskoot Wim, Mastrobattista Enrico. Addressing the cold reality of mRNA vaccine stability. J. Pharm. Sci. 2021;110(3):997–1001. doi: 10.1016/j.xphs.2020.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CureVac says its COVID-19 vaccine can be stored at standard refrigerator temperature. https://www.pmlive.com/pharma_news/curevac_says_its_covid-19_vaccine_can_be_stored_at_standard_refrigerator_temperature_1356911. Accessed March 20, 2021.
- CureVac's COVID-19 vaccine candidate, CVnCoV, suitable for standard fridge temperature logistics. https://www.curevac.com/en/2020/11/12/curevacs-covid-19-vaccine-candidate-cvncov-suitable-for-standard-fridge-temperature-logistics/. Accessed March 20, 2021.
- Danaee Padideh, Rouches Mason, Wiley Michelle, Deng Dezhong, Huang Liang, Hendrix David. bprna: largescale automated annotation and analysis of rna secondary structure. Nucl. Acids Res. 2018;46(11):5381–5394. doi: 10.1093/nar/gky285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamie Ducharme. Why you may not be able to get pfizer’s frontrunner covid-19 vaccine. Time magazine, Nov 2020.
- David K Duvenaud, Dougal Maclaurin, Jorge Iparraguirre, Rafael Bombarell, Timothy Hirzel, Alán Aspuru-Guzik, and Ryan P Adams. Convolutional networks on graphs for learning molecular fingerprints. in: Advances in neural information processing systems, pages 2224–2232, 2015.
- Esteban Ortiz-Ospina Max Roser, Hannah Ritchie and Joe Hasell. Coronavirus pandemic (covid-19). Our World in Data, 2020. https://ourworldindata.org/coronavirus. [DOI] [PMC free article] [PubMed]
- Fabre A.-L., Colotte M., Luis A., Tuffet S., Bonnet J. An efficient method for long-term room temperature storage of rna. European J. Hum. Genet. 2014;22(3):379–385. doi: 10.1038/ejhg.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frasconi P., Gori M., Sperduti A. A general framework for adaptive processing of data structures. IEEE Trans. Neural Networks. 1998;9(5):768–786. doi: 10.1109/72.712151. [DOI] [PubMed] [Google Scholar]
- Giulini M., Potestio R. A deep learning approach to the structural analysis of proteins. Interface focus. 2019;9(3):20190003. doi: 10.1098/rsfs.2019.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohin M., Fournier E., Dufort I., Sirard M.A. Discovery, identification and sequence analysis of RNAs selected for very short or long poly A tail in immature bovine oocytes. Mol. Hum. Reprod. 2014;20(2):127–138. doi: 10.1093/molehr/gat080. [DOI] [PubMed] [Google Scholar]
- Gori, M., Monfardini, G. and Scarselli, F., 2005, July. A new model for learning in graph domains. in: Proceedings. 2005 IEEE International Joint Conference on Neural Networks, 2005. (Vol. 2, pp. 729-734). IEEE.
- Will Hamilton, Zhitao Ying, and Jure Leskovec. Inductive representation learning on large graphs. in: Advances in neural information processing systems, pages 1024–1034, 2017.
- Hamilton W.L., Ying R., Leskovec J. Proceedings of the 31st International Conference on Neural Information Processing Systems. 2017. December. Inductive representation learning on large graphs; pp. 1025–1035. [Google Scholar]
- He Kaiming, Zhang Xiangyu, Ren Shaoqing, Sun Jian. Proceedings of the IEEE conference on computer vision and pattern recognition. 2016. Deep residual learning for image recognition; pp. 770–778. [Google Scholar]
- Hinton Geoffrey E, Salakhutdinov Ruslan R. Reducing the dimensionality of data with neural networks. science. 2006;313(5786):504–507. doi: 10.1126/science.1127647. [DOI] [PubMed] [Google Scholar]
- Hochreiter Sepp, Schmidhuber Jürgen. Long short-term memory. Neural Comput. 1997;9(8):1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- Information for healthcare professionals on pfizer BioNTech COVID-19 vaccine. UK department of Health and social care. Accessed March 20, 2021.
- International Federation of Pharmaceutical Manufacturers & Associations, 2020. THE COMPLEX JOURNEY OF A VACCINE. The Steps Behind Developing a New Vaccine. [online] Switzerland: IFPMA, pp.1-6. Available at: <https://www.ifpma.org/wp-content/uploads/2019/07/IFPMA-ComplexJourney-2019_FINAL.pdf> [Accessed 22 March 2021].
- Jackson N.A.C., Kester K.E., Casimiro D., Gurunathan S., DeRosa F. The promise of mRNA vaccines: a biotech and industrial perspective. npj Vaccines. 2020;5(1) doi: 10.1038/s41541-020-0159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyanathan M., Afkhami S., Smaill F., Miller M.S., Lichty B.D., Xing Z. Immunological considerations for COVID-19 vaccine strategies. Nature Rev. Immunol. 2020;20(10):615–632. doi: 10.1038/s41577-020-00434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Yang H., Ji W., Wu W., Chen S., Zhang W., Duan G. Virology, epidemiology, pathogenesis, and control of COVID- 19. Viruses. 2020;12:372. doi: 10.3390/v12040372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juvin P. Adult Vaccinations. Springer; Cham: 2019. Complexity of Vaccine Manufacture and Supply; pp. 1–5. [Google Scholar]
- Keshavarzi Arshadi A., Webb J., Salem M., Cruz E., Calad-Thomson S., Ghadirian N., Collins J., Diez-Cecilia E., Kelly B., Goodarzi H., Yuan J.S. Artificial intelligence for COVID-19 drug discovery and vaccine development. Front. Artificial Intelligence. 2020;3:65. doi: 10.3389/frai.2020.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederik P. Kingma and Jimmy Ba. Adam: A method for stochastic optimization, 2017.
