

Abstract
α7 nicotinic acetylcholine receptors (nAChRs) are widely expressed in the brain where they promote fast cholinergic synaptic transmission and serve important neuromodulatory functions. However, their high permeability to Ca2+ also predisposes them to contribute to disease states. Here, using transfected HEK-tsa cells and primary cultured hippocampal neurons from male and female rats, we demonstrate that two proteins called Ly6h and NACHO compete for access to α7 subunits, operating together but in opposition to maintain α7 assembly and activity within a narrow range that is optimal for neuronal function and viability. Using mixed gender human temporal cortex and cultured hippocampal neurons from rats we further show that this balance is perturbed during Alzheimer's disease (AD) because of amyloid β (Aβ)-driven reduction in Ly6h, with severe reduction leading to increased phosphorylated tau and α7-mediated neurotoxicity. Ly6h release into human CSF is also correlated with AD severity. Thus, Ly6h links cholinergic signaling, Aβ and phosphorylated tau and may serve as a novel marker for AD progression.
SIGNIFICANCE STATEMENT One of the earliest and most persistent hypotheses regarding Alzheimer's disease (AD) attributes cognitive impairment to loss of cholinergic signaling. More recently, interest has focused on crucial roles for amyloid β (Aβ) and phosphorylated tau in Alzheimer's pathogenesis. Here, we demonstrate that these elements are linked by Ly6h and its counterpart, NACHO, functioning in opposition to maintain assembly of nicotinic acetylcholine receptors (nAChRs) within the physiological range. Our data suggests that Aβ shifts the balance away from Ly6h and toward NACHO, resulting in increased assembly of Ca2+-permeable nAChRs and thus a conversion of basal cholinergic to neurotoxic signaling.
Keywords: Alzheimer's disease, amyloid β, Ly6, NACHO, nAChRs, neurotoxicity
Introduction
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels that mediate fast synaptic transmission by the endogenous neurotransmitter ACh. These ionotropic receptors mediate important physiological functions including autonomic signaling in sympathetic and parasympathetic ganglia (Xu et al., 1999; David et al., 2010), release of circulating epinephrine by the adrenal medulla (Fenwick et al., 1982; Kidokoro et al., 1982; Petrovic et al., 2010), skeletal muscle contraction (Tintignac et al., 2015), long-term potentiation (Lagostena et al., 2008; Nakauchi and Sumikawa, 2012; Criscuolo et al., 2015), regulation of arousal (Léna et al., 2004; Criscuolo et al., 2015), and anti-inflammatory responses (Borovikova et al., 2000; Bernik et al., 2002; Wang et al., 2003; Li et al., 2011). nAChRs are also known to be dysregulated in several disease states. For example, the reinforcing and withdrawal effects of the addictive drug nicotine are thought to involve upregulation of α4, α6 and β2-containing nAChRs (Picciotto et al., 1998; Tapper et al., 2004; Maskos et al., 2005; Nashmi et al., 2007; Pons et al., 2008; Govind et al., 2012; Liu et al., 2012; Akers et al., 2020), whereas Alzheimer's disease (AD) conditions are associated with changes in levels of α7 nAChRs (Burghaus et al., 2000; Counts et al., 2007; Ikonomovic et al., 2009; Liu et al., 2013; Ren et al., 2020). In the latter case in particular, however, the underlying regulatory mechanisms and their causal relation to disease pathology are poorly understood.
New opportunities to explore these mechanisms have arisen with the recent discovery of two classes of molecules that regulate signaling by nAChRs. The first class consists of the ER-resident protein NACHO, which shifts the equilibrium of nAChR subunits from mononomeric to oligomeric complexes (Gu et al., 2016; Matta et al., 2017). Since only fully assembled, pentameric receptors are delivered to the cell surface at high efficiency, the presence of NACHO enhances and the absence of NACHO limits the number of nAChRs available to respond to ACh. The second class of regulatory proteins consists of select members of the Ly6 family. These proteins have a single domain consisting of a small, three-fingered motif (Miwa et al., 2006; Galat et al., 2008; Lyukmanova et al., 2011; Wu et al., 2016; Vasilyeva et al., 2017). Most Ly6 proteins are anchored to organellar membranes and to the outer leaflet of the cell surface by glycosylphosphatidylinositol (GPI; Vasilyeva et al., 2017). This feature allows them to regulate properties of nAChRs during different stages of biogenesis. One such property is the availability of nAChRs at the plasma membrane (Puddifoot et al., 2015; Wu et al., 2015), which is an important determinant of the neuromodulatory influence of ACh and the efficacy of exogenous agonists of nAChRs, including the addictive drug, nicotine. The mechanism by which at least several Ly6 proteins regulate this property has yet to be determined.
Here, we define the functional relationship between NACHO and the Ly6 family member Ly6h. We find that NACHO enhances and Ly6h reduces assembly of Ca2+-permeable α7 nAChRs in hippocampal pyramidal neurons. This antagonistic relationship appears to be critical for maintaining α7 signaling within a tight range that permits cholinergic modulation of hippocampal function while simultaneously limiting cytotoxic damage caused by excess Ca2+ influx. Importantly, we demonstrate that this balance is likely shifted toward excitotoxicity and neurodegeneration in AD because of amyloid β (Aβ)-driven downregulation of neuroprotective Ly6h.
Materials and Methods
Primary culture of hippocampal neurons
Hippocampi were dissected from mixed gender P2-3 Sprague Dawley rat pups as previously described (Puddifoot et al., 2015). Cells were plated in dishes coated with poly-L-lysine (Sigma) and mouse laminin (Life Technologies) and cultured in Neurobasal medium (Life Technologies) supplemented with 1% fetal bovine serum (Hyclone), 2% B-27 (Life Technologies), 0.5% L-glutamine (Sigma), and 0.5% penicillin/streptomycin (Mediatech) and maintained at 37°C with 5% CO2. Cytosine arabinoside (Sigma) was added at 2 d in vitro (DIV) to inhibit glial proliferation. For treatment with Aβ (1–42 peptide; Cellmano Biotech), peptide was prepared to enrich for the fibrillar form (Liu et al., 2013). Specifically, peptide was reconstituted in DMSO according to manufacturer's instructions, diluted in PBS to a concentration of 200 μm, frozen in 50 µl aliquots for long-term storage, then thawed, diluted to 100 µm in PBS and incubated at 37°C for at least 96 h before use. Peptide was applied to cells at a final concentration of 100 nm. Cells were infected with adenovirus at 4 DIV in culture medium, which was replaced with fresh conditioned medium after 6–8 h. For imaging experiments, cells were plated on glass coverslips coated with poly-D lysine (Sigma) and mouse laminin (Life Technologies) at 25% density. Detailed information on resources is listed in Table 1.
Table 1.
Key reagents and resources
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GFP | Life Technologies | A11122 |
| Mouse monoclonal anti-V5 | Life Technologies | R960-25 |
| Mouse monoclonal anti-Myc | Santra Cruz Biotechnologies | Sc-40 |
| Mouse monoclonal anti-actin | Millipore Sigma | MAB1501 |
| Mouse monoclonal anti-GluR1 | Millipore Sigma | MAB2263 |
| Rabbit polyclonal anti-TMEM35 | Millipore Sigma | HPA048583 |
| Mouse monoclonal anti-Ly6h | Novus Biologicals | NBP224405 |
| Mouse monoclonal anti-PHF-1 | Dr. Peter Davies | |
| Tau5 monoclonal anti-total Tau | Abcam | Ab80579 |
| Bacterial and virus strains | ||
| Virapower adenovirus | Life Technologies | K4940-00 |
| Biological samples | ||
| Normal and AD brain tissue and CSF | Shiley–Marcos ADRC | http://adrc.ucsd.edu/index.html |
| Neonatal Sprague Dawley rat brain tissue | Harlan | |
| Chemicals, peptides, and recombinant proteins | ||
| CF640R-αBTX | Biotium | #00004 |
| ACh chloride | Sigma | A2661-25G |
| Nicotine ditartrate | R&D Systems | 3546 |
| PNU-120596 | R&D Systems | 249810 |
| Metyllycaconitive citrate | R&D Systems | 1029 |
| Aβ1–42 peptide | Cellmano Biotech | A-1423 |
| Fura-2 AM | Life Technologies | F1221 |
| Calcein blue AM | EBioscience | 65-0855-39 |
| Ethidium homodimer | Biotium | 89139-052 |
| Critical commercial assays | ||
| Cytotox 96 LDH assay | Promega | G1780 |
| Experimental models: cell lines | ||
| HEKtsa | Dr. Carol Deutsch, University of Pennsylvania | |
| 293A | Life Technologies | R705-05 |
| Oligonucleotides | ||
| Ly6 h-shRNA-S: 5′-GATCCCCGGACTGCTGCGAGAAAGATTTTTCAAGAGAAAATCTTTCTCGCAGCAGTCCTTTTTTGGAAA-3′ | IDT | |
| Ly6 h-shRNA-AS: 5′-AGCTTTCCAAAAAAGGACTGCTGCGAGAAAGATTTTCTCTTGAAAAATCTTTCTCGCAGCAGTCCGGG-3′ | IDT | |
| NACHO-shRNA-S: 5′-GATCCCCAGGCTCAGCAAGGATGCCTACAGTGAGATTTCAAGAGAATCTCACTGTAGGCATCCTTGCTGAGCCTTTTTTTGGAAA-3′ | IDT | |
| NACHO-shRNA-AS: 5′-AGCTTTTCCAAAAAAAGGCTCAGCAAGGATGCCTACAGTGAGATTCTCTTGAAATCTCACTGTAGGCATCCTTGCTGAGCCTGGG-3′ | IDT | |
| Scrambled control-S: 5′-GATCCCCGATACGTGCGCAATGAATAGGACGTCAGCTTCAAGAGAGCTGACGTCCTATTCATTGCGCACGTATCTTTTTTGGAAA-3′ | IDT | |
| Scrambled control-AS: 5′-AGCTTTTCCAAAAAAGATACGTGCGCAATGAATAGGACGTCAGCTCTCTTGAAGCTGACGTCCTATTCATTGCGCACGTATCGGG-3′ | IDT | |
| Recombinant DNA | ||
| α7-pcDNA3.1 | Dr. Henry Lester, Caltech | |
| α7-YFP-pcDNA3.1 | H. Lester lab, Pasadena, CA | |
| Ly6 h-pcDNA3.1 | Puddifoot et al. (2015) | |
| Ly6 h-Myc-pcDNA3.1 | Puddifoot et al. (2015) | |
| NACHO-pcDNA3.1 | NM_026239.2 | |
| V5-NACHO-pcDNA3.1 | This paper | |
| TN-XXL | Yamauchi et al. (2011) | |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ/FIJI | https://imagej.net/Fiji |
Cell lines
HEK-tsa cells were cultured and transfected as previously described (Puddifoot et al., 2015). Briefly, cells were maintained in culture medium consisting of DMEM (Mediatech) supplemented with 10% fetal bovine serum (Life Technologies), 1% penicillin/streptomycin (Mediatech) and 1% L-glutamine (Life Technologies) at 37°C and 5% CO2. Cells were transfected using X-tremeGene HP reagent (Roche) at a ratio of 2 μl reagent:1ug DNA in Optimem medium (Life Technologies)
Human samples
Samples of human temporal cortex were obtained from the Shiley–Marcos Alzheimer's Disease Research Center (ADRC) Neuropathology Core at University of California San Diego. Information about samples is shown in Table 2. Tissue samples were homogenized in RIPA lysis buffer (150 mm NaCl, 25 mm HEPES pH 7.5, 1% Triton X-100, 0.1% SDS, and 1% sodium deoxycholate) supplemented with Complete protease inhibitor and PhosStop phosphatase inhibitor (Roche). Pooled CSF samples were obtained from the ADRC and equal volumes were loaded for Western blot analysis.
