

Abstract
We report a synthetic route to BODIPY-cholesterol conjugates in which the key steps are Suzuki or Liebeskind-Srogl cross-coupling of cholesterol phenyl moieties 11, 12, and 13 with structurally diverse BODIPY scaffolds. All conjugates feature single-bonded and hydrophobic linkages between the fluorophore and sterol devoid of heteroatoms. Using HeLa cells, we show that these BODIPY-cholesterol analogs can be used simultaneously with the parent BODIPY-cholesterol for cell imaging and flow cytometry. The BODIPY-cholesterol analogs exhibit a similar cellular localization in HeLa cells and show similar cholesterol efflux properties from THP1 cells to HDL acceptors. These results demonstrate that the red-shifted BODIPY-cholesterol analogs behave in a manner similar to unlabeled cholesterol and are useful probes for simultaneous visualization of intracellular cholesterol pools and for monitoring cholesterol efflux from cells to extracellular acceptors.
Keywords: BODIPY-cholesterol, confocal microscopy, fluorescence imaging, Liebeskind-Srogl coupling, Suzuki coupling
Graphical Abstarct
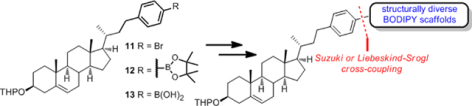
We report a convergent synthetic route to versatile BODIPY-cholesterol conjugates in which the key steps are Suzuki or Liebeskind-Srogl cross-coupling of cholesterol phenyl moieties 11, 12, and 13 with structurally diverse BODIPY scaffolds. The BODIPY-cholesterol analogs described were used to enable the simultaneous tracking of different cellular cholesterol pools by confocal microscopy and flow cytometry.
Introduction
Cholesterol, an important constituent of mammalian plasma membranes and plasma lipoproteins, plays fundamental roles in diverse biological processes, such as membrane permeability, lateral lipid organization, signal transduction, and membrane trafficking.1 A detailed understanding of cholesterol-sphingolipid microdomains remains a challenge. Therefore, the development of spectroscopically observable lipid analogs that faithfully reflect the properties of the native cholesterol would provide useful tools to investigate the molecular properties of lipid rafts and other membrane-associated properties, as well as the movement of cholesterol between different cell types and cholesterol pools.2
An ideal cholesterol probe would exhibit properties similar to those of cholesterol in partitioning between the liquid-disordered phase and the liquid-ordered phase of model membranes and plasma membranes of cells, would only minimally induce a tilt in the steroid nucleus with respect to the bilayer normal, and would possess photophysical properties (such as long wavelength emission and photostability) suitable for use in live cell fluorescence microscopy of cholesterol-rich membrane domains. Although many fluorescent cholesterol analogs have been prepared3, only one compound, referred to here as boron dipyrromethene difluoride (BODIPY)-cholesterol (1) in which C-24 of the cholesterol aliphatic chain is linked directly to a BODIPY moiety, has been found to exhibit a similar physical behavior to cholesterol. Probe 1, in which the linkage between the BODIPY moiety and the sterol is devoid of oxygen atoms, has already been successfully employed in many biological4–9 and photophysical investigations.10–14 Other fluorescent sterols do not partition efficiently into liquid-ordered lipid phase.13
In the context of a broader program directed toward the synthesis of BODIPY-cholesterol conjugates,10 we wished to incorporate a diverse array of substituted BODIPY scaffolds into cholesterol, with a special interest in developing red-shifted analogs of BODIPY-cholesterol. We report here the preparation of BODIPY-cholesterol analogs bearing aromatic groups (phenyl and thiophene) conjugated to the BODIPY moiety at different positions. Although our previous synthesis of BODIPY-cholesterol conjugates from pyrroles and acid chlorides has been accomplished successfully,10 the synthetic routes are lengthy or impractical and not amenable to wide structural modification. We developed a novel method for the rapid assembly of cholesterol analogs with substituted BODIPY fluorophores of highly structural versatility (highlighted in red in Scheme 1) through a non-heteroatom and single-bonded linkage. This method proceeds by the convergent construction between post-functionalized BODIPY cores and cholesterol-phenyl moieties via either Suzuki15 or Liebeskind-Srogl cross-coupling.16
Scheme 1.
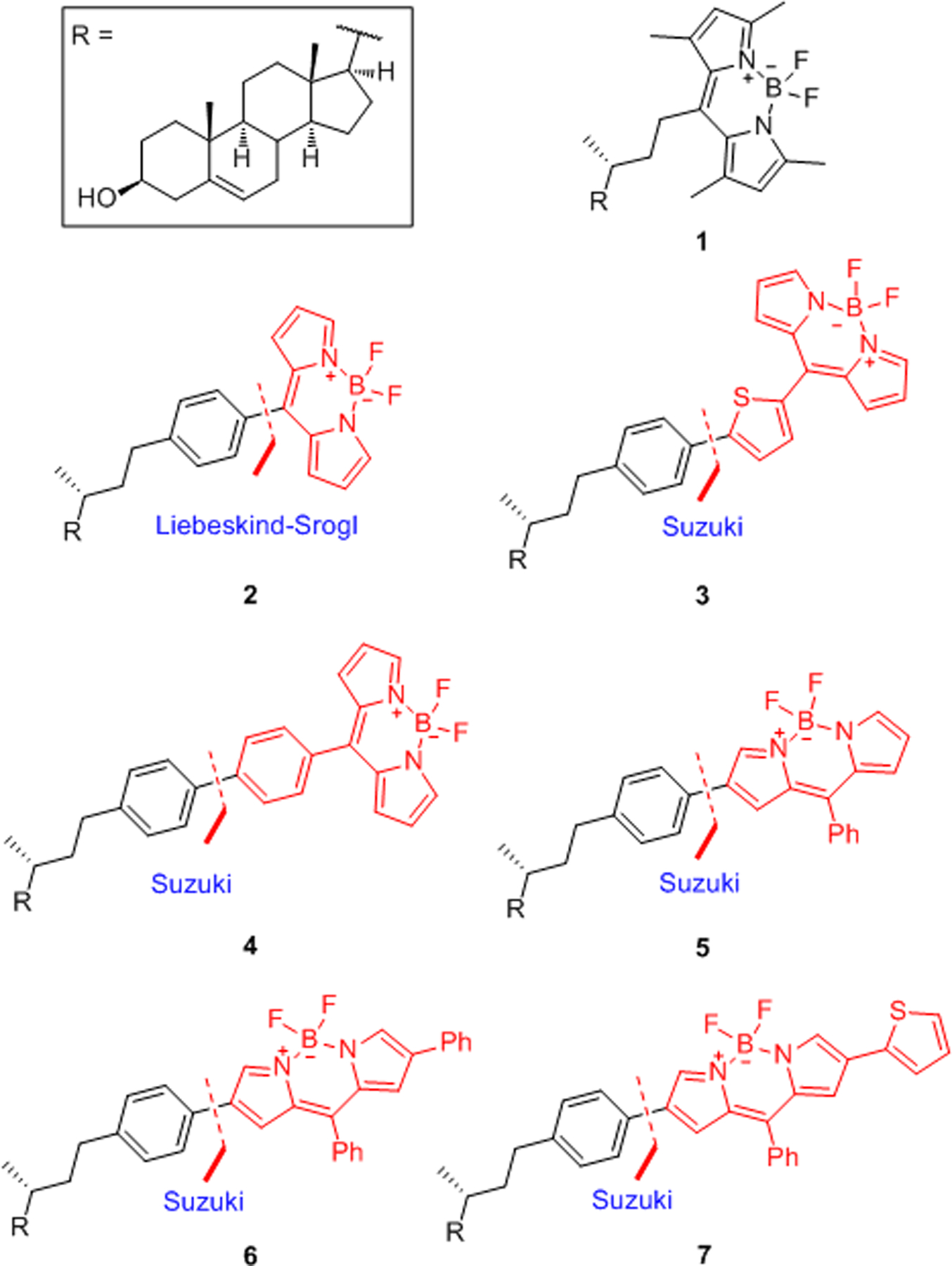
Structures of BODIPY-cholesterol conjugates 1-7.
Results and Discussion
Chemistry
The synthesis of BODIPY-cholesterol conjugate 2 is outlined in Scheme 2. Alcohol 9 was prepared on a gram scale by a modification of a known procedure.10 Reduction of THP protected carboxylic acid 8 with lithium aluminum hydride was achieved in THF at reflux within 30 min, and crude alcohol 9 was purified by recrystallization from EtOAc. This protocol was scaled up to 15 mmol. Dess-Martin oxidation of alcohol 9 provided the corresponding aldehyde17, which was immediately subjected to Wittig olefination to produce terminal alkene 10, which was recrystallized from EtOAc. Thus the gram-scale synthesis of 10 from commercially available bisnorcholenic acid (8) was achieved in 63% yield (over four steps) without column chromatography.
Scheme 2.
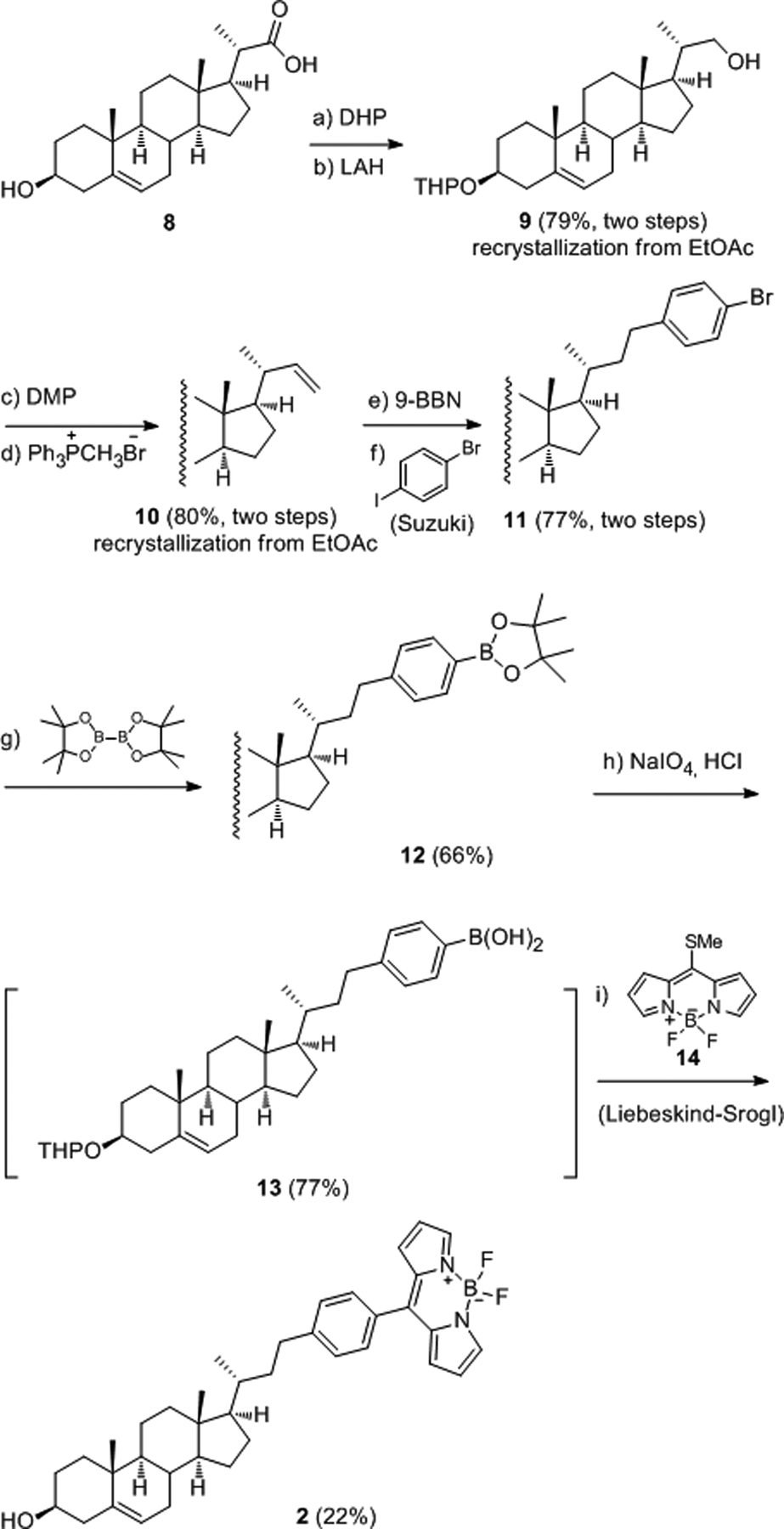
Synthesis of 2 via B-alkyl Suzuki coupling and Liebeskind-Srogl coupling. Detailed reaction conditions and reagents: a) 3,4-dihydro-2H-pyran, p-TsOH, THF, rt, 24 h; b) LiAlH4, THF, reflux, 30 min; c) Dess-Martin periodinane, NaHCO3, CH2Cl2, rt, 1h; d) Ph3P+CH3Br‒, n-BuLi, THF, −78 °C, 1 h; e) 9-BBN, PhMe/THF (1:1), 85 °C, 20 min; f) 1-bromo-4-iodobenzene, Pd(dppf)Cl2∙CH2Cl2, 3 M aqueous NaOH solution, 85 °C, 1 h; g) Pd2(dba)3, DavePhos, bis(pinacolato)diboron, KOAc, 1,4-dioxane, microwave heating at 110 °C, 30 min; h) NaIO4, HCl, THF/H2O (4:1), rt, overnight; i) 14, CuTC, Pd2(dba)3, TFP, THF, microwave heating at 100 °C, 30 min.