- Kipf, T.N. and Welling, M., 2016. Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907.
- Lazarus J.V., Ratzan S.C., Palayew A., Gostin L.O., Larson H.J., Rabin K., Kimball S., El-Mohandes A. A global survey of potential acceptance of a COVID-19 vaccine. Nature Med. 2020:1–4. doi: 10.1038/s41591-020-1124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong-Hyun Lee. Pseudo-label: The simple and efficient semi-supervised learning method for deep neural networks. In Workshop on challenges in representation learning, ICML, volume 3, 2013.
- Peter Loftus, Jared Hopkins, and Bojan Pancevski. Moderna and pfizer are reinventing vaccines, starting with covid. Wall Street Journal, Nov 2020.
- Ronny Lorenz, Stephan H Bernhart, Christian Höner Zu Siederdissen, Hakim Tafer, Christoph Flamm, Peter F Stadler, and Ivo L Hofacker, 2011, Viennarna package 2.0. Algorithms for molecular biology, 6(1):26. [DOI] [PMC free article] [PubMed]
- Rune Lyngsø and Christian Pedersen, 2000, Rna pseudoknot prediction in energy-based models. Journal of computational biology : a journal of computational molecular cell biology, 7:409–27. [DOI] [PubMed]
- Metzker M.L. Sequencing technologies—the next generation. Nature Rev. Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Moderna announces longer shelf life for its COVID-19 vaccine candidate at refrigerated temperatures. https://www.businesswire.com/news/home/202 01116005606/en/. Accessed March 20, 2021.
- Muneer A., Ali R.F., Fati S.M., Naseer S. COVID-19 recognition using self-supervised learning approach in three new computed tomography databases. J. Hunan Univ. Natural Sci. 2021;48(9) [Google Scholar]
- Naseer S., Ali R.F., Muneer A., Fati S.M. IAmideV-deep: Valine amidation site prediction in proteins using deep learning and pseudo amino acid compositions. Symmetry. 2021;13(4):560. [Google Scholar]
- Naseer S., Ali R.F., Fati S.M., Muneer A. iNitroY-Deep: computational identification of nitrotyrosine sites to supplement carcinogenesis studies using deep learning. IEEE Access. 2021;9:73624–73640. [Google Scholar]
- OpenVaccine. Openvaccine: Covid-19 mrna vaccine degradation prediction. Stanford University, Eterna, Sept 2020.
- Pardi Norbert, Hogan Michael J., Porter Frederick W., Weissman Drew. mRNA vaccines—a new era in vaccinology. Nature Rev. Drug Discov. 2018;17(4):261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qaid T.S., Mazaar H., Alqahtani M.S., Raweh A.A., Alakwaa W. Deep sequence modelling for predicting COVID-19 mRNA vaccine degradation. PeerJ Comput. Sci. 2021;7 doi: 10.7717/peerj-cs.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seetin Matthew G, Kladwang Wipapat, Bida John P, Das Rhiju. RNA Folding. Springer; 2014. Massively parallel rna chemical mapping with a reduced bias map-seq protocol; pp. 95–117. [DOI] [PubMed] [Google Scholar]
- Singhal, A., 2020. Application and Comparison of Deep Learning Methods in the Prediction of RNA Sequence Degradation and Stability. arXiv preprint arXiv:2011.05136.
- Subtelny A.O., Eichhorn S.W., Chen G.R., Sive H., Bartel D.P. Poly (A)-tail profiling reveals an embryonic switch in translational control. Nature. 2014;508(7494):66–71. doi: 10.1038/nature13007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The cold truth about COVID-19 vaccines. https://www.genengnews.com/news/ the-cold-truth-about-covid-19-vaccines/. Accessed March 22, 2021.
- Laurens van der Maaten and Geoffrey Hinton. Visualizing data using t-sne. Journal of machine learning research, 9(Nov):2579–2605, 2008.
- van Hoof A., Parker R. Messenger rna degradation: beginning at the end. Curr. Biol. 2002;12(8):R285–R287. doi: 10.1016/s0960-9822(02)00802-3. [DOI] [PubMed] [Google Scholar]
- Wang J., Peng Y., Xu H., Cui Z., Williams R.O. The COVID-19 vaccine race: challenges and opportunities in vaccine formulation. AAPS PharmSciTech. 2020;21(6):1–12. doi: 10.1208/s12249-020-01744-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters Kyle E, Lucks Julius B. RNA Structure Determination. Springer; 2016. Mapping rna structure in vitro with shape chemistry and next-generation sequencing (shape-seq) pp. 135–162. [DOI] [PubMed] [Google Scholar]
- Zichao Yan, William L Hamilton, and Mathieu Blanchette, 2020, Graph neural representational learning of RNA secondary structures for predicting RNA-protein interactions. Bioinformatics, 36(Supplement_1):i276–i284. [DOI] [PMC free article] [PubMed]
- Zhang Y., Ma Z.F. Impact of the COVID-19 pandemic on mental health and quality of life among local residents in Liaoning Province, China: a cross-sectional study. Int. J. Environ. Res. Public Health. 2020;17:2381. doi: 10.3390/ijerph17072381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The proposed model source code is available on https://gitlab.com/hackshields/rna-paper, and the dataset used in this study also available on https://www.kaggle.com/c/stanford-covid-vaccine. We expect researchers to add new ideas to take this model in interesting directions.