Table 2.
AD scores from individual tissue samples
| Sample ID | Age | Gender | Diagnosis | AD score |
|---|---|---|---|---|
| X6783 | 84 | M | Normal | 1 |
| X5709 | 94 | M | Normal | 1 |
| X5687 | 84 | M | Normal | 1 |
| X5302 | 83 | F | Normal | 1 |
| X5788 | 92 | M | AD | 2 |
| X5776 | 96 | M | AD | 2 |
| X5749 | 83 | M | AD-severe | 3 |
| X5738 | 85 | F | AD-severe | 3 |
| X5680 | 84 | M | AD | 2 |
| 4996 | 91 | M | Normal | 1 |
| 4954 | 76 | M | Normal | 1 |
| 4942 | 83 | M | Normal | 1 |
| 4870 | 63 | F | Normal | 1 |
| 4689 | 79 | F | Normal | 1 |
| 5665 | 79 | M | AD | 2 |
| 5661 | 97 | F | AD-severe | 3 |
| 5626 | 83 | M | AD | 2 |
| 5047 | 69 | M | AD | 2 |
| 4971 | 102 | M | AD | 2 |
| 5114 | 87 | M | Normal | 1 |
| 5070 | 97 | F | Normal | 1 |
| 5049 | 102 | F | Normal | 1 |
| 5006 | 69 | M | Normal | 1 |
| 5800 | 91 | F | AD-severe | 3 |
| 5795 | 81 | M | AD-severe | 3 |
| 5759 | 87 | F | AD-severe | 3 |
| 5546 | 93 | F | Normal | 1 |
| 5515 | 73 | F | Normal | 1 |
| 5517 | 86 | M | Normal | 1 |
| 5529 | 81 | M | Normal | 1 |
| 5844 | 96 | F | Normal | 1 |
| 5722 | 91 | F | AD-severe | 3 |
| 5779 | 73 | M | AD-severe | 3 |
| 5695 | 86 | F | AD-severe | 3 |
| 5797 | 81 | M | AD-severe | 3 |
| 5823 | 96 | F | AD-severe | 3 |
| 5589 | 94 | F | Normal | 1 |
| 5567 | 86 | M | Normal | 1 |
| 5510 | 92 | M | Normal | 1 |
| 5655 | 82 | F | Normal | 1 |
| 5367 | 94 | F | AD-severe | 3 |
| 5831 | 86 | M | AD-severe | 3 |
| 5789 | 89 | M | AD-severe | 3 |
| 5788 | 92 | M | AD-severe | 3 |
| 5707 | 82 | M | AD-severe | 3 |
Postmortem samples of temporal cortex from human subjects were diagnosed as normal, mild AD (AD), or severe AD (AD-severe). To facilitate correlation with Ly6h expression, subjects were assigned a corresponding AD score based on diagnosis with 1 = normal, 2 = mild AD, and 3 = severe AD. Gender and age of each subject are also listed.
α7 activity assays
HEKtsa cells were transiently transfected with mouse α7, human Ric3, and the Ca2+-activated FRET reporter TN-XXL, with or without Ly6h and NACHO as previously described (Puddifoot et al., 2015; Wu et al., 2015). Twenty-four hours after transfection, cells were re-plated in clear bottom, black 96-well dishes coated with poly-D-Lysine (Sigma); 48 h after transfection, culture media was removed and replaced with artificial CSF (ACSF; 121 mm NaCl, 5 mm KCl, 26 mm NaHCO3, 1.2 mm NaH2PO4H2O, 10 mm glucose, 5 mm HEPES, 2.4 mM CaCl2, and 1.3 mM MgCl2; pH 7.4) supplemented with 10 μm PNU-120596 (R&D Systems) to block desensitization. Following 30 min of preincubation in PNU, FRET-based measurements of α7 activation by nicotine were assayed as described (Yamauchi et al., 2011; Puddifoot et al., 2015). Endogenous α7 activity was measured in cultured primary rat hippocampal neurons at 14 DIV and 10 d postadenovirus infection. Cells were plated in clear bottom black 96-well dishes coated with poly-L-lysine (Sigma) and mouse laminin (Life Technologies). 30 min before assay, culture medium was removed and replaced with 100 µl fura-2 (dissolved in DMSO + 0.5 mm pluronic F127, Life Technologies) in ABP (1× ACSF + 1% BSA, 2.5 mm probenecid; Sigma) and incubated at 37°C. Following fura-2 preincubation, cells were washed 2× with 100 µl ABP to remove excess fura-2 and replaced with 100 µl ABP + 10 μm PNU-120596. Cells were incubated with PNU for 30 min at 37°C. α7 activation by addition of ACh (Acros Organics) was measured using a Flexstation (Molecular Devices) set to read 340/510 and 380/510 (cutoff 495) with 90-s reads at 3.9-s intervals. Following ACh stimulation, maximum and minimum readings were obtained by sequential addition of ionomycin and EGTA (Sigma). Peak 340/380 ratios were normalized to Max/Min and used to calculate dose–response curves using Prism 7 (GraphPad); 100 nm methyllycaconitine (R&D Systems) was included with the PNU incubation to demonstrate α7-specificity.
Western blotting, surface protein biotinylation, and immunoprecipitations
HEKtsa cells and primary hippocampal neurons were lysed in SDS lysis buffer (10 mm Tris, pH 7.5, 100 mm NaCl, 5 mm EDTA, 1% Triton X-100, and 0.05% SDS) supplemented with Complete protease inhibitor and PhosStop phosphatase inhibitor (Roche). Protein levels in lysates were measured using BCA kit (Pierce) and run on 4–12% Tris-glycine gels (Life Technologies). Antibodies used for Western blotting were: rabbit anti-GFP, mouse anti-V5 (Life Technologies), mouse anti-Myc (Santa Cruz Biotechnologies), mouse anti-actin and mouse anti-GluR1 (Millipore-Sigma), mouse anti-Ly6h (Novus Biologicals), rabbit anti-TMEM35 (Sigma Prestige), PHF-1 (gift of Peter Davies), and Tau5 (total Tau, Abcam). Protein levels in Western blot were quantified by pixel density measurement using Image J software. Cell surface biotinylation and immunoprecipitations were performed as previously described (Puddifoot et al., 2015), with the biotinylated α-bungarotoxin (BTX; Biotium) used to immunoprecipitate α7 pentamers.
Adenovirus shRNA knock-down in primary cultured hippocampal neurons
H1-pTrip-EF1a-eGFP shRNA entry vector was constructed by subcloning the EF1a-EGFP cassette from pTRIP-du-EF1A-EGFP lentiviral vector (gift from L.VanAelst, Cold Spring Harbor labs) into the entry vector mENTRY (gift from J. Ni, Harvard) at the HindIII/XhoI sites. The H1 promoter was inserted upstream of this cassette between the SpeI/HindIII sites. Annealed oligos containing shRNA sequences were inserted between BamHI and BglII. Adenovirus constructs were generated by gateway recombination with the shRNA entry vector and pADplDest (Life Technologies). Adenovirus was produced by transfecting Pac1-digested adenovirus vector into 293A cells (Life Technologies) using X-tremegene HP reagent (Sigma) and amplified for virus harvesting according to the manufacturer's protocol (ViraPower, Life Technologies). Purified adenovirus was aliquoted and flash frozen in liquid nitrogen and stored at −80°C. Infection of primary cultured neurons was performed at 4 DIV in neuron culture media (see above) at a concentration of ∼2–8 × 108 OPU (optical particle unit; measured at OD 260) per cm2 culture depending on cell density. After a 4- to 6-h incubation with adenovirus, infection media was replaced with fresh conditioned media.
Cell death assays
For PNU-120596-mediated cell death, rat primary hippocampal neurons were plated in 24-well dishes coated with poly-D-lysine (Sigma) and laminin (Life Technologies). At 8 DIV, 10 μm PNU (R&D Systems) or and equal volume of DMSO with or without 100 nm methyllycaconatine (R&D Systems) was added to the culture media for 3 h; 3 × 50 µl aliquots of culture medium was removed for lactate dehydrogenase (LDH) assay (see below). Cells were stained with 5 μm calcein-blue AM (EBioscience) and 2 μm ethidium homodimer-1 (Biotium) in prewarmed live cell imaging solution (LCIS; 140 mm NaCl, 2.5 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, and 20 mm HEPES; pH 7.4) for 25 min at room temperature in a dark chamber. Dyes were removed and replaced with fresh prewarmed LCIS and imaged on a Zeiss Axios Observer fluorescent light microscope at 20× magnification and 10 fields per well were imaged. Live and dead cells were counted and the average for each condition was normalized to DMSO control. For Ly6h-knock-down and NACHO-knock-down cell death assays, neurons were cultured in 48-well dishes coated with poly-D-lysine (Sigma) and laminin (Life Technologies). Cells were infected with adenovirus at four DIV and maintained in a sterile chamber with dampened tissue to maintain evenness of humidity and prevent unequal volume loss in the dish through evaporation. To measure the time course of cell death, all but 200 µl of media was removed from duplicate wells for each condition 20 h before LDH assay (see below). PNU-toxicity was performed on adenovirus infected neurons at 14 DIV, 10 d after infection as described above. H2O2 toxicity was performed on adenovirus infected neurons at 11 DIV, 7 d postinfection. Conditioned media was collected and stored at 4°C and replaced with 300 ml of ACSF (121 mm NaCl, 5 mm KCl, 26 mm NaHCO3, 1.2 mm NaH2PO4-H2O, 10 mm glucose, 5 mm HEPES pH 7.4, 2.4 mm Ca2+, and 1.3 mm Mg2+) with 0.1% bovine serum albumin with or without 50 mm H2O2. Cells were exposed H2O2 to for 2 h at 37°C and then returned to 250-µl fresh conditioned media. LDH assay was performed as described below 20–22 h after H2O2 exposure. Hypoxia-induced cell death was performed on adenovirus infected neurons at 10 DIV, 6 d after infection. Conditioned media was collected and stored at 4°C and replaced with 250-µl LCIS and placed in a hypoxia chamber flooded with 95% nitrogen/5% CO2 for 10 min. The sealed chamber was placed at 37°C for 2 h after which the cells were returned to normal oxygen in fresh conditioned media. Controls cells were treated similarly but without hypoxia/glucose starvation. LDH assay was performed as described below 15 h after hypoxia/glucose starvation. Following cell toxicity challenge, all but 200 µl of culture media per well was removed in preparation for LDH assay 20–24 h before LDH measurements, which was performed from duplicate wells using the CytoTox 96 LDH assay (Promega) as described. Briefly, 50 µl of media was removed from the cells and placed in clear, flat-bottomed 96-well dishes; 50 µl of reconstituted CytoTox96 reagent was added to the media and incubated for 30 min at room temperature in a dark chamber. The reaction was stopped by addition of 50-µl stop reagent and 490 nm absorbance was read using a TECAN plate reader. Remaining cells were lysed in 150 µl of growth media by addition of 15-µl 10× lysis solution and incubated for 45 min at 37°C. Total LDH was read from 50 µl of cell lysates as above. Percent cell death was calculated as LDH (released)/MaxLDH.
