

Abstract
Introduction:
Adrenocortical carcinoma is an aggressive cancer with a poor prognosis. Long noncoding RNAs are differentially expressed in cancer patients and contribute to cellular homeostasis, survival, and metastasis. We hypothesize that our novel C-terminal Hsp90 inhibitor KU758 can effectively target adrenocortical carcinoma cells and favorably alter long noncoding RNA expression.
Methods:
Cell viability after KU758 treatment was measured in the adrenocortical carcinoma cell lines SW13, RL251, and NCI-H295R by MTS assay. Cellular mobility and metastatic potential after Hsp90 inhibition was measured through migration, invasion, and aggregate formation assays. β-catenin activity in NCI-H295R cells was determined by immunofluorescence and polymerase chain reaction. Long non-coding RNA expression was determined by polymerase chain reaction array after Hsp90 inhibition.
Results:
KU758 is selective for adrenocortical carcinoma cells with IC50 values of 0.6 to 2.4 μM. KU758 treatment can effectively reduce migration, invasion, and aggregate formation in NCI-H295R and SW13 cells. β-catenin activity is decreased after treatment with KU758. Treatment with KU758 is associated with overall statistically significant upregulation of long noncoding RNA expression, including the tumor suppressor GAS5, which is implicated in the β-catenin and mammalian target of rapamycin pathways in adrenocortical carcinoma.
Conclusion:
The novel C-terminal Hsp90 inhibitor KU758 is effective in the treatment of adrenocortical carcinoma cells and can significantly alter long noncoding RNA expression for tumor suppression.
Introduction
Adrenocortical carcinoma (ACC) is a rare but clinically aggressive endocrine cancer with an estimated incidence of approximately 1 to 2 people per million.1 Prognosis varies widely based on the patient stage at diagnosis, with 5-year survival rates ranging from 65% for localized disease to under 10% for metastatic disease.2 Despite advances in early diagnosis, changes in treatment paradigms, and improved understanding of molecular mechanisms, adrenal cancer patients continue to have a high risk of disease recurrence and survival is often less than 1 year for patients with stage IV disease.3 The combination of mitotane with multidrug cytotoxic chemotherapy (a platinum agent [cisplatin or carboplatin], etoposide, and doxorubicin, [ie, the “Italian Protocol”]) remains the most widely used treatment for advanced adrenal cancer but has a significant toxicity profile, which often limits its administration and duration of benefits.1 The clinical aggressiveness of this disease combined with the distinct lack of treatment options underscores the great need for the development of novel therapies for patients with ACC.
ACC has also been associated with many hereditary tumor syndromes, including Beckwith-Wiedemann syndrome and Multiple Endocrine Neoplasia Type 1, suggesting an important molecular/genetic role in the pathogenesis of this disease.4 Additionally, many sporadic ACC tumors are associated with key genetic markers, including mammalian target of rapamycin (mTOR) pathway dysregulation, IGF2 (insulin growth factor 2) overexpression, and β-catenin signaling activation.5,6 Extensive work has demonstrated that these pathways may be promising targets for treating adrenal cancer.
Heat shock chaperone proteins (Hsp90 and Hsp70), as part of a heterochaperone complex, play an important role in the folding and conformational maintenance of a diverse set of client proteins involved in cancer initiation and progression,7 including kinase proteins critical to the mTOR, IGF2, and β-catenin pathways. To date, pharmacological inhibition of Hsp90 has shown promising results in phase I/II clinical trials for various cancers.8 All Hsp90 inhibitors tested in the clinic bind to the N-terminal adenosine triphosphate binding site of Hsp90. However, N-terminal Hsp90 inhibitors, including 17-N-allylamino-17-demethoxygeldanamycin (17-AAG), 17-dimethylaminoethylamino-17-demethoxygeldanamycin, and others, have all had significant limitations, including side effects related to the induction of the pro-survival heat shock response, as well as dose-limiting toxicities including gastrointestinal and hepatotoxicity, preventing their development into U.S. Food and Drug Administration approved monotherapy anticancer agents.8 To overcome the shortcomings of N-terminal Hsp90 inhibitors, our group has looked at other mechanisms to inhibit Hsp90 and heterochaperone complex function through targeting the carboxyterminal binding site of the Hsp90 protein. The first small molecule identified to bind this target was novobiocin, but it had a poor inhibitory effect on tumors with IC50 values in many cancers well over 500 μM. We have since utilized our medicinal chemistry applications to develop several novel, more potent C-terminal Hsp90 inhibitors and have evaluated their efficacy in targeting several cancers in both in vitro and in vivo models.9–13
A possible mechanism by which some drugs, including Hsp90 inhibitors, may be able to exert its therapeutic effects is through differential expression of long noncoding RNAs (lncRNAs). LncRNAs belong to a class of RNA transcripts of more than 200 nucleotides that correspond to DNA regions that do not code for protein. As regulators of transcription, mRNA processing, and protein activity, lncRNAs are increasingly being identified for their ability to function as tumor suppressors or oncogenes.14 Specifically in adrenocortical cancer, lncRNA transcriptome analysis of ACC patients compared to the normal adrenal cortex demonstrated differential expression of lncRNAs, including lncRNAs that are known oncogenes or tumor suppressors.15 This supports the possibility that key lncRNAs may be targeted in ACC and be a possible mechanism by which anticancer therapies exert their specific effects.
In the present study, we demonstrate that our novel CT-Hsp90i, KU758, can effectively target ACC cells, and we evaluate possible mechanisms through which KU758 exerts its anticancer effects in this tumor as a possible novel therapeutic strategy for ACC.
Methods
Cell lines and reagents
Validated ACC cell lines SW13, RL251, and NCI-H295R were grown in appropriate culture medium. Briefly, NCI-H295R cells were grown in 1:1 Dulbecco’s Modified Eagle Media (DMEM):F12 nutrient mixture (Thermo Fisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA), and ITS (Thermo Fisher Scientific, Waltham, MA). ACC cell lines SW13 and RL251 were grown in DMEM supplemented with 10% fetal bovince serum (FBS) and 1% penicillin/streptomycin. Human lung fibroblast cell line MRC5 (ATCC) cells were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Novel C-terminal Hsp90 inhibitor KU758 (Fig 1) was a gift from Dr. Brian S. J. Blagg (University of Notre Dame, IN), and the N-terminus Hsp90 inhibitor 17-AAG was obtained from Sigma-Aldrich (St. Louis, MO).
Fig 1.