With the key intermediate 10 in hand, we embarked on the investigation of conditions for hydroboration of 10 and the subsequent Suzuki cross-coupling with 1-bromo-4-iodobenzene.18 Hydroboration of 10 with 5 equiv of 9-BBN in THF at rt proved to be exceedingly slow, and considerable starting material was present even after 24 h. However, when the reaction of 10 with 5 equiv of 9-BBN was carried out in toluene/THF (1:1) at 85 °C, the starting material disappeared completely within 15–20 min. Suzuki cross-coupling gave rise to phenyl bromide 11 in 77% yield. Hydroboration under these harsh conditions occurred chemoselectively at the less hindered C22-C23 double bond even in the presence of a C5-C6 double bond.19 The cross-coupling reaction with 1,4-dibromobenzene provided 11 in low isolated yield (32%). It is noteworthy that our initial shorter synthetic route to 2 involved B-alkyl Suzuki cross-coupling of 10 with BODIPY-containing phenyl bromide 1620 (see the structure of 16 in Scheme 3) (Pd(dppf)Cl2, 3 M NaOH, PhMe/THF, 75 °C); unfortunately, this reaction failed to afford the desired coupling product. The fluorescence disappeared within 30 min, presumably because BODIPY dyes are highly base sensitive.21
Scheme 3.
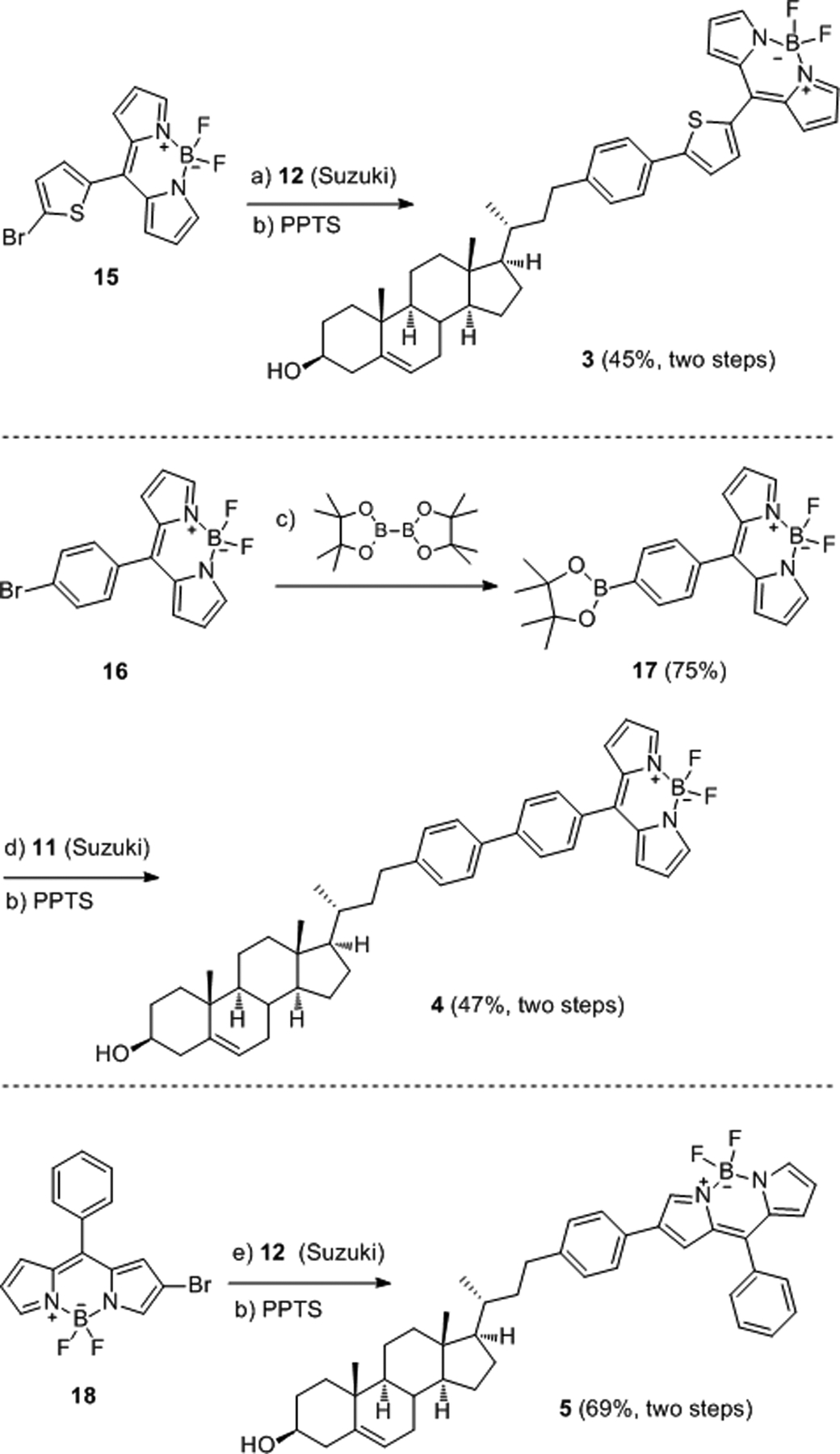
Synthesis of 3, 4, and 5. Detailed reaction conditions and reagents: a) 12, Pd(dppf)Cl2∙CH2Cl2, K2CO3, PhMe/H2O (2:1), microwave heating 120 °C, 30 min; b) PPTS, MeOH/CH2Cl2 (2:1), reflux, 2 h; c) Pd2(dba)3, DavePhos, bis(pinacolato)diboron, KOAc, 1,4-dioxane, microwave heating at 110 °C, 30 min; d) 11, Pd(dppf)Cl2∙CH2Cl2, K2CO3, PhMe/H2O (2:1), microwave heating at 120 °C, 30 min; e) 12, Pd2(dba)3, DavePhos, Cs2CO3, 1,4-dioxane/H2O (10:1), microwave heating at 80 °C, 30 min.
Lithium-halogen exchange of 4-bromophenyl 11 and in situ quenching with trimethyl borate provided the desired boronic acid 13 in very low yield (~20%) because protodebromination was highly competitive.22 Fortunately, palladium-catalyzed borylation of 11 with bis(pinacolato)diboron (B2Pin2)23 provided pinacol boronate esters 12 in 66% yield. Oxidative cleavage of 12 with NaIO4 produced arylboronic acid 13 in 77% yield. Liebeskind-Srogl cross-coupling16 between 13 and 1424 then delivered the expected BODIPY-cholesterol conjugate 2 in 22% yield. Meanwhile, removal of the THP ether group was observed in the same pot, presumably as a result of the acidity of the boronic acid moiety in 13.25
The syntheses of BODIPY-cholesterol conjugates 3, 4, and 5 are outlined in Scheme 3. The Suzuki cross-coupling between pinacol boronic ester 12 and 1520 followed by acidic deprotection of the THP group furnished the desired cholesterol conjugate 3 in 45% yield. Our initial attempt to achieve borylation of 15 (with B2Pin2, Pd2(dba)3, DavePhos, KOAc, 1,4-dioxane, microwave heating at 110 °C for 30 min)23 led to homocoupling (32% yield) and protodebromination (48% yield). In contrast, borylation of 16 under same conditions delivered pinacol boronic ester 17 in 75% yield, which was, in turn, subjected to Suzuki cross-coupling with 11; after THP deprotection with PPTS in MeOH/CH2Cl2 (2:1), 4 was obtained in 47% yield.
It is already known that when BODIPY conjugates contain a phenyl substituent at the meso-position, the absorption and emission wavelengths are not altered significantly compared to those of the parent BODIPY chromophore; however, the introduction of extended conjugation at the α- or β-position of the BODIPY moiety do cause a significant red shift in the excitation and emission wavelengths.26 Initially, the incorporation of the sterol-phenyl moiety into the β-position of BODIPY was attempted by Pd(dppf)Cl2-catalyzed Suzuki cross-coupling between 1827 and 12; however, this reaction resulted in only 20% conversion of 12. When Pd2(dba)3/DavePhos was employed, 12 was completely consumed, and upon removal of the THP group, the final product 5 was obtained in 69% yield over two steps.
As shown in Scheme 4, Suzuki cross-coupling of 18 with phenylboronic acid afforded the fluorescent dye 19, which was regioselectively brominated28 to generate 20. Then, Suzuki cross-coupling between 20 and 12 followed by THP deprotection gave conjugate 6 in 30% yield over two steps. It was reported that the incorporation of a thiophene moiety into BODIPY dyes provides red-shifted absorption and emission maxima.29–30 Thus, BODIPY-cholesterol conjugate 7 was prepared in a similar manner.
Scheme 4.