Immunohistochemistry and imaging
Cultured neurons were grown on glass coverslips coated with poly-D-lysine and mouse laminin at 25% density. Samples were collected for staining at 11–14 DIV. For surface α-BTX labeling, CF-640R-conjugated BTX (Biotium) was added to culture media at 5 mg/ml and incubated at 37°C for 30 min. Samples were washed 3× in PHEM buffer (60 mm PIPES, pH 6.9, 25 mm HEPES, 10 mm EGTA, and 2 mm MgCl2•6H2O) and fixed as described below. For permeabilized cells staining, cells were rinsed 1× in PHEM wash buffer (60 mm PIPES, pH 6.9, 25 mm HEPES, 10 mm EGTA, 2 mm MgCl2•6H2O, and 0.1% Triton X-100), and fixed in 4% formaldehyde/PHEM for 10 min at room temperature. Cells were permeabilized in PHEM-T buffer (60 mm PIPES, pH 6.9, 25 mm HEPES, 10 mm EGTA, 2 mm MgCl2•6H2O, and 0.5% Triton X-100) for 5 min at room temperature and then washed and incubated in blocking buffer (150 mm NaCl, 20 mm Tris-HCl, pH 7.4, 0.1% Triton X-100, and 2% bovine serum albumin) for 1 h at room temperature. Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C. Antibodies used for immunostaining were: rabbit anti-GFP (Life Technologies), mouse anti-Ly6h (Novus Biologicals), rabbit anti-TMEM35 (Sigma Prestige), and PHF-1 (gift of Peter Davies). Cells were imaged using a Nikon A1R HD confocal with a four-line (405, 488, 561, and 640 nm) LUN-V laser engine and a DU4 detector using bandpass and longpass filters for each channel (450/50, 525/50, 595/50, and 700/75), mounted on a Nikon Ti2-E using a Plan-Apo 20 × 0.75 NA objective. Images stacks were acquired in galvano scanning mode with unidirectional scanning, and Z-steps of 0.975 µm. To avoid cross-talk between fluorphores, Z-stacks were acquired of the DAPI, AlexaFluor488 and AlexaFluor 633 or αBTX-640R in a channel series. Scans were made at 2 s per frame, and the pinhole was set at 1 Airy unit.
Statistical analyses
Statistical comparisons were performed using GraphPad Prism version 8. One-way ANOVA with Holm–Sidak's multiple comparison post hoc test was used to calculate p values for experimental versus control groups, unless otherwise indicated. Exact p values are reported in figure legends unless p < 0.0001. Data are presented as mean ± SEM.
Results
Ly6h and NACHO function in opposition to regulate α7 nAChR activity
Our lab and others previously identified proteins that regulate Ca2+-permeable α7 nAChRs (Puddifoot et al., 2015; Gu et al., 2016). Two of these proteins, Ly6h and NACHO, suppress and enhance delivery of α7 nAChRs to the cell surface, thus leading to reduced and potentiated signaling, respectively, by the endogenous neurotransmitter ACh. To determine whether the opposing functions of Ly6h and NACHO normally coexist in the same cells, we knocked down each protein in primary cultured hippocampal neurons using adenovirus-delivered shRNAs. To confirm the efficiency of knock-down, we Western blotted lysates of infected neurons and found that our shRNAs reduced Ly6h and NACHO protein expression by 65–75% at 7 d postadenoviral delivery (Fig. 1A,B). Under similar conditions we then assayed for knock-down effects on activity of α7 nAChRs. In these experiments we blocked receptor desensitization with the α7-selective positive allosteric modulator, PNU-120596, to measure steady-state Ca2+ fluxes through α7 receptors, and we used the ratiometric Ca2+ indicator, fura-2, to measure average cellular responses. Consistent with Ly6h and NACHO functioning together in the same population of neurons, we found that maximal ACh-induced Ca2+ influxes through α7 nAChRs were enhanced by knock-down of Ly6h and suppressed by knock-down of NACHO (Fig. 1C,D). Ca2+ responses were mediated by α7 nAChRs since pretreatment with the α7-selective antagonist methylycaconitine (MLA) blocked all response to agonist (data not shown). To confirm that Ly6h and NACHO are responsible for opposing effects on α7 nAChRs in the same cells we immunolabeled Ly6h and NACHO in cultured hippocampal neurons and examined the results by confocal microscopy. As expected, we found that all neurons co-expressed both regulatory proteins (Fig. 1E).
Figure 1.
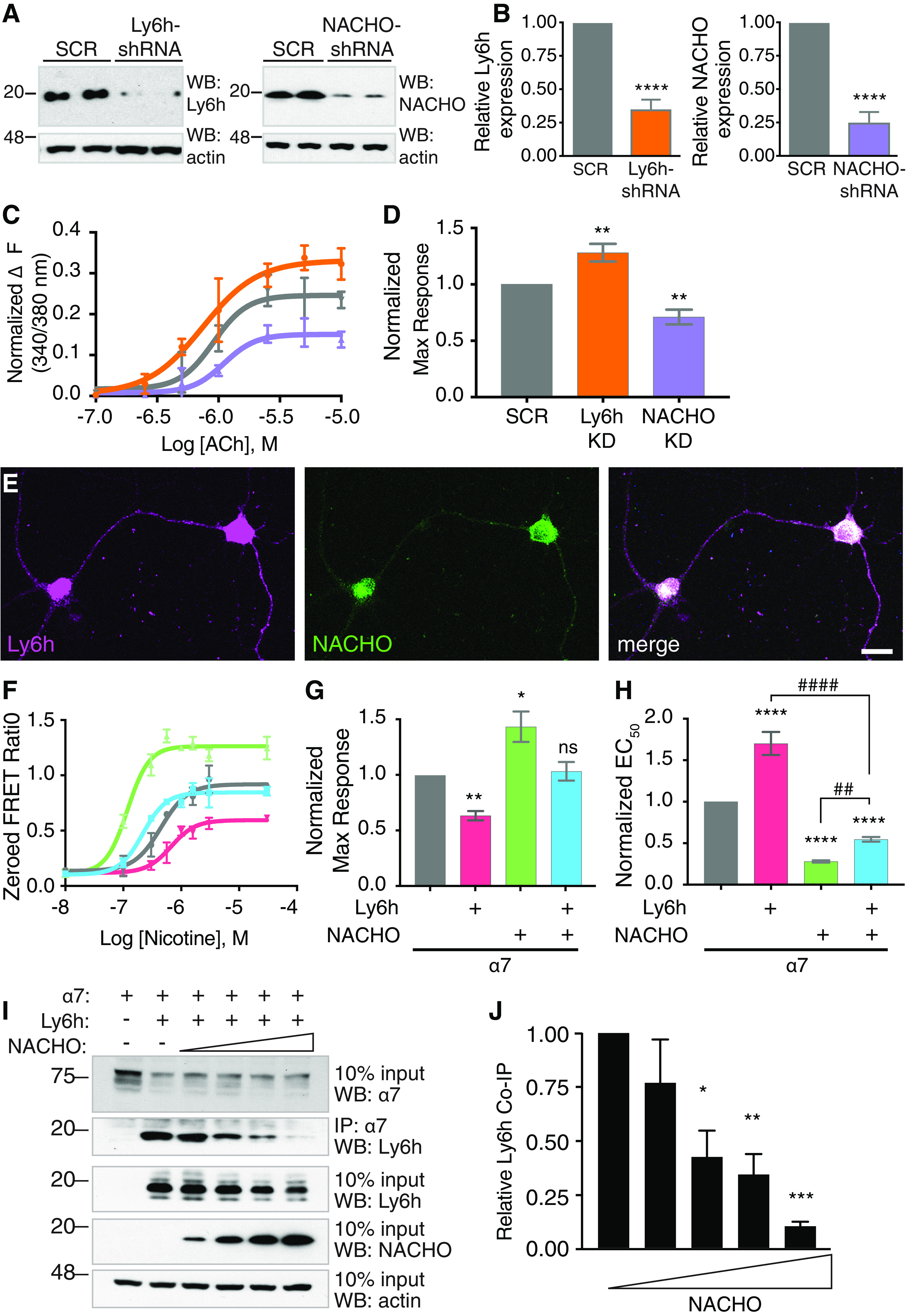
Competition between Ly6h and NACHO for access to receptor subunits causes opposing effects on activity of α7 nAChRs. A, Western blot of lysates from hippocampal neurons 7 d postinfection with adenovirus expressing scrambled control (SCR), Ly6h-shRNA, or NACHO-shRNA. Top panels, Immunoblots with anti-Ly6h (left) and anti-NACHO (right) antibodies. Bottom panels, Immunoblots with anti-actin antibody. B, Quantification of Ly6h (orange) and NACHO (purple) normalized to actin and plotted relative to the corresponding values for scrambled controls. Values are mean ± SEM, N ≥ 19 for each condition; ****p < 0.0001 by one-way ANOVA with Holm–Sidack's multiple comparison post hoc test compared with scrambled control. C, Representative concentration-response curves for ACh-induced Ca2+ influx through α7 nAChRs in primary cultured hippocampal neurons following shRNA knock-down of Ly6h (orange), NACHO (purple), or scrambled control (SCR, gray; error bars = SEM from triplicate measurements in an individual experiment). D, Average maximum response from repeated experiments as in A (mean ± SEM, N = 8); **p = 0.0093 for Ly6h-shRNA versus SCR and **p = 0.0059 for NACHO-shRNA versus SCR by one-way ANOVA with Holm–Sidack's multiple comparison test. E, Primary cultured hippocampal neurons immunostained against Ly6h (magenta) and NACHO (green). F, Representative concentration-response curves for activation of α7 nAChRs by nicotine in transiently transfected HEKtsa cells. The control response in the absence of Ly6h and NACHO is shown in gray. Other curves are from responses in the presence of Ly6h (red), NACHO (green) or both (blue; error bars = SEM from triplicate measurements in an individual experiment). Average maximum response (G) and EC50 (H) normalized to α7 alone (mean ± SEM, N = 8); *p = 0.125, **p = 0.0031, ****p < 0.0001, ##p = 0.0063, ####p < 0.0001 by one-way ANOVA with Holm–Sidack's multiple comparison test. I, Representative immunoblot of Ly6h co-immunoprecipitated with α7-YFP in the presence of increasing amounts of NACHO in transfected HEKtsa cells. Second panel shows complexes between Ly6h and α7 decrease as NACHO increases. J, Quantification of Ly6h complexed with α7 as a function of increasing NACHO expression (N = 5). Levels of co-immunoprecipitated Ly6h were normalized to levels of actin in the input; *p = 0.0049, **p = 0.0024, ***p = 0.0002 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test.