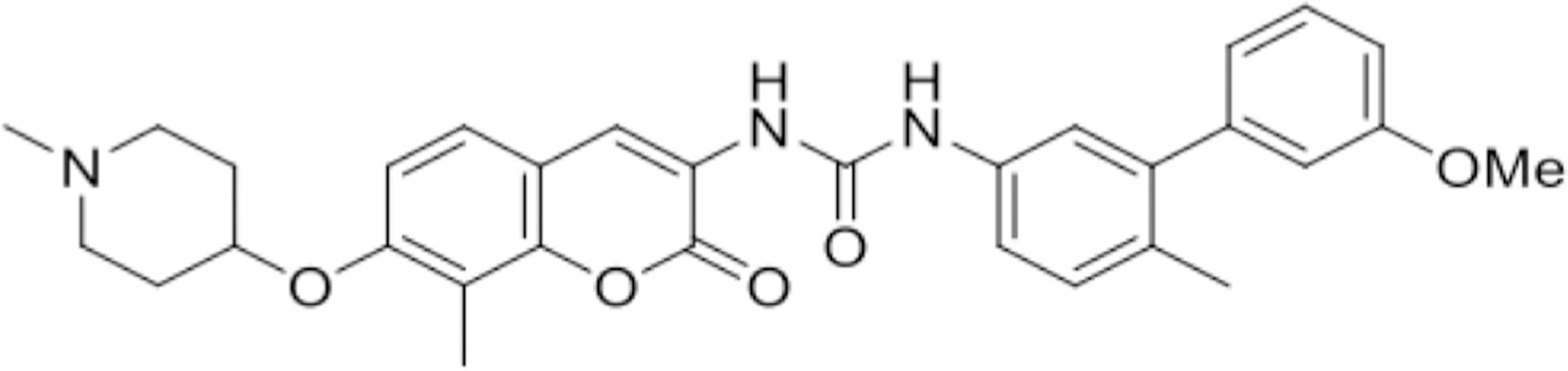
Chemical structure of novel C-terminal Hsp90 inhibitor KU758.
Cell proliferation assay
Equal numbers of non-cancerous fibroblast MRC5 and adrenocortical cancer NCI-H295R, SW13, and RL251 cells were plated in 96-well plates. Cells were then treated with serial concentrations of KU758 over 9 orders of magnitude. Following 72 hours of treatment, cell viability was assessed by the colorimetric CellTiter96 Aqueous MTS assay (Promega, Madison, WI) as per the manufacturer’s protocol. Absorbance at 490 nm was recorded on a BioTek Synergy 2 plate reader (BioTek Instruments, Inc, Winooski, VT) after the addition of MTS reagent, and cytotoxicity was expressed as a percentage of control untreated cells. The half-maximal inhibitory concentrations (IC50) were calculated using GraphPad Prism (GraphPad Company, San Diego, CA).
Western blot analysis
SW13 cells grown to 60% to 80% confluence were treated with 1.0 μM KU758 and 1.0 μM 17-AAG for 48 hours. Post treatment cells were collected, lysed, and proteins were quantified using bovine serum albumin protein assay (Thermo Fischer Scientific, Waltham, MA). Equal amounts of proteins were then separated on a sodium dodecyl sulfate–polyacrylamide gel electrophoresis, transferred onto a nitrocellulose membrane, and immunoblotted for Hsp70 protein. Actin was used as a loading control. Image J analysis was used for quantification of Western blots. Hsp70 protein signal intensity after treatment with DMSO, 1.0 μM KU758, and 1.0 μM 17-AAG was determined after normalization with actin signal intensity.
Trans-well migration and invasion assay
For the cell migration and invasion assays, NCI-H295R and SW13 cells were suspended in serum free medium with drug treatment (0.5–4.0 μM KU758 or 1 μM 17-AAG) as indicated. Equal amounts of cells were placed into the upper wells of 8-micron polycarbonate Boyden chambers (Corning, NY)). The wells were coated with 100 μL of 300 μg/mL reduced growth factor Matrigel (Corning, NY) for the invasion assay, and the standard chambers were used for the migration assay. The chambers were incubated for 24 hours with 10% FBS containing culture medium in the lower chamber as a chemo attractant. The invaded cells at the bottom of the chamber were fixed in 2% paraformaldehyde, stained with 1% crystal violet in 20% methanol for 20 minutes, washed, photographed, and counted using an EVOS microscope (Thermo Fisher Scientific, Waltham, MA) after the removal of noninvaded cells using a cotton swab.
Aggregate formation assay
NCI-H295R and SW13 cells were plated in ultralow attachment 6-well tissue culture plates (Corning, NY) and then treated with varying concentrations of KU758 (0.5–2.0 μM) or 17-AAG (1.0 μM) in complete MammoCult (human) medium (STEMCELL Technologies, Inc, Vancouver, Canada). Aggregates were photographed using an EVOS microscope after 48 hours.
mRNA isolation and real-time (RT) polymerase chain reaction (PCR)
RNA from the ACC cell lines NCI-H295R and SW13 after treatment with 2.0 μM KU758 for 24 hours was prepared using Qiagen RNA isolation kit (Qiagen Sciences, Hilden, Germany). Approximately 1 μg of RNA was reverse transcribed using the Superscript RT kit from Thermo Fisher Scientific (Waltham, MA). Quantitative PCR was performed in a one-step real-time polymerase chain reaction machine using gene specific primer sets as published. Relative gene expression levels were calculated using the delta delta CT method after normalization with internal glyceraldehyde 3-phosphate dehydrogenase (GAPDH) controls.
Immunofluorescence
NCI-H295R cells were treated with 1.0 to 2.0 μM KU758 or 1.0 μM 17-AAG for 24 hours. The cells were then fixed in paraformaldehyde for 15 minutes, washed with phosphate-buffered saline (PBS), and incubated with 5% normal goat serum for 1 hour for blocking. After washing, the fixed slides were then incubated overnight with anti-β-catenin primary antibody (Thermo Fisher Scientific, Waltham, MA). Cells were then washed with PBS and incubated with secondary goat anti-mouse Alexa Fluor 488 antibody (Cell Signaling Technology, Danvers, MA) for 2 hours and washed with PBS. For nuclear counterstaining, cells were incubated with DAPI. Expression of β-catenin was observed with an EVOS microscope with fixed exposure settings for all samples.
LncRNA expression analysis
NCI-H295R cells were treated with 1.0 to 2.0 μM KU758 or 1.0 μM 17-AAG for 24 hours. Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and measured by Nano Drop spectrophotometer (Thermo Fisher Scientific, Waltham, MA). 800 ng of RNA was transcribed to cDNA using the RT2 First Strand Kit (Qiagen, Hilden, Germany) and lncRNA expression was determined using SYBR Green quantitative PCR and the RT2 lncRNA PCR Array Human Cancer Pathway Finder (Qiagen, Hilden, Germany).
Statistical analysis
To determine the significance between different treatments two-tailed, unpaired Student’s t test was used. For the lncRNA PCR array, data analysis was performed through the Data Analysis Center from Qiagen (Hilden, Germany). Data are presented as mean values with error bars denoting standard deviation. P values less than .05 were considered statistically significant. All experiments were repeated in triplicate to ensure accuracy.
Results
KU758 is effective in reducing cell viability and proliferation of ACC cells without induction of the heat shock response
The effect of KU758 treatment on the ACC cell lines NCI-H295R, SW13, and RL251 and human lung fibroblast cell line MRC5 was determined by MTS assay. The half maximal inhibitory concentration (IC50) was determined from dose-response curves and reported in Fig 2. Treatment with KU758 for 72 hours had minor effects on fibroblast cell viability (IC50 = 4.39μM); however, KU758 was potent in ACC cells, with IC50 values ranging from 0.609 to 2.41 μM. Hsp70 expression after treatment with N-terminal and C-terminal Hsp90 inhibitor was evaluated by Western blot after treating SW13 cells with 1.0 μM 17-AAG and KU758, respectively. As seen in Fig 3, treatment with KU758 does not cause an upregulation in the heat shock response compared to vehicle (DMSO) only, while treatment with 17-AAG does cause a significant upregulation in Hsp70 levels.
