

Abstract
Ribosome profiling, first developed in 2009, is the gold standard for quantifying and qualifying changes to translation genome-wide [1]. Though first designed and optimized in vegetative budding yeast, it has since been modified and specialized for use in diverse cellular states in yeast, as well as in bacteria, plants, human cells, and many other organisms ([1], reviewed in [2, 3]). Here we report the current ribosome profiling protocol used in our lab to study genome-wide changes to translation in budding yeast undergoing the developmental process of meiosis [4, 5]. We describe this protocol in detail, including the following steps: collection and flash freezing samples, cell lysis and extract prep, sucrose gradient centrifugation and monosome collection, RNA extraction, library preparation, and library quality control. Almost every step presented here should be directly applicable to performing ribosome profiling in other eukaryotic cell types or cell states.
Keywords: Translation, Meiosis, Yeast, Ribosome profiling, Gene expression, Cellular development
1. Introduction
Ribosome profiling is a quantitative assay used to report transcriptome-wide measurements of translation in a given cellular context ([1], reviewed in [2, 3]). This method, when performed in conjunction with mRNA sequencing, allows global identification of the transcriptional and translational regulatory changes between multiple cellular states ([1], reviewed in [2, 3]). The developmental process of meiosis is a context that depends on extensive gene regulation ([4, 5], reviewed in [6, 7]). This specialized cell division remodels diploid cells into specialized haploid gametes, called spores in yeast (reviewed in [6, 7]). The elaborate gene expression program that drives meiosis includes many cases of translation-level control, most of which were identified by ribosome profiling [4, 5, 8–11]. Interestingly, during meiosis, translation control coordinates the expression levels of sets of mRNAs, as well as changes the identities of some translated products (ie. extended proteins, short proteins, and proteins initiated from non-AUG codons) [4, 12]. Our lab has used ribosome profiling to elucidate both of these types of changes to translation in meiosis, and continues to use the ribosome profiling protocol for these purposes [4, 5]. Here we describe, in detail, our current protocol for using ribosome profiling to study changes to translation, in both vegetatively growing and meiotic yeast cells.
Successful ribosome profiling relies upon a rapid sample collection strategy to avoid perturbing the cellular state that you wish to measure, as well as a strategy (pharmacological agents, for example) to halt ribosomes at the precise location on the mRNA that was being translated at the time of cell collection ([1], reviewed in [2, 3]). For traditional ribosome profiling, cycloheximide (CHX), is used to non-specifically stall elongating ribosomes [1]. This allows for collection of ribosome protected fragments along the entire coding regions of translated genes, although it can also introduce positional artifacts [13–21, 25]. Our standard protocol uses a brief CHX pre-treatment followed by rapid filtration and flash freezing of cells to maintain and measure the most accurate in vivo ribosomal positions [1, 4]. We note however, that it is possible, and sometimes beneficial, to exclude a translation inhibitor pre-treatment of cells to avoid positional artifacts, depending on the downstream analysis desired. The disadvantage of excluding a translation inhibitor pre-treatment of cells is that ribosomes will continue to elongate between filtration and flash freezing which may result in the runoff of ribosomes from the 5’ end of open reading frames (ORFs) and a loss of the corresponding data for these regions ([1, 21, 22, 25], reviewed in [2]). However, it is well known that translation inhibitor pre-treatments of cells can elicit technical artifacts in the precise ribosomal positions within coding regions. Furthermore, pre-treatment of cells with translation inhibitors can also cause biological changes to gene expression by inducing expression of ribosome biogenesis genes as a reaction to decreased translation [1, 4, 15, 17, 21–23]. Thus, one must weigh all the benefits and disadvantages of using a translation inhibitor pre-treatment when deciding to use one or not, depending on the specific desired downstream analyses to be performed.
Following rapid collection by filtration, cells are lysed cryogenically and the resulting extract is treated with RNAse I. The digested cell extract is then run on a sucrose gradient to isolate single ribosomes that protect ~30 nt mRNA fragments (also referred to as ribosome footprints (FPs)). Next, total RNA is extracted from the monosome fraction and gel-based size selection is used to enrich for ribosome-protected mRNA fragments from the pool of mostly ribosomal RNA (rRNA). It is these fragments that are taken through a series of enzymatic steps to make a sequencing library [1, 4, 24].
Our standard protocol uses linker ligation in order to generate libraries out of the pool of mRNA FPs [13]. Though this method is often used for studies focused on gene expression quantification, it should be noted that ligation can produce bias in the FPs sequenced based on the nucleotide identity of the 3’ end of the FP. Thus, for studies focused on codon level resolution of translation characteristics, care should be taken to use the most unbiased approaches possible in library preparation [22, 24, 25]. One alteration that can be used to alleviate linker ligation bias is to use a library of linker oligos, with random nucleotides on their 5’ end [22, 25]. Similarly, using a library of reverse transcription primers with varied nucleotides on their ends can help minimize any bias introduced during the following circularization step [25]. Following reverse transcription, linear sequencing libraries are created from the circular libraries using the minimal number of PCR cycles required to generate the desired amount of material.
After sequencing, the fragments are aligned to the yeast transcriptome to find the corresponding position of each ribosome. Footprints are then quantified per transcript to measure the amount of translation occurring on each transcript, or viewed on a genome browser or subjected to metagene analysis to look for changes to the regions being translated [1, 13, 23]. Comparison to a matched RNAseq sample prepared in parallel is used to determine whether gene expression changes are exerted at the transcriptional or translational level ([1, 4, 5], reviewed in [14]). The protocol described here, can be applied to yeast cells in a variety of cellular states with only minor differences, based primarily on the total levels of translation, and thus the amount of FPs collected in each state.
2. Materials
2.1. Media
Media should be made with water from a MilliQ filtration system and sterilized prior to storing. Described percent compositions represent weight/volume calculations.
YPD: 1% yeast extract, 2% bacto peptone, 2% dextrose.
BYTA: 1% yeast extract, 2% bacto tryptone, 1% potassium acetate, 1.02% potassium phthalate.
SPO: 2% potassium acetate, 40 mg/L adenine, 40 mg/L uracil, 20 mg/L histidine, 20 mg/L leucine, 20 mg/L tryptophan, pH to 7.0 using acetic acid, bring to final volume.
YPG plates: 1% yeast extract, 2% bacto peptone, 3% glycerol, 2% agar.
YPD 4% plates: 1% yeast extract, 2% bacto peptone, 4% dextrose, 2% agar.
2.2. Base reagents and materials
All reagents and solutions should be prepared to be nuclease-free. Hazardous materials should be disposed of and used with proper safety precautions as specified by regulations. Reagents should be stored at room temperature unless otherwise indicated. Solutions are made in water unless otherwise noted.
3 L sterile flasks for yeast culture.
Cycloheximide: 50 mg/ml in ethanol (500X), store at −20°C, dispose of cycloheximide hazardous waste by following the appropriate regulations and handle all materials containing cycloheximide with gloves, (see Note 1).
Nuclease free water.
1M Tris-HCl (pH 7.0).
1M Tris-HCl (pH 8.0).
10 mM Tris-HCl (pH 7.0).
10 mM Tris-HCl (pH 8.0).
2 M KCl.
1 M MgCl2.
20% Triton X-100.
20 and 22 gauge needles.
50 mL plastic, screw cap, nuclease-free tubes.
2 mL screw cap, nuclease-free tubes.
1.5 mL non-stick, nuclease-free tubes.
0.5 mL non-stick, nuclease-free tubes.
Metal spatulas.
Liquid Nitrogen (LN2): Note that LN2 can cause cryogenic burns, frostbite, and can displace oxygen. It should be stored according to established safety regulations, handled using cryogloves, and in a well-ventilated area.
Sytrofoam box: Ideally at least 6 inches each in height, width, and depth.
Plastic rack for 50 mL tubes (fits within styrofoam box).
Tube rack to hold ultracentrifuge tubes (small 15 mL tube racks work).
Filter membranes, 0.45 μm pore size, cellulose nitrate, to be used with the glass filtration apparatus for yeast cell harvesting, (see Note 2).
Metal tweezers.
Metal tongs to hold mixer mill chambers.
Cryogloves.
12 mL open-top, polyclear ultracentrifuge tubes and short rubber caps, (see Note 3).
1 M DTT: store at −20°C, handle on ice.
Ultra pure sucrose.
10 mL syringes with long metal tip attachments and SW 41 Ti marker block.
RNAse I: 100 U/μl, store at −20°C, handle on ice.
Acid-phenol:Chloroform:Isoamyl alcohol (125:24:1), pH 4.5, store at 4°C. Note that acid-phenol is hazardous, dispose of and use according to established safety regulations.
Chloroform, note that chloroform is hazardous, dispose of and use according to established safety regulations.
20% Sodium dodecyl sulfate (SDS).
3 M NaOAc, pH 5.5.
Isopropanol.
80% Ethanol, ice cold, store at −20°C.
GlycoBlue, store at −20°C, (see Note 4).
15% TBE-Urea polyacrylamide gels, store at 4°C, (see Note 5).
10% TBE-Urea polyacrylamide gels, store at 4°C, (see Note 5).
8% TBE polyacrylamide gels, store at 4°C, (see Note 5).
TBE-Urea sample buffer 2X store at 4°C.
SYBR Gold, store at −20°C, Note that SYBR Gold is hazardous, dispose of and use according to established safety regulations, (see Note 6).
1X TBE: 89 mM Tris, 89 mM Borate, 2 mM EDTA.
10 bp Ladder (optional).
Single-use razors or scalpels.
Centrifuge tube filters, cellulose acetate, 0.45 μm pore size.
SUPERase·In (20 U/μl), store at −20°C, handle on ice.
T4 Polynucleotide Kinase New England Biolabs (NEB), store at −20°C, handle on ice, (see Note 7).
Truncated T4 RNA ligase 2 (NEB), store at −20°C, handle on ice, (see Note 7).
Oligo Clean & Concentrator kit (Zymo Research), (see Note 8).
3 M NaCl.
0.5 M EDTA.
10% Tween 20.
MyOne Streptavidin C1 Dynabeads, (Thermo Fischer), store at 4°C, (see Note 9).
20X SSC: 3M Sodium Chloride, 300 mM Sodium Citrate.
1 M NaOH.
1 M HCl.
dNTPs: 10 mM dATP, 10 mM dCTP, 10 mM dGTP, 10 mM dTTP, store at −20°C, handle on ice.
Superscript III reverse transcriptase (Invitrogen), store at −20°C, handle on ice, (see Note 7).
CircLigase II ssDNA ligase (Epicentre), store at −20°C, handle on ice, (see Note 7).
1 mM ATP, store at −20°C, handle on ice.
Phusion DNA polymerase 2,000 U/mL (NEB), store at −20°C, handle on ice, (see Note 7).
6X gel loading dye for non-denaturing polyacrylamide gels.
High sensitivity D1000 ScreenTape (Agilent Technologies), (see Note 10).
High Sensitivity D1000 Reagents (Agilent Technologies), (see Note 10).
2.3. Non-standard equipment or facilities required
30°C shaker for yeast cultures.
Glass filtration apparatus for yeast cell harvesting, (see Note 2).
Mixer mill that can be cryogenically operated with 50 mL stainless steel cannisters, (see Note 11).
Refrigerated centrifuge (fits 50 mL tubes).
Refrigerated microcentrifuge.
Ultracentrifuge with Beckman SW 41 Ti rotor and buckets.
Gradient station, (see Note 3).
UV monitor for gradient station, (see Note 3).
Light box for gel cutting.
Magnet rack for 1.5 mL tubes to use for steps involving Dynabeads.
Agilent BioAnalyzer, TapeStation, or equivalent, (see Note 10).
Illumina HiSeq 4000.
2.4. Buffers and solutions
All buffers listed are to be prepared in water unless otherwise noted.