Given these results, we hypothesized that Ly6h and NACHO have opposing cell biological functions that sum together to maintain signaling by α7 nAChRs within a narrow range. To test this hypothesis, we measured the isolated and combined effects of each auxiliary subunit on α7 nAChR signaling in transfected HEKtsa cells, which do not normally express any of these proteins. As expected, we found that Ly6h alone reduced and NACHO alone potentiated the maximum response of α7 nAChRs to agonist. Furthermore, when both regulatory subunits were combined, the net effect was similar to what we measured in control cells lacking both subunits (Fig. 1F–H).
Two simple scenarios could account for the ability of Ly6h and NACHO to limit each other's effects. In the first scenario, Ly6h and NACHO could regulate α7 nAChRs independently, but their limited expression could prevent either auxiliary protein from exerting a dominant effect. In the second scenario Ly6h and NACHO could exert opposing effects if at least one regulatory protein limited access of the other to α7 subunits. In both scenarios the net Ca2+ response would reflect the summed effects of Ly6h and NACHO on separate pools of receptor subunits. However, in the second scenario the two auxiliary proteins would have mutually exclusive effects on individual α7 subunits. To distinguish between these possibilities we tested whether NACHO could reduce interactions between Ly6h and α7 nAChRs, which are known to form stable complexes (Puddifoot et al., 2015). To carry out this experiment we held Ly6h and α7 at constant levels and varied the amount of NACHO that was introduced into HEKtsa cells. We then immunoprecipitated α7 subunits and Western blotted for the presence of complexed Ly6h. We found that increasing amounts of NACHO caused a reciprocal reduction in complex formation between Ly6h and α7 (Fig. 1I,J). This result is consistent with scenario two and suggests that NACHO prevents Ly6h from binding to α7 subunits.
Ly6h inhibits and NACHO promotes assembly and surface expression of α7 nAChRs
What is the mechanism responsible for the opposing effects of Ly6h and NACHO on α7 nAChRs? One recent study indicates that NACHO functions as a chaperone to enhance multimerization of nAChRs (Gu et al., 2016). However, it is unclear what factor normally limits this process and thus requires NACHO to overcome it. We hypothesized that in hippocampal neurons this factor is Ly6h, which competes with NACHO to maintain assembly and ultimately signaling of α7 nAChRs within a physiological range. We tested this hypothesis by measuring fluorescent-labeled αBTX bound to the surface of hippocampal neurons in which we manipulated levels of Ly6h and NACHO by adenoviral-mediated knock-down. αBTX binds with high selectivity and affinity to mature, pentameric α7 nAChRs, thus serving as a proxy for the terminal state of assembly during receptor biogenesis (Drisdel and Green, 2000; Huang et al., 2013). Consistent with our hypothesis, we found that αBTX binding was enhanced significantly in neurons in which we knocked down Ly6h (Fig. 2A, center panels, B). We cannot rule out the additional possibility that the changes in αBTX binding that we measured are caused by changes in expression of α7 receptors. However, this distinction cannot be easily addressed in neurons since a reliable antibody is currently unavailable for recognition of native α7 subunits.
Figure 2.
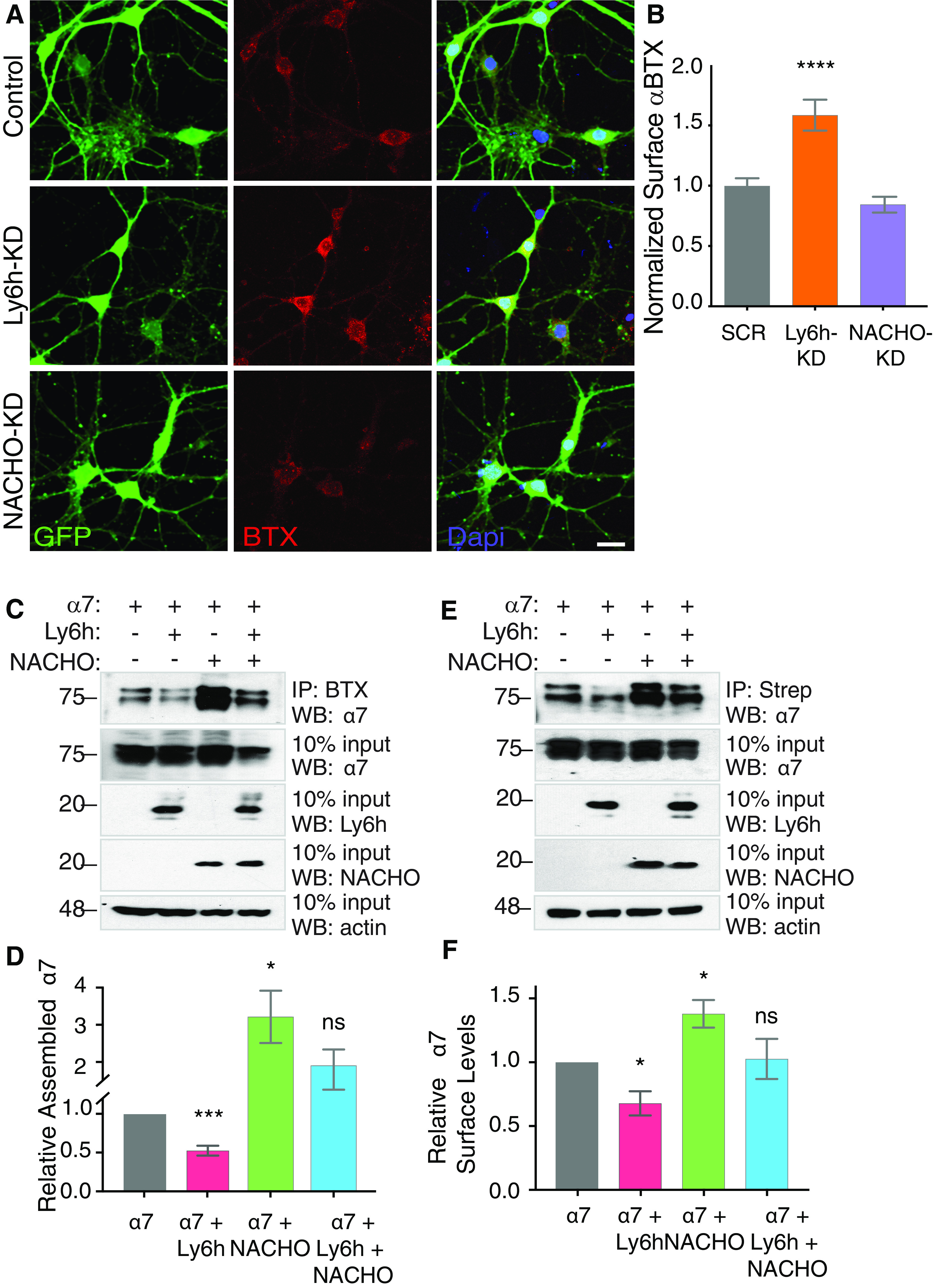
Ly6h inhibits and NACHO promotes pentameric assembly and surface expression of α7 nAChRs. A, Fluorescent images of cultured primary hippocampal neurons stained with anti-GFP (green, left panels) and fluorescently labeled αBTX (red, center panels), and merges with Dapi-labeled nuclei (blue, right panels). B, Quantification of αBTX fluorescence intensity in adenovirus positive cells (green) normalized to average values in scrambled control (N > 70 cells each); ***p < 0.001 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test. C, Representative immunoblot of αBTX-immunoprecipitated pentameric α7-YFP in transiently transfected HEKtsa cells. Top panel, αBTX immunoprecipitation followed by Western blotting with anti-GFP to detect pentameric α7. Middle panels, 10% input of total cell lysates Western blotted with anti-GFP to detect total α7, anti-Myc to detect Ly6h expression, and anti-V5 to detect NACHO expression. Bottom panel, 10% input total cell lysates Western blotted with anti-actin as loading control. D, Average levels of αBTX-immunoprecipitated receptor normalized to total immunoprecipitated receptor relative to control conditions with α7 alone (N = 10); *p = 0.0383, ***p = 0.001 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test. E, Representative immunoblot of streptavidin immunoprecipitations of biotinylated surface α7-YFP in transiently transfected HEK-tsa cells. Top panel, Streptavidin immunoprecipitation followed by Western blotting with anti-GFP to detect surface α7. Middle panels, 10% input of total cell lysates Western blotted with anti-GFP to detect total α7, anti-Myc to detect Ly6h expression, and anti-V5 to detect NACHO expression. Bottom panel, 10% input total cell lysates Western blotted with anti-actin as loading control. F, Average α7 surface levels of streptavidin-immunoprecipitated receptor normalized to total immunoprecipitated receptor relative to control conditions with α7 alone (N = 7); *p = 0.0166 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test.
To address this shortcoming in a context in which we could control and measure expression of each protein, we transfected HEKtsa cells with YFP-tagged α7. We extracted pentameric receptors with αBTX and then quantified them by Western blotting with an anti-GFP antibody. Under these control conditions we were clearly able to detect subunits of fully assembled α7 nAChRs (Fig. 2C). Furthermore, these levels were reduced with the addition of Ly6h and enhanced with the addition of NACHO to transfection mixtures, with intermediate levels of fully assembled receptors present when Ly6h and NACHO were included together (Fig. 2C,D). Importantly, levels of α7 subunits in total cell lysates were unaffected by the different transfection conditions. Thus, the opposing effects of Ly6h and NACHO on assembly of α7 nAChRs likely underlies the opposing effects of the same proteins on the magnitude of Ca2+ signaling through α7 nAChRs in hippocampal neurons.