Fig 2.
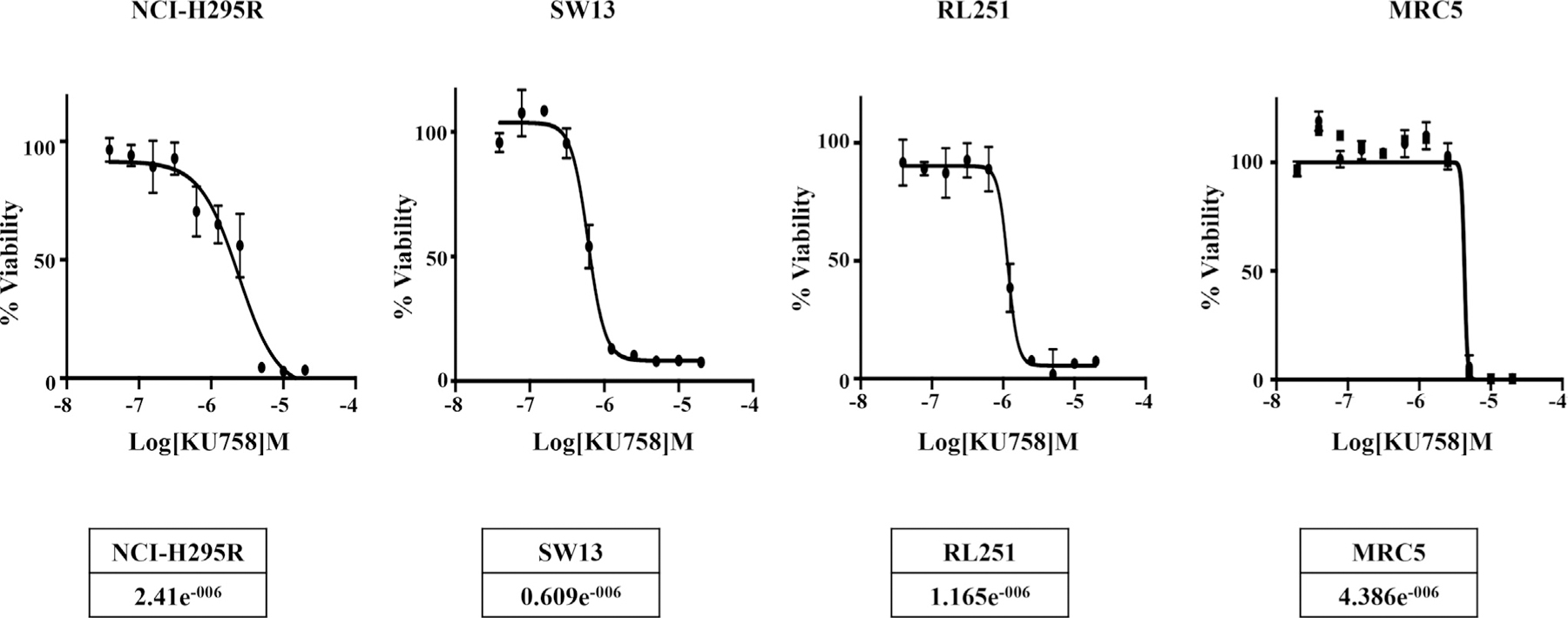
ACC cell lines NCI-H295R, SW13, and RL251 and fibroblast cell line MRC5 were treated with varying concentrations of KU758 and the viability of cells was measured using MTS assay. The IC50 values are shown. ACC, adrenocortical carcinoma.
Fig 3.
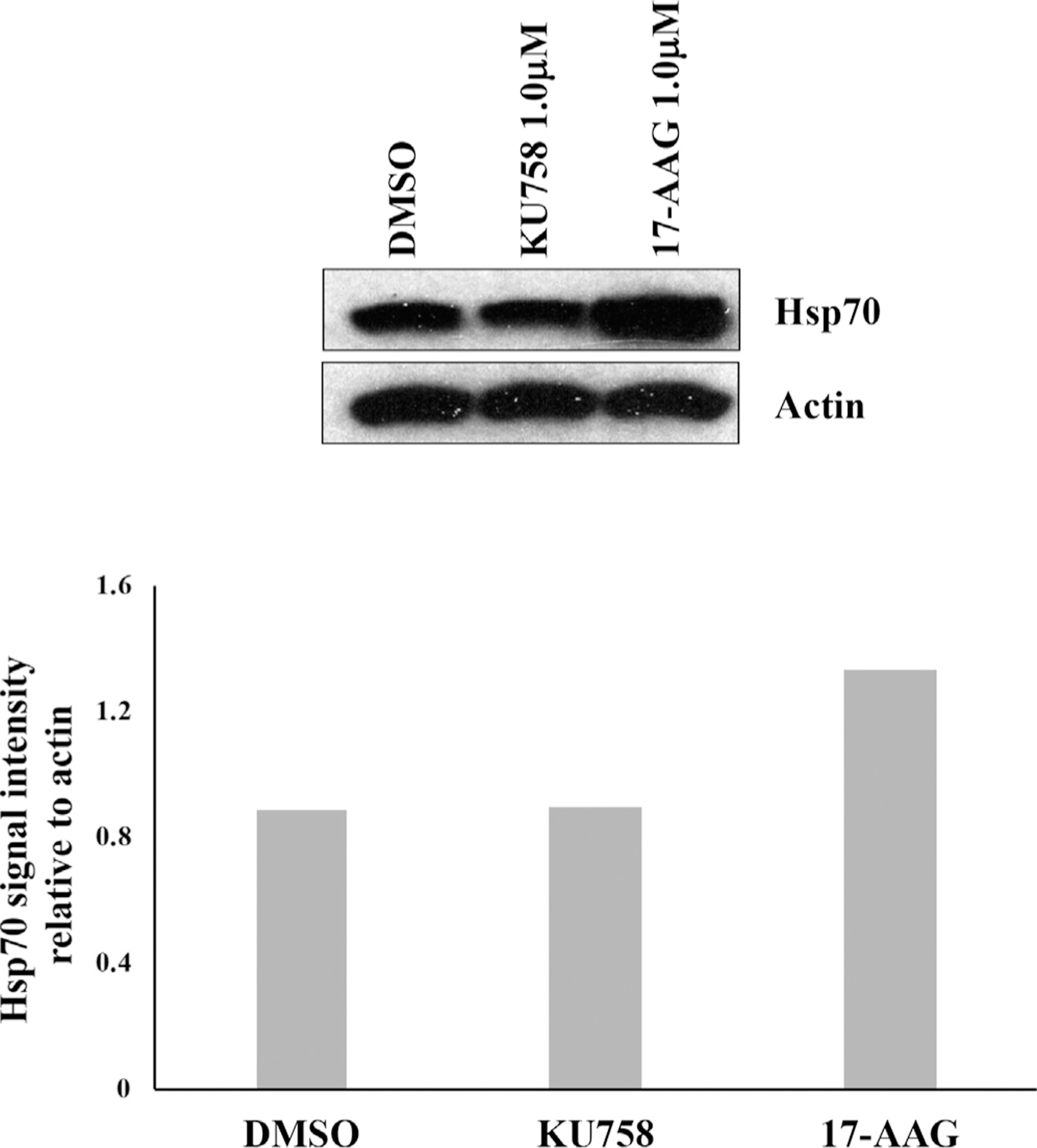
Treatment of SW13 cells with 1.0 μM KU758 does not cause upregulation of Hsp70 (heat shock response) compared to control (DMSO only). In contrast, treatment of SW13 cells with 1.0 μM 17-AAG causes induction of the heat shock response. Hsp70 signal intensity relative to actin is shown.