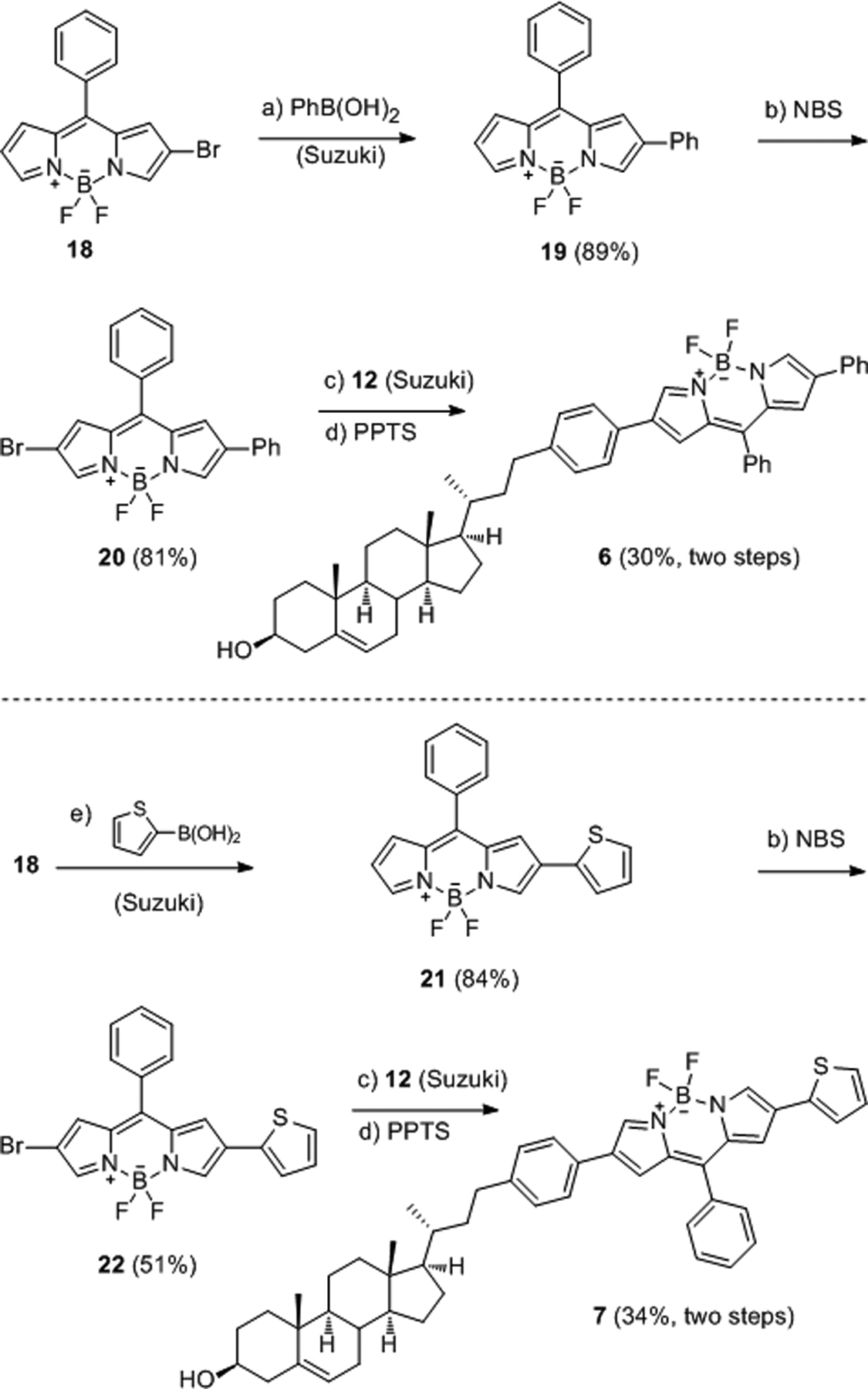
Synthesis of 6 and 7. Detailed reaction conditions and reagents: a) PhB(OH)2, Pd2(dba)3, DavePhos, Cs2CO3, 1,4-dioxane/H2O (10:1), microwave heating at 80 °C, 30 min; b) NBS, DMF/CH2Cl2 (1:1), 70 °C, 30 min; c) 12, Pd2(dba)3, DavePhos, Cs2CO3, 1,4-dioxane/H2O (10:1), microwave heating at 100 °C, 30 min; d) PPTS, MeOH/CH2Cl2 (2:1), reflux, 2 h; e) 2-thienylboronic acid, Pd2(dba)3, DavePhos, Cs2CO3, 1,4-dioxane/H2O (10:1), microwave heating at 80 °C, 30 min.
The photoimages of analogs 2-7 in methanol taken under normal room illumination and UV light are shown in Figure 1. The distinct emission color enabled us to distinguish them with the naked eye. It is noteworthy that conjugates 5, 6, and 7 are significantly red shifted in their excitation and emission maxima relative to the widely used BODIPY-cholesterol (1). Table 1 shows that probes 2 and 4 have very similar photophysical behavior as 1 (λex 495 nm, λem 507 nm),11 but have higher extinction coefficients However, the excitation maxima of 5, 6, and 7 are red-shifted by 46, 94, and 83 nm, respectively, and the emission maxima of 5, 6, and 7 are red-shifted by 108, 121, and 106 nm, respectively, relative to those of 1. Thus the excitation and emission spectra cover a broad range of the visible spectrum.
Figure 1.

Photoimages of 2-7 at 10 μM in methanol.
Table 1.
Spectroscopic Data in Methanol
| Compd | λmax[a] (abs), nm | ε, M−1cm−1 | λem,[b] nm |
|---|---|---|---|
| 2 | 495 | 35,700 | 511 |
| 3 | 508 | 40,000 | 542 |
| 4 | 497 | 48,600 | 514 |
| 5 | 541 | 24,300 | 615 |
| 6 | 589 | 30,000 | 638 |
| 7 | 578 | 17,900 | 613 |
Absorption spectra were recorded at 7.0 μM at 25 °C.
Emission spectra were recorded at 0.2 μM.
Biology
The utility of these newly described conjugated BODIPY-cholesterol analogs depends on their ability to mimic unmodified cholesterol. To address the potential suitability of these new conjugated BODIPY-cholesterol analogs as cellular probes for cholesterol, we first compared their cell distribution with that of the widely used BODIPY-cholesterol (compound 1).5, 10, 31 HeLa cells, a commonly used mammalian cell line, were incubated with 25 μM fluorescent sterol complexed with methyl-β-cyclodextrin (mβCD) to achieve labeling of the plasma membrane, as previously described.5 Compounds 2 and 4-7 labeled the plasma membrane as well as intracellular membranes in a manner, that was nearly identical to unmodified BODIPY-cholesterol (1) (Fig. 2). Compound 3 was tested as well, but we were unable to detect uptake of this compound in cells. This could be due to reduced fluorescence as seen in Figure 1 or due to decreased cellular uptake. Figure 2 also demonstrates that, as expected, compounds 5, 6, and 7 have a distinct spectral excitation and emission with no observable overlap with that of the unmodified BODIPY-cholesterol analog (1).
Figure 2.
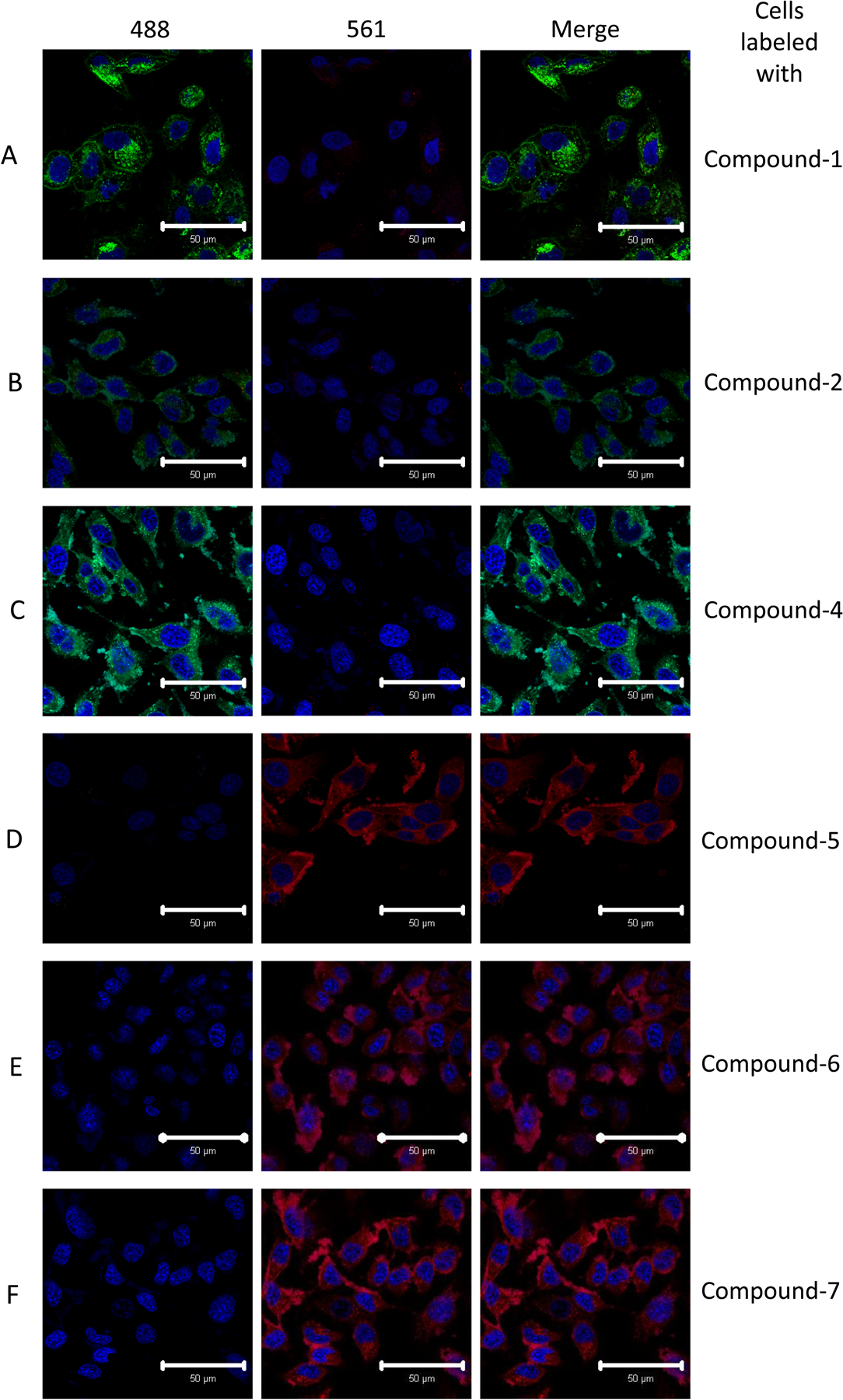
Uptake of BODIPY-cholesterol analogs by HeLa cells. HeLa cells were loaded with each BODIPY-cholesterol analog using mβCD as a donor. A-F show representative images of the localization of fluorescent cholesterol in cells loaded with compounds 1, 2, 4, 5, 6, and 7, respectively. Nuclei were labeled with Hoechst 33342 and are shown in blue. Columns are labeled with emission spectra (nm) of the laser used to acquire the image. Images were acquired using a 40X objective on a confocal microscope. Scale bar indicates 50 μm. Images are representative of three experiments.