If our interpretation is correct, then we expect Ly6h and NACHO to have parallel effects on both assembly and surface expression of α7 nAChRs. To examine the latter, we first biotinylated the surfaces of HEKtsa cells transfected with YFP-tagged α7 subunits. Then we immunoprecipitated labeled proteins with streptavidin and Western blotted for the presence of α7 using an anti-GFP antibody. Since nAChRs must fully assemble to escape the endoplasmic reticulum and traffic to the cell surface (Wang et al., 2002), levels of anti-GFP immunoreactivity in our experimental conditions are thus proportional to levels of α7 nAChRs at the plasma membrane. Consistent with our expectations, under control conditions we found a low but detectable presence of surface-biotinylated α7 protein (Fig. 2E). Furthermore, this level was reduced by the addition of Ly6h and enhanced by the addition of NACHO to the transfection mixture, with an intermediate level resulting from addition of both regulatory subunits (Fig. 2E,F). Combined with our previous results, these data strongly suggest that Ly6h and NACHO exert opposing effects on assembly of α7 subunits, which cause proportionate changes in surface expression and ultimately in amplitude of Ca2+ signaling by functional α7 nAChRs in neurons.
Sustained activation of α7 nAChRs sensitizes neurons to premature cell death
Why are Ly6h and NACHO, which have opposing functions, expressed together in the same neurons? We hypothesized that together these proteins maintain ACh-induced Ca2+ signaling through α7 receptors within a range that balances two opposing influences: the need for plasticity, which favors receptor assembly, and the need to avoid neurotoxicity, which favors obstacles to receptor assembly. Ample evidence already suggests that α7 nAChRs are important for plastic processes that may contribute to cognition (Gu et al., 2012; Lozada et al., 2012; Stoiljkovic et al., 2016; Townsend et al., 2016; Shenkarev et al., 2020). Therefore, we focused on testing the other aspect of our hypothesis, i.e., that high levels of functional α7 nAChRs must be avoided to prevent neurotoxicity. To address this idea, we allowed ambient choline in culture media to activate receptors at low levels while blocking desensitization with PNU. Then after 3 h of chronic activation we used two different assays to measure cell death. First we stained primary cultured hippocampal neurons with calcein AM, which fluoresces when taken up by intact cells, and with ethidium homodimer-1, which selectively labels nuclei of dead cells (Andree et al., 2019). In the second assay, we measured release of LDH from dead neurons into culture media (Wang et al., 2020). In both cases, we found that sustained activation of α7 nAChRs by PNU increased cell death (Fig. 3A–C). These effects were clearly because of α7 nAChRs as they were blocked by the α7-selective competitive antagonist, MLA (Ward et al., 1990). Our results also suggest that the extraordinarily fast desensitization of α7 nAChRs (tau < 10 ms; Bouzat et al., 2008; Puddifoot et al., 2015) may have evolved at least in part to limit sustained Ca2+ entry and the neurotoxicity that appears to result from it.
Figure 3.

Reducing Ly6h sensitizes and reducing NACHO protects neurons from cytotoxic challenges. A, Fluorescent images of cultured primary hippocampal neurons stained with calcein blue-AM and ethidium homodimer-1 (red, with white arrows) after a 3-h exposure to 10 μm PNU-120596 (PNU) or vehicle control (DMSO) with or without 100 nm MLA. B, Quantification of cell death after PNU/MLA exposure. At least 10 fields were counted per condition per experiment (N = 7 experiments); ****p < 0.0001 by one-way ANOVA with Holm–Sidak's multiple comparison test. C, Quantification of LDH released into media following drug induction as a fraction of total LDH released after lysing all cells. Values were normalized to those of DMSO controls (N = 7). Data are represented as mean ± SEM, ***p = 0.0009 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test. D, Fractional cell death as measured in C after shRNA knock-down of Ly6h (orange) versus scrambled control (SCR, black). Each condition was measured in the absence (solid line) versus the presence of MLA (dashed line; N > 16); ****p < 0.0001 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test for % cell death at 16 d with Ly6h-Sh versus SCR (no MLA), and #p = 0.0004 by extra sum-of-squares F test for the slopes of Ly6h-Sh –MLA versus +MLA. E–G, Cell death was measured as in C, D following shRNA knock-down of scrambled controls (SCR, gray), Ly6h (orange), or NACHO (purple) in primary hippocampal neurons. Measurements were made (E) after 3-h exposure to DMSO vehicle control or 10 μm PNU (N = 8), (F) 24 h after 1-h exposure to saline control or H2O2 (N = 4), and (G) 15 h after 2-h exposure to saline control or hypoxia (N = 5). Data are represented as mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test.
Next, we asked how Ly6h and NACHO modify α7-mediated cell death. Since Ly6h reduces and NACHO promotes assembly of α7 nAChRs, we hypothesized that decreasing the Ly6h:NACHO ratio would be neurotoxic, whereas increasing the ratio would be neuroprotective. To test this hypothesis, we first measured LDH release from cultured hippocampal neurons infected with either our Ly6h shRNA or our scrambled control shRNA. As predicted, knock-down of Ly6h increased basal neuronal death by nearly 30%. Furthermore, addition of MLA to the culture medium at the time of infection slowed this effect, indicating it was caused by enhanced α7 activity (Fig. 3D).
To further test our hypothesis, we manipulated Ly6h and NACHO levels in hippocampal neurons under three conditions, each of which presented neurotoxic challenges. First, we disinhibited receptors by blocking their desensitization with PNU. As expected based on our prior results, we found that PNU increased cell death. Furthermore, we found that this effect was enhanced by knock-down of Ly6h and suppressed by knock-down of NACHO (Fig. 3E). Thus, Ly6h protects against and NACHO contributes to neurotoxicity caused by sustained activation of α7 nAChRs.
A more commonly used method of assaying for neuronal cell death is exposure to hydrogen peroxide (H2O2), a reactive oxygen species that is thought to function as a signaling molecule at low levels but as an oxidative stressor at high levels under certain pathophysiological conditions (Lennicke and Cochemé, 2020). Consistent with the latter, we found that acute exposure to H2O2 caused a nearly 50% increase in cell death in scrambled control infected neurons. Importantly, we found that knock-down of Ly6h enhanced and knock-down of NACHO reduced this toxicity (Fig. 3F). We also tested our cultured neurons with another physiologically relevant challenge; transient exposure to hypoxia/glucose starvation, which mimics conditions of ischemia/reperfusion seen in brain injuries such as stroke. Using this assay, we also observed elevated cytotoxicity in scrambled control-infected neurons and a further enhancement of cell death in neurons in which Ly6h was knocked down (Fig. 3G). In these conditions knock-down of NACHO was not able to protect neurons, suggesting that additional mechanisms that bypass activation of α7 nAChRs might be sufficient to upregulate cell death in response to hypoxia. Regardless, in all three conditions we tested, manipulating the levels of Ly6h or NACHO had neurotoxic or neuroprotective effects, respectively. Collectively these results support our hypothesis that the Ly6h:NACHO ratio is crucial for keeping assembly of α7 receptors, and thus Ca2+-influx in response to ACh, within a tight physiological range, with deleterious consequences resulting from dysregulation, particularly if the upper range of receptor efficacy is enhanced.
Chronic exposure to Aβ causes downregulation of Ly6h
The cytotoxic effects of excessive Ca2+ signaling are well-established and likely contribute to many neurologic disorders (Guerra-Álvarez et al., 2015; Giorgi et al., 2018). Since we found that Ly6h buffers against excessive signaling through Ca2+-permeable α7 nAChRs, we hypothesized that Ly6h may be reduced in certain neurodegenerative diseases. The most common neurodegenerative conditions are tauopathies, which involve hyperphosphorylation of the microtubule associated protein tau, leading to its aggregation into neurofibrillary tangles. To test whether Ly6h buffers against hyperphosphorylation of tau, we first knocked down Ly6h in cultured hippocampal neurons with our viral shRNA and measured phospho-tau (p-tau) by Western blotting. We found that p-tau levels increased by over 100% in neurons depleted of Ly6h relative to control neurons (Fig. 4A,B). In addition, we stained neurons with a p-tau-specific antibody and measured resulting levels of immunofluorescence. Consistent with data from Western blot, we measured ∼60% increase in p-tau signal in neurons depleted of Ly6h relative to controls (Fig. 4C,D). In both experiments we also knocked down NACHO with our viral shRNA. Although we did not measure any resulting change in p-tau by Western blotting (Fig. 4B), we were able to discern a small but significant decrease by staining (Fig. 4D). Thus, Ly6h and NACHO may respectively buffer against and facilitate phosphorylation of tau.
Figure 4.

Reducing Ly6h increases phosphorylation of tau. A, Representative immunoblots of hippocampal neuronal lysates after shRNA knock-down of Ly6h, NACHO, or scrambled control (SCR). Top panel, Anti-PHF1 for pTau (S396/404). Second panel, Anti-Tau5 for total tau. Third panel, Anti-Ly6h. Bottom panel, Anti-actin for loading control. B, Average pTau levels normalized to total tau relative to the average in SCR condition (N = 8). Data are represented as mean ± SEM; *p = 0.0295 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test. C, Fluorescent images of primary hippocampal neurons stained with anti-PHF1 for pTau (top panels, red) and anti-GFP (merged, bottom panels) 7 d after infection with adenoviral shRNAs. D, Quantification of pTau fluorescence in knock-down conditions relative to those in scrambled controls (N > 85 cells each); **p = 0.0045, ****p < 0.0001 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test.
The most common tauopathy, AD, is distinguished by concomitant accumulation of plaques composed of Aβ protein (Stancu et al., 2019). To determine whether a functional relationship exists between Aβ and Ly6h, we first treated cultured hippocampal neurons with a peptide fragment of Aβ (Aβ1–42) that is believed to be pathogenic (Steiner et al., 2018). Following exposure to Aβ1–42 for various durations, cells were lysed and Western blotted with an antibody against Ly6h. While exposure times of up to 4 d led to no significant change in Ly6h levels, we found that 7 d of treatment caused Ly6h protein levels to decrease by ∼50% without affecting levels of other membrane proteins such as GluR1. Furthermore, this decrease in Ly6h expression required activation of α7 nAChRs since simultaneous addition of MLA completely blocked the response (Fig. 5A,B).
Figure 5.

Chronic exposure to Aβ reduces Ly6h levels. A, Representative immunoblots from primary hippocampal neuronal lysates after 7 d exposure to 100 nm Aβ1–42 or DMSO vehicle control with or without 100 nm MLA. Western blot with anti-Ly6h (top), anti-GluR1 (center), and anti-actin (bottom) antibodies. B, Quantification of Ly6h levels normalized to actin relative to the average in vehicle control (N > 16); ****p < 0.0001 by one-way ANOVA with Holm–Sidak's multiple comparison post hoc test. C, Representative immunoblots from temporal cortex of age-controlled human brains with or without AD. Blots were probed with anti-Ly6h and anti-actin antibodies. D, Quantification of Ly6h (normalized to actin) levels from brains of age-matched control and AD temporal cortex (mean ± SEM, N = 23 each); **p = 0.0034 by unpaired t test. E, Correlation plot of Ly6h protein levels and disease severity where 1 = normal, 2 = mild AD, and 3 = severe AD. R2=0.2204 and p = 0.0011 by extra sum-of-squares F test for non-zero slope. F, Immunoblot of Ly6h in pooled CSF samples from patients with low, medium, or high AD scores.