KU758 decreases migration and invasion of ACC cells
To examine whether Hsp90 inhibition decreases invasive potential of adrenal cancer cells, we analyzed cellular migration and invasion of NCI-H295R and SW13 cells after treatment with varying concentrations of KU758 and 17-AAG. As seen in Fig 4, both NCI-H295R and SW13 cells exhibit a dose-dependent decrease in both invasion and migration with KU758 treatment. For NCI-H295R cells, there is a significant reduction in migration by 32% with 1.0 μM KU758 and nearly total (98%) reduction in migration with 2.0 μM KU758 compared to vehicle (DMSO) only (P < .01). Treatment with 1.0 μM 17-AAG causes a 23% reduction in migration of NCI-H295R compared to DMSO (P = .05), which is slightly less than seen with 1.0 μM KU758 (P = .22). There is a similar dose-dependent decrease in NCI-H295R invasion after Hsp90 inhibition, with 65% reduction at even the lowest concentration of KU758, and 100% inhibition of invasion at 2.0 μM KU758 compared to DMSO (P < .01). By comparison, 1.0 μM 17-AAG causes a reduction in invasion by only 45% compared to DMSO, which is significantly less than the reduction in invasion seen after treatment with 1.0 μM KU758 (P = .03). Although SW13 cells are more sensitive to KU758 treatment based on cell viability assay, relatively higher concentrations are required for inhibition of migration and invasion compared to NCI-H295R cells. At a concentration of 2.0 μM KU758, there is a 79% reduction in migration and 86% reduction in invasion (P < .01 versus DMSO) and total inhibition of migration and invasion at a concentration of 4.0 μM KU758. Treatment of SW13 cells with 1.0 μM 17-AAG resulted in a 40% reduction of both invasion and migration (P = .01 versus DMSO); there is no difference found in reduction of invasion/migration after treatment with 1.0 μM 17-AAG compared to 1.0 μM KU758.
Fig 4.

Migration and invasion of NCI-H295R and SW13 cells decreases after treatment with KU758 in a dose-dependent manner with total inhibition of migration and invasion at 2.0 μM and 4.0 μM KU758 in NCI-H295R and SW13 cells, respectively. Migration and invasion after treatment of ACC cells with 1.0 μM 17-AAG are shown for comparison. ACC, adrenocortical carcinoma.
Treatment of ACC cells with KU758 decreases aggregate formation
Aggregate formation is implicated in the recurrence and metastasis of cancer. Therefore, to evaluate the efficacy of the novel Hsp90 inhibitor KU758 in targeting aggregate formation, the ACC cell lines NCI-H295R and SW13 were treated with varying concentrations of KU758 from 0.5 μM to 2.0 μM. As seen in Fig 5, when ACC cells are treated with KU758, there is a dose-dependent decrease in aggregate formation with near total inhibition at doses of both 1.0 μM and 2.0 μM KU758 in SW13 and NCI-H295R cells. However, when cells were treated with the N-terminal Hsp90 inhibitor 17-AAG at a concentration of 1.0 μM, there is comparable inhibition of aggregate formation in NCI-H295R cells only.
Fig 5.
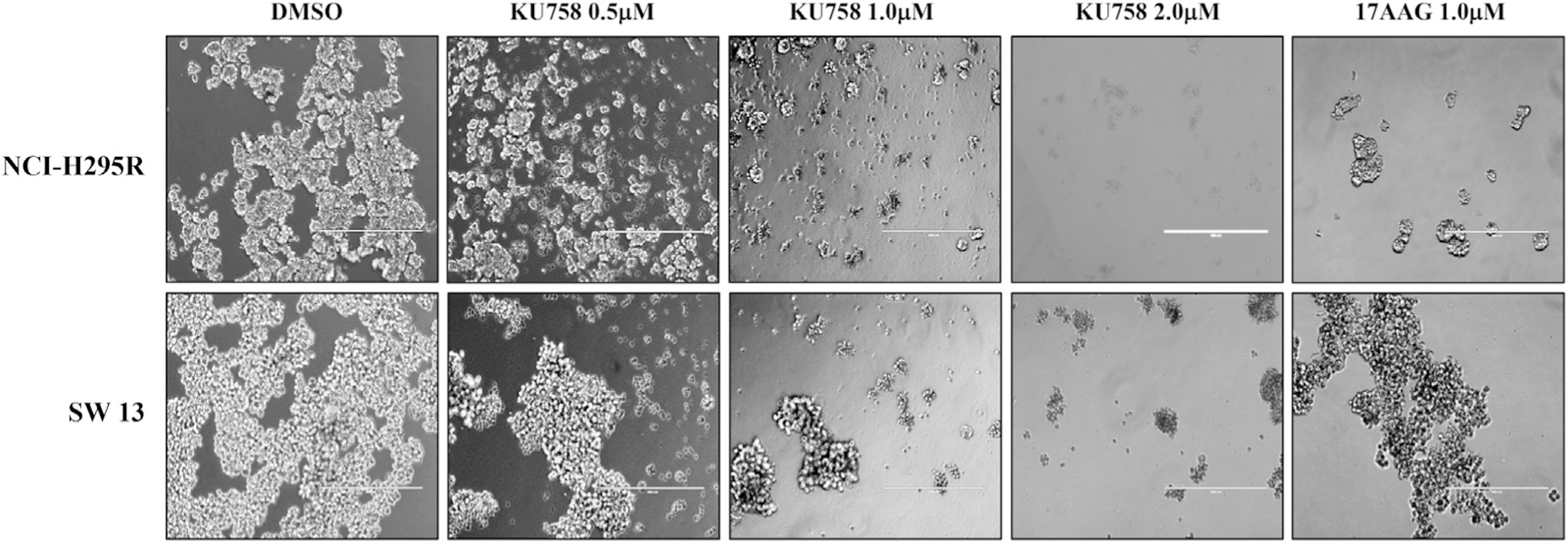
Treatment of NCI-H295R and SW13 with KU758 decreases aggregate formation in a dose dependent manner with near total inhibition at doses of both 1.0 μM and 2.0 μM KU758 in SW13 and NCI-H295R cells. Aggregate formation after treatment of ACC cells with 1.0 μM 17-AAG is shown for comparison. ACC, adrenocortical carcinoma.
