

Abstract
There is a growing interest for viral vector-free chimeric antigen receptor (CAR) T-cells due to its ability to kill cancer cells without adverse side effects. A potential avenue for manufacturing viral-vector free CAR T-cells is to utilize mRNA electroporation. One of the major concerns with mRNA electroporated CAR T-cells is the shorter cytotoxic lifespan of a few days, which is insufficient or not ideal for therapy. To better understand this issue and develop a potential solution, this study focused on examining the translation of electroporated mRNA to CAR molecules, time dependent degradation of CAR molecules and cytotoxicity produced by CAR T-cells on cancer cells. It was found that the initial expression of CAR molecules dictates the cytotoxicity. Initial CAR expression could be controlled by the experimental parameters such as electroporation time and mRNA concentration in the electroporation buffer. Experiments were carried out using a novel two-step electroporation that allows for controlled and uniform transfection of T-cells. These technical advancements and subsequent findings could provide a viable path for producing CAR T-cells with longer cytotoxic lifespans.
Graphical Abstract
Introduction
Cell-based therapies, especially Chimeric Antigen Receptor T-cells (CAR T-cells), have shown great potential as a therapeutic strategy for cancers such as Acute Lymphoblastic Leukemia (ALL) [1]. The United States (US) Food and Drug Administration (FDA) has recently approved CAR T-cells for treating ALL patients [2]. CAR T-cells are ex vivo-engineered T-cells that express CAR molecules on their surfaces [2–3]. The CAR molecule is an antibody-derived binding domain that selectively recognizes the target tumor cell’s surface antigen and is directly linked to a signaling domain (such as TCRξ) to provide T-cell stimulation [2–3]. Once CAR T-cells are infused back into the patient, conjugation of the antigen (from the tumor cell) and CAR molecules triggers T-cell mediated cytotoxicity and kills the tumor cell. Currently, almost all CAR gene transfer into T-cells is achieved using a retrovirus or lentivirus. The use of viruses could be risky because the insertion mutation utilized to induce the immune response risks the development of tumorigenesis [4–6]. Recent reports indicate serious adverse events such as cytokine release syndrome, neurological toxicity, and on-target/off-tumor toxicity after administering CAR T-cells engineered by viral gene transfer [4–6]. In addition, when viral DNA is integrated into the host cell genome, it could create a potential for insertional mutagenesis [4–6].
mRNA-based CAR T-cell manufacturing could address these issues as it employs viral vector-free CAR T-cell technology that can produce transient expression of CARs [7–8]. Also, clinical-grade mRNA production costs less than viral vector-based CAR T-cells. However, the issue with CAR T-cells manufactured with mRNA is the significantly shorter lifespan of the cytotoxicity [7–9]. Typically, transfected mRNA molecules transiently express CAR molecules and produce cytotoxicity for up to one week, which is not ideal for therapy [7–9]. From a therapeutic standpoint, the patient would require very frequent infusions of CAR T-cells, which result in unpredictable outcomes, high cost, and lengthy clinical visits. Ideally, to address the personalized therapeutic needs, a CAR T-cell with significantly longer cytotoxicity than a week (or preferably tunable cytotoxicity) is needed. If this problem is not addressed, the therapeutic use of CAR T-cells manufactured using mRNA will not become a reality. The objective of this study was to investigate potential avenues for increasing cytotoxic lifespans of mRNA-based CAR T-cells or make the CAR T-cells more potent for cancer cells. Our study was based on the recent results reported by others on a potential relationship between mRNA concentration in T-cells after transfection, corresponding CAR expression, and the lifespan of cytotoxicity toward target cancer cells [9, 28].
Electroporation is commonly used to transfect T-cells with mRNA molecules [7–8]. For example, Birkholtz et al., have demonstrated that T-cells electroporated with mRNA molecules produced CAR molecules targeting ErbB2 and CEA immunoreceptors. These CAR T-cells were able to selectively kill cancer cells that express ErbB2 and CEA antigens [9]. The cell electroporation can be performed using either DC electric potential pulses or AC potentials [7, 8, 10]. When DC pulses are used, T-cells are exposed to short, external DC electric field pulses (< 1ms). In contrast, low frequency AC electric potentials (<1000 Hz) are also used to transfect cells by electroporation. Fundamentally, AC or DC electric fields induce transmembrane electric fields in the T-cell membranes. When induced transmembrane electric field values exceed the threshold electric potential value of 1V, reversible pores are formed on T-cell membranes [11]. mRNA molecules suspended in the cell buffer flow into the T-cells through pores by Brownian motion or a combination of Brownian motion and electrophoresis [12]. Traditional two macro electrode-based electroporators or miniaturized versions of two-microelectrode based electroporation devices have been utilized in T-cell transfection experiments. However, current electroporation methods used for T-cell transfection cannot be used for experiments in this study. Below, we provide a summary of the technical issues.