To determine whether the Aβ-driven downregulation of Ly6h in cell culture is reflected by parallel changes in AD brains, we obtained tissue samples of temporal cortex from AD and age-matched control patients (Table 2). After Western blotting these samples using our antibody against Ly6h, we found that AD patients had significantly lower levels of Ly6h than in control patients (Fig. 5C,D). But perhaps more importantly, we also found that Ly6h levels were inversely correlated with disease severity (Fig. 5E). The Aβ-induced reduction in Ly6h (Fig. 5) and the resulting sensitization of neurons to premature cell death following direct depletion of Ly6h (Fig. 3) suggest that Ly6h might also represent a novel biomarker of AD progression. Since anchoring to the outer leaflet of the plasma membrane might allow Ly6h to be released enzymatically or as exosomes, we hypothesized that changes in this molecule might be detectable in the CSF. To test this hypothesis, we obtained pooled CSF from AD patients at different stages of disease progression and assayed for the presence of Ly6h by Western blotting. We found that Ly6h is readily detectable in CSF from patients with early AD, mildly detectable in patients with mild-moderate AD, and practically undetectable in CSF from patients at late stages of AD (Fig. 5F). Collectively, these data suggest that the level of Ly6h from patient CSF may be a useful indicator of pathologic severity in the progression of AD. Such a correlation is also likely to be a direct assessment of neurotoxicity since Ly6h is predominantly found in neurons and not glia (data not shown).
Discussion
Opposing functions of Ly6h and NACHO ensure balanced signaling by α7 nAChRs
Here, we demonstrate that Ly6h and NACHO function antagonistically to maintain amplitude of signaling by neuronal α7 nAChRs within an optimal range. The mechanism is straightforward and elegant. Ly6h binds to and retards assembly of α7 subunits into functional pentameric receptors, whereas NACHO displaces Ly6h, thus disinhibiting receptor assembly. Essentially Ly6h and NACHO function as anti-chaperone and chaperone, respectively, with the relative abundance of the two proteins determining the net effect on expression of α7 nAChRs at the cell surface. Notably, Ly6h forms stable complexes with α7 subunits, whereas NACHO does not (Fig. 1I,J; Gu et al., 2016; Matta et al., 2017). Thus, it remains to be determined whether NACHO's ability to displace Ly6h from α7 subunits is because of weak binding of NACHO to a site to which Ly6h might otherwise occupy, to sequestration of Ly6h by NACHO, or to an unknown effector of NACHO serving one of these roles.
In addition to functioning as an anti-chaperone, Ly6h has a second inhibitory effect as well, which is to increase the effective concentration of agonist required to activate α7 nAChRs, presumably because of competitive antagonism at the cell surface (Fig. 1H; Puddifoot et al., 2015). Our interpretation of this second effect has recently been confirmed experimentally by Moriwaki et al. (2020). Although these authors concluded that Ly6h mediates its inhibitory effects entirely at the cell surface, their experiments were conducted in the presence of NACHO, which, as we have shown here, overcomes and thus masks the inhibitory effects of Ly6h on receptor assembly. Thus, our current results and the recent findings by Moriwaki et al. (2020) reinforce our hypothesis that Ly6h acts as a bifunctional inhibitor of α7 nAChRs by retarding subunit oligomerization and acutely antagonizing receptors at the cell surface.
Interestingly, the effects of Ly6h and NACHO on the amplitude of α7-mediated Ca2+ signaling seem to cancel out, which raises the question of why both regulators evolved at all. We hypothesize that two opposing selection pressures favored the evolution of Ly6h and NACHO over the absence of both proteins. On the one hand, high α7 expression is potentially dangerous since, as we have shown here, sustained receptor activation kills neurons. Thus, the ability of Ly6h to buffer against this possibility by limiting both assembly of nAChR subunits and the activity of fully functional receptors would help ensure neuronal survival (a homolog of Ly6h, Lynx1, may have related functions (Miwa et al., 2006; Kobayashi et al., 2014; Nichols et al., 2014). On the other hand, signaling through Ca2+-permeable α7 nAChRs has been shown to facilitate synaptic plasticity and memory formation (Gu et al., 2012; Lozada et al., 2012; Stoiljkovic et al., 2016; Townsend et al., 2016; Lykhmus et al., 2019; Sadigh-Eteghad et al., 2020), so NACHO's ability to counterbalance some of the inhibitory effects of Ly6h and perhaps of other Ly6 proteins also likely promotes fitness. Overall, we propose that the opposing effects of Ly6h and NACHO help maintain activity of α7 nAChRs within a range that balances both competing selective pressures to optimize brain function (Fig. 6A).
Figure 6.

Models for regulation α7 nAChRs by Ly6h and NACHO. A, Relationship between cell function and Ca2+ influx through α7 nAChRs. Neurons require α7 nAChRs for proper cognitive function. However, too much Ca2+ influx causes cell death. Thus, levels of α7 nAChRs must be tightly regulated by the opposing functions of Ly6h and NACHO to maintain Ca2+ signaling within the physiological range. B, Ly6h reduces and NACHO enhances assembly of α7 nAChRs. In an unknown process that requires α7 activity, Αβ causes Ly6h levels to be reduced, thus allowing NACHO's pro-assembly function to dominate and more α7 nAChRs to be deposited at the cell surface. Basal cholinergic signaling now causes excessive Ca2+ influx and thus increased neurotoxicity.
The relation between Ly6h dysfunction and AD
The inverse correlation we found between levels of Ly6h and AD severity in human temporal cortex suggests a strong link exists between Ly6h function and AD neuropathology. In support of this in vivo relationship our in vitro data show that Ly6h connects several important aspects of AD. First, Aβ, a putative driver of this disease, causes downregulation of Ly6h. Second, strong reduction in Ly6h upregulates phosphorylated tau, a histopathological marker and correlate of neuronal loss in AD. Third, strong reduction in Ly6h causes degeneration of hippocampal neurons, an early hallmark of disease pathogenesis.
Interestingly, Aβ-driven reduction in Ly6h requires activation of α7 nAChRs since the induced change could be blocked by the α7-selective antagonist, MLA. This result is consistent with previous findings that Aβ can bind to and activate α7 nAChRs (Wang et al., 2000a,b; Liu et al., 2001, 2013; Pettit et al., 2001; Dineley et al., 2002). Paradoxically, reduction in Ly6h also disinhibits assembly of α7 nAChRs. Thus, α7 appears to lie both upstream and downstream of Ly6h. This property is a hallmark of homeostatic regulation. As a result, we propose that Aβ coopts homeostatic control of α7 by Ly6h, leading to downregulation of the latter and thus upregulation of α7 beyond the physiological range in which Ca2+ influxes can be appropriately buffered. In this scenario the threshold for neurotoxic activation of α7 would be expected to drop, thus causing signaling by endogenous ACh to sensitize neurons to additional insults that precipitate premature cell death. A model of this scenario is depicted in Figure 6B.
Several types of studies support this model. First, ablation of the gene encoding α7 has been shown to ameliorate synaptic impairment and memory deficits in a mouse model of AD, consistent with a role for α7 in disease phenotypes (Dziewczapolski et al., 2009). Second, although results vary, some studies have shown that Aβ can cause acute downregulation but chronic upregulation of α7 currents in rodent neurons, consistent with homeostatic compensation (Liu et al., 2001, 2013, 2015). Third, other studies have shown that α7 nAChRs are elevated in proportion to soluble Aβ in temporal cortex (Hellström-Lindahl et al., 2004), to Aβ plaque density in entorhinal cortex (Perry et al., 2000), and to AD diagnostic scoring criteria in superior frontal cortex (Ikonomovic et al., 2009). To some extent these results are to be expected since neurotransmitter receptors are often upregulated to compensate for reduced presynaptic activity, and reduced brainstem cholinergic signaling to the hippocampus and neocortex is one of the early hallmarks of AD. However, our results suggest that any such compensatory effect is further driven by a putative causative agent of disease pathogenesis, namely Aβ, through downregulation of Ly6h.
Since α7 nAChRs have been reported to localize to presynaptic terminals of some neurons (Jones and Wonnacott, 2004; Grilli et al., 2006; Gu et al., 2012; Cheng and Yakel, 2014), it will be interesting to determine whether the upregulation we observe for these receptors in pyramidal neurons also occurs in basal forebrain cholinergic neurons (BFCNs). Indeed, this effect might underlie cell death in the latter at the earliest stages of disease pathology and thus contribute to cognitive deficits known to be caused by reduced cholinergic signaling. Consistent with this hypothesis, Aβ-sensitive α7-containing nAChRs are present in BFCNs (Liu et al., 2009); chronic Aβ exposure causes upregulation of α7-containing nAChRs in a cholinergic cell line; and Aβ-induced neurotoxicity in these cells can be blocked by the α7-selective antagonist MLA (Liu et al., 2015).
While it is difficult to extrapolate directly from our results to interventional strategies in AD, several points are worth considering. First, AD currently affects ∼6 million people in the US alone, and treatment options are limited (Alzheimer's Association, 2019). Current FDA-approved drugs treat only cognitive symptoms, not disease progression, and the effects of these drugs decrease with time (Sun et al., 2008). Thus, there is a clear need to identify new molecular targets for therapeutic intervention, particularly to prevent loss of synapses and neurodegeneration. Second, current FDA-approved drugs for AD fall into two classes, both of which affect signaling by ACh. One class consists of memantine, which is best known as an uncompetitive NMDA receptor antagonist (Chen et al., 1992) but is an even more potent inhibitor of α7 nAChRs (Aracava et al., 2005). Another class of drugs consists of acetylcholinesterase inhibitors (AChEIs) such as donepezil, galantamine, and rivastigmine. AChEIs elevate synaptic levels of ACh and thus facilitate activation of AChRs (both nicotinic and muscarinic), which compensates for reduced cholinergic signaling from degenerating neurons in the basal forebrain (Joe and Ringman, 2019). Our results suggest that although AChEIs may provide short-term cognitive benefits, by activating α7 nAChRs these drugs may exacerbate neurotoxicity over the long-term.
Intriguingly, we also found that Ly6h levels are detectable in human CSF by Western blot at levels that are inversely proportional to Aβ load. This result was unexpected since a GPI moiety is believed to tether most Ly6 proteins to the outer leaflet of the plasma membrane (Vasilyeva et al., 2017). However, it is possible that in certain conditions Ly6h is liberated into tissue fluid. We hypothesize that the decrease in Ly6h that we measured with more severe AD pathology is caused by loss of the neurons that express it. However, an important caveat is that the CSF we analyzed was pooled from multiple patients, so it is possible that certain individuals skewed our results. On the other hand, Ly6h was recently identified by mass spectrometry as among 21 proteins that are most significantly changed in CSF of AD patients relative to age-matched controls (Park et al., 2020). Thus, in this context our data suggest that Ly6h may be a novel biomarker of AD that could be collected from CSF of living patients to monitor disease progression.