Treatment of ACC cells with KU758 downregulates β-catenin signaling
Sequencing of ACC tumors has shown frequent mutations in the β-catenin gene CTNNB1 and activation of the Wnt/β-catenin pathway has been associated with higher tumor stage and decreased overall and disease-free survival. Therefore, to evaluate the effect of Hsp90 inhibition on the β-catenin pathway, NCI-H295R cells were treated with 1.0 to 2.0 μM KU758 for 24 hours. Cells were fixed with paraformaldehyde, and β-catenin levels were determined by immunofluorescence. As seen in Fig 6, treatment with KU758 causes a significant decrease in total cellular β-catenin levels. Additionally, the ACC cell lines NCI-H295R and SW13 were treated with 2.0 μM KU758 for 24 hours and mRNA expression levels of the β-catenin pathway dependent proteins Axin2 and c-MET were determined by RT-PCR. As shown in Fig 7, treatment with KU758 is associated with a 56% and 11% decrease in Axin2 levels in NCI-H295R and SW13 cells compared to vehicle (DMSO) only, respectively (P = .03 for NCI-H295R). Axin2 is a known target of the Wnt/β-catenin pathway, and expression of Axin2 is induced by Wnt signaling. Additionally, KU758 treatment is associated with a 72% and 25% decrease in c-MET expression compared to DMSO in NCI-H295R and SW13 cells, respectively (P < .01 for NCI-H295R). C-MET is important in noncanonical (nonWnt) mediated activation of β-catenin. Notably, the decrease in Axin2 and c-MET expression is greater with KU758 treatment of NCI-H295R cells compared to SW13 cells.
Fig 6.
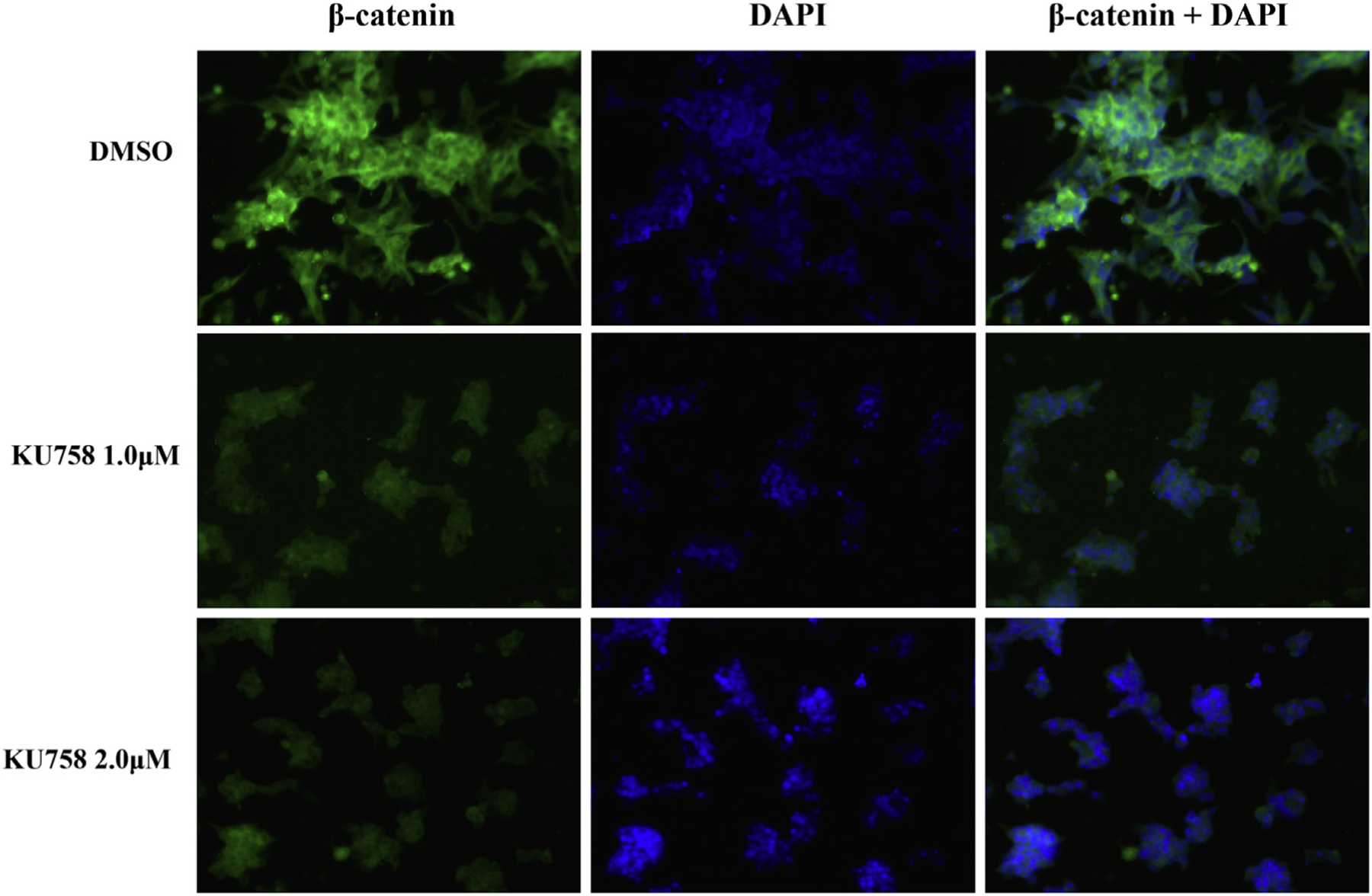
Treatment of NCI-H295R cells with KU758 decreases total cellular β-catenin levels by immunofluorescence in a dose dependent manner. Green staining = β-catenin; Nuclear blue staining = DAPI. (Color version of figure is available online.)
Fig 7.