Polysome lysis buffer: 20 mM Tris-HCl pH 8.0, 140 mM KCl, 1.5 mM MgCl2, 100 μg/mL cycloheximide, 1% (v/v) Triton X-100, do not thaw and re-freeze buffer.
Polysome gradient buffer: 20 mM Tris-HCl pH 8.0, 140 mM KCL, 5 mM MgCl2, 100 μg/mL cycloheximide, 500 μM DTT, 20 U/mL SUPERase·In, make fresh on ice.
10% Sucrose: 10% sucrose (w/v) in polysome gradient buffer, make fresh.
50% Sucrose: 50% sucrose (w/v) in polysome gradient buffer, make fresh.
Dynabead B&W buffer (2X): 10 mM Tris-HCl pH 7.5, 1 mM EDTA, 2 M NaCl, 0.01% Tween 20, (see Note 9).
Dynabead B&W buffer (1X): 5 mM Tris-HCl pH 7.5, 500 μM EDTA, 1 M NaCl, 0.01% Tween 20, (see Note 9).
Dynabead Solution A: 100 mM NaOH, 50 mM NaCl, (see Note 9).
Dynabead Solution B: 100 mM NaCl, (see Note 9).
2.5. Oligonucleotides
Example sequences for sizing oligos are given, however the specific sequence of these oligos is not critical. The 28-mer RNA Oligo and 31-mer RNA oligos described here are unpublished and were designed by Gloria Brar and Nick Ingolia. If sequences are changed for the ligated oligo, note that the reverse transcription primer must be designed to amplify the sequence used. Oligos for rRNA subtraction were designed by Nick Ingolia and Gloria Brar [13]. The linker and amplification primers described here were designed and validated by Calvin Jan and their sequences generously shared with our lab. Alternative linker and primer sequences can be used for library preparation, but they must be validated prior to use to ensure high efficiency of ligation and amplification, respectively. For all oligonucleotides listed that include specialized modifications, we described the modifications present and listed the modification codes used by IDT. Equivalent modifications by other oligonucleotide providers may likely be substituted, however in all cases, any oligonucleotides used must be confirmed to be highly efficient and unbiased for sequencing library preparation. We dilute all oligos in water to the stated concentrations and store stocks at −20°C.
28-mer RNA control oligo: 5’AGUCACUUAGCGAUGUACACUGACUGUG/3Phos/3’, oligo has a 3’ phosphate (3Phos) and can be used for FP sizing and as a positive control and sizing guide throughout library preparation, 10 μM, oligo is PAGE purified.
31-mer RNA control oligo: 5’AUGUACACGGAGUCGAGCACCCGCAACGCGA/3Phos/3’, oligo has a 3’ phosphate (3Phos) and is used for FP sizing, 10 μM, oligo is PAGE purified.
Linker: 5’/5rApp/GATCGGAAGAGCACACGT/3ddC/3’, oligo is 5’ adenylated (5rApp), terminated using a 3’ dideoxycytidine (3ddC), DNA, and is ligated onto FPs for library prep, 20 μM, oligo is HPLC purified.
asDNA1b: 5’/biosg/GATCGGTCGATTGTGCACC3’, DNA, 5’ biotin (biosg) with the standard linker from IDT (C6), and HPLC purified, used for rRNA subtraction from S. cerevisiae FPs, 20 pmol/µL.
asDNA2b: 5’/biosg/CCGCTTCATTGAATAAGTAAAGAAAC3’, DNA, 5’ biotin (biosg) with the standard linker from IDT (C6), and HPLC purified, used for rRNA subtraction of S. cerevisiae FPs, 20 pmol/µL.
asDNA3b: 5’/biosg/GACGCCTTATTCGTATCCATCTATA3’, DNA, 5’ biotin (biosg) with the standard linker from IDT (C6), and HPLC purified, used for rRNA subtraction of S. cerevisiae FPs, 20 pmol/µL.
RT primer: 5’/5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT/iSp18/GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC3’, DNA, internal spacer 18 from IDT (iSp18), this primer is used to reverse transcribe linker ligated RNA FPs, 20 μM, oligo is PAGE purified.
PCR F primer: 5’AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT3’, DNA, used to amplify all FP samples, 10 μM, oligo is PAGE purified.
Barcoding primers: 5’CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACG3’, oligo is PAGE purified, reverse primer used to amplify and barcode samples with Illumina barcodes in place of “XXXXXXXX”, we use the following barcodes: D701 index ATTACTCG, D702 index TCCGGAGA, D703 index CGCTCATT, D704 index GAGATTCC, D705 index ATTCAGAA, D706 index GAATTCGT, D707 index CTGAAGCT, D708 index TAATGCGC, D709 index CGGCTATG, D710 index TCCGCGAA, D711 index TCTCGCGC, D712 index AGCGATAG. Example sequence of full primer sequence with the D701 index, 5’CAAGCAGAAGACGGCATACGAGATCGAGTAATGTGACTGGAGTTCAGACG3’.
3. Methods
Carry out all methods at room temperature unless otherwise noted.
3.1. Yeast growth and sporulation conditions (see Note 18)
Meiotic samples:
Day 1: Thaw a fresh patch of diploid yeast from the desired glycerol stock onto a YPG plate at ~5 pm, Grow patch at 30°C for ~16 h.
Day 2: Patch yeast from the YPG plate to a YPD 4% plate at ~9 am, grow for ~8 h at 30°C. At ~5 pm transfer a scoop of yeast into 10 mL of YPD liquid media. Grow cells, with shaking, for either ~24 h, at room temperature or ~16 h at 30°C.
Day 3: At ~5 pm measure the OD600 of the YPD culture. Dilute the culture such that it is in the accurate range of the spectrophotometer (1:20 usually works well), before measuring. Start a 100 mL culture of BYTA at 0.25 OD600, grow overnight (12–16 h) at 30°C, with shaking.
Day 4: At ~9 am measure the OD600 of the BYTA culture, calculate the amount of culture volume needed to start an SPO culture of 200 mL at a density of 1.9 OD600 (note that this is an excess quantity for one ribosome profiling sample). Pellet cells in BYTA (1,100–2,000 rcf for 2–2.5 minutes). Discard the supernatant and resuspend cells with sterile MilliQ water (wash should be greater than the volume of culture pelleted). Pellet cells in water (1,100–2,000 rcf for 2–2.5 minutes) and discard the supernatant. Resuspend pellet in 200 mL of SPO and move to a 3 L flask, shake culture at 30°C and record the time.
Take samples for staging (to assess DNA content, spindle morphology, etc.) as necessary, (see Note 13). Ten minutes prior to the desired time of sample collection, measure 150 mL of SPO culture and transfer the excess volume to a new flask to monitor meiotic progression in parallel. Continue to shake both at 30°C. Proceed to Section 3.2 “Cell harvesting and preparing polysome lysis buffer”.
Vegetative exponential phase samples:
Day 1: Thaw yeast and start an overnight culture exactly as described for the meiotic sample protocol (steps 1–2).
Day 2: Measure the OD600 of the culture, calculate the amount of culture volume needed to start a 450 mL culture at a cell density of 0.15 OD600. Pipette the calculated volume of yeast into a 3 L flask with 450 mL of YPD. Place culture on a shaker and incubate at 30°C until grown to ~0.6 OD600. Measure growth periodically by taking and recording the OD600. As cell density nears 0.6 OD600 (mid-exponential phase), move on to Section 3.2 “Cell harvesting and preparing polysome lysis buffer”.
3.2. Cell harvesting and preparing polysome lysis buffer (see Notes 19-20)
Label and prepare one 50 mL tube per sample by piercing 3–4 holes in the cap with a needle, and label one 2 mL tube/sample for the matched RNAseq sample (see Note 12).
Set up the filtration apparatus. Connect a filter flask to a vacuum pump, place a filter piece into the flask, and lay the nitrocellulose membrane on top. Place the collection beaker over the filter and attach it to the filter piece with a clasp.
Fill a styrofoam box with LN2 and place a 50 mL tube rack inside. Cool one large and one small metal spatula in liquid nitrogen with the handles sticking out. Fill the first labeled 50 mL tube with about 20–30 mL LN2 and leave uncapped, within the box of LN2. Uncap the 2 mL tube for the same sample. See Fig. 1 for an example filtration apparatus and representative images of cell collection from the filter.
Take CHX stock (50 mg/mL) out of the freezer on ice. If CHX has precipitated out of solution, vortex until fully dissolved and then return to ice, (see Note 15). Turn the vacuum pump on.
Add CHX to 100 μM in the first culture, (300 µL of 50 mg/mL stock for the 150 mL meiotic culture, 900 µL of 50 mg/mL stock for 450 mL of YPD culture), return shaking for 30 seconds, (see Note 15). Carry flask quickly to filter apparatus and pour culture into collection container. Place flask down and as media is almost completely filtered, remove the clasp and collection top and begin scraping cells horizontally off the filter with the large cooled spatula. Scrape ~90% of the cells into a mound on the metal spatula. Plunge the cells, and if needed, the entire spatula directly into the LN2-filled conical. Quickly recover the small patch of cells leftover into the 2 mL tube with the smaller cooled spatula, cap and drop into LN2.
Place the 50 mL sample tube with LN2 upright to allow venting from the cap in a −80°C freezer and store the 2 mL tube there as well. Allow LN2 to completely evaporate off before moving on to sample lysis.
Dispose of the used membrane and rinse or switch the collection container. Repeat steps 2–7 for each remaining sample.
When sample collection is complete, rinse all the glassware and spatulas with MilliQ water and allow to dry.
Next, make 5 mL of polysome lysis buffer (enough for two samples at 2–2.5 mL/sample). Mix 4.28 mL of water, 100 µL 1M Tris-HCl (pH 8.0), 350 µL 2M KCL, 7.5 µL 1M MgCl2, 10 µL 50 mg/mL CHX, and 250 µL 20% Triton-X 100 in a 50 mL tube on ice.
Label and prepare one 50 mL tube per sample exactly as done previously for sample tubes in steps 1 and 3. Uncap tubes, ensure buffer is mixed, and slowly dispense 2 mL of polysome lysis buffer directly into the LN2 within each 50 mL tube. Small 5 mL serological pipettes work well to dispense buffer at a controlled rate into the LN2 such that it freezes in droplets before contacting the sides of the tube. It is important that the frozen buffer is not stuck to the tubes for later use. Cap tubes with pierced caps and allow LN2 to freely evaporate while tubes are sitting upright in the −80°C freezer.
When LN2 has completely evaporated off both samples and buffer aliquots, you can move on to Section 3.3 “Yeast cell lysis”. Alternatively, samples can be stored at −80°C indefinitely.
Fig. 1.