Footnotes
Acknowledgements: We thank Jeffrey Metcalf and Sara Shuldberg from the Rissman lab and Shiley-Marcos Alzheimer's Disease Research Center for providing human brain tissue and CSF; Nicole Purcell and Joan Brown for providing rat tissue for primary neuronal cultures; Eric Griffis and Daphne Bindels from the Nikon Imaging Center at University of California San Diego for advice with microscopy; and the late Dr. Peter Davies for donating antibody PHF-1. This work was supported by National Institutes of Health Grants R01 GM125080 to WJJ and P30 AG062429 to RAR.
The authors declare no competing financial interests.
References
- Akers AT, Cooper SY, Baumgard ZJ, Casinelli GP, Avelar AJ, Henderson BJ (2020) Upregulation of nAChRs and changes in excitability on VTA dopamine and GABA neurons correlates to changes in nicotine-reward-related behavior. eNeuro 7:ENEURO.0189-20.2020. 10.1523/ENEURO.0189-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Association (2019) 2019 Alzheimer's disease facts and figures. Alzheimer's Dementia 15:321–387. [Google Scholar]
- Andree KC, Abali F, Oomens L, Passanha FR, Broekmaat JJ, Kraan J, Mendelaar PAJ, Sleijfer S, Terstappen L (2019) Self-seeding microwells to isolate and assess the viability of single circulating tumor cells. Int J Mol Sci 20:477. 10.3390/ijms20030477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aracava Y, Pereira EFR, Maelicke A, Albuquerque EX (2005) Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-D-aspartate receptors in rat hippocampal neurons. J Pharmacol Exp Ther 312:1195–1205. 10.1124/jpet.104.077172 [DOI] [PubMed] [Google Scholar]
- Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, Sudan S, Czura CJ, Ivanova SM, Tracey KJ (2002) Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med 195:781–788. 10.1084/jem.20011714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405:458–462. 10.1038/35013070 [DOI] [PubMed] [Google Scholar]
- Bouzat C, Bartos M, Corradi J, Sine SM (2008) The interface between extracellular and transmembrane domains of homomeric Cys-loop receptors governs open-channel lifetime and rate of desensitization. J Neurosci 28:7808–7819. 10.1523/JNEUROSCI.0448-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghaus L, Schütz U, Krempel U, de Vos RA, Jansen Steur EN, Wevers A, Lindstrom J, Schröder H (2000) Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Brain Res Mol Brain Res 76:385–388. 10.1016/s0169-328x(00)00031-0 [DOI] [PubMed] [Google Scholar]
- Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA (1992) Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci 12:4427–4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Yakel JL (2014) Presynaptic α7 nicotinic acetylcholine receptors enhance hippocampal mossy fiber glutamatergic transmission via PKA activation. J Neurosci 34:124–133. 10.1523/JNEUROSCI.2973-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts SE, He B, Che S, Ikonomovic MD, DeKosky ST, Ginsberg SD, Mufson EJ (2007) Alpha7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch Neurol 64:1771–1776. 10.1001/archneur.64.12.1771 [DOI] [PubMed] [Google Scholar]
- Criscuolo C, Accorroni A, Domenici L, Origlia N (2015) Impaired synaptic plasticity in the visual cortex of mice lacking alpha7-nicotinic receptor subunit. Neuroscience 294:166–171. 10.1016/j.neuroscience.2015.03.022 [DOI] [PubMed] [Google Scholar]
- David R, Ciuraszkiewicz A, Simeone X, Orr-Urtreger A, Papke RL, McIntosh JM, Huck S, Scholze P (2010) Biochemical and functional properties of distinct nicotinic acetylcholine receptors in the superior cervical ganglion of mice with targeted deletions of nAChR subunit genes. Eur J Neurosci 31:978–993. 10.1111/j.1460-9568.2010.07133.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Bell KA, Bui D, Sweatt JD (2002) Beta-amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem 277:25056–25061. 10.1074/jbc.M200066200 [DOI] [PubMed] [Google Scholar]
- Drisdel RC, Green WN (2000) Neuronal alpha-bungarotoxin receptors are alpha7 subunit homomers. J Neurosci 20:133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziewczapolski G, Glogowski CM, Masliah E, Heinemann SF (2009) Deletion of the alpha 7 nicotinic acetylcholine receptor gene improves cognitive deficits and synaptic pathology in a mouse model of Alzheimer's disease. J Neurosci 29:8805–8815. 10.1523/JNEUROSCI.6159-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick EM, Marty A, Neher E (1982) A patch-clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. J Physiol 331:577–597. 10.1113/jphysiol.1982.sp014393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galat A, Gross G, Drevet P, Sato A, Ménez A (2008) Conserved structural determinants in three-fingered protein domains. FEBS J 275:3207–3225. 10.1111/j.1742-4658.2008.06473.x [DOI] [PubMed] [Google Scholar]
- Giorgi C, Marchi S, Pinton P (2018) The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol 19:713–730. 10.1038/s41580-018-0052-8 [DOI] [PubMed] [Google Scholar]
- Govind AP, Walsh H, Green WN (2012) Nicotine-induced upregulation of native neuronal nicotinic receptors is caused by multiple mechanisms. J Neurosci 32:2227–2238. 10.1523/JNEUROSCI.5438-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grilli M, Raiteri L, Patti L, Parodi M, Robino F, Raiteri M, Marchi M (2006) Modulation of the function of presynaptic alpha7 and non-alpha7 nicotinic receptors by the tryptophan metabolites, 5-hydroxyindole and kynurenate in mouse brain. Br J Pharmacol 149:724–732. 10.1038/sj.bjp.0706914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Lamb PW, Yakel JL (2012) Cholinergic coordination of presynaptic and postsynaptic activity induces timing-dependent hippocampal synaptic plasticity. J Neurosci 32:12337–12348. 10.1523/JNEUROSCI.2129-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, Matta JA, Lord B, Harrington AW, Sutton SW, Davini WB, Bredt DS (2016) Brain α7 nicotinic acetylcholine receptor assembly requires NACHO. Neuron 89:948–955. 10.1016/j.neuron.2016.01.018 [DOI] [PubMed] [Google Scholar]
- Guerra-Álvarez M, Moreno-Ortega AJ, Navarro E, Fernández-Morales JC, Egea J, López MG, Cano-Abad MF (2015) Positive allosteric modulation of alpha-7 nicotinic receptors promotes cell death by inducing Ca(2+) release from the endoplasmic reticulum. J Neurochem 133:309–319. 10.1111/jnc.13049 [DOI] [PubMed] [Google Scholar]
- Hellström-Lindahl E, Mousavi M, Ravid R, Nordberg A (2004) Reduced levels of Abeta 40 and Abeta 42 in brains of smoking controls and Alzheimer's patients. Neurobiol Dis 15:351–360. 10.1016/j.nbd.2003.11.024 [DOI] [PubMed] [Google Scholar]
- Huang S, Li SX, Bren N, Cheng K, Gomoto R, Chen L, Sine SM (2013) Complex between α-bungarotoxin and an α7 nicotinic receptor ligand-binding domain chimaera. Biochem J 454:303–310. 10.1042/BJ20130636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Wecker L, Abrahamson EE, Wuu J, Counts SE, Ginsberg SD, Mufson EJ, DeKosky ST (2009) Cortical α7 nicotinic acetylcholine receptor and β-amyloid levels in early Alzheimer disease. Archiv Neurol 66:646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe E, Ringman JM (2019) Cognitive symptoms of Alzheimer's disease: clinical management and prevention. BMJ 367:l6217. [DOI] [PubMed] [Google Scholar]
- Jones IW, Wonnacott S (2004) Precise localization of alpha7 nicotinic acetylcholine receptors on glutamatergic axon terminals in the rat ventral tegmental area. J Neurosci 24:11244–11252. 10.1523/JNEUROSCI.3009-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidokoro Y, Miyazaki S, Ozawa S (1982) Acetylcholine-induced membrane depolarization and potential fluctuations in the rat adrenal chromaffin cell. J Physiol 324:203–220. 10.1113/jphysiol.1982.sp014107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Parker RL, Wright AP, Brahem H, Ku P, Oliver KM, Walz A, Lester HA, Miwa JM (2014) Lynx1 supports neuronal health in the mouse dorsal striatum during aging: an ultrastructural investigation. J Mol Neurosci 53:525–536. 10.1007/s12031-014-0352-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagostena L, Trocme-Thibierge C, Morain P, Cherubini E (2008) The partial alpha7 nicotine acetylcholine receptor agonist S 24795 enhances long-term potentiation at CA3-CA1 synapses in the adult mouse hippocampus. Neuropharmacology 54:676–685. 10.1016/j.neuropharm.2007.11.016 [DOI] [PubMed] [Google Scholar]
- Léna C, Popa D, Grailhe R, Escourrou P, Changeux JP, Adrien J (2004) Beta2-containing nicotinic receptors contribute to the organization of sleep and regulate putative micro-arousals in mice. J Neurosci 24:5711–5718. 10.1523/JNEUROSCI.3882-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennicke C, Cochemé HM (2020) Redox signalling and ageing: insights from Drosophila. Biochem Soc Trans 48:367–377. 10.1042/BST20190052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Zhou XD, Kolosov VP, Perelman JM (2011) Nicotine reduces TNF-α expression through a α7 nAChR/MyD88/NF-ĸB pathway in HBE16 airway epithelial cells. Cell Physiol Biochem 27:605–612. 10.1159/000329982 [DOI] [PubMed] [Google Scholar]