To further demonstrate that the conjugated BODIPY-cholesterol analogs behave in a manner similar to BODIPY-cholesterol, HeLa cells were loaded simultaneously with compound 1 and either compound 5, 6, or 7. As expected, compounds 5, 6, and 7 display a nearly identical labeling of cells as compound 1, as can be seen by the yellow color in the overlay image after simultaneous incubation of both cholesterol analogs (Fig. 3). Moreover, the degree of overlap for the fluorescence signal from compound 1 with the fluorescence signals from compounds 5, 6, and 7 was determined by a co-localization analysis, and the Pearson coefficients were found to be 0.78 ± 0.06, 0.82 ± 0.07, and 0.85 ± 0.02, respectively. The spectral characteristics of 2 and 4 are nearly identical to those of compound 1, and thus prevent the simultaneous imaging with compound 1, but as seen in Figure 2 labeling by these compounds appears very similar to compound 1. To further validate that the BODIPY-cholesterol analogs behave in a manner similar to cholesterol we compared these analogs to filipin, a well-known cytochemical stain that specifically binds to free cholesterol.32 We observed that there was a high degree of overlap between the BODIPY-cholesterol analogs and the labeling of intracellular cholesterol by filipin, with the Pearson coefficients ranging from 0.50 (for compound 4) to 0.72 (for compound 5) (see Supplementary Fig. 1). Furthermore, the BODIPY-cholesterol analog’s fluorescence signal is distinct from that of the free BODIPY moiety in compounds 16, 18, and 21 (Supplementary Fig. 2).
Figure 3.
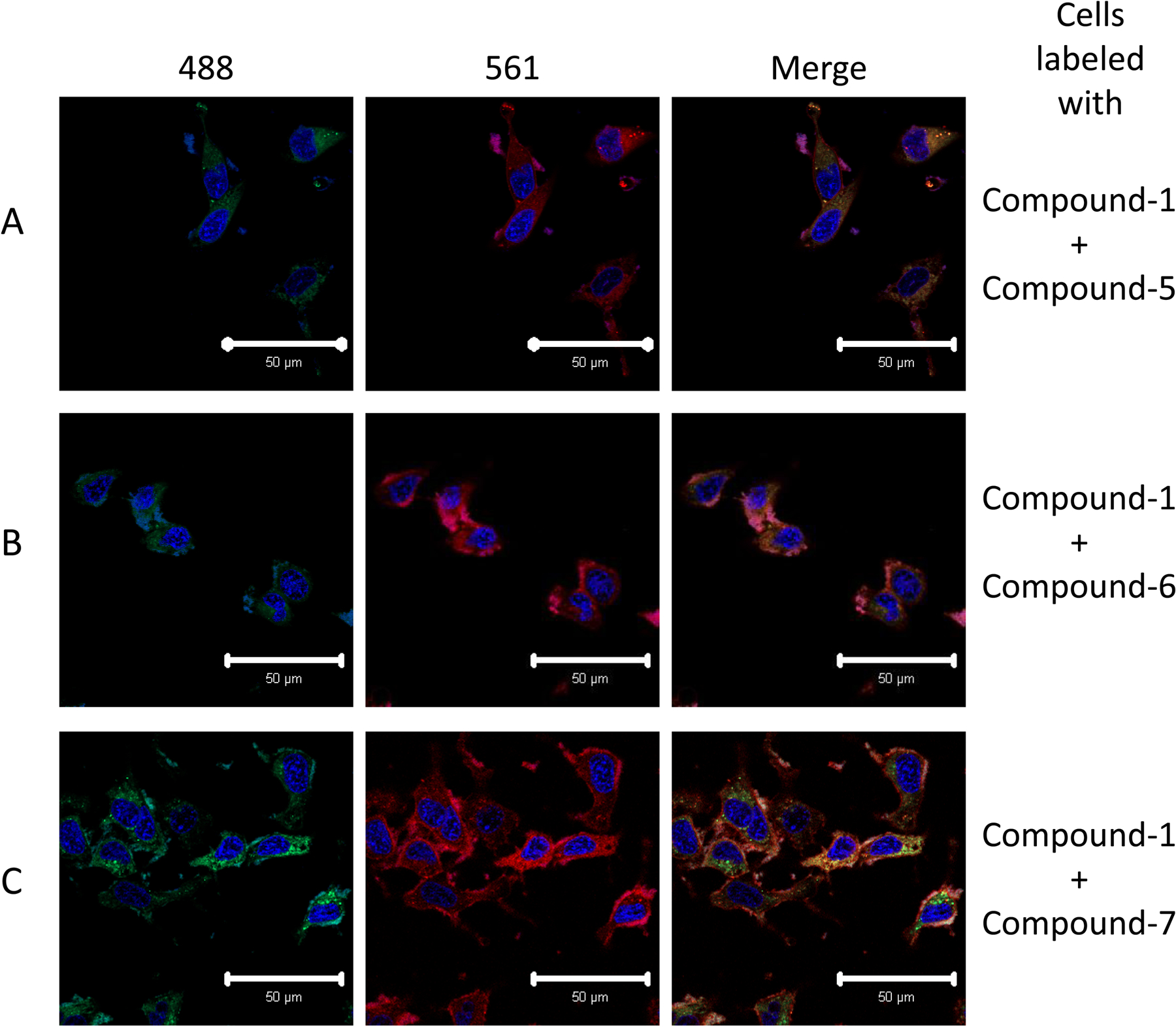
BODIPY-cholesterol analogs have similar cellular distributions. HeLa cells were loaded with 25 μM compound 1 and an equal amount of compound 5, 6, or 7 as indicated. Compound 1 co-localized with compounds 5, 6, and 7 as indicated by the yellow color in the merged image (A-C, respectively). Nuclei were labeled with Hoechst 33342 and are shown in blue. Columns are labeled with emission spectra (nm) of the laser used to acquire the image. Images were acquired using a 40X objective on a confocal microscope.The scale bar indicates 50 μm. Images are representative of three experiments.
Recently, Sankaranarayanan et al.5 demonstrated in J774 cells that BODIPY-cholesterol underwent efflux at an almost identical rate to HDL as [3H]-cholesterol. Therefore, we sought to determine if the cellular efflux of our newly synthesized BODIPY-cholesterol analogs is similar to that of compound 1. Using the monocytic THP-1 cell line and the method described by Sankaranarayanan et al.5, we observed that all of the tested compounds similarly underwent efflux in a dose-dependent manner to HDL (0–500 μg/mL, Fig. 4). Because the signal was relatively weak using a conventional plate reader, we quantified the amount of residual fluorescence associated with the cells by flow cytometry, which yielded a robust signal. However, this prevented examination of compounds 6 and 7, because the excitation and emission spectra of these compounds were incompatible with the flow cytometer used in the present study.
Figure 4.
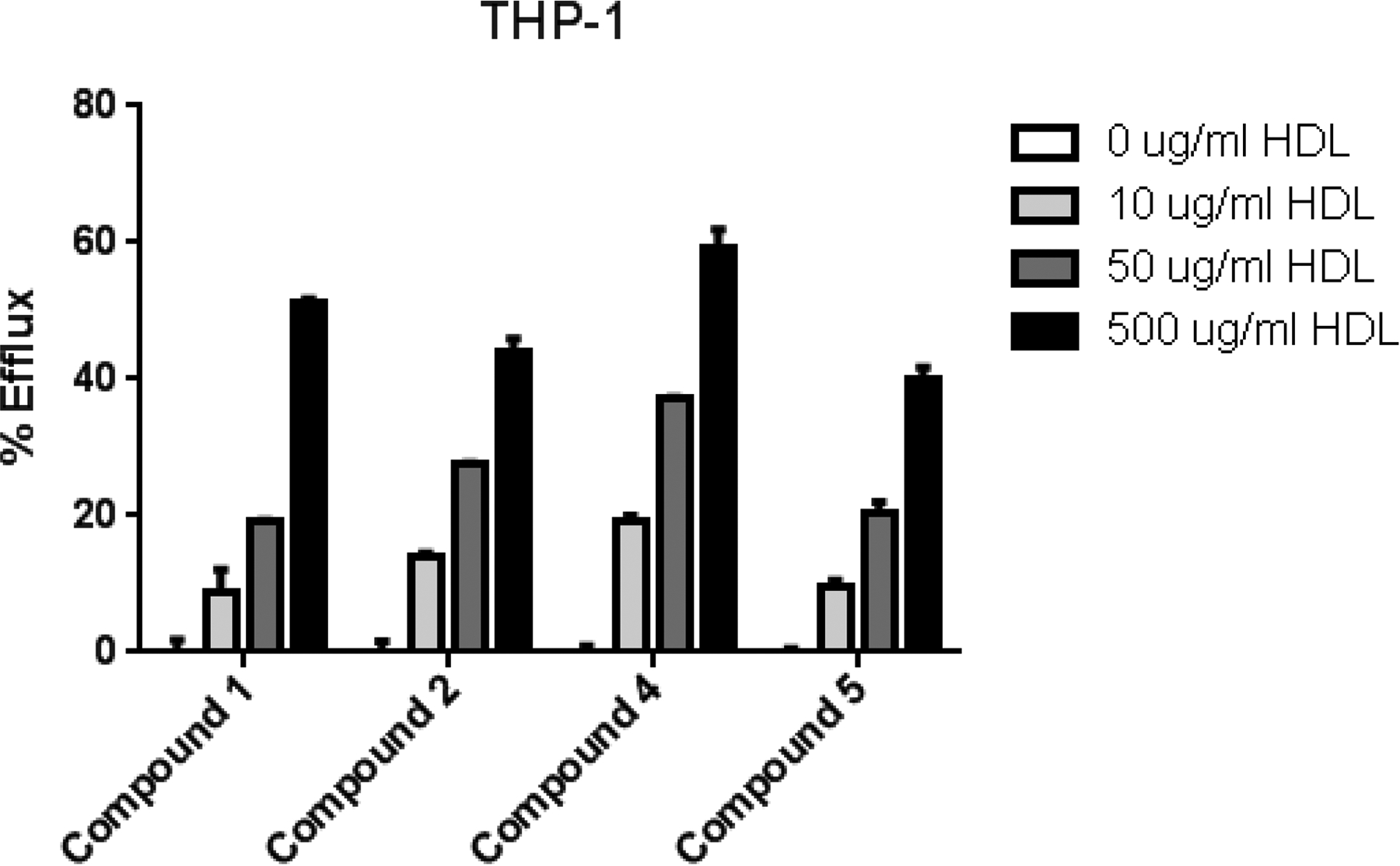
Dose-dependent efflux of conjugated BODIPY-cholesterol analogs to HDL. Compounds 1, 2, 4, and 5 all exhibited efflux in a dose-dependent manner to HDL from THP-1 cells. Results represent the mean % efflux ± SEM (n = 3).
To further demonstrate the potential utility of these new BODIPY-cholesterol analogs to simultaneously monitor multiple cellular cholesterol pools, we used confocal microscopy to detect two different BODIPY-cholesterol conjugates in HeLa cells (compound 1 and either compound 5, 6, or 7). HeLa cells were first incubated with acetylated LDL (ac-LDL) to promote the rapid cellular uptake into the lysosomal and endosomal compartments by the scavenger receptor;33 and then the cells were briefly incubated with BODIPY-cholesterol (1) in the presence of serum to primarily label the plasma membranes of cells. Loading of cells with ac-LDL resulted in a distinct labeling of the endosomal and lysosomal compartments (Fig. 5A). This pattern of staining was distinct from the staining we observed after incubation of cells with compounds 5, 6, and 7, which were primarily localized to the plasma membrane (Fig. 5 B, C, and D). The dual labeling we observed demonstrates that the distinct spectral properties of the different BODIPY-cholesterol analogs may be used to simultaneously visualize multiple cholesterol pools.