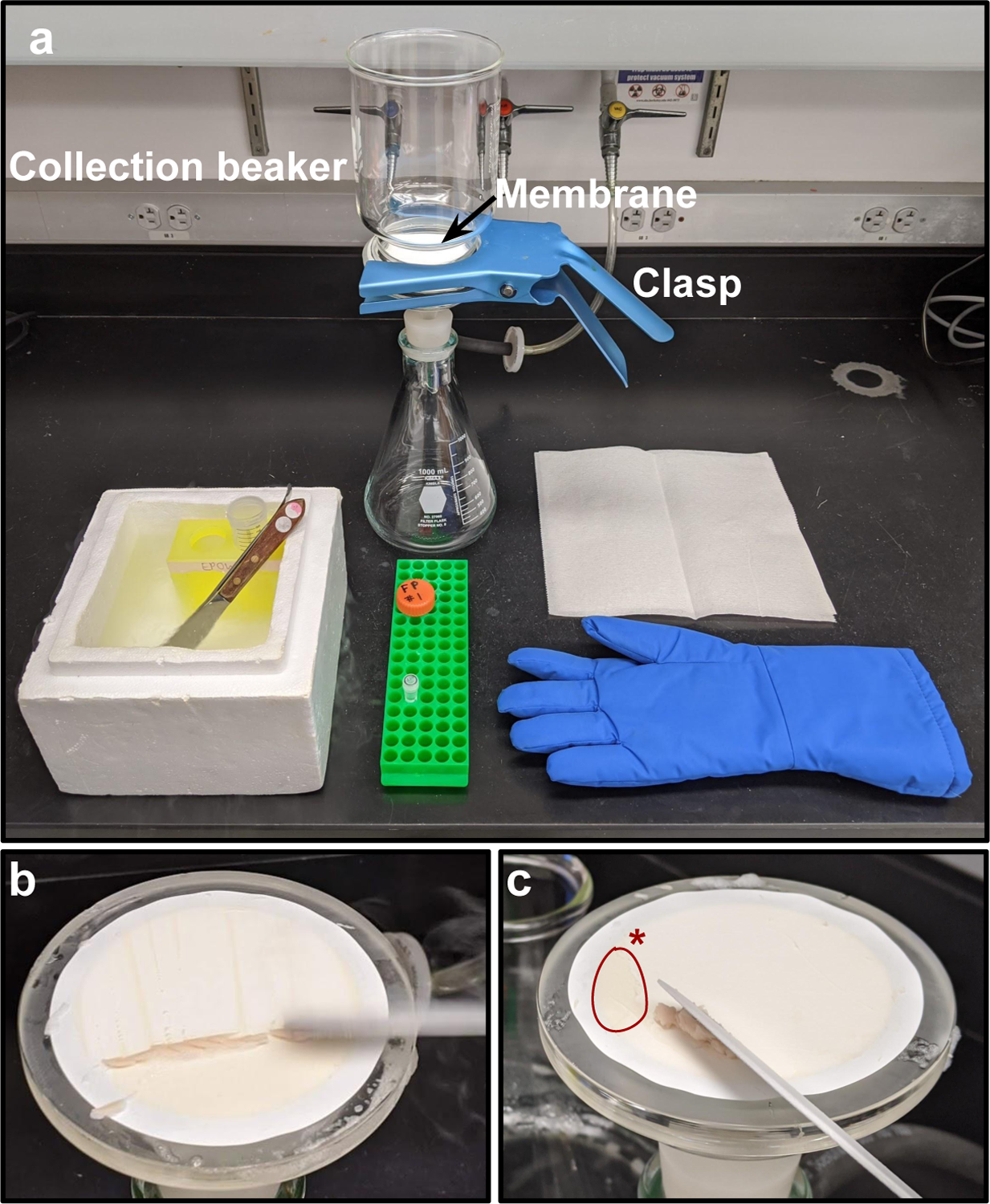
The set up for ribosome profiling/mRNAseq sample collection. (a) A typical bench-top set up in preparation for filtration. A filter flask is connected to a vacuum pump as shown. The filter piece is placed into the filter flask with a cellulose nitrate membrane (labeled) laid on top. Next, the collection beaker (labeled) is placed on top of the filter piece and over the membrane and held in place using a clasp (labeled). A styrofoam box is close by and contains LN2, two cooled metal spatulas (one large, one small), and a labeled uncapped 50 mL tube with LN2 inside (with a labeled and pierced cap nearby). A labeled 2 mL tube is nearby for RNAseq sample collection. A cryoglove is used to protect your hands from the cooled metal spatulas and LN2. (b) An image of a LN2 cooled metal spatula horizontally scraping yeast into a large pile for ribosome profiling collection. (c) A small patch of un-scraped yeast is circled as it is left for RNAseq sample collection, the larger heap of yeast will be scraped directly into LN2 in the 50 mL tube for ribosome profiling.
3.3. Yeast cell lysis (see Notes 11 and 21)
Record which sample will go in each mill chamber prior to starting, place chambers and balls into a LN2 filled styrofoam box using tongs, place a small 50 mL tube rack into the box as well. Let chambers cool until boiling stops (wait until large bubbles have ceased and only small ones persist).
Collect each sample/buffer tube from the freezer and place into the LN2 to prevent thawing. Loosen pellets by softly hitting each tube with metal tongs and pour one aliquot of the polysome lysis buffer pellets prepared in “Cell harvesting and preparing polysome lysis buffer” step 8 into each tube with a frozen sample pellet.
Use metal tongs to remove both halves of chamber one from LN2 and place onto a paper towel on the bench top. Pour out any liquid LN2 from both chambers and place the ball into the large chamber. Pour sample one into the large half of chamber one, lightly hit the bottom of the overturned tube to dislodge any stuck buffer or cell pellet. Close the chamber and place back into LN2 to re-cool. Repeat this process with chamber two and sample two. Keep labeled sample tubes and caps nearby.
Turn on the mixer mill. Set the frequency to 15 Hz, and breaking rounds to 3 minutes. Pull up knobs on mixer mill holders and twist to keep them open. Make sure that holders are fully loosened with the side wheels.
Remove chambers from LN2 with tongs, and loosen one quarter of a turn. Place chambers into the holders and use the side wheels to tighten them into place. Once mostly tightened, close the knobs on the holders and tighten for 1–2 more clicks. Be sure that the chamber is properly sealed within the holder and do not over tighten the wheels.
Press start and record which side of the mill each chamber was in as well as the orientation of the chambers. When the round is complete, lift knobs on the chamber holder and twist to hold open, loosen wheels, remove chambers, tighten chambers and return them to LN2 to re-cool.
Repeat steps 5–6 two more times such that each sample has had three total rounds of breaking, alternating the side and orientation that each chamber is on each round. During the last round of breakage, pre-cool two spatulas (one per sample).
While chambers are re-cooling, place the empty sample tube from sample one un-capped into LN2 to re-cool. Dump out any LN2 within the tube prior to collecting sample.
Remove chamber one from LN2, tap the chamber firmly on the top and the sides with metal tongs, this should dislodge most of the cell powder to the bottom of the large chamber. Place the chamber onto a paper towel on the bench top and unscrew the small half of chamber and remove vertically.
Use a cooled spatula to scoop cell powder from the top chamber into the open conical. When most of the powder is gone from the top chamber, transfer the ball into this half and proceed to scoop the rest of the powder from the larger half of the chamber. When complete, cap sample, and store indefinitely at −80°C, move the used chambers aside and repeat steps 8–10 with sample two.
When you are done with both chambers, rinse them with hot water and then distilled water. Make sure to remove the silicone rings and wash the entire chamber thoroughly. Spray chambers with methanol and allow to air dry. If multiple rounds of milling are to be done using the same chambers, be sure to let them dry completely between use. Move forward to Section 3.4 “Extract preparation”.
3.4. Extract preparation (see Note 22)
Label two non-stick 1.5 mL tubes and six screw cap nuclease free 2 mL tubes per sample, place tubes on ice. Pre-heat a water bath to 30°C and cool both a large centrifuge with 50 mL adaptors and a microfuge for 1.5 mL tubes to 4°C.
Hold tubes of cell powder in a 30°C water bath and swirl until thawed. Quickly move the tubes to ice and proceed immediately to step 3.
Spin samples for 5 minutes at 3,000 rcf at 4°C.
Move supernatant to 1.5 mL tubes on ice (~2 mL total for each sample), and spin for 10 minutes at 20,000 rcf at 4°C.
Take the supernatant into a screw cap 2 mL tube (combine duplicate tubes), avoiding unwanted material (see Note 22). Briefly vortex each sample and dispense into 200 µL aliquots in the screw cap tubes on ice. Mix 5 µL of each leftover sample with 495 µL of 10 mM Tris pH 7, to make a 1:100 dilution. Freeze sample aliquots in LN2 and store at −80°C, or keep two aliquots per sample on ice if proceeding directly to Section 3.5 “Footprint isolation by sucrose gradient”.
Measure the 260 nm absorbance of the diluted samples with a nanodrop. Record the A260 units and calculate the total A260 units present in each undiluted aliquot. If a diluted sample has a A260 of greater than 5 such that there are more than 100 A260 units in the total aliquot of 200 µL, do not use the whole aliquot for digestion. Split the aliquot to keep the A260 units within the range of 20 and 100 A260 units per digestion (see Note 22 for sample calculations).
Store extract aliquots at −80°C indefinitely or move forward to Section 3.5 “Footprint isolation by sucrose gradient”.
3.5. Footprint isolation by sucrose gradient (see Note 23)
Note that the directions below apply to the BioComp Gradient Master and BioRad Economonitor and may differ for other gradient collection set-ups.
Cool the SW 41 Ti rotor, paired buckets, and ultracentrifuge to 4°C. Thaw two extract aliquots per sample and keep on ice (one for digestion and footprint isolation and one for “mock” digestion to ensure the quality of the extract).
Make 50 mL of polysome gradient buffer by mixing 1 mL 1M Tris pH 8.0, 3.5 mL 2M KCL, 250 µL 1M MgCl2, 100 µL 50 mg/mL CHX, 25 µL 1M DTT, 50 µL SUPERase·In, and 45.08 mL of water (enough for 4 gradients).
Make 30 mL of 10% sucrose. Mix 3 g of ultra pure sucrose with 27.8 mL of polysome gradient buffer in a 50 mL tube. Make 30 mL of 50% sucrose, mix 14.9 g of ultra pure sucrose with 20.8 mL of polysome gradient buffer in a 50 mL tube. Shake vigorously until fully dissolved (usually ~15–30 minutes, using a lab shaker).
Calculate the amount of RNAse I (100 U/µL) to add to each sample for digestion. Add 10 U RNAse I per A260 unit of extract. For example, if a sample contains 30 A260 units total in the 200 µL aliquot, add 3 µL of RNAse I.
Add RNAse I to “cut” sample tubes and mix by gently flicking tubes. Set up “mock” digested samples by adding an equal volume of SUPERase·In, in place of RNAse I. Incubate for 1 hour at room temperature with slight mixing. When the hour is complete place tubes onto ice.
Take out a 12 mL ultracentrifuge tube for each “cut” or “mock” digested sample, clean tubes with compressed air to remove any debris.
Place a tube in the marker block and mark a line along the upper edge of the block, repeat for all tubes. Collect sucrose solutions from shaker and allow all bubbles to rise to the surface before use.
Fill a 10 mL serological pipette with ~6 mL of 10% sucrose, slowly dispense sucrose into the bottom of an ultracentrifuge tube until it reaches the marked line. Pull ~6.3 mL of 50% sucrose into a 10 mL syringe, wipe the outside of the metal syringe tip, and place the tip of the syringe at the bottom of the tube. Slowly inject 50% sucrose until it reaches the marked line. As the interface between the layers rises, move the tip of the syringe such that it is always just below the interface and remove carefully to avoid disruption of the interface. Repeat for all tubes.
Place a rubber cap onto each tube, lower the cap at an angle with the side of the cap with the hole being the last to lower. Take note of where each cap hole is by marking the side of the tube.
Turn on the gradient station and make 7–47% sucrose gradients for SW 41 Ti rotor buckets for tubes with short caps (81.5° tilt, rotating at 16 rpm, for 2 minutes). Ensure the platform is leveled and place tubes in holder. Run the program.
Retrieve the cooled SW 41 Ti buckets and uncap them while the program is running.
Carefully remove tubes from the gradient station. Remove caps by raising the side of the cap with the hole (near the marking) first at an angle. Wipe the outside of each tube carefully to remove any sucrose. It is very important sucrose does not get into the rotor buckets or threading along the bucket lids. Place each tube into a bucket, loosely cap the buckets, and re-cool for 15 minutes at 4°C. Soak rubber caps in water until you are ready to wash them.
Pulse spin down “cut” and “mock” digested samples and return to ice. Retrieve the cooled gradients and remove the caps to the rotor buckets. Make a list of which samples will go into each bucket. Load each sample onto the top of the respective gradient by slowly pipetting the sample against the wall of the tube, just over the gradient, until the entire sample is floating across the top. Repeat with all samples.