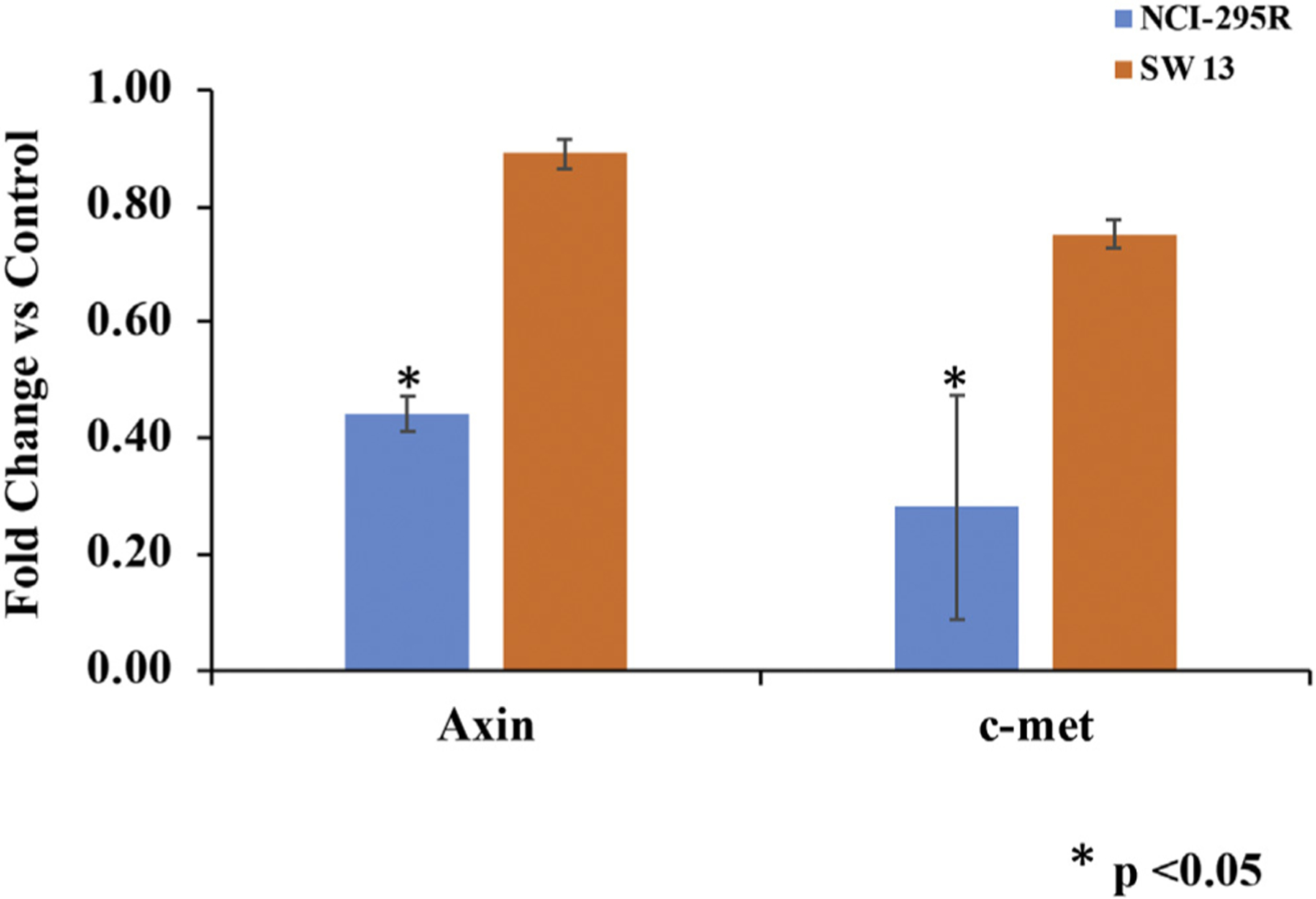
Treatment of ACC cells with 2.0 μM KU758 is associated with a 56% and 11% decrease in Axin2 levels in NCI-H295R and SW13 cells compared to control (DMSO only), respectively (P = .03 for NCI-H295R). KU758 treatment is associated with a 72% and 25% decrease in c-MET expression compared to control in NCI-H295R and SW13 cells, respectively (P < .01 for NCI-H295R). ACC, adrenocortical carcinoma.
Treatment of ACC cells with KU758 results in differential lncRNA expression
LncRNAs have been shown to be differentially expressed in adrenal cancer and have important roles in the regulation of cellular proliferation and survival. Therefore, to evaluate the effect of Hsp90 inhibition on lncRNA expression, NCI-H295R cells were treated with 1.0 to 2.0 μM KU758 or 1.0 μM 17-AAG for 24 hours. LncRNA expression analysis was determined by lncRNA cancer pathway profiler array from Qiagen, Hilden, Germany. As shown in Fig 8, treatment of adrenal cancer cells with KU758 is associated with overall upregulation of lncRNA expression, while treatment with 17-AAG is associated with overall downregulation of lncRNA expression. Specifically, treatment with KU758 is associated with statistically significant (greater than 2-fold, P < .05) upregulation of the tumor suppressors ADAMTS9-AS2, CAHM, GAS5, RMST, and ZFAS1, and the oncogenes BCAR4, SPRY4-IT1, CBR3-AS1, HIF1A-AS1, HULC, LINC00887, and LUCAT1. Treatment with 17-AAG is associated with statistically significant upregulation of the oncogenes BCAR4 and BLACAT1; downregulation of the tumor suppressors GACAT1, LINC00261, and TSIX; and downregulation of the oncogene, LSINCT5.
Fig 8.
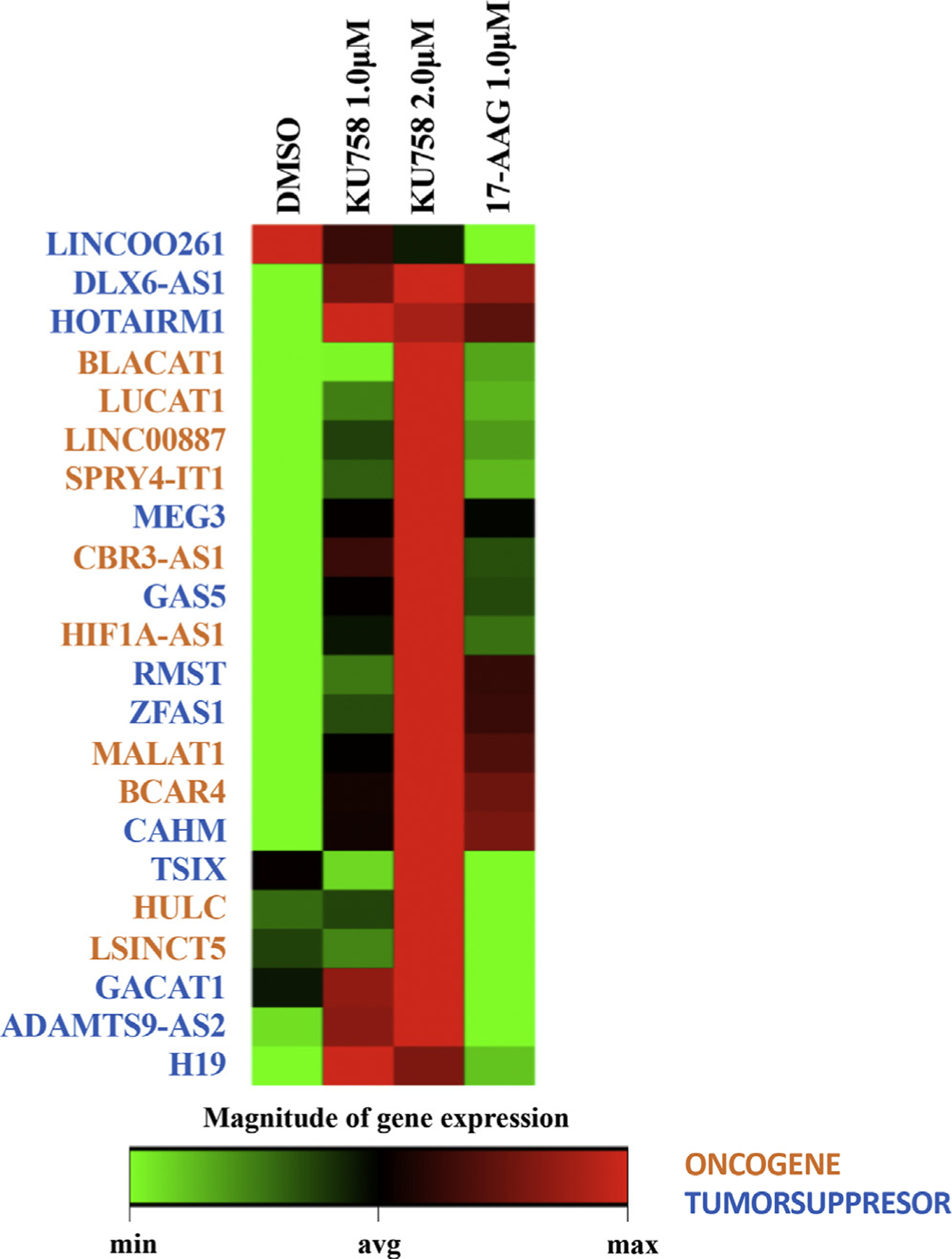
Treatment of NCI-H295R cells with KU758 causes statistically significant (greater than 2-fold, P < .05) upregulation of the lncRNAs ADAMTS9-AS2, CAHM, GAS5, RMST, ZFAS1, BCAR4, SPRY4-IT1, CBR3-AS1, HIF1A-AS1, HULC, LINC00887, and LUCAT1. Treatment with 17-AAG is associated with statistically significant upregulation of BCAR4 and BLACAT1 and downregulation of GACAT1, LINC00261, TSIX, and LSINCT5. Treatment of NCI-H295R cells with Hsp90 inhibition results in upregulation of MEG3, DLX6-AS1, H19, GAS5, HOTAIRM1, and MALAT1. Oncogenes are denoted in orange and tumor suppressors are denoted in blue. (Color version of figure is available online.)