Figure 5.
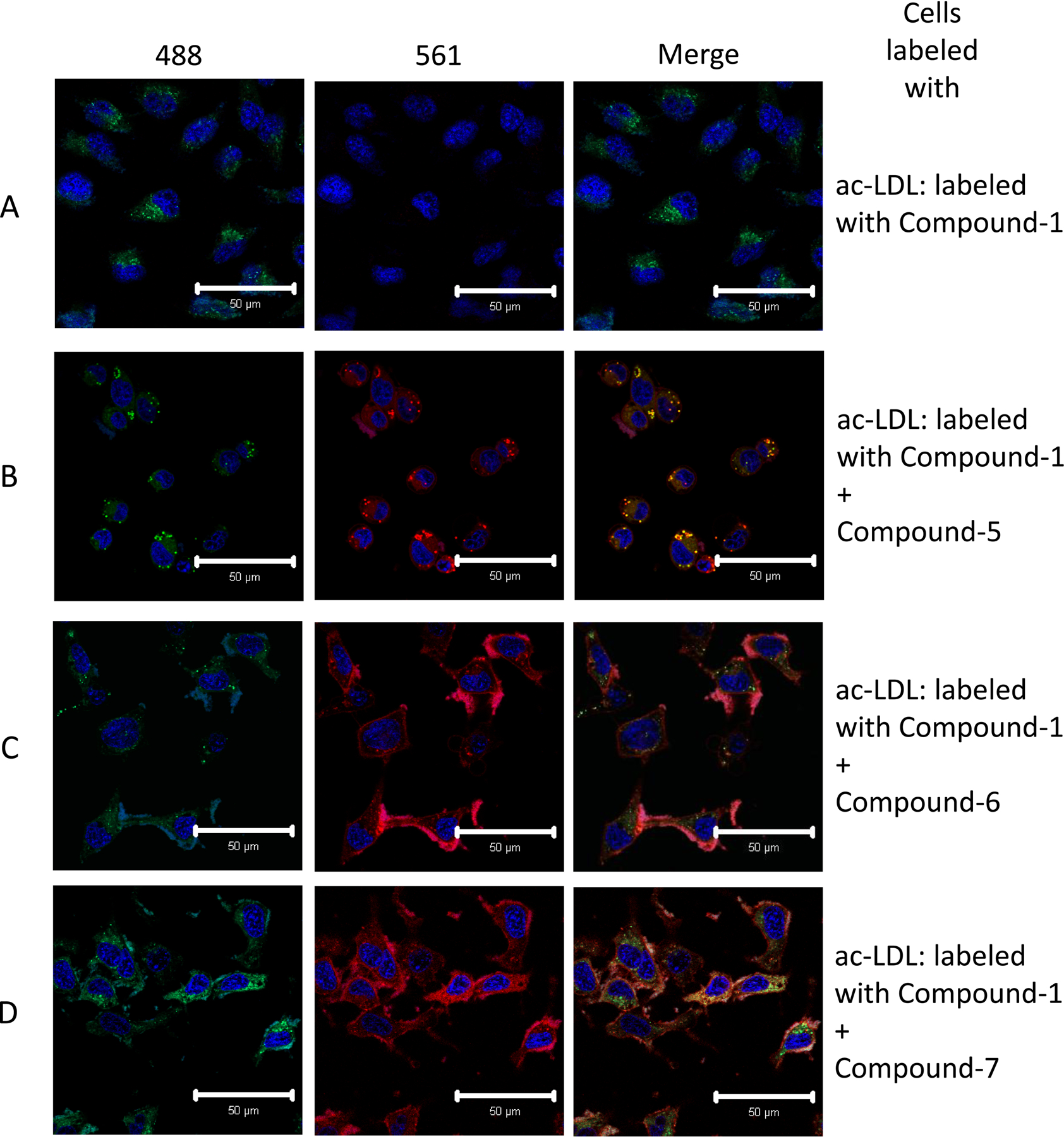
Multiple cholesterol pools labeled with BODIPY-cholesterol. HeLa cells were incubated with ac-LDL labeled with compound 1, followed by incubation with mβCD loaded with compound 5, 6, or 7. Confocal analysis demonstrated that ac-LDL is localized to lysosomal and endocytic compartments, whereas compounds 5-7 stain cell-surface membranes (B-D, respectively). Nuclei were labeled with Hoechst 33342 and are shown in blue. Columns are labeled with emission spectra (nm) of the laser used to acquire the image. Images were acquired using a 40X objective on a confocal microscope. Images are representative of three experiments.
Conclusion
Although our convergent synthetic sequence is still compromised by the low yields of the cross-coupling steps, it nevertheless provides a rapid and broadly applicable access to structurally diverse BODIPY-cholesterol conjugates due to the high functional group compatibility and ease of product separation. The synthetic strategies presented here permit other substituents to be incorporated by coupling post-functionalized BODIPY dyes with cholesterol phenyl moieties 11, 12, or 13. The results from the labeling of HeLa cells and the cellular cholesterol efflux from THP-1 cells are promising and suggest that the cell permeable, red-shifted BODIPY-cholesterol analogs such as described in this paper may enable the simultaneous tracking of different cellular cholesterol pools by confocal microscopy and flow cytometry, and can be used on living cells unlike filipin.
Experimental Section
Chemistry
General remarks:
All reactions were carried out under a dry N2 atmosphere using oven-dried glassware and magnetic stirring. The solvents were dried before use as follows: Toluene, THF and Et2O were heated at reflux over sodium benzophenone ketyl; CH2Cl2 was dried over CaH2. Anhydrous DMF was used directly as purchased. Commercially available reagents were used without further purification unless otherwise noted. Aluminum TLC sheets (silica gel 60 F254) of 0.2-mm thickness were used to monitor the reactions. The spots were visualized with short wavelength UV light or by charring after spraying with a solution prepared from one of the following solutions: phosphomolybdic acid (5.0 g) in 95% EtOH (100 mL); p-anisaldehyde solution (2.5 mL of p-anisaldehyde, 2 mL of AcOH, and 3.5 mL of conc. H2SO4 in 100 mL of 95% EtOH); or KMnO4 solution (1.5 g of KMnO4 and 1.5 g of K2CO3 in 100 mL of 0.1% NaOH). Flash chromatography was carried out with silica gel 60 (230–400 ASTM mesh). NMR spectra were obtained on a 400-MHz or 500-MHz spectrometer. Chemical shifts were referenced on residual solvent peaks: CDCl3 (δ = 7.26 ppm for 1H NMR and 77.00 ppm for 13C NMR), CD2Cl2 (δ = 5.32 ppm for 1H NMR and 53.84 ppm for 13C NMR). High-resolution mass spectra were acquired by electrospray ionization. Fluorescence quantum yields were calculated in ethanol as previously described34 with rhodamine 6G in ethanol as a standard (ΦF 0.86) as the standard.
Phenylbromide 11:
A flame-dried round-bottomed flask was charged with 789 mg (1.91 mmol) of alkene 1035 and 20 mL of dry toluene. A 0.5 M solution of 9-BBN in THF (20 mL, 10.0 mmol) was added via syringe, and the flask was then fitted with a reflux condenser and placed under N2 atmosphere. The solution was brought to 80–85 °C with the aid of a preheated oil bath. The resulting clear solution was stirred at this temperature for approximately 15–20 min. The heating bath was removed and the reaction was cooled to rt, and 3 M aqueous NaOH (10 mL, 30.0 mmol) was added. After the reaction was stirred vigorously for 20 min, 1-bromo-4-iodobenzene (1.62 g, 5.73 mmol) and Pd(dppf)Cl2∙CH2Cl2 (108 mg, 0.132 mmol) were added. The reaction mixture was stirred at 85 °C for 1 h, cooled to rt, poured into brine, and extracted with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. To the residue was added 100 mL of hexane and 2 mL of ethanolamine, and the mixture was stirred at rt for 30 min to precipitate the 9-BBN/ethanolamine complex. The solid was removed by filtration through a pad of Celite, which was washed with hexane. After the filtrate was concentrated under reduced pressure, the crude residue was purified by flash chromatography (hexanes/Et2O 20:1 to 10:1) to furnish product 11 (835 mg, 77%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 0.66 (s, 3H), 0.81–1.76 (m, 27H), 1.78–1.93 (m, 4H), 1.93–2.05 (m, 2H), 2.16–2.24 (m, 0.5 H), 2.30–2.45 (m, 2.5 H), 2.59–2.68 (m, 1H), 3.44–3.57 (m, 2H), 3.89–3.95 (m, 1H), 4.69–4.74 (m, 1H), 5.32–5.38 (m, 1H), 7.02–7.06 m, 2H), 7.36–7.40 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 11.8, 18.6, 19.4, 20.0, 20.1, 20.99, 21.01, 24.2, 25.4, 27.9, 28.2, 29.6, 31.22, 31.25, 31.82, 31.87, 31.92, 35.5, 36.7, 37.1, 37.4, 38.0, 38.7, 39.7, 40.2, 42.3, 50.06, 50.09, 55.8, 56.7, 62.8, 62.9, 75.9, 96.8, 96.9, 119.1, 121.4, 121.5, 130.1, 131.2, 140.8, 141.0, 142.3; ESI-HRMS (M+NH4)+ calcd for C34H53BrNO2+ 586.3254, found 586.3247.
Boronic pinacol ester 12:
A mixture of 11 (108 mg, 0.190 mmol), Pd2(dba)3 (4.4 mg, 4.8 μmol), DavePhos (9.0 mg, 0.0229 mmol), bis(pinacolato)diboron (144 mg, 0.570 mmol), and KOAc (67 mg, 0.683 mmol) in 1,4-dioxane (2 mL) was stirred and heated by microwave irradiation (110 °C) for 30 min. The reaction mixture was allowed to cool to rt. The reaction solution was then filtered through a thin pad of Celite (eluting with EtOAc) and the filtrate was concentrated under reduced pressure. The crude material was purified via flash chromatography (hexanes/Et2O 20:1, 10:1, 8:1) on silica gel to provide product 12 (77 mg, 66%) as white solid, together with the side product of protodebromination (16 mg, 17%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 0.66 (s, 3H), 0.81–1.76 (m, 39 H), 1.79–2.06 (m, 6H), 2.16–2.24 (m, 0.5H), 2.31–2.39 (m, 1.5H), 2.43–2.51 (m, 1H), 2.66–2.74 (m, 1H), 3.45–3.58 (m, 2H), 3.89–3.95 (m, 1H), 4.70–4.74 (m, 1H), 5.33–5.38 (m, 1H), 7.16–7.21 (m, 2H), 7.70–7.75 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 11.8, 18.6, 19.3, 19.99, 20.07, 20.99, 21.01, 24.2, 24.8, 25.4, 27.9, 28.1, 29.6, 31.22, 31.25, 31.82, 31.86, 32.7, 35.6, 36.70, 36.74, 37.1, 37.4, 38.0, 38.7, 39.7, 40.2, 42.3, 50.06, 50.10, 55.8, 56.7, 62.8, 62.9, 75.93, 75.95, 83.5, 96.8, 96.9, 121.4, 121.5, 127.8, 134.8, 140.8, 141.0, 146.9; ESI-HRMS (M+NH4)+ calcd for C40H61BNaO4+ 639.4562, found 639.4564.