Balance opposing buckets (1 and 4, 2 and 5, 3 and 6) to within 10 mg of each other. Use 10% sucrose to carefully add weight to the top of the gradient of the lighter of the paired buckets and include the caps when balancing each bucket.
Screw lids onto buckets tightly, avoid contacting sucrose with lids. Hang buckets on their respective positions of the rotor, and carefully place the rotor into the ultracentrifuge. Always hang empty buckets (without ultracentrifuge tubes within them) on the rotor when spinning less than 6 gradients, and always make sure to balance the rotor properly.
Set up a run that is cooled to 4°C, lasts for 3 hours, and spins at 35,000 rpm.
Start ultracentrifugation, ensure the centrifuge reaches proper speed and temperature. It will take 5–10 minutes for the vacuum to fully engage and for the temperature and speed to adjust. While the spin is running, rinse all equipment used to set up the gradients including the gradient caps and syringe tips, and wipe the gradient holder with a wet paper towel. Also label two 2 mL screw cap tubes for each “cut” sample to use for monosome collection.
After the spin is completed, remove the rotor and place buckets into holders, enter the spin into the ultracentrifuge log, and turn off the ultracentrifuge.
Turn on the gradient station, UV lamp, and computer. Place a collection tip onto the gradient piston and position output tubing into a waste container. Set the voltage rate on UV lamp to between −2.0 and + 2.0 to achieve maximum detection range.
Carefully remove the first “mock” digested gradient from the bucket using tweezers, snap tube into the top piece of the gradient holder, ensure it is properly inserted and can spin freely before removing support from the bottom of the tube. Place the tube and top into the cylindrical gradient holder and spin to lock into place. Place the holder onto the platform and spin to lock into place with the window facing you.
On the gradient panel, select “fractionate”, lower the piston slowly until it comes just into contact with the top of the fraction. Choose “Rset” to mark the position of the top of the gradient, and select “Singl”. Set the following parameters on the gradient station panel, 0.2 mm/sec “Speed”, between 75 and 80 mm “Dist”, and 1 fraction for “Numb”.
Press “start” on the gradient station and start recording on the UV monitor. You can either collect all polysome fractions for downstream analysis or discard them into a waste container if not needed.
Watch profile on the computer screen. You should first see a large spike of material at the top of the tube representing free mRNA and other cellular material that absorbs at 260nm. Once that material has cleared you should see much smaller peaks representing the small (40S) ribosome subunit and the large (60S) ribosome subunit. If your resolution is good, you may see a peak in between the two, which might represent small ribosome scanning species. Following the 60S peak, you will see a very large peak corresponding to the monosome, followed by periodic peaks representing sequentially more ribosomes occupying a single mRNA transcript (disome, trisome, etc). At the end of the gradient you will see a large accumulation of material that is not periodically separated into peaks and may represent cellular material other than polysomes. See Fig. 2a for an example uncut polysome trace from high-quality extract.
Once data for the uncut sample is recorded, save the trace. Return the piston to the top position. Remove the finished tube and replace with a new ultracentrifuge tube full of MilliQ water. Rinse the tubing by collecting water through the piston, raise the piston again and drive the piston through the empty tube to move air through the tubing.
Repeat steps 20–24 with your cut samples. You should still expect to see peaks representing the 40S and 60S ribosome subunits, although they may be less distinct than in your “mock” sample. The monosome peak should be much larger than in the “mock” sample and it often smears into the 60S subunit peak in the “cut” sample. Be careful to collect only material from the monosome and not the 60S peak. See Fig. 2b for a “cut” polysome example trace. Collect the monosome fraction into the labeled screw cap 2 mL tubes and place on ice. Repeat collection for remaining “cut” samples.
Flash freeze collected monosome fractions in screw cap tubes, be sure to not overfill the tubes as the sucrose will expand while frozen and may break the tubes.
Be sure to properly clean the gradient station after use. This includes rinsing the tubing with water, drying it by running air through, and wiping down the entire gradient station surface with a wet paper towel. Any rack, bucket, lid, or ultracentrifuge tube holder used should be thoroughly rinsed with water and air-dried. Turn off all equipment. Move forward to Section 3.6 “RNA extraction from monosome fraction” or store monosome fractions indefinitely at −80°C.
Fig. 2.
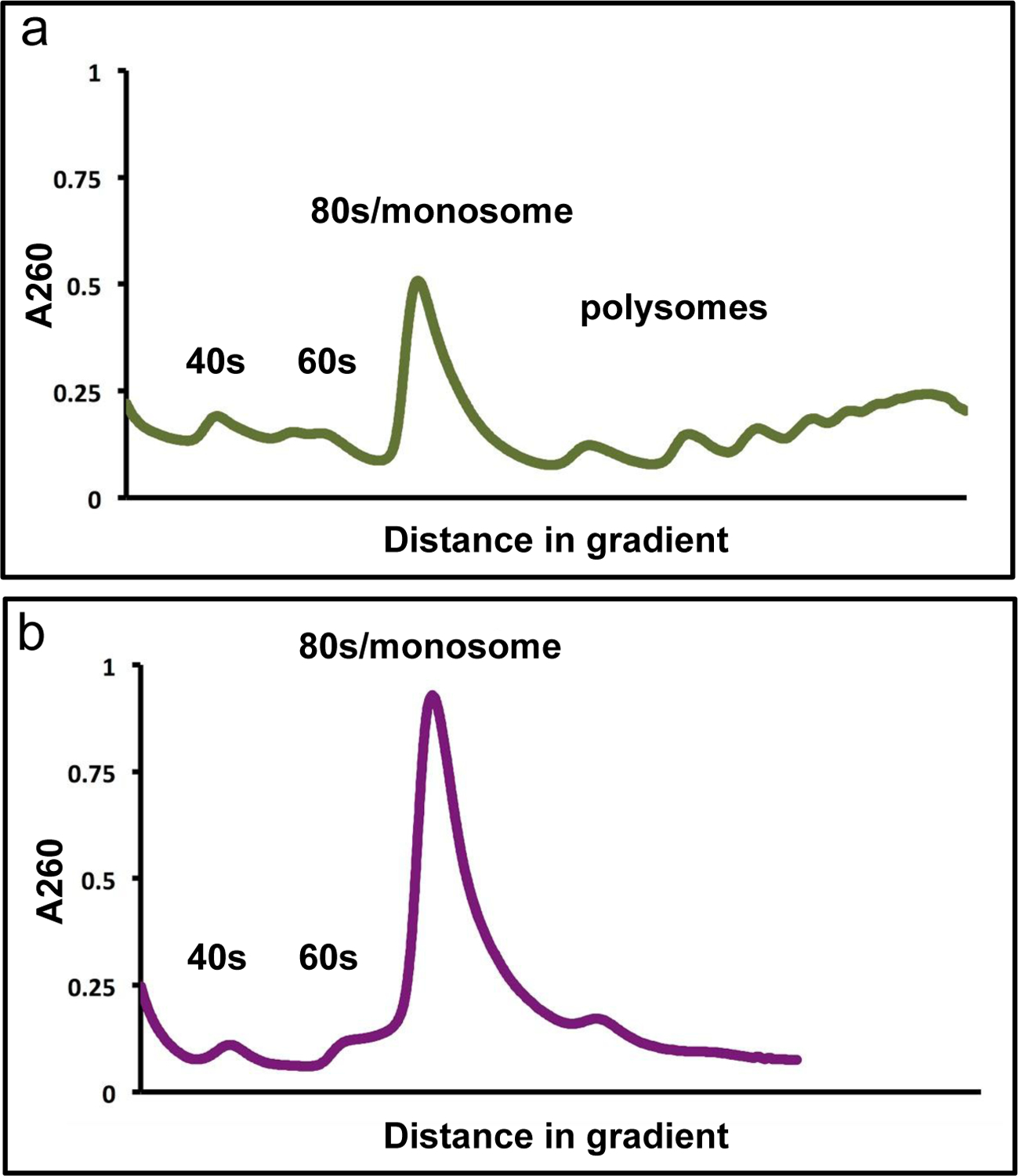
The A260 traces of non-digested and digested cell extract sucrose gradients. (a) The A260 polysome profile of non-digested cell extract with high quality. The top (and leftmost) spike of free mRNAs and other cell material is cropped out for clarity. Major ribosome species peaks are labeled. It is possible to see a small peak in between the 40S and 60S peak that we believe may represent scanning 40S subunits with additional initiation factors. (b) The A260 profile of digested cell extract, major ribosome species peaks are labeled. The polysome peaks in a digested sample should ideally be non-existent. Here there is a small disome peak suggesting a low level of incomplete polysome digestion, which is typical. The monosome peak is very large and runs close to the 60S peak in the digested samples as shown. When collecting the monosome fraction for FP isolation, exclude any 60S or disome peak.
3.6. RNA extraction from monosome fraction (see Note 24)
All steps described are to be carried out at room temperature unless otherwise noted.
Heat one thermomixer to 65°C and keep one thermomixer at room temperature.
Prepare the following sets of labeled tubes using non-stick 1.5 mL tubes: two tubes/sample with 750 µL of acid-phenol pre-warmed to 65°C, two tubes/sample with 40 µL of 20% SDS on ice, two tubes/sample with 700 µL of acid-phenol at room temperature, two tubes/sample with 600 µL of chloroform at room temperature, two tubes/sample with 40 µL of 3M NaOAc.
Thaw monosome fractions on ice and vortex well before use. Dispense 1.4 mL of each sample into two tubes (700 µL to each), each containing 40 µL of 20% SDS on ice. Heat tubes at 65°C until the SDS dissolves. Next, add the SDS/sample mixtures to 750 µL of pre-heated (65°C) acid-phenol. Incubate for 5 minutes at 65°C with vigorous mixing. To prevent caps from popping open during incubation, place a flat plastic object (eg. the lid of a box of micropipette tips) over the sample lids and tape on firmly.
Chill samples for 5 minutes on ice, then spin for 2 minutes at 20,000 rcf.
Recover the aqueous layer (~600 µL) and add to new tubes containing 700 µL of acid phenol at room temperature. Mix well and incubate for 5 minutes with vigorous mixing at room temperature. Spin the tubes for 2 minutes at 20,000 rcf.
Recover the aqueous layer (~475 µL) and add to the tube with 600 µL of chloroform, mix well. Incubate for 30 seconds at room temperature with vigorous mixing. Spin for 2 minutes at 20,000 rcf.
Move the aqueous layer (~360 µL) to a new tube containing 40 µL of 3M NaOAc, add 1 volume of isopropanol, and 2.5 µL of GlycoBlue, vortex.
Chill at −20°C for at least 30 minutes, or longer/overnight if desired.
Cool a microfuge to 4°C. Spin samples for 30 minutes at 20,000 rcf, at 4°C. After spin, remove the supernatant and wash the RNA pellet in 750 µL of 80% ice-cold ethanol.
Pulse spin samples at 20,000 rcf at 4°C and discard the supernatant. Pulse spin to collect any residual ethanol and remove all liquid.
Air-dry the pellet and resuspend in 5 µL of 10 mM Tris pH 7. Pipette up and down to fully re-suspend the RNA, pulse spin, and combine samples from duplicate tubes. Move forward to Section 3.7 “FP size selection”.
3.7. FP size selection
Prepare a 15% TBE-Urea gel in 1X TBE, pre-run gel at 200V for 15 minutes, (see Note 5 and Note 16). Rinse wells with 1X TBE to remove urea.
Add 10 µL of 2X urea sample buffer to each RNA sample and mix well.
Prepare 2 lanes worth of both the 28-mer and 31-mer RNA Oligos in 1X urea sample buffer by mixing 2 µL of each 10 μM stock with 20 µL of 2X urea sample buffer and 18 µL of water. Mix 1 µL of the 10bp ladder with 9 µL of water and 10 µL of 2X urea sample buffer to make 1 lane of ladder (optional).