Additionally, treatment of NCI-H295R cells with Hsp90 inhibition results in differential expression of several lncRNAs that have been demonstrated to be implicated in the pathogenesis of adrenal cancer. Treatment with either KU758 or 17-AAG resulted in upregulation of tumor suppressors (MEG3 [1.24–1.48 fold], DLX6-AS1 [1.19–1.25 fold], H19 [1.05–1.44 fold], GAS5 [1.37–2.04 fold]), =and HOTAIRM1 [1.27–1.37 fold]), and the oncogene MALAT1 (1.27–1.53 fold).
Discussion
While no chemotherapy regimen results in a cure for patients with advanced ACC, mitotane alone or in combination with other cytotoxic agents like the Italian protocol remain the standard of care option to slow down the progression of disease and improve (albeit transiently) patient outcomes.16 To date, there have not been any clinical trials dedicated to the evaluation of Hsp90 inhibitors in the treatment of ACC. However, several client proteins chaperoned by Hsp90 such as IGF-1R, survivin, and steroid hormone receptors are important in the molecular pathogenesis of ACC.17 Therefore, it is expected that Hsp90 inhibition could, in effect, downregulate many of the pathways important in the proliferation of ACC. Importantly in our study, we demonstrate that the novel CT-Hsp90i, KU758, is effective at inducing cellular death in all 3 ACC cell lines tested at potent doses, regardless of steroidogenic potential of the different cell lines. In addition to decreasing cell viability and proliferation, treatment with KU758 decreased migration, invasion, and aggregate formation of ACC cells in a dose-dependent manner, indicating that Hsp90 inhibition is blocking processes related to metastatic spread of disease.
A major limiting factor of utilizing N-terminal Hsp90 inhibitors for cancer treatment is that the concentration of drug required to induce client protein degradation is the same concentration that is needed to induce the heat shock response, resulting in the upregulation of Hsp70 and other pro-survival cellular processes. To overcome this effect and maintain the anticancer/proapoptotic effect, higher doses of Hsp90 inhibitors are required, leading to eventual dose-limiting toxicity.18 Thus, our approach to inhibit Hsp90 at the C-terminal dimerization domain as opposed to the N-terminal ATPase domain is an attractive strategy, as C-terminal inhibition still targets client protein degradation but does not upregulate Hsp70 or induce the pro-survival heat shock response.19
The Wnt/β-catenin pathway is vital in tumorigenesis due to the activation of downstream transcription factors and target genes such as survivin, vascular endothelial growth factor, Axin2, and Cyclin D that have important implications for the regulation of cellular survival.20 Typically, β-catenin is present at very low levels in cells due to its formation of a complex with glycogen synthase kinase-3β, adenomatous polyposis coli protein, and Axin, which leads to its degradation by ubiquitination. However, in ACC, there is frequent mutation of the β-catenin gene (CTNNB1), allowing for an increase in cytoplasmic and nuclear levels of β-catenin and activation of pro-survival pathways.20 Mutations in the β-catenin gene are present in more than 30% of both adrenocortical adenoma and ACC patient samples, and the presence of β-catenin mutations was associated with decreased survival and higher tumor stage.20,21 Similarly, in in vitro studies, inhibition of β-catenin can decrease tumor growth and reverse epithelial to mesenchymal transition.22 Therefore, it is important to note that Hsp90 inhibition with the novel C-terminal inhibitor KU758 was associated with decreased expression of β-catenin by immunofluorescence, indicating suppression of this pro-survival pathway. Axin2, also known as conduction, participates in negative feedback regulation of the β-catenin pathway. Axin2 has been shown to be a direct target of the Wnt/β-catenin pathway, and therefore, β-catenin signaling is known to directly induce the transcription of Axin2. Axin2 levels have been used successfully to quantify β-catenin activity in a variety of cancer models, including colorectal and hepatobiliary tumors. Therefore, the decrease in Axin2 expression after treatment of SW13 and NCI-H295R cells with KU758 supports the finding that C-terminal Hsp90 inhibition suppresses the β-catenin pathway. Notably, there is also a decrease in c-MET expression with KU758 treatment. c-MET is important in noncanonical (nonWnt mediated) activation of β-catenin, and this has been shown to be important in the pathogenesis of β-catenin dependent cancers.23 Taken as a whole, these findings suggest that Hsp90 inhibition with KU758 may exert its anti-tumor effects on ACC cells in part through suppression of the β-catenin pathway.
LncRNAs are increasingly recognized for their role in the pathogenesis of various tumors. In our experiments, treatment of ACC cells with Hsp90 inhibitors had a significant effect on lncRNA expression. Interestingly, although KU758 and 17-AAG both function as Hsp90 inhibitors, they are associated with very different effects on lncRNA expression, suggesting that C-terminal versus N-terminal Hsp90 inhibition has quite different effects at the transcriptional level. KU758 treatment is associated with significant upregulation of 12 different lncRNAs, including both oncogenes and tumor suppressors. Conversely, 17-AAG treatment resulted in significant upregulation of several oncogenes and downregulation of several tumor suppressors, leading to an overall balance favoring a proliferative effect on the cell rather than an inhibitory effect. Though lncRNAs are still in the early stages of characterization, they have been implicated in multiple types of cellular stress including the heat shock response, which has been associated with N-terminal but not C-terminal Hsp90 inhibitors.24 Taken together, these findings suggest that the differential expression of lncRNAs between adrenal cancer cells treated with KU758 versus 17-AAG may be in part due to the induction or lack of induction each drug has on the heat shock response.