Analog 2:
Boronic acid pinacol ester 12 (70 mg, 0.114 mmol) and NaIO4 (73 mg, 0.341 mmol) were stirred in 5 mL of a 4:1 mixture of THF and water for 30 min, at which time aqueous HCl (1 M, 80 μL, 0.080 mmol) was added to the suspension. The reaction mixture was stirred overnight at rt. The reaction mixture was diluted with water (5 mL) and extracted with EtOAc. The combined extracts were washed with water and brine, dried over Na2SO4, filtered, and concentrated to provide boronic acid 13 (47 mg, 77%). The product was not purified but was used immediately after preparation. Boronic acid 13 (10.8 mg, 20.5 μmol), Pd2(dba)3 (1.8 mg, 2.0 μmol), tris(2-furyl)phosphine (1.6 mg, 7.0 μmol), copper(I) thiophene-2-carboxylate (11 mg, 0.58 mmol), and thiomethyl-BODIPY 1424 (9 mg, 0.038 mmol) were placed in a sealed tube, and anhydrous THF (2 mL) was added. This suspension was stirred and heated by microwave irradiation (100 °C) for 30 min. The solution was evaporated to dryness under reduced pressure and diluted with saturated aqueous NaHCO3 solution. The aqueous solution was extracted with CH2Cl2. The dried organic layer was evaporated under reduced pressure and the resulting oil was purified by flash chromatography on silica gel (EtOAc/hexane 1:5, 4:1, to 3:1) to afford product 2 (2.7 mg, 22%) as a red powder. ΦF 0.04; 1H NMR (500 MHz, CDCl3) δ 0.70 (s, 3H), 0.79–2.08 (m, 28H), 2.20–2.34 (m, 2H), 2.53–2.62 (m, 1H), 2.76–2.84 (m, 1H), 3.49–3.58 (m, 1H), 5.34–5.38 (m, 1H), 6.53–6.57 (m, 2H), 6.95–6.99 (m, 2H), 7.31–7.35 (m, 2H), 7.47–7.51 (m, 2H), 7.93 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 11.9, 18.7, 19.4, 21.1, 24.3, 28.2, 31.6, 31.9, 32.5, 35.7, 36.5, 37.2, 37.9, 39.7, 42.3, 42.4, 50.1, 55.8, 56.7, 71.8, 118.3, 121.7, 128.5, 129.4, 130.7, 131.1, 131.6, 134.9, 140.7, 143.7, 146.9; ESI-HRMS (M+Na)+ calcd for C38H47BF2N2NaO+ 619.3648, found 619.3652.
Boronic pinacol ester 17:
A mixture of 1620 (40 mg, 0.115 mmol), Pd2(dba)3 (5.0 mg, 5.5 μmol), DavePhos (9.0 mg, 0.023 mmol), bis(pinacolato)diboron (67 mg, 0.264 mmol), and KOAc (43 mg, 0.438 mmol) were placed in a sealed tube, and anhydrous 1,4-dioxane (2 mL) was added. The reaction mixture was stirred and heated by microwave irradiation (110 °C) for 30 min. The reaction mixture was allowed to cool to rt. The reaction solution was then filtered through a thin pad of Celite (eluting with EtOAc) and the filtrate was concentrated under reduced pressure. The crude material was purified via flash chromatography (hexanes/EtOAc 8:1) on silica gel to provide product 17 (34 mg, 75%) as a red solid. 1H NMR (500 MHz, CDCl3) δ 1.39 (s, 12H), 6.53–6.56 (m, 2H), 6.90–6.94 (m, 2H), 7.55–7.59 (m, 2H), 7.92–7.98 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 24.9, 84.3, 118.6, 129.7, 131.5, 134.6, 134.8, 136.2, 144.2; ESI-HRMS (M−) calcd for C21H22B2F2N2O2− 394.1848, found 394.1851.
Analog 3:
A mixture of BODIPY derivative 153 (19 mg, 0.538 mmol), boronic acid pinacol ester 12 (7 mg, 11.4 μmol), Pd(dppf)Cl2∙CH2Cl2 (1.8 mg, 2.2 μmol), and K2CO3 (19.5 mg, 0.0684 mmol) were placed in a sealed tube, and PhMe/H2O (1.5 mL, 2:1) was added. The reaction mixture was stirred and heated by microwave irradiation (120 °C) for 30 min. The reaction mixture was allowed to cool to rt, poured into saturated aqueous NH4Cl solution, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in hexane/EtOAc (3:1) and filtered through a short pad of silica gel (eluting with hexane/EtOAc 3:1). The filtrate was concentrated under reduced pressure to provide the crude coupling product. A solution of the crude coupling product and PPTS (7.0 mg, 0.028 mmol) in CH2Cl2/MeOH (2 mL/4 mL) was heated at reflux until the reaction was complete (2 h, monitored by TLC using hexane/EtOAc, 2:1). The reaction mixture was allowed to cool to rt, poured into an aqueous solution of saturated NaHCO3, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified via flash chromatography (hexanes/EtOAc 3:1, 2:1, 1:1) on silica gel to provide product 3 (3.5 mg, 45% over two steps) as a red solid. ΦF 0.08; 1H NMR (500 MHz, CD2Cl2) δ 0.71 (s, 3H), 0.85–1.67 (m, 22H), 1.70–2.09 (m, 6H), 2.16–2.30 (m, 2H), 2.50–2.59 (m, 1H), 2.71–2.80 (m, 1H), 3.43–3.52 (m, 1H), 5.35–5.38 (m, 1H), 6.63–6.66 (m, 2H), 7.28–7.32 (m, 2H), 7.43–7.46 (m, 2H), 7.51 (d, J = 3.9 Hz, 1H), 7.62–7.67 (m, 3H), 7.91 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 11.9, 18.7, 19.4, 21.1, 24.3, 28.2, 31.6, 31.9, 32.4, 35.6, 36.5, 37.2, 37.9, 39.8, 42.3, 2.4, 50.1, 55.8, 56.7, 71.8, 118.3, 121.7, 123.9, 125.3, 126.1, 128.2, 129.0, 129.2, 130.3, 131.1, 133.2, 134.0, 134.6, 140.7, 143.3, 144.9, 151.7; ESI-HRMS (M+Na)+ calcd for C42H49BF2N2NaOS+ 701.3526, found 701.3532.
Analog 4:
A mixture of 11 (90 mg, 0.158 mmol), boronic acid pinacol ester 17 (34 mg, 0.086 mmol), Pd(dppf)Cl2∙CH2Cl2 (7.0 mg, 8.6 μmol), and K2CO3 (106 mg, 0.767 mmol) were placed in a sealed tube, and PhMe/H2O (3 mL) was added. The reaction mixture was stirred and heated by microwave irradiation (120 °C) for 30 min. The reaction mixture was allowed to cool to rt, poured into saturated aqueous NH4Cl solution, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in hexane/EtOAc (3:1) and filtered through a short pad of silica gel (eluting with hexane/EtOAc 3:1). The filtrate was concentrated under reduced pressure to provide the crude coupling product. A solution of the crude coupling product and PPTS (30 mg, 0.119 mmol) in CH2Cl2/MeOH (5 mL/10 mL) was heated at reflux until the reaction was complete (2 h, monitored by TLC using hexane/EtOAc, 2:1). The reaction mixture was allowed to cool to rt, poured into an aqueous solution of saturated NaHCO3, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified via flash chromatography (hexanes/EtOAc 3:1, 2:1, 1:1) on silica gel to provide product 4 (27 mg, 47%) as a red solid. ΦF 0.02; 1H NMR (500 MHz, CD2Cl2) δ 0.71 (s, 3H), 0.85–1.70 (m, 22H), 1.75–2.09 (m, 6H), 2.18–2.33 (m, 2H), 2.52–2.61 (m, 1H), 2.74–2.83 (m, 1H), 3.44–3.52 (m, 1H), 5.36–5.39 (m, 1H), 6.59–6.64 (m, 2H), 7.05–7.10 (m, 2H), 7.32–7.37 (m, 2H), 7.61–7.65 (2H), 7.67–7.71 (m, 2H), 7.77–7.81 (m, 2H), 7.95 (s, 2H); 13C NMR (100 MHz, CD2Cl2) δ 12.1, 18.9, 19.6, 21.5, 24.7, 28.6, 0.1, 32.1, 32.3, 32.6, 36.2, 36.9, 37.7, 38.6, 40.2, 42.73, 42.77, 50.6, 56.3, 57.2, 72.1, 118.9, 121.8, 127.2, 127.4, 129.5, 131.7, 132.0, 132.7, 135.3, 137.3, 141.4, 144.13, 144.17, 144.3, 147.8; 13C NMR (125 MHz, CDCl3) δ 11.9, 18.7, 19.4, 21.1, 24.3, 28.2, 31.6, 31.9, 32.2, 35.7, 36.5, 37.2, 38.1, 39.8, 42.3, 42.4, 50.1, 55.8, 56.7, 71.8, 118.5, 121.7, 126.9, 127.1, 129.1, 131.1, 131.5, 132.4, 134.9, 136.9, 140.7, 143.7, 143.8, 143.9, 147.2; ESI-HRMS (M+Na)+ calcd for C44H51BF2N2NaO+ 695.3962, found 695.3967.
Analog 5:
A mixture of 12 (40 mg, 0.0649 mmol), Pd2(dba)3 (3.4 mg, 3.7 μmol), DavePhos (9.4 mg, 0.0239 mmol), 1827 (45 mg, 0.130 mmol), and Cs2CO3 (121 mg, 0.371 mmol) were placed in a sealed tube, and 1,4-dioxane/H2O (2.2 mL, 10:1) was added. The reaction mixture was stirred and heated by microwave irradiation (80 °C) for 30 min. The reaction mixture was allowed to cool to rt, poured into brine, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in hexane/EtOAc (3:1) and filtered through a short pad of silica gel (eluting with hexane/EtOAc 3:1). The filtrate was concentrated under reduced pressure to provide the crude coupling product. A solution of the crude coupling product and PPTS (49 mg, 0.195 mmol) in CH2Cl2/MeOH (5 mL/10 mL) was heated at 55 °C until the reaction was complete (1 h, monitored by TLC using hexane/EtOAc, 2:1). The reaction mixture was allowed to cool to rt, poured into an aqueous solution of saturated NaHCO3, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified via flash chromatography (hexanes/EtOAc 3:1, 2:1, 1:1) on silica gel to provide product 5 (30 mg, 69% over two steps) as a purple solid. ΦF 0.02; 1H NMR (500 MHz, CDCl3) δ 0.66 (s, 3H), 0.81–1.75 (m, 23 H), 1.79–1.90 (m, 3H), 1.92–2.07 (m, 2H), 2.19–2.33 (m, 2H), 2.41–2.50 (m, 1H), 2.64–2.73 (m, 1H), 3.48–3.56 (m, 1H), 5.33–5.36 (m, 1H), 6.54–6.56 (m, 1H), 6.91–6.94 (m, 1H), 7.07 (s, 1H), 7.15–7.20 (m, 2H), 7.40–7.46 (m, 2H), 7.53–7.64 (m, 5H), 7.94 (s, 1H), 8.28 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 11.9, 18.7, 19.4, 21.0, 24.2, 28.2, 31.6, 31.8, 32.3, 35.6, 36.5, 37.2, 38.0, 39.7, 42.2, 42.3, 50.0, 55.8, 56.7, 71.8, 118.3, 121.6, 125.3, 128.5, 128.9, 129.8, 130.5, 130.7, 131.2, 133.8, 134.5, 135.0, 135.6, 140.7, 142.3, 143.1, 143.7, 146.7; ESI-HRMS (M+Na)+ calcd for C44H51BF2N2NaO+ 695.3966, found 695.3962.