Denature all samples, control oligos, and ladder in 1X urea sample buffer by incubating at 80°C for two minutes, and then placing directly on ice.
Pulse spin the samples and load the gel. Always flank each FP RNA sample with a 28-mer and a 31-mer oligo control lane on either side. Do not run actual samples in adjacent lanes as cross-contamination may occur.
Run the gel for 65 minutes at 200V, (see Note 5). While the gel is running prepare a 0.5 mL tube for each sample by piercing a hole in the bottom with a 20 gauge needle and nesting this tube into a non-stick 1.5 mL tube. Pre-heat a thermomixer to 70°C. Image the gel as described in Section 3.8 “Gel staining and imaging”. After imaging, cut out the size selected footprint samples as described below in step 7 (see Note 25).
For each lane of ribosome footprint sample, cut out all the material that falls within the size range of ~28–31nt. Make cuts just below the 28-mer oligo and just over the 31-mer oligo to ensure the full range of FP sizes are collected. Place the excised gel pieces into the labeled 0.5 mL tubes with holes, nested within the 1.5 mL tubes. Excise and extract one of the 28-mer RNA oligo lanes, this can be used as a sizing control later in the protocol. See Fig. 3 for an example FP size selection gel. Move forward to Section 3.9 “Gel extraction” and use RNA-specific instructions. Resuspend samples in 10 µL of water and the 28-mer control oligo in 14.5 µL of 10mM Tris-HCl. After extraction, proceed to Section 3.10 “rRNA subtraction”.
Fig. 3.
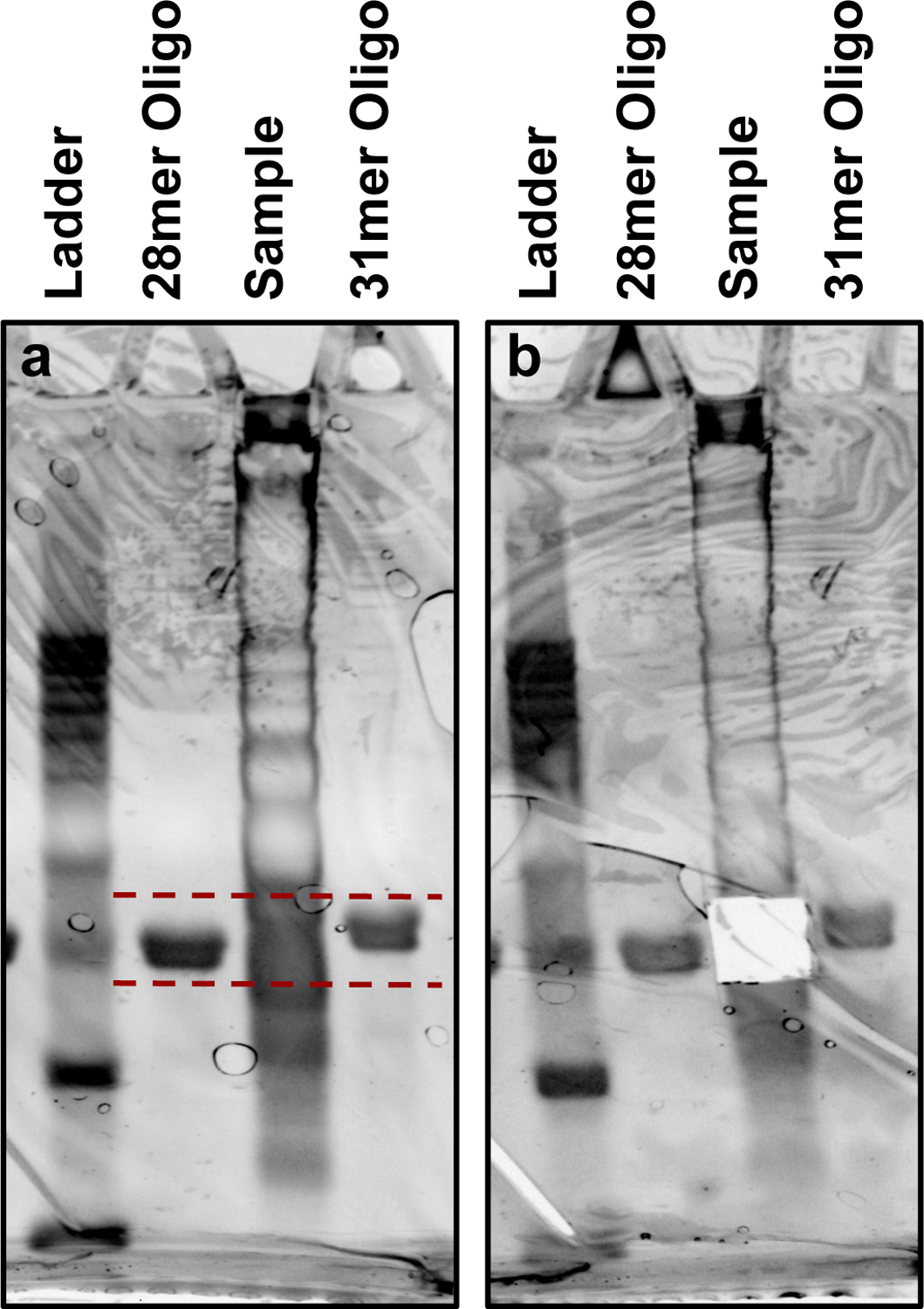
An example image of a size selection gel for collecting FPs. (a) The gel prior to the excision of FP samples is shown. The size range to be collected is marked and encompasses the range between and including the 28-mer and 31-mer RNA oligo controls. Diffuse banding seen along the sample lane represents digested rRNA fragments extracted from the monosome sample. A 10 bp DNA ladder is shown for clarity here, but is unnecessary for FP sizing. (b) The size selection gel post-excision of FP sample is shown to highlight the selected sizes.
3.8. Gel staining and imaging (see Notes 16 and 25)
Mix 6 µL of SYBR Gold with 60 mL of 1X TBE (you can reuse the TBE directly from the gel box).
Carefully remove the gel from its plastic casing and place into the SYBR gold and 1X TBE mixture, incubate with low shaking for 5 minutes.
Place the gel onto clean Saran wrap and image. Carry the gel on the Saran wrap to the light box and proceed to gel cutting protocol for each specific procedure.
3.9. Gel extraction (see Note 26)
Place excised gel pieces into the 0.5 mL tubes, and spin the nested tubes for 3 minutes at 20,000 rcf to force the gel through the hole. Repeat spin if gel did not break through on the first attempt. When most of the gel has been broken through the hole, collect any unbroken material in the 0.5 mL tube and move it to the 1.5 mL tube.
Add 700 µL of water to the gel pieces and incubate for 10 minutes at 70°C with vigorous shaking.
Use a cut p1000 tip to transfer gel mixture to a centrifuge tube filter column.
Centrifuge for 3 minutes at 20,000 rcf to recover the elution (free of gel debris). Transfer this eluate to a new non-stick tube.
Add 2 µL of Glycoblue, 78 µL of 3M NaOAc for RNA, or 78 µL of 3M NaCl for DNA, for precipitation.
Add 780 µL of isopropanol. Vortex and precipitate for at least 30 minutes (overnight works well) at −20°C.
Cool a microfuge to 4°C and spin samples at 20,000 rcf for 30 minutes at 4°C to pellet the DNA or RNA.
Remove the supernatant and add 1 mL of ice-cold 80% ethanol. Invert several times to wash pellet and pulse spin at 4°C and 20,000 rcf.
Carefully remove the supernatant, pulse spin to collect the residual ethanol and remove the remaining liquid. Air dry pellet for about 10 minutes or for as long as it takes for ethanol to evaporate off.
Resuspend in either 10 mM Tris-HCl pH 8 for DNA, or pH 7 for RNA, or water if proceeding to Section 3.10 “rRNA subtraction”, making sure to use the correct volume for the next procedure.
3.10. rRNA subtraction (see Notes 9, 14, and 28)
Perform rRNA subtraction on all FP samples. It is not necessary to perform rRNA subtraction on the 28-mer Oligo control sample, and to minimize reagent costs, we typically do not. Instead, you may place this aside to use as a control in the dephosphorylation and linker ligation steps.
Prepare a 1:10 dilution of asDNA2b and asDNA3b by mixing 1.5 µL of each 20 μM stock oligo with 13.5 µL of water (makes 15 µL enough for 2 subtractions). Make all of the MyOne C1 Dynabead solutions described in Section 2.4 “Buffers and solutions”.
Add the following to the tubes with 10 µL of RNA samples: 5 µL of asDNA1b (20 pmoles/µL), 6 µL of 1:10 asDNA2b (2 pmoles/µL), 5 µL of 1:10 asDNA3b (2 pmoles/µL), and 3 µL of 20X SSC.
Mix well, and incubate samples at 80°C for two minutes, then place directly back on ice.
Add 1 µL of SUPERase·In to each tube and mix.
Incubate for 15 minutes at 37°C with low shaking, place back on ice for at least 5 minutes.
While samples are shaking, prepare MyOne Streptavidin C1 Dynabeads by doing the following: vortex the MyOne C1 Streptavidin Dynabeads to resuspend and take 150 µL of beads/sample into a non-stick tube; wash beads three times in 150 µL of 1X Dynabead B&W buffer using inversion to mix and magnetic rack placement to pellet; wash beads two times in 150 µL of Dynabead Solution A; wash beads two times in 150 µL of Dynabead Solution B. Finally resuspend beads in 30 µL of 2X Dynabead B&W buffer/sample and dispense into aliquots of 30 µL in each tube (1 per sample).
Pulse spin samples to collect at bottom of tubes and add all (30 µl) to the Dynabeads. Incubate at room temperature for 15 minutes with low shaking.
Place tubes on the magnet rack and collect the supernatant (60 µL) to a new 1.5 mL non-stick tube on ice. Add 468 µL of water, 70 µL of 3M NaOAc, and 2 µL of GlycoBlue. Vortex to mix and add 600 µL of isopropanol, vortex again.
Incubate samples at −20°C for at least 30 minutes.
Pellet RNA by spinning for 20,000 rcf for 30 minutes at 4°C, remove supernatant and wash pellets by adding 1 mL of ice-cold 80% ethanol and by inverting the tubes several times. Pulse spin samples at 4°C and 20,000 rcf.
Remove supernatant carefully, pulse spin samples to remove any remaining ethanol and air dry pellet until ethanol has evaporated ~10 minutes. Resuspend pellet in 14.5 µL of 10 mM Tris-HCl pH 7 and proceed to Section 3.11 “Dephosphorylation and linker ligation”.
3.11. Dephosphorylation and linker ligation (see Note 27)
Resuspend rRNA subtracted RNA FPs and the gel-extracted 28-mer RNA oligo control in 14.5 µL of Tris-HCl pH 7. Move 14 µL of sample to a new non-stick tube for dephosphorylation reactions.
Set up a dephosphorylation reaction for each sample and control by adding the following, 2 µL of 10X T4 PNK Buffer, 2 µL of (10U/µL) T4 PNK, and 2 µL of SUPERase·In. Pipette up and down to mix well and incubate for 1 hour at 37°C, with mixing.
Prepare the linker ligation reactions in the same tubes by adding 14 µL of 50% w/v PEG-8000, 2 µL of 10X T4 RNA ligase buffer, 1 µL of 20 μM pre-adenylated Linker oligo, 2 µL of T4 Rnl2(tr) K227Q (200 U/µL), and 1 µL of water. Mix well by pipetting up and down and incubate for 3 hours at 22°C, with mixing.
Purify ligations on an Oligo Clean & Concentrator column as described, (see Note 8). Add 10 µL of water to bring sample volume up to 50 µL. Add 100 µL of Oligo Binding Buffer, mix well. Add 400 µL of 100% ethanol, mix well, and load onto a Zymo-Spin Column in a collection tube.
Spin the column at 12,000 rcf for 30 seconds and discard the flow-through.