- Liu L, Zhao-Shea R, McIntosh JM, Gardner PD, Tapper AR (2012) Nicotine persistently activates ventral tegmental area dopaminergic neurons via nicotinic acetylcholine receptors containing α4 and α6 subunits. Mol Pharmacol 81:541–548. 10.1124/mol.111.076661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Kawai H, Berg DK (2001) beta -Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc Natl Acad Sci USA 98:4734–4739. 10.1073/pnas.081553598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Huang Y, Xue F, Simard A, DeChon J, Li G, Zhang J, Lucero L, Wang M, Sierks M, Hu G, Chang Y, Lukas RJ, Wu J (2009) A novel nicotinic acetylcholine receptor subtype in basal forebrain cholinergic neurons with high sensitivity to amyloid peptides. J Neurosci 29:918–929. 10.1523/JNEUROSCI.3952-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Xie X, Lukas RJ, St John PA, Wu J (2013) A novel nicotinic mechanism underlies β-amyloid-induced neuronal hyperexcitation. J Neurosci 33:7253–7263. 10.1523/JNEUROSCI.3235-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Xie X, Emadi S, Sierks MR, Wu J (2015) A novel nicotinic mechanism underlies β-amyloid-induced neurotoxicity. Neuropharmacology 97:457–463. 10.1016/j.neuropharm.2015.04.025 [DOI] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z, Berg DK (2012) Glutamatergic synapse formation is promoted by α7-containing nicotinic acetylcholine receptors. J Neurosci 32:7651–7661. 10.1523/JNEUROSCI.6246-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykhmus O, Kalashnyk O, Uspenska K, Skok M (2019) Positive allosteric modulation of alpha7 nicotinic acetylcholine receptors transiently improves memory but aggravates inflammation in LPS-treated mice. Front Aging Neurosci 11:359. 10.3389/fnagi.2019.00359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyukmanova EN, Shenkarev ZO, Shulepko MA, Mineev KS, D'Hoedt D, Kasheverov IE, Filkin SY, Krivolapova AP, Janickova H, Dolezal V, Dolgikh DA, Arseniev AS, Bertrand D, Tsetlin VI, Kirpichnikov MP (2011) NMR structure and action on nicotinic acetylcholine receptors of water-soluble domain of human LYNX1. J Biol Chem 286:10618–10627. 10.1074/jbc.M110.189100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos U, Molles BE, Pons S, Besson M, Guiard BP, Guilloux JP, Evrard A, Cazala P, Cormier A, Mameli-Engvall M, Dufour N, Cloëz-Tayarani I, Bemelmans AP, Mallet J, Gardier AM, David V, Faure P, Granon S, Changeux JP (2005) Nicotine reinforcement and cognition restored by targeted expression of nicotinic receptors. Nature 436:103–107. 10.1038/nature03694 [DOI] [PubMed] [Google Scholar]
- Matta JA, Gu S, Davini WB, Lord B, Siuda ER, Harrington AW, Bredt DS (2017) NACHO mediates nicotinic acetylcholine receptor function throughout the brain. Cell Rep 19:688–696. 10.1016/j.celrep.2017.04.008 [DOI] [PubMed] [Google Scholar]
- Miwa JM, Stevens TR, King SL, Caldarone BJ, Ibanez-Tallon I, Xiao C, Fitzsimonds RM, Pavlides C, Lester HA, Picciotto MR, Heintz N (2006) The prototoxin lynx1 acts on nicotinic acetylcholine receptors to balance neuronal activity and survival in vivo. Neuron 51:587–600. 10.1016/j.neuron.2006.07.025 [DOI] [PubMed] [Google Scholar]
- Moriwaki Y, Kubo N, Watanabe M, Asano S, Shinoda T, Sugino T, Ichikawa D, Tsuji S, Kato F, Misawa H (2020) Endogenous neurotoxin-like protein Ly6H inhibits alpha7 nicotinic acetylcholine receptor currents at the plasma membrane. Sci Rep 10:11996. 10.1038/s41598-020-68947-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakauchi S, Sumikawa K (2012) Endogenously released ACh and exogenous nicotine differentially facilitate long-term potentiation induction in the hippocampal CA1 region of mice. Eur J Neurosci 35:1381–1395. 10.1111/j.1460-9568.2012.08056.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA (2007) Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci 27:8202–8218. 10.1523/JNEUROSCI.2199-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols WA, Henderson BJ, Yu C, Parker RL, Richards CI, Lester HA, Miwa JM (2014) Lynx1 shifts α4β2 nicotinic receptor subunit stoichiometry by affecting assembly in the endoplasmic reticulum. J Biol Chem 289:31423–31432. 10.1074/jbc.M114.573667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SA, Jung JM, Park JS, Lee JH, Park B, Kim HJ, Park JH, Chae WS, Jeong JH, Choi SH, Baek JH (2020) SWATH-MS analysis of cerebrospinal fluid to generate a robust battery of biomarkers for Alzheimer's disease. Sci Rep 10:7423. 10.1038/s41598-020-64461-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry E, Martin-Ruiz C, Lee M, Griffiths M, Johnson M, Piggott M, Haroutunian V, Buxbaum JD, Nãsland J, Davis K, Gotti C, Clementi F, Tzartos S, Cohen O, Soreq H, Jaros E, Perry R, Ballard C, McKeith I, Court J (2000) Nicotinic receptor subtypes in human brain ageing, Alzheimer and Lewy body diseases. Eur J Pharmacol 393:215–222. 10.1016/s0014-2999(00)00064-9 [DOI] [PubMed] [Google Scholar]
- Petrovic J, Walsh PL, Thornley KT, Miller CE, Wightman RM (2010) Real-time monitoring of chemical transmission in slices of the murine adrenal gland. Endocrinology 151:1773–1783. 10.1210/en.2009-1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel JL (2001) beta-Amyloid(1-42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci 21:RC120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, Fuxe K, Changeux JP (1998) Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 391:173–177. 10.1038/34413 [DOI] [PubMed] [Google Scholar]
- Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, Changeux JP, Maskos U, Fratta W (2008) Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci 28:12318–12327. 10.1523/JNEUROSCI.3918-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddifoot CA, Wu M, Sung R-J, Joiner WJ (2015) Ly6h regulates trafficking of alpha7 nicotinic acetylcholine receptors and nicotine-induced potentiation of glutamatergic signaling. J Neurosci 35:3420–3430. 10.1523/JNEUROSCI.3630-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren JM, Zhang SL, Wang XL, Guan ZZ, Qi XL (2020) Expression levels of the α7 nicotinic acetylcholine receptor in the brains of patients with Alzheimer's disease and their effect on synaptic proteins in SH-SY5Y cells. Mol Med Rep 22:2063–2075. 10.3892/mmr.2020.11253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadigh-Eteghad S, Vatandoust SM, Mahmoudi J, Rahigh Aghsan S, Majdi A (2020) Cotinine ameliorates memory and learning impairment in senescent mice. Brain Res Bull 164:65–74. 10.1016/j.brainresbull.2020.08.010 [DOI] [PubMed] [Google Scholar]
- Shenkarev ZO, Shulepko MA, Bychkov ML, Kulbatskii DS, Shlepova OV, Vasilyeva NA, Andreev-Andrievskiy AA, Popova AS, Lagereva EA, Loktyushov EV, Koshelev SG, Thomsen MS, Dolgikh DA, Kozlov SA, Balaban PM, Kirpichnikov MP, Lyukmanova EN (2020) Water-soluble variant of human Lynx1 positively modulates synaptic plasticity and ameliorates cognitive impairment associated with α7-nAChR dysfunction. J Neurochem 155:45–61. 10.1111/jnc.15018 [DOI] [PubMed] [Google Scholar]
- Stancu IC, Ferraiolo M, Terwel D, Dewachter I (2019) Tau interacting proteins: gaining insight into the roles of tau in health and disease. Adv Exp Med Biol 1184:145–166. 10.1007/978-981-32-9358-8_13 [DOI] [PubMed] [Google Scholar]
- Steiner H, Fukumori A, Tagami S, Okochi M (2018) Making the final cut: pathogenic amyloid-β peptide generation by γ-secretase. Cell Stress 2:292–310. 10.15698/cst2018.11.162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiljkovic M, Kelley C, Nagy D, Hurst R, Hajós M (2016) Activation of α7 nicotinic acetylcholine receptors facilitates long-term potentiation at the hippocampal-prefrontal cortex synapses in vivo. Eur Neuropsychopharmacol 26:2018–2023. 10.1016/j.euroneuro.2016.11.003 [DOI] [PubMed] [Google Scholar]
- Sun Y, Lai MS, Lu CJ, Chen RC (2008) How long can patients with mild or moderate Alzheimer's dementia maintain both the cognition and the therapy of cholinesterase inhibitors: a national population-based study. Eur J Neurol 15:278–283. 10.1111/j.1468-1331.2007.02049.x [DOI] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA (2004) Nicotine activation of alpha4* receptors: sufficient for reward, tolerance, and sensitization. Science 306:1029–1032. 10.1126/science.1099420 [DOI] [PubMed] [Google Scholar]
- Tintignac LA, Brenner HR, Rüegg MA (2015) Mechanisms regulating neuromuscular junction development and function and causes of muscle wasting. Physiol Rev 95:809–852. 10.1152/physrev.00033.2014 [DOI] [PubMed] [Google Scholar]
- Townsend M, Whyment A, Walczak JS, Jeggo R, van den Top M, Flood DG, Leventhal L, Patzke H, Koenig G (2016) α7-nAChR agonist enhances neural plasticity in the hippocampus via a GABAergic circuit. J Neurophysiol 116:2663–2675. 10.1152/jn.00243.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyeva NA, Loktyushov EV, Bychkov ML, Shenkarev ZO, Lyukmanova EN (2017) Three-finger proteins from the Ly6/uPAR family: functional diversity within one structural motif. Biochemistry (Mosc) 82:1702–1715. 10.1134/S0006297917130090 [DOI] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421:384–388. 10.1038/nature01339 [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, Davis CB, Shank RP (2000a) Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem 75:1155–1161. 10.1046/j.1471-4159.2000.0751155.x [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB (2000b) beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem 275:5626–5632. 10.1074/jbc.275.8.5626 [DOI] [PubMed] [Google Scholar]
- Wang JM, Zhang L, Yao Y, Viroonchatapan N, Rothe E, Wang ZZ (2002) A transmembrane motif governs the surface trafficking of nicotinic acetylcholine receptors. Nat Neurosci 5:963–970. 10.1038/nn918 [DOI] [PubMed] [Google Scholar]
- Wang K, Ru J, Zhang H, Chen J, Lin X, Lin Z, Wen M, Huang L, Ni H, Zhuge Q, Yang S (2020) Melatonin enhances the therapeutic effect of plasma exosomes against cerebral ischemia-induced pyroptosis through the TLR4/NF-κB pathway. Front Neurosci 14:848. 10.3389/fnins.2020.00848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JM, Cockcroft VB, Lunt GG, Smillie FS, Wonnacott S (1990) Methyllycaconitine: a selective probe for neuronal alpha-bungarotoxin binding sites. FEBS Lett 270:45–48. 10.1016/0014-5793(90)81231-c [DOI] [PubMed] [Google Scholar]
- Wu M, Puddifoot CA, Taylor P, Joiner WJ (2015) Mechanisms of inhibition and potentiation of α4β2 nicotinic acetylcholine receptors by members of the Ly6 protein family. J Biol Chem 290:24509–24518. 10.1074/jbc.M115.647248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Liu CZ, Joiner WJ (2016) Structural analysis and deletion mutagenesis define regions of QUIVER/SLEEPLESS that are responsible for interactions with shaker-type potassium channels and nicotinic acetylcholine receptors. PLoS One 11:e0148215. 10.1371/journal.pone.0148215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou CN, Patrick J, Role L, De Biasi M, Beaudet AL (1999) Megacystis, mydriasis, and ion channel defect in mice lacking the alpha3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci USA 96:5746–5751. 10.1073/pnas.96.10.5746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi JG, Nemecz A, Nguyen QT, Muller A, Schroeder LF, Talley TT, Lindstrom J, Kleinfeld D, Taylor P (2011) Characterizing ligand-gated ion channel receptors with genetically encoded Ca2++ sensors. PLoS ONE 6:e16519. [DOI] [PMC free article] [PubMed] [Google Scholar]