Differential expression of lncRNAs has been shown between ACC cells and normal adrenal cortex cells.15 This is supported by our findings that demonstrated upregulation of several lncRNAs between 1.25 to 2.04-fold with KU758 treatment and 1.05 to 1.37 fold with 17-AAG treatment for oncogenes like MALAT1 and tumor suppressors such as MEG3, DLX6-AS1, H19, GAS5, and HOTAIRM1. Of note, GAS5, which is downregulated in ACCs, is significantly upregulated with 2 μM KU758 treatment (P < .05). GAS5 has been well-characterized for its vital role in multiple types of cancer and has prognostic implications in hepatocellular carcinoma and breast cancer by promoting apoptosis and decreasing invasion and migration of tumor cells.25 Additionally, GAS5 and H19 have both been shown to be involved in the β-catenin and mTOR pathways, which are frequently modulated in ACC.25 Upregulating these downregulated lncRNA tumor suppressors with KU758 treatment could provide a possible mechanism by which our CT-Hsp90i suppresses β-catenin activity.
In conclusion, our findings demonstrate that C-terminal Hsp90 inhibition with our novel compound, KU758, potently targets ACC cells leading to inhibition of growth, migration, invasion, endothelial-to-mesenchymal transition, and induction of apoptosis. Our evaluation of the effect of CT-Hsp90i KU758 on expression and modulation of specific lncRNAs suggests that this inhibitor’s effect on ACCs may be in part through the downregulation of the β-catenin pathway. While these early results show promise for CT-Hsp90 inhibitors like KU758 as a novel anti-cancer strategy for ACCs, further studies will be required to better characterize the mechanistic effects of individual lncRNAs and translate these effects at the protein level and then to heterogeneous tumors in vivo before these drugs can be applied to help patients clinically.
Funding/Support
This work was funded in part by grant funding from the National Institutes of Health (R01 CA216919 [MSC and BJB]; R01CA213566 [MSC and BJB] R01 CA120458 [MSC and BJB] T32 CA009672 [MSC and TW]), the Coller Surgical Society Research Fellowship (TW), and the University of Michigan Department of Surgery.
Footnotes
Conflict of interest/Disclosures
The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article.
References
- 1.Allolio B, Fassnacht M. Clinical review: adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91:2027–2037. [DOI] [PubMed] [Google Scholar]
- 2.The American Cancer Society. Survival rates for adrenal cancer, 2015. https://www.cancer.org/cancer/adrenal-cancer/detection-diagnosis-staging/survival-by-stage.html. Accessed February 10, 2019.
- 3.Ayala-Ramirez M, Jasim S, Feng L, et al. Adrenocortical carcinoma: clinical outcomes and prognosis of 330 patients at a tertiary care center. Eur J Endocrinol. 2013;169:891–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koch CA, Pacak K, Chrousos GP. The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J Clin Endocrinol Metab. 2002;87:5367–5384. [DOI] [PubMed] [Google Scholar]
- 5.De Martino MC, Feelders RA, de Herder WW, et al. Characterization of the mTOR pathway in human normal adrenal and adrenocortical tumors. Endocr Relat Cancer. 2014;21:601–613. [DOI] [PubMed] [Google Scholar]
- 6.Mohan DR, Lerario AM, Hammer GD. Therapeutic targets for adrenocortical carcinoma in the genomics era. J Endocr Soc. 2018;2:1259–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiosis G, Neckers L. Tumor selectivity of Hsp90 inhibitors: the explanation remains elusive. ACS Chem Biol. 2006;1:279–284. [DOI] [PubMed] [Google Scholar]
- 8.Yuno A, Lee MJ, Lee S, et al. Clinical evaluation and biomarker profiling of Hsp90 inhibitors. Methods Mol Biol. 2018;1709:423–441. [DOI] [PubMed] [Google Scholar]
- 9.Zhao J, Zhao H, Hall JA, et al. Triazole containing Novobiocin and Biphenyl Amides as Hsp90 C-terminal inhibitors. MedChemComm. 2014;5:1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subramanian C, Kovatch KJ, Sim MW, et al. Novel C-terminal heat shock protein 90 inhibitors (KU711 and Ku757) are effective in targeting head and neck squamous cell carcinoma cancer stem cells. Neoplasia. 2017;19:1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White PT, Subramanian C, Zhu Q, et al. Novel HSP90 inhibitors effectively target functions of thyroid cancer stem cell preventing migration and invasion. Surgery. 2016;159:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byrd KM, Subramanian C, Sanchez J, et al. Synthesis and biological evaluation of Novobiocin core analogues as Hsp90 inhibitors. Chemistry. 2016;22: 6921–6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen SM, Mukerji R, Samadi AK, et al. Novel C-terminal Hsp90 inhibitor for head and neck squamous cell cancer (HNSCC) with in vivo efficacy and improved toxicity profiles compared with standard agents. Ann Surg Oncol. 2012;19(Suppl 3):S483–S490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018;109:2093–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glover AR, Zhao JT, Ip JC, et al. Long noncoding RNA profiles of adrenocortical cancer can be used to predict recurrence. Endocr Relat Cancer. 2015;22:99–109. [DOI] [PubMed] [Google Scholar]
- 16.Megerle F, Herrmann W, Schloetelburg W, et al. Mitotane monotherapy in patients with advanced adrenocortical carcinoma. J Clin Endocrinol Metab. 2018;103:1686–1695. [DOI] [PubMed] [Google Scholar]
- 17.Sbiera S, Kendl S, Weigand I, Sbiera I, Fassnacht M, Kroiss M. Hsp90 inhibition in adrenocortical carcinoma: limited drug synergism with mitotane. Mol Cell Endocrinol. 2019;480:36–41. [DOI] [PubMed] [Google Scholar]
- 18.Kim YS, Alarcon SV, Lee S, et al. Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem. 2009;9:1479–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis RE, Zhang Z, Blagg BSJ. A scaffold merging approach to Hsp90 C-terminal inhibition: synthesis and evaluation of a chimeric library. Medchemcomm. 2017;8:593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tissier F, Cavard C, Groussin L, et al. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005;65:7622–7627. [DOI] [PubMed] [Google Scholar]
- 21.Gaujoux S, Grabar S, Fassnacht M, et al. β-catenin activation is associated with specific clinical and pathologic characteristics and a poor outcome in adrenocortical carcinoma. Clin Cancer Res. 2011;17:328–336. [DOI] [PubMed] [Google Scholar]
- 22.Salomon A, Keramidas M, Maisin C, Thomas M. Loss of β-catenin in adrenocortical cancer cells causes growth inhibition and reversal of epithelial-to-mesenchymal transition. Oncotarget. 2015;6:11421–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell R, Childs M, Maibach R, et al. HGF/c-Met related activation of β-catenin in hepatoblastoma. J Exp Clin Cancer Res. 2011;30:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valadkhan S, Valencia-Hipolito A. lncRNAs in stress response. Curr Top Microbiol Immunol. 2016;394:203–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang T, Wang M, Huang B, et al. Long noncoding RNAs in the mTOR signaling network: biomarkers and therapeutic targets. Apoptosis. 2018;23: 255–264. [DOI] [PubMed] [Google Scholar]