Add 750 µL of DNA Wash Buffer to the column, spin at 12,000 rcf for 30 seconds and discard the flow-through.
Spin the column for 1 minute at 20,000 rcf.
Place the column in a new 1.5 mL non-stick tube and add 10.5 µL of water directly to the column matrix. Spin at 12,000 rcf for 30 seconds to elute the RNA.
Add 10 µL of 2X urea sample buffer to each sample and control and mix well.
Denature all samples and control lanes by incubating at 80°C for 2 minutes then placing back on ice.
Prepare a 10% TBE-Urea gel by placing in 1X TBE and pre-running the gel for 15 minutes at 200V, (see Note 5 and Note 16).
Rinse urea out of the wells with 1X TBE and load the gel, 20 µL/lane. Do not place different samples in adjacent lanes, use a control sizing oligo or an empty lane between them. Run the gel at 200V for 50 minutes, (see Note 5). While the gel is running prepare a 0.5 mL tube for each sample by piercing a hole in the bottom with a 20 gauge needle and placing this tube into a non-stick 1.5 mL tube. Pre-heat a thermomixer to 70°C. Image the gel as described in Section 3.8 “Gel staining and imaging”. After imaging, cut out the linker ligated footprint samples as described in step 13.
Excise the larger band representing samples that now have a linker. There will be a large lower band with un-ligated linker that should be avoided. Use the linker ligated 28-mer RNA Oligo as a sizing guide if the samples are low concentration and hard to visualize on the gel. Cut out and gel extract the linker ligated 28-mer Control Oligo and use as a positive control and sizing guide for Section 3.12 “Reverse transcription”.
Proceed to Section 3.9 “Gel extraction” and follow directions for RNA samples. Resuspend the pellets in 10.5 µL of 10 mM Tris-HCl pH 7 and proceed to Section 3.12 “Reverse transcription”.
3.12. Reverse transcription (see Note 14)
Use the linker-ligated 28-mer control as a positive control and sizing guide for the reverse transcription steps.
Take 10μL of precipitated RNA samples and controls in 10 mM Tris-HCl pH 7 and move to a new 1.5 mL non-stick tube.
Add the following to each tube, 3.28 µL of 5X FS buffer, 0.82 µL of 10 mM dNTPs, 0.5 µL of the 20 μM RT primer, mix well.
Denature samples for 2 minutes at 80°C, place directly on ice.
Add 0.5 µL of SUPERase·In and 0.82 µL of 0.1M DTT to each sample, mix well.
Add 0.82 µL of Superscript III to each tube. Incubate at 48°C for 30 minutes with low mixing.
Add 1.8 µL of 1M NaOH to each tube and mix well to hydrolyze any remaining RNA template.
Incubate at 98°C for 20 minutes with low mixing. Use a thermomixer with a thermo top to prevent samples from evaporating out of the tube. Glycoblue will turn pink.
Add 1.8 µL of 1M HCl and mix well to neutralize samples.
Place tubes on ice and add 20 µL of 2X urea sample buffer.
Denature samples at 95°C for 3 minutes prior to loading, then place on ice. Note that each sample will be split between two gel lanes (20 µL each lane).
Pre-run a 10% TBE-Urea gel at 200V for 15 minutes, (see Note 5). Rinse the wells with 1X TBE before loading to remove urea.
Load the gel with 20 µL/lane. Do not place different samples in adjacent wells, leave an empty well or run a sizing oligo between them.
Run samples for 65 minutes at 200V and proceed to stain and visualize gel as described in Section 3.8 “Gel imaging and staining”, (see Note 5). While the gel is running prepare a 0.5 mL tube for each sample by piercing a hole in the bottom with a 20 gauge needle and placing this tube into a non-stick 1.5 mL tube. Pre-heat a thermomixer to 70°C.
Use the 28-mer RNA Oligo that has been linker ligated and carried through the RT reaction as a sizing guide. Cut out and gel-extract the band corresponding to RT elongated product and avoid any lower bands corresponding to leftover RT primer or non-ligated linker. The footprint bands from each sample can run slightly higher than the control, as footprints include a mixture of sizes, with 28nt as the lowest size selected. See Fig. 4 for an example of a gel showing samples after reverse transcription, and indicating which material to collect from the gel.
Proceed to Section 3.9 “Gel extraction” and use directions for precipitating DNA. Following extraction, resuspend pellets in 15.5 µL of 10 mM Tris-HCl pH 8 and proceed to Section 3.13 “Circularization”.
Fig. 4.
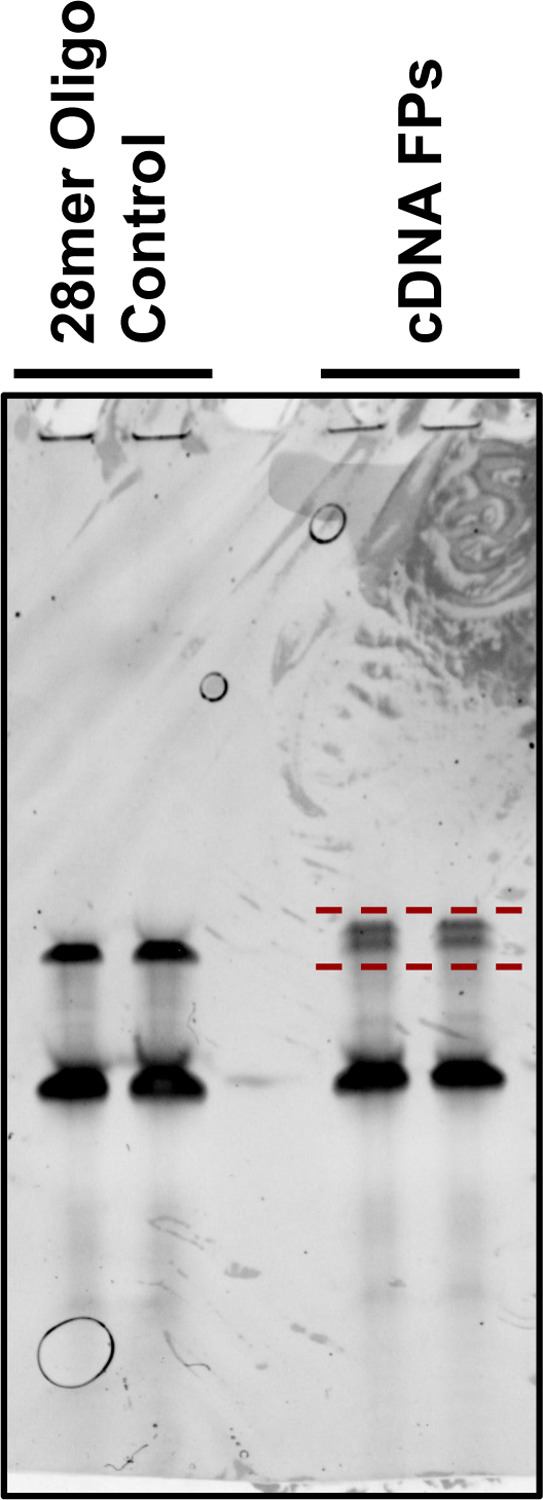
An example image of a size selection gel to collect linker ligated and reverse transcribed FP samples. The 28-mer oligo lanes represent 28-mer oligo that was dephosphorylated, ligated to the linker, and reverse transcribed as a positive control and a sizing guide. FP samples can run slightly larger than the 28-mer control and occasionally show banding. The size range that should be excised from the gel is marked and includes the larger sets of bands in each lane, and roughly matches the size of the 28-mer control.
3.13. Circularization (see Note 14)
After resuspending pellets from RT gel extraction in Tris-HCl pH 8, move 15 µL of each sample to PCR tubes.
Prepare circularization reactions by adding 2 µL of CircLigase II 10X reaction buffer, 1 µL of 1 mM ATP, and 1 µL of 50 mM MnCl2 to each sample, mix well.
Add 1 µL of CircLigase II to each sample, mix well.
Incubate at 60°C for 1 hour, and then 80°C for 10 minutes to heat inactivate the enzyme. Put tube on ice if proceeding directly to Section 3.14 “PCR amplification”, or store at −20°C indefinitely.
3.14. PCR amplification (see Notes 14 and 29-30)
Prepare PCR reactions for each sample by mixing the following in a PCR tube: 3.34 µL of 5X HF Phusion buffer, 0.34 µL of 10 mM dNTPs, 0.8 µL of 10 μM PCR F primer, 0.8 µL of 10 μM barcoding primer (SPECIFIC TO EACH SAMPLE), 10.4 µL of water, and 0.16 µL of Phusion polymerase.
Pipette 1 µL of each circularized sample into the corresponding PCR tube, mix well.
Perform PCR with the following steps: A. 98°C for 30s, B. 98°C for 10s, C. 60°C for 10s, D. 72°C for 10s, and repeat steps B-D for the desired number of cycles for each sample, E. hold at 4°C indefinitely.
Add 3.4 µL of 6X DNA loading dye to each PCR tube and mix well.
Prepare a 8% TBE gel in 1X TBE buffer and pre-run the gel for 15 minutes at 180V, (see Note 5).
Load samples on gel (20 µL/sample) and run the gel for 55 minutes at 180V, (see Note 5). While the gel is running prepare a 0.5 mL tube for each sample by piercing a hole in the bottom with a 22 gauge needle and placing this tube into a non-stick 1.5 mL tube. Pre-heat a thermomixer to 70°C.
Proceed to Section 3.8 “Gel staining and imaging”. Then excise the amplified sequencing libraries as described below in step 8.
Excise the PCR amplified sequencing library bands as described. See Fig. 5 and reference notes for directions on how to select the proper number of PCR cycles to amplify each sample library. Excise the discrete bands that correspond to the size of the major product in the 28-mer control lane. Leave behind any large smeary bands, and bands corresponding to excess primers or empty vector that has been carried through the library preparation and shows up in smaller bands (see Fig. 5).
Proceed to Section 3.9 “Gel extraction” and follow directions for a DNA sample, resuspend pellets in 10 µL of 10 mM Tris-HCl pH 8. Proceed to Section 3.15 “Bioanalysis for quality control assessment”.
Fig. 5.
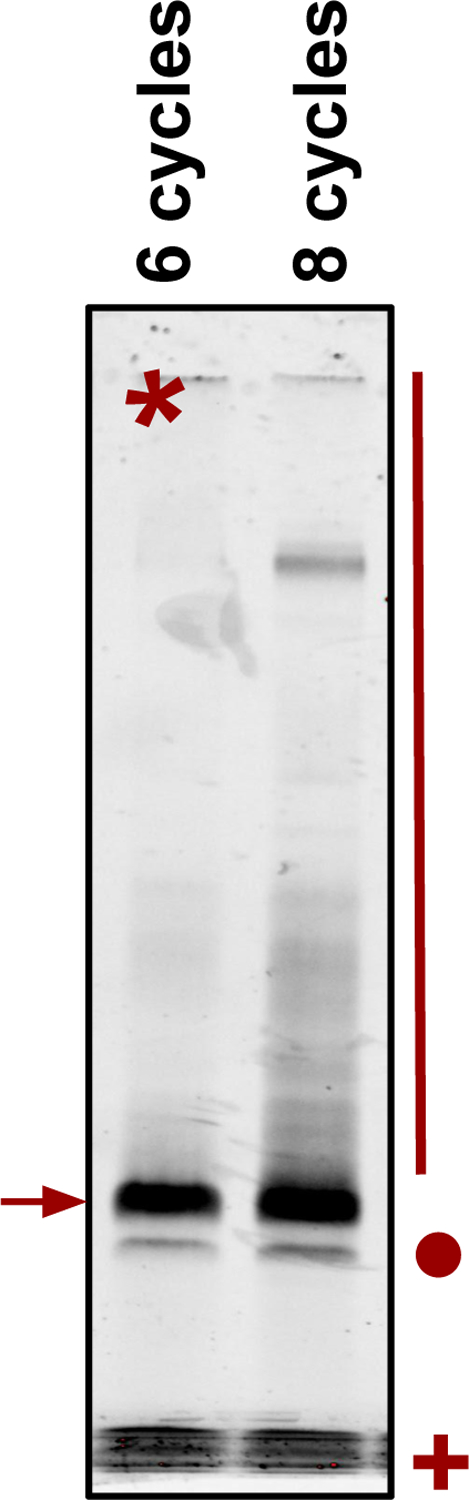
An example image of a size selection gel to isolate PCR amplified and multiplexed samples. Two lanes are shown with the same sample amplified for different numbers of cycles. The desired product is the brightest band and is marked with an arrow. For this sample, 6 cycles of amplification provided robust linear amplification with low levels of undesirable background products, as shown by the higher amounts of smear (marked with a red line) in the lane with 8 cycles of amplification. Take care to also exclude any material representing amplified empty vector (shown with a red circle), or excess primer from the amplification step (shown with a red + sign).