BODIPY derivative 19:
A mixture of 18 (100 mg, 0.289 mmol), Pd2(dba)3 (6.6 mg, 0.0072 mmol), DavePhos (11.4 mg, 0.029 mmol), phenylboronic acid (70 mg, 0.574 mmol), and Cs2CO3 (312 mg, 0.958 mmol) were placed in a sealed tube, and 1,4-dioxane/H2O (2.2 mL, 10:1) was added. The reaction mixture was stirred and heated by microwave irradiation (80 °C) for 30 min. The reaction mixture was allowed to cool to rt, poured into brine, and extracted with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in hexane/EtOAc (3:1) and filtered through a short pad of silica gel (eluting with hexane/EtOAc 3:1). The filtrate was concentrated under reduced pressure to provide crude coupling product 1936 (89 mg, 89%), which was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 6.57 (dd, J = 1.6, 4.2 Hz, 1H), 6.95 (d, J = 4.1 Hz, 1H), 7.11 (s, 1H), 7.26–7.30 (m, 1H), 7.35–7.40 (m, 2H), 7.50–7.64 (m, 2H), 7.97 (s, 1H), 8.30 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 118.5, 125.4, 125.7, 127.6, 128.5, 128.9, 130.4, 130.8, 131.5, 132.5, 133.7, 134.3, 135.1, 135.5, 142.0, 144.1, 146.9; ESI-HRMS (M+Na)+ calcd for C21H15BF2N2Na+ 367.1189, found 367.1191.
BODIPY derivative 20:
A mixture of 19 (64 mg, 0.186 mmol) and NBS (43 mg, 0.242 mmol) in anhydrous DMF/CH2Cl2 (4 mL, 1:1) was stirred at 70 °C for 30 min, and the solvents were removed under reduced pressure. The residue was purified via flash chromatography (hexanes/EtOAc 20:1, 15:1, to 10:1) on silica gel to provide product 20 (50 mg, 64%) as a purple solid. 1H NMR (500 MHz, CDCl3) δ 6.90 (s, 1H), 7.15 (s, 1H), 7.29–7.33 (m, 1H), 7.36–7.41 (m, 2H), 7.48–7.68 (m, 7H), 7.81 (s, 1H), 8.35 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 125.5, 126.8, 128.0, 128.7, 129.0, 130.1, 130.37, 130.41, 131.2, 131.9, 133.3, 134.4, 135.3, 136.0, 142.0, 144.3, 146.5; ESI-HRMS (M+Na)+ calcd for C21H14BBrF2N2Na+ 445.0297, found 445.0301.
Analog 6:
A mixture of 12 (25 mg, 0.0405 mmol), Pd2(dba)3 (3.7 mg, 4.1 μmol), DavePhos (6.3 mg, 0.016 mmol), 20 (25 mg, 0.0591 mmol), and Cs2CO3 (66 mg, 0.203 mmol) were placed in a sealed tube, and 1,4-dioxane/H2O (2.2 mL, 10:1) was added. The reaction mixture was stirred and heated by microwave irradiation (100 °C) for 30 min. The reaction mixture was allowed to cool to rt, poured into brine, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in hexane/EtOAc (3:1) and filtered through a short pad of silica gel (eluting with hexane/EtOAc 3:1). The filtrate was concentrated under reduced pressure to provide the crude coupling product. A solution of the crude coupling product and PPTS (30 mg, 0.119 mmol) in CH2Cl2/MeOH (5 mL/10 mL) was heated at 55 °C until the reaction was complete (1 h, monitored by TLC using hexane/EtOAc, 2:1). The reaction mixture was allowed to cool to rt, poured into an aqueous solution of saturated NaHCO3, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified via flash chromatography (hexanes/EtOAc 3:1, 2:1, 1:1) on silica gel to provide product 6 (9.0 mg, 30% over two steps) as a violet solid. ΦF 0.06; 1H NMR (500 MHz, CDCl3) δ 0.68 (s, 3H), 0.86–1.63 (m, 22H), 1.67–1.76 (m, 1H), 1.80–1.90 (m, 3H), 1.94–2.06 (m, 2H), 2.19–2.33 (m, 2H), 2.43–2.51 (m, 1H), 2.65–2.74 (m, 1H), 3.48–3.57 (m, 1H), 5.34–5.37 (m, 1H), 7.06–7.11 (m, 2H), 7.16–7.20 (m, 2H), 7.26–7.31 (m, 1H), 7.35–7.41 (m, 2H), 7.42–7.46 (m, 2H), 7.51–7.55 (m, 2H), 7.57–7.62 (m, 2H), 7.63–7.68 (m, 3H), 8.27–8.31 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 11.9, 18.7, 19., 21.1, 24.3, 28.2, 31.6, 31.9, 32.3, 35.6, 36.4, 37.2, 38.0, 39.8, 42.3, 42.4, 50.1, 55.8, 56.7, 71.8, 121.7, 125.29, 125.38, 125.41, 127.5, 128.6, 128.95, 128.96, 129.8, 130.5, 130.8, 132.6, 133.8, 134.1, 134.6, 135.7, 135.9, 140.7, 141.6, 142.5, 143.2, 146.2; ESI-HRMS (M+Na)+ calcd for C50H55BF2N2NaO+ 771.4276, found 771.4287.
BODIPY derivative 21:
A mixture of 18 (105 mg, 0.303 mmol), Pd2(dba)3 (6.0 mg, 6.6 μmol), DavePhos (13 mg, 0.033 mmol), 2-thienylboronic acid (96 mg, 0.75 mmol), and Cs2CO3 (453 mg, 1.39 mmol) were placed in a sealed tube, and 1,4-dioxane/H2O (2.2 mL, 10:1) was added. The reaction mixture was stirred and submitted to microwave irradiation (80 °C) for 30 min. The reaction mixture was allowed to cool to rt, poured into brine, and extracted with EtOAc. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in hexane/EtOAc (3:1) and filtered through a short pad of silica gel (eluting with hexane/EtOAc 3:1). The filtrate was concentrated under reduced pressure to provide crude coupling product 2137 (85 mg, 84%), which was used in the next step without further purification. 1H NMR (500 MHz, CDCl3) δ 6.55–6.59 (m, 1H), 6.93–6.97 (m, 2H), 7.01–7.05 (m, 1H), 7.15–7.18 (m, 1H), 7.20–7.23 (m, 1H), 7.54–7.66 (m, 5H), 7.97 (s, 1H), 8.19 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 118.7, 123.3, 124.3, 124.7, 127.9, 128.2, 128.6, 130.4, 130.9, 131.7, 133.6, 135.2, 135.7, 141.5, 144.4, 146.8; ESI-HRMS (M+Na)+ calcd for C19H13BF2N2NaS+ 373.0756, found 373.0758.
BODIPY derivative 22:
A mixture of 21 (24 mg, 0.0685 mmol) and NBS (18 mg, 0.101 mmol) in anhydrous DMF/CH2Cl2 (2 mL, 1:1) was stirred at 70 °C for 30 min, and the solvents were removed under reduced pressure. The residue was purified via flash chromatography (hexanes/EtOAc 10:1, 8:1, to 5:1) on silica gel to provide product 2238 (15 mg, 51%) as a violet solid. 1H NMR (400 MHz, CDCl3) δ 6.57–6.60 (m, 1H), 6.87 (s, 1H), 6.89–6.91 (m, 1H), 6.95–6.99 (m, 2H), 7.52–7.66 (m, 5H), 7.98 (s, 1H), 8.09 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 110.8, 119.1, 123.4, 124.5, 127.1, 128.6, 130.4, 130.7, 131.0, 132.2, 133.6, 135.1, 135.4, 137.5, 140.6, 145.2, 147.1; ESI-HRMS (M+Na)+ calcd for C19H12BBrF2N2NaS+ 452.9842, found 452.9846.
Analog 7:
A mixture of 12 (14 mg, 0.023 mmol), Pd2(dba)3 (2.1 mg, 2.3 μmol), DavePhos (3.6 mg, 9.2 μmol), 22 (15 mg, 0.035 mmol), and Cs2CO3 (45 mg, 0.138 mmol) were placed in a sealed tube, and 1,4-dioxane/H2O (2.2 mL, 10:1) was added. The reaction mixture was stirred and heated by microwave irradiation (100 °C) for 30 min. The reaction mixture was allowed to cool to rt, poured into brine, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The residue was dissolved in hexane/EtOAc (3:1) and filtered through a short pad of silica gel (eluting with hexane/EtOAc 3:1). The filtrate was concentrated under reduced pressure to provide the crude coupling product. A solution of the crude coupling product and PPTS (40 mg, 0.159 mmol) in CH2Cl2/MeOH (5 mL/10 mL) was heated at 55 °C until the reaction was complete (1 h, monitored by TLC using hexane/EtOAc, 2:1). The reaction mixture was allowed to cool to rt, poured into an aqueous solution of saturated NaHCO3, and extracted with CH2Cl2. The combined organic extracts were dried over Na2SO4 and concentrated under reduced pressure. The crude material was purified via flash chromatography (hexanes/EtOAc 3:1, 5:2, 2:1) on silica gel to provide product 7 (6.0 mg, 34% over two steps) as a deep green solid. ΦF 0.01; 1H NMR (500 MHz, CDCl3) δ 0.68 (s, 3H), 0.86–1.63 (m, 22H), 1.67–1.76 (m, 1H), 1.80–1.90 (m, 3H), 1.94–2.06 (m, 2H), 2.19–2.33 (m, 2H), 2.43–2.51 (m, 1H), 2.65–2.74 (m, 1H), 3.48–3.57 (m, 1H), 5.34–5.37 (m, 1H), 6.57 (dd, J = 1.8, 4.1 Hz. 1H), 6.94–6.96 (m, 2H), 7.13 (d, J = 3.7 Hz, 1H), 7.16–7.21 (m, 3H), 7.47–7.51 (m, 2H), 7.55–7.66 (m, 5H), 7.96 (s, 1H), 8.21 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 11.8, 18.7, 19.4, 21.1, 24.3, 28.2, 31.6, 31.9, 32.3, 35.7, 36.5, 37.2, 38.0, 39.8, 42.3, 42.4, 50.1, 55.8, 56.7, 71.8, 110.0, 118.6, 121.7, 123.3, 124.2, 124.3, 125.5, 128.6, 128.9, 130.5, 130.9, 131.4, 131.6, 133.7, 134.5, 135.4, 140.7, 141.6, 143.2, 143.5, 144.3, 146.7; ESI-HRMS (M+Na)+ calcd for C48H53BF2N2NaOS+ 777.3840, found 777.3846.