3.15. Bioanalysis for quality control assessment (see Note 10)
Use an Agilent Bioanalyzer or TapeStation to access the quality of the sequencing libraries prior to submission. We use the Agilent High Sensitivity D1000 ScreenTape and TapeStation to quantify and observe the sizes of our sequencing libraries as described below.
Allow all reagents to equilibrate at room temperature for 30 minutes, and vortex buffer and ladder before use.
Make a 1:2 dilution of each sample by mixing 1.5 µL of sample with 1.5 µL of 10 mM Tris-HCl pH 8.
Mix 2 µL of the High Sensitivity D1000 Sample Buffer with 2 µL of the High Sensitivity D1000 Ladder. Prepare samples by mixing 2 µL of the 1:2 dilution with 2 µL of the High Sensitivity D1000 Sample Buffer.
Spin down samples and ladder, then vortex at 2000 rpm for 1 minute.
Pulse spin to collect sample at the bottom of the tube.
Load samples into the Agilent 4200 TapeStation instrument, place the ladder in position A1.
Select the required sample positions on the 4200 TapeStation Controller Software.
Specify the position of the ladder, click Start and specify a filename with which to save the results.
If samples look as expected, submit to your sequencing core following their submission guidelines. Sequence samples using a 50 nt single read run on an HiSeq4000. Use data for your preferred downstream analysis (see Note 17).
Acknowledgements
We thank Nick Ingolia, the Ingolia lab, and Ze Cheng for technical assistance and helpful conversations. We thank Calvin Jan for sharing validated linker and oligo sequences.
Footnotes
Notes
Only use cycloheximide that is certified for treatment of live cells. We use the Biotechnology Performance Certified cycloheximide from Sigma.
Choose a glass filtration apparatus that will yield rapid filtration of the entire sample collected, and the proper size of corresponding filter membranes. We use a 90mm glass filtration unit from Fisher Scientific, and 90mm cellulose nitrate filter membranes from Whatman.
Our protocol uses the SW 41 Ti ultracentrifuge rotor and the Gradient Master gradient station from BioComp. We find the Seton polyclear ultracentrifuge tubes the most reliable in this system, however other tubes may be used as long as they are compatible with the SW 41 Ti rotor and the gradient station used. We use the BioRad EM-1 Economonitor as the UV monitor at our gradient station; other monitors may be suitable if they are compatible with the gradient station used.
Use of a coprecipitant is optional, but can be necessary if sample quantity is low. We prefer to use GlycoBlue as a coprecipitant, as it not only improves precipitation efficiency, but also allows for easy visualization of small nucleic acid RNA pellets.
We use 8cm x 8cm pre-cast gels that fit in the Life Technologies mini gel tank. The recommended voltage and length of run listed in our directions reflects conditions that yield good separation in this context. If an alternate size of gel is used, the voltage or length of run needed to achieve good separation may vary.
Other high sensitivity nucleic acid stains could work, we prefer SYBR Gold because it is highly sensitive and can be used to rapidly stain gels after electrophoresis.
For most enzymes needed in this protocol, we specified the exact enzyme and provider we typically use. Other suppliers of the same or similar enzymes may work in some cases, but enzyme preparations vary substantially between companies and we have not verified any alternatives to those listed here.
We use the Oligo Clean & Concentrator kit from Zymo Research to purify and concentrate linker ligated mRNA footprints prior to gel electrophoresis size selection. Other kits or methods that will reliably purify and elute small RNA fragments may be suitable for this purpose. If an alternate kit is used, follow the corresponding manufacturers protocol.
Magnetic streptavidin beads are used in this protocol to remove rRNA fragments by pulling down biotinylated antisense rRNA oligos. We use the MyOne Streptavidin C1 Dynabeads (and corresponding recommended buffers) as they are optimized for use with nucleic acids and have a high binding capacity. Other types of magnetic streptavidin beads may be suitable for this purpose however we have not confirmed the efficiency of rRNA pulldowns using alternate beads and buffers.
We use an Agilent TapeStation for library quality control. If another means of quality control is used, substitute the listed tape station reagents with reagents suitable for your preferred quality control analysis and follow the manufacturer recommended instructions.
We use the Retsch MM400 to cryogenically mill ribosome profiling samples. Similar cryogenic mills should also work as long as they can fit similar volumes of sample. Directions here are for the Retsch MM400 and may not be applicable to other instruments.
Much of the power of ribosome profiling relies on its ability to distinguish whether changes to gene expression are occurring as a result of transcriptional or translational control. In order to determine this, one must perform an mRNA sequencing (RNAseq) experiment in parallel. We describe how to collect a matched sample for RNAseq in the cell harvesting step of this protocol. In our experience, extracting RNA directly from a frozen cell pellet (rather than from the extract used for RNAse treatment and ribosome profiling) minimizes the 3’ end bias that can occur when mRNA is PolyA-selected directly from cell extract. Our RNAseq protocol uses hot acid phenol extraction of RNA, followed by PolyA-selection to enrich the sequencing sample for mRNAs, rather than rRNAs and tRNAs which are highly abundant. We next use alkaline-based mRNA fragmentation and select mRNA fragments that have a similar size distribution to FPs, by cutting a slice from a 15% TBE-Urea gel. These mRNA fragments are then carried through the same library preparation protocol as described in detail here for FP samples (dephosphorylation, linker ligation, reverse transcription, circularization, and PCR amplification/multiplexing). The only modification of these steps of the protocol to process mRNA rather than FP samples is the approach for enrichment of the fragments of choice (rRNA subtraction vs polyA selection), and thus library prep can be done in parallel for RNAseq and FP samples. We note that PolyA-selection is known to produce bias towards mRNAs with longer PolyA tails and also produces an enrichment for 3’ mRNA ends for samples, due to baseline degradation. It is possible (and often valuable) to perform total RNA sequencing in the absence of PolyA-selection to avoid bias, however this will result in a vast majority of sequencing reads (>90%) being dedicated to ribosomal and transfer RNAs, rather than mRNA. rRNA depletion kits are an alternative option, although these will also produce some biases in sequencing data yielded.
Ribosome profiling relies on taking bulk measurements of the translation occurring within a large population of cells at a given time. During a meiotic experiment, it is important to validate that the population at the time of sample collection largely represents the cellular state you wish to measure. Part of this consideration includes confirming that the population is well synchronized during the experiment. Lack of synchrony during a meiotic experiment can lead to measurements of gene expression that are inaccurate or misleading, as the total expression levels will represent an average of each genes expression over the spread of cellular states present in the population, rather than the measurement of each genes expression during one particular state. There are many ways to confirm the cellular state and assess synchrony during a meiotic experiment, and staging methods can vary from those that give only broad information about meiotic completion (eg. by assessing the dynamics of tetrad formation by light microscopy), to specialized approaches for detailed assessments of the morphology of a specific cellular compartment. One very reliable method used to determine the synchrony and staging of a meiotic culture at discreet time points uses DAPI staining to visualize chromosomal DNA. This allows one to assess when cells undergo the two chromosomal divisions. To use this method, take samples at defined intervals during the meiotic experiment. Fix the samples (most fixation methods will work, we use formaldehyde fixation) and then permeabilize the membranes (most permeabilization methods will work, we use an alcohol- or detergent-based method). Add DAPI stain and visualize on a fluorescent microscope. As the cells progress through meiosis they will undergo two nuclear divisions. You can determine what percentage of the population is within each stage of meiosis as determined by how many nuclear masses are visualized and the shape of those masses. To gain information on the stages of meiosis prior to the nuclear divisions, or more detailed analysis of the stages during the nuclear divisions, fix samples in accordance with your favorite immunofluorescence microscopy protocol and stain for beta-tubulin and DAPI. The meiotic spindle goes through discreet morphological changes prior to the nuclear first division occurring, and this approach provides also provides more detailed assessment of nuclear division stages (eg. anaphase I, metaphase II, etc) .
Many of the steps described include adding small volumes of multiple components to the same tube at the same time. It can be helpful to make a master mix in some of these cases, but if you do, you will want to make master mix for an extra sample so that you do not run out due to loss of material on pipette tips during transfer.
As discussed in the introduction to this method, pre-treatment of cells with cycloheximide is optional. Collect cells with no cycloheximide pre-treatment if, for example, quantitative codon-specific positional information within gene bodies is critical to downstream analyses.
Preventing RNAse degradation of samples (post RNAse I digestion) is of extreme importance. Before running or imaging any gels with RNA samples we suggest rinsing the gel chamber with an RNAse decontaminant (RNAse AWAY, for example).
For more instructions on how to process and analyze the resulting sequencing data from your experiment, see Ingolia et al., 2012 [13].
Synchronous sporulation requires a shift to post-diauxic growth prior to sporulation induction, it is best that overnight cultures grow to saturation (>10 OD600 in YPD, and >5 OD600 in BYTA). Sporulation requires respiration, and sporulation media includes a non-fermentable carbon source, while lacking nitrogen and fermentable carbon sources. It is important that there is no carryover of nutrients from the BYTA culture, so the yeast must be washed well with sterile water prior to re-suspension in SPO. Thaw yeast from frozen stocks on glycerol plates to select for and confirm cells are competent for respiration prior to use. It is also important to ensure the yeast are properly aerated prior to and during sporulation. To do this, grow all cultures in flasks that can hold at least 10X the volume of the actual culture and confirm shaking is at least 280 RPM for small orbital and 260 RPM for large orbital shakers. For meiosis experiments, it is useful to confirm the culture sporulated homogeneously using light microscopy. To check the staging at time of sample collection, fixed samples can be analyzed for DAPI staining or immunofluorescence. For vegetative exponential phase growth cultures, it is important that the yeast have time to recover from their saturated state after overnight growth. Allow cultures to double at least three times on day of collection to ensure cells have fully transitioned back to exponential growth.
Note that exponentially growing cells in rich media contain the most ribosomes and overall translation levels of any conditions known in yeast. This results in polysome profiles that contain more disomes, trisomes, etc in “mock” (non-RNAse-digested) samples than seen in other conditions. Meiotic cells have a translation rate of ~50% that of exponentially growing cells (Brar lab, unpublished data) and thus polysome levels are low compared to monosome levels. It is important to have an idea of the “normal” polysome traces expected for “mock” samples under a given condition to assess quality of samples to be used for ribosome profiling. Also note that samples with lower translation levels will result in fewer ribosome footprints than exponentially growing cells and thus bands on gels for these samples may be faint and amounts of oligos and enzymes added to reactions may need to be adjusted if FP quantities differ dramatically from the types of samples described here. It is also possible to collect more cells in conditions with lower translation to address the issue of low FP quantity.
It is important to work efficiently and quickly after CHX addition such that each sample has the same length of CHX treatment. Minimizing the time spent collecting cells will also help to avoid any gene expression changes that are secondary effects from CHX treatment, or from cells being removed from the culture media. Be careful not to rip the membrane as you are collecting cells following filtration as this will substantially slow your ability to scrape the yeast off. We recommend rapidly scraping most of the yeast (90%) into a single pile, rapidly scraping that pile into the LN2 with the large spatula, then collecting the remaining patch (10%) into the 2 mL tube with another spatula, followed by immediate flash freezing. Note that polysome lysis buffer must be transferred in drops to samples ahead of time, such that all LN2 has evaporated off before mixer milling but that samples containing polysome lysis buffer pellets can be stored indefinitely at −80°C. We recommend using 2 mL of polysome lysis buffer for cell collections of the quantities described here. However, you may adjust the volume of lysis buffer and the number of cells collected to make the concentration of the resulting extract higher or lower, based on analysis of extract from a pilot experiment.
Cryomilling requires requires a lot of LN2, so it is best to start with a full styrofoam box as well as a backup dewar in case you need to refill the box during breaking. It is important that samples do not thaw during breaking. To prevent this, everything that will touch the samples is pre-cooled in LN2. Wear two sets of latex or nitrile gloves underneath a pair of cryo-gloves to protect your hands from the caustic temperature of LN2 and exposed materials. Closed chambers should not be outside of LN2 for more than 5 minutes at a time, and open chambers should not be out of LN2 for more than 3 minutes. When collecting powder, it is important to collect as much as you can as fast as you can. Do not continue to collect small amounts of extract if the chamber has been outside of LN2 for more than 3 minutes – directions here include material in excess, such that gathering most of the cell powder will provide a sufficient amount of extract for multiple aliquots. If it is difficult to get enough cell powder out of the chambers using only 2 mL of polysome lysis buffer, increase the lysis buffer and or cell number as necessary to increase material or use smaller (10 mL stainless steel cannisters). Small spatulas with curved ends work well for collecting cell powder. Chambers can become very tight from rotation during breaking cycles, to prevent chambers from locking shut, loosen and re-tighten the chamber each time it is removed or placed into LN2. It is also important to keep the chamber halves in their matched pairs. If only one sample is to be lysed, place an empty chamber (no ball) on the opposing side to balance the mill.
Keep thawed extract on ice as much as possible and minimize the overall time the extract is thawed until it is flash frozen. After the final spin, there will be a pellet, a hazy layer just above the pellet, and a top layer of unwanted material floating on the top of the supernatant. Be careful to collect only the middle layer of supernatant representing cleared cell extract and minimize collection of other materials. Pipetting slowly and using multiple rounds of smaller volume collections works best. We collect ~75% of the volume of our input and leave behind material to avoid collecting non-desired material. To calculate the number of A260 units present in each sample aliquot, first multiply the diluted measurement by 100. This represents the A260 units that would be present for 1 mL of undiluted sample. Each aliquot is 200 µL so multiply the A260 units present in 1 mL of sample by 0.2. An example calculation is shown below.
Sample 1 (1:100) = 1.3 A260 units/mL
Sample 1 = 1.3 A260 * 100 = 130 A260 units/mL
Sample 1 (aliquot) = (130 A260 units/mL)* 0.200 mL = 26 A260 units
While preparing the ultracentrifuge tubes check each tube for and discard any tubes with cracks. Once the sucrose gradients are made, take great care to minimize the amount of movement they are subjected to and keep them in a stable rack. While fractionating, it is important that the collection tip of the gradient station holds a full seal and is not mixing the gradient as it descends. It is helpful to remove the collection tip, rinse it, and press it firmly against a smooth flat surface to extend the sides prior to collecting each sample. The total volume of the digested monosome peak will be larger than needed for making a ribosome profiling library and may be more than 2 mL, it is useful however to collect the entire fraction and keep it frozen as backup material.
The volume of the digested monosome peak is too large to extract in a single 1.5 mL tube, the samples should be very well mixed and can be aliquoted into smaller scale RNA extractions (two extractions of 700 µL monosome fraction is usually more than enough material). Leftover monosome fraction can be re-frozen and kept as backup material in case something fails downstream. Hot acid phenol extractions release gas and pressure will build in the tubes during mixing. Opening tubes periodically to release pressure can prevent tubes from popping open during the extraction. Work in the hood, with goggles, and change gloves frequently to protect yourself from hot acid phenol. Wear all recommended personal protective equipment and follow all regulations for use of hot phenol and disposal of phenol- chloroform mixtures. The aqueous layer containing RNA should always be on top, using this extraction protocol.
The same gel staining and imaging process is used throughout this protocol four times. For all gels, limit the amount of time that passes after running the gel, until cutting out the desired bands, as diffusion can occur. Place gels on clean Saran wrap for ease of imaging. For any gels prior to PCR amplification (footprint size selection, post-linker ligation, and post-RT), do not run different samples adjacent to each other. Always run an empty lane or a control oligo in between. This, in combination with using new single use scalpels to cut out each sample will help to prevent cross-contamination. Post-PCR amplification, when unique barcodes are included for each sample, samples can be run adjacent to each other and cut with the same scalpel. It is useful to excise and gel extract one lane of the 28-mer control RNA oligo from the FP size selection gel. This oligo can be carried through the subsequent enzymatic library preparation steps as a control sample and can additionally serve as a sizing guide during each subsequent gel extraction. Always record a new image of the gel post excision, or make a marking on the original image to show the material that was taken through to the next steps of the protocol.
This procedure is used four times in the protocol with only minor modifications. The first variation is the size of the hole used to break the gel, use a 20 gauge needle for the 15% and 10% TBE-Urea gels, and a 22 gauge needle for the 8% gels. The second is the salt used to precipitate the extracted materials. For any step prior to reverse transcription, follow directions for RNA. For any step after reverse transcription, follow directions for DNA. The third is the solution used to resuspend the final pellet, for DNA pellets (post-RT) use 10 mM Tris-HCl pH 8, for RNA pellets (prior to RT) use 10 mM Tris-HCl pH 7.
It is possible to use either gel-extracted or fresh 28-mer RNA oligo as a control for dephosphorylation and linker ligation. To use fresh oligo, mix 1 µL of 10 μM oligo with 13 µL of water. To use gel-extracted oligo, treat as the rest of the samples. Do not add ATP to the reaction. T4 PNK will dephosphorylate ATP and phosphorylate itself instead of dephosphorylating the FPs if ATP is added.
This protocol is optimized to remove three rRNA fragments found in great abundance in ribosomal footprint samples in the original ribosome profiling paper [1]:
RDN25 734–760 (Here referred to as “rRNA#1”, 5’AAGAGGTGCACAATCGACCGATCCTG3’)
RDN25 2502–2528 (Here referred to as “rRNA#2”, 5’TAGTTTCTTTACTTATTCAATGAAGCGG3’)
RDN25 3167–3193 (Here referred to as “rRNA#3”, 5’AATATAGATGGATACGAATAAGGCGTC3’)
rRNA subtraction will greatly lower the amount of rRNA present in the final footprint samples for sequencing, however, it is common to still have between 30–50% of the total “FP” sequencing reads align to rRNA. This pulldown protocol was optimized on untailed, radiolabeled RNA to maximize rRNA pull-down and minimize non-specific loss of material. Modifications such as increasing the concentration of biotinylated antisense oligos, variation in hybridization buffer conditions, variations in hybridization temperature, increased ratio of dynabeads to biotinylated antisense oligos, decreased volume of hybridization/dynabead incubations, and the absence of Tween in the dynabead buffers did not yield any increases to rRNA subtraction in our hands.
Mix circularized samples thoroughly prior to use. Choose a unique indexing primer for each sample, this will allow you to sequence all the samples on the same sequencing lane. Keep detailed notes of which sample gets which index. Estimate the number of amplification cycles you will use for each sample based on the amount of material seen in the RT gel. If samples were bright on the reverse transcription gel, they are likely concentrated and will not need many cycles of PCR (try 6 or 8 cycles). If the samples are dim on the gel, try 10 or 12 cycles. It is important to try at least two amplification cycle choices per sample to ensure that product is amplifying linearly. After imaging the gel, re-amplify any samples that have large non-desired products with fewer cycles. Re-amplify any samples in which the main product band is not bright using more cycles. See Fig. 5 for example images.
If samples are outside the range of quantification with a 1:2 dilution, try a higher dilution and re-analyze. Footprint samples should have a single major peak corresponding to the size of the full length of any FPs plus the extensions of the linker, RT primer, and PCR primers. If the size is not correct re-amplify and extract again. Do not submit samples for sequencing if they do not pass the quality control analysis at this step.
References
- 1.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS (2009) Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science (80- ) 324:218 LP–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingolia NT, Hussmann JA, Weissman JS (2019) Ribosome profiling: Global views of translation. Cold Spring Harb Perspect Biol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brar GA, Weissman JS (2015) Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol 16:651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brar G a., Yassour M, Friedman N, et al. (2012) High-Resolution View of the Yeast Meiotic Program Revealed by Ribosome Profiling. Science (80- ) 335:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng Z, Otto GM, Powers EN, et al. (2018) Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172:910–923.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitchell AP (1994) Control of meiotic gene expression in Saccharomyces cerevisiae. Microbiol. Rev 58:56–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neiman AM (2011) Sporulation in the budding yeast Saccharomyces cerevisiae. Genetics 189:737–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berchowitz LE, Gajadhar AS, van Werven FJ, et al. (2013) A developmentally regulated translational control pathway establishes the meiotic chromosome segregation pattern. Genes Dev [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlile TM, Amon A (2008) Meiosis I Is Established through Division-Specific Translational Control of a Cyclin. Cell 133:280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LE B, Kabachinski G, MR W, et al. (2015) Regulated Formation of an Amyloid-like Translational Repressor Governs Gametogenesis. Cell 163:406–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin L, Zhang K, Xu Y, et al. (2015) Sequestration of mRNAs Modulates the Timing of Translation during Meiosis in Budding Yeast. Mol Cell Biol 35:3448–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollerer I, Higdon A, Brar GA (2018) Strategies and Challenges in Identifying Function for Thousands of sORF-Encoded Peptides in Meiosis. Proteomics 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ingolia NT, Brar GA, Rouskin S, et al. (2012) The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7:1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ingolia NT (2016) Ribosome Footprint Profiling of Translation throughout the Genome. Cell 165:22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santos DA, Shi L, Tu BP, Weissman JS (2019) Cycloheximide can distort measurements of mRNA levels and translation efficiency. Nucleic Acids Res 47:4974–4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duncan CDS, Mata J (2017) Effects of cycloheximide on the interpretation of ribosome profiling experiments in Schizosaccharomyces pombe /631/337 /631/337/574 /38/91 /38/39 article. Sci Rep 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Z, Brar GA (2019) Global translation inhibition yields condition-dependent de-repression of ribosome biogenesis mRNAs. Nucleic Acids Res 47:5061–5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerashchenko MV, Gladyshev VN (2014) Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hussmann JA, Patchett S, Johnson A, et al. (2015) Understanding Biases in Ribosome Profiling Experiments Reveals Signatures of Translation Dynamics in Yeast. PLoS Genet 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Requião RD, de Souza HJA, Rossetto S, et al. (2016) Increased ribosome density associated to positively charged residues is evident in ribosome profiling experiments performed in the absence of translation inhibitors. RNA Biol 13:561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lareau LF, Hite DH, Hogan GJ, Brown PO (2014) Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. Elife 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinberg DE, Shah P, Eichhorn SW, et al. (2016) Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep 14:1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kearse MG, Goldman DH, Choi J, et al. (2019) Ribosome queuing enables non-AUG translation to be resistant to multiple protein synthesis inhibitors. Genes Dev 33:871–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGlincy NJ, Ingolia NT (2017) Transcriptome-wide measurement of translation by ribosome profiling. Methods 126:112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lecanda A, Nilges BS, Sharma P, et al. (2016) Dual randomization of oligonucleotides to reduce the bias in ribosome-profiling libraries. Methods 107:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]