Biology
Materials:
BODIPY-cholesterol (1) was synthesized as described previously10 and was also purchased from Avanti Polar Lipids (Alabaster, AL). The BODIPY-cholesterol analogs were dissolved in ethanol at a final concentration of 1 mg/mL. For labeling of cells, BODIPY-cholesterol analogs were complexed with mβCD (Sigma-Aldrich, St. Louis, MO) to achieve a final concentration of 25 μM in the labeling media. Cells were incubated with the cell labeling media for 1 h.
Cell culture:
All cell culture reagents were from Invitrogen (Grand Island, NY) unless otherwise indicated. HeLa and THP-1cells were acquired from ATCC (Manassas, VA). HeLa and THP-1 cells were cultured as recommended by ATCC.
Acetylation of LDL and loading ac-LDL with BODIPY-cholesterol analogs:
LDL was isolated by density gradient as previously described.39 LDL was acetylated in the following manner. LDL was adjusted to 2 mg of protein/mL and supplemented with 10 μM EDTA (Sigma-Aldrich). LDL was placed in an ice-water bath and an equal volume of aqueous saturated sodium acetate solution was added. While mixing, acetic anhydride was added in 2-μL aliquots over 1 h until 1.5 times the mass of LDL had been added. After the reaction, LDL was extensively dialyzed against PBS (Invitrogen). Before use, LDL was sterilized by filtration through a 0.2-μm filter (Millipore, Billerica, MA). To load ac-LDL with BODIPY-cholesterol analogs, ac-LDL was incubated for 1 h with 1 μg of the BODIPY-cholesterol analog in ethanol; the final concentration of ethanol was kept below 0.1%. Unincorporated BODIPY-cholesterol analog was removed using a spin filter (Millipore).
Cholesterol efflux to HDL:
Cholesterol efflux was carried out as previously described5 with modifications. THP-1 cells were loaded for 1 h with BODIPY-cholesterol pre-complexed with mβCD. After incubation, the cells were washed three times with PBS and resuspended in serum-free media containing 0.2% fatty acid free BSA. The THP-1 cells (100,000) were then incubated with HDL isolated from healthy controls by density gradient. After 6 h of incubation, the cells were immediately placed on ice until analysis. Levels of fluorescence were analyzed using a LSR II Flow Cytometer (BD Biosciences, San Jose, CA). For the efflux assay, 10,000 events were collected and analyzed with FCS express (De Novo Software, Los Angeles, CA). Percent efflux was determined by calculating the mean fluorescent intensity of the cells after HDL incubation divided by the mean fluorescence intensity of cells not treated with HDL.
Microscopy and dual-labeling experiments:
HeLa cells were grown on chambered coverglass (Nunc, Rochester, NY). Cells were incubated with ac-LDL loaded with the BODIPY-cholesterol analogs for 4 h and then were washed with PBS. Fresh media with a different BODIPY-cholesterol analog was added to cells for 1 h, followed by 2 washes with PBS. To label DNA, Hoechst 33342 (5 μg/mL, Invitrogen) was added for the last 15 min of incubation. In some experiments, cells where fixed with 4% formaldehyde following labeling of cells with the BODIPY-cholesterol analogs. Cells where then stained with 120 μg/mL filipin (Polysciences, Warrington, PA). After labeling, the cells were imaged at 40X with a Zeiss LSM 510 confocal microscope (Carl Zeiss Microscopy, Thornwood, NY), and the images were analyzed using Zen 2009 software (Carl Zeiss). To investigate whether the BODIPY-cholesterol analogs are co-localized with filipin, confocal microscopy images were analyzed in 3D with Imaris software v7.7 (Bitplane, Zurich, Switzerland). Quantification of co-localization was assessed in 3D, and pixel codistribution was calculated for green or red and blue staining patterns. Co-localized pixels were displayed as a 2-color histogram (scattergram-fluorogram). 2D fluorograms represent quantification of the co-localization as distribution of pairs of pixel intensities (aligned to a diagonal, perfect co-localization, randomly scattered, or toward one channel in the case of lack of co-localization). In addition, Pearson coefficients in the co-localized volume were computed and compared (1, perfect correlation; 0, no correlation; −1, perfect inverse correlation).
Supplementary Material
Acknowledgements
This work was supported in part by National Institutes of Health Grant HL-083187. We thank Alyssa Reimer for the synthesis of 9 on a large scale. Confocal Microscopy was performed in the NHLBI light microscopy core facility, and we thank Daniela Malide for assistance with the co-localization analysis.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org.
References
- [1].Gimpl G, Gehrig-Burger K, Steriods 2011, 76, 216. [DOI] [PubMed] [Google Scholar]
- [2].Marks DL, Bittman R, Pagano RE, Histochem. Cell Biol 2008, 130, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maxfield FR, Wüstner D, Methods Cell Biol. 2012, 108, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Holtta-Vuori M, Uronen R-L, Repakova J, Salonen E, Vattulainen I, Panula P, Li Z, Bittman R, Ikonen E, Traffic 2008, 9, 1839. [DOI] [PubMed] [Google Scholar]
- [5].Sankaranarayanan S, Kellner-Weibel G, de la Llera-Moya M, Phillips MC, Asztalos BF, Bittman R, Rothblat GH, J. Lipid Res 2011, 52, 2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Martel C, Li W, Fulp B, Platt AM, Gautier EL, Westerterp M, Bittman R, Tall AR, Chen SH, Thomas MJ, Kreisel D, Swartz MA, Sorci-Thomas MG, Randolph GJ, J. Clin. Invest 2013, 123, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bidet M, Joubert O, Lacombe B, Ciantar M, Nehmé R, Mollat P, Brétillon L, Faure H, Bittman R, Ruat M, Mus-Veteau I, PLoS One 2011, 6, e23834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Walters JW, Anderson JL, Bittman R, Pack M, Farber SA, Chem Biol. 2012, 19, 913. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [9].Crowley JT, Toledo AM, LaRocca TJ, Coleman JL, London E, Benach JL, PLoS Pathog. 2013, 9, e1003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li Z, Mintzer E, Bittman R, J. Org. Chem 2006, 71, 1718. [DOI] [PubMed] [Google Scholar]
- [11].Ariola FS, Li Z, Cornejo C, Bittman R, Heikal AA, Biophys. J 2009, 96, 2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lund FW, Lomholt MA, Solanko LM, Bittman R, Wüstner D, BMC Biophys. 2012, 5, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sezgin E, Levental I, Grzybek M, Schwarzmann G, Mueller V, Honigmann A, Belov VN, Eggeling C, Coskun U, Simons K, Schwille P, Biochim. Biophys. Acta 2012, 1818, 1777. [DOI] [PubMed] [Google Scholar]
- [14].Wüstner D, Solanko L, Sokol E, Garvik O, Li Z, Bittman R, Korte T, Herrmann A, Chem. Phys. Lipids 2011, 164, 221. [DOI] [PubMed] [Google Scholar]
- [15].For a review of Suzuki cross-coupling, see:; Miyaura N, Suzuki A, Chem. Rev 1995, 95, 2457. [Google Scholar]
- [16].For a review of Liebeskind-Srogl cross-coupling, see:; Prokopcova H, Kappe CO, Angew. Chem., Int. Ed 2009, 48, 2276. [DOI] [PubMed] [Google Scholar]
- [17].Visbal G, Alvarez-Aular A, Synth. Commun 2004, 34, 533. [Google Scholar]
- [18].For a review of the B-alkyl Suzuki cross-coupling reaction, see:; Chemler SR, Trauner D, Danishefsky SJ, Angew. Chem., Int. Ed 2001, 40, 4544. [DOI] [PubMed] [Google Scholar]
- [19].Giannis A, Heretsch P, Sarli V, Stößel A, Angew. Chem., Int. Ed 2009, 48, 7911. [DOI] [PubMed] [Google Scholar]
- [20].Lager E, Liu J, Aguilar-Aguilar A, Tang BZ, Pena-Cabrera E, J. Org. Chem 2009, 74, 2053. [DOI] [PubMed] [Google Scholar]
- [21].Han J, Gonzalez O, Aguilar-Aguilar A, Peña-Cabrera E, Burgess K, Org. Biomol. Chem 2009, 7, 34. [DOI] [PubMed] [Google Scholar]
- [22].The formation of the corresponding phenol was also observed, but it was avoided by degassing with nitrogen.
- [23].Billingsley KL, Barder TE, Buchwald SL, Angew. Chem., Int. Ed 2007, 46, 5359. [DOI] [PubMed] [Google Scholar]
- [24].Goud TV, Tutar A, Biellmann J-F, Tetrahedron 2006, 62, 5084. [Google Scholar]
- [25].Boric acid has been used for deprotection of the THP group; see:; Gigg J, Gigg R, J. Chem. Soc . C 1967, 431. [Google Scholar]; We found that the Liebeskind-Srogl cross coupling between 14 and the tributyltin counterpart of 13 did not lead to the removal of the THP group.
- [26].Li Z, Bittman R, J. Org. Chem 2007, 72, 8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jiao L, Pang W, Zhou J, Wei Y, Mu X, Bai G, Hao E, J. Org. Chem 2011, 76, 9988. [DOI] [PubMed] [Google Scholar]
- [28].Hayashi Y, Yamaguchi S, Cha WY, Kim D, Shinokubo H, Org. Lett 2011, 13, 2992. [DOI] [PubMed] [Google Scholar]
- [29].Rihn S, Retailleau P, Bugsaliewicz N, Nicola AD, Ziessel R, Tetrahedron Lett. 2009, 50, 7008. [Google Scholar]
- [30].Zhang X, Yu H, Xiao Y, J. Org. Chem 2012, 77, 669. [DOI] [PubMed] [Google Scholar]
- [31].Hölttä-Vuori M, Uronen RL, Repakova J, Salonen E, Vattulainen I, Panula P, Li Z, Bittman R, E. Ikonen Traffic 2008, 9, 1839. [DOI] [PubMed] [Google Scholar]
- [32].Severs NJ, “Cholesterol cytochemistry in cell biology and disease,” in Subcellular Biochemistry, Vol. 28, Bittman R, ed., Plenum Press, 1997, pp 477–505. [DOI] [PubMed] [Google Scholar]
- [33].Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW J. Biol. Chem 2002, 277, 49982. [DOI] [PubMed] [Google Scholar]
- [34].Williams ATR, Winfield SA, Miller JN, Analyst 1983, 108, 1067. [Google Scholar]
- [35].Eignerova B, Dracinsky M, Kotora M, Eur. J. Org. Chem 2008, 4493. [Google Scholar]
- [36].18 and 19 have the same Rf value on TLC (hexane/EtOAc 5:1), but 18 is red, and 19 is purple.
- [37].18 and 21 have the same Rf value on TLC (hexane/EtOAc 4:1), but 18 is red, and 21 is violet.
- [38].21 and 22 have the same Rf value on TLC (hexane/EtOAc 4:1) as well as the same color.
- [39].Havel RJ, Eder HA, Bragdon JH J. Clin. Invest 1955, 34, 1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.