In the traditional electroporation devices, cell sample is pipetted between electrodes, and generally, cells are randomly dispersed between electrodes. Electrically, cell sample is treated as non-conductive spheres suspended in a conductive buffer solution. When an electric potential is applied between electrodes, the distribution of electric potential in the cell sample is perturbed by the presence of non-conductive spheres, producing higher and lower electric potentials based on the cell density and the distribution of cells between the electrodes. The local electric potentials produce induced transmembrane potential in the cell membranes, which control the important electroporation parameters, such as pore formation, diameter, and the number of pores. Uneven distribution of induced electric potentials in the cell sample produces uncontrollable and untunable levels of mRNA levels in the cell sample [13–15]. Another issue with the uneven induced transmembrane potentials is the low transfection efficiency and cell viability values. Studies have reported that transfection efficiency (percentage transfected in the sample) with current electroporation devices was about 40% or less [9]. If the induced transmembrane potential is greater than 1V (or electric field of 108 V/m across 10 nm cell membrane), then there is significant, irreversible damage to the cell membrane resulting cell death [11]. Poor cell viability values of approximately 40% were reported for the T-cells after electroporation [9]. Number of other electroporation approaches, including single-cell electroporation and flow through electroporation methods in microfluidic channels have also been investigated. Briefly, in single-cell electroporation, cells flow as single files in microfluidics channels, and single cells are electroporated when they pass through electrode(s) [16–17]. Buie et al. have developed flow through microfluidics device with tapered microfluidics channel to determine the best electroporation conditions (external electric fields needed) for a cell sample. The tapered microfluidics channel produces a range of electric field values to accommodate various cell types. Depending on the location of the channel, where electroporation takes place, optimum electric field value can be experimentally determined [17]. Although these techniques are excellent additions to the electroporation, there are number of technical issues that need to answer before application in CAR T-cell manufacturing. Briefly, these issues are: transfection assays may take a few hours and lengthy exposure of T-cells and mRNA molecules to room temperature could lower the viability of cells and degrade mRNA molecules. Transfection of T-cells with tunable levels of mRNA is difficult. In addition, cell loss and contamination could also occur during infusion into the microfluidics device [16,17].
Susil et al. utilized finite element modeling and calculated the electric fields in the vicinities of the cell strands (or single files of cells) and sheets of cells. The study demonstrated that orientation of cell strands or sheets with respect to the direction of an external electric field is critical to the production of induced transmembrane potential in cells [13]. Moreover, cell-strands that are located parallel to the external electric field direction significantly disturb the local electric fields near cells and suppress the induced transmembrane potential values, which is also known as shadowing effect. As a result, significant variations of induced transmembrane potential values between individual cells take place. In contrast, external electric fields applied in the perpendicular direction to the cell strands potentiate the induced membrane potential of cells. Similar results for the induced transmembrane electric fields in the suspensions of cells were reported by Pavlin et al., and Mistani et al. [14−15]. Therefore, producing cell strands or single-file of cells per electrode provides a potential avenue for the production of controlled and tunable transfection of T-cells with mRNA. Based on the successful early results reported by others, as our first step, we designed and utilized interdigitated microelectrodes manufactured on glass substrates to produce single-file T-cells patterns per electrode pair (one single file T-cell pattern per pair of electrodes) and subsequently electroporation of patterned T-cells by AC or DC electric fields applied perpendicular to the single file cells. In addition, for tunable and uniform electroporation of all the cells in single file cell patterns, single file cell patterns should locate in the uniform electric field regions produced by the interdigitated microelectrodes. Typically, cells patterns should be produced between each electrode pair to expose the cells to uniform electric fields. The rationale for two-step electroporation was that cell patterning was needed to produce uniform and tunable electric fields across every single T-cell that is in the single file, eliminating the adverse effects outlined above. Additionally, equal, uniform, and easily controllable DC pulse electric fields or AC electric fields could be produced across all the cells that are in the single file patterns, enabling the ability to control the transfection level. Also, there is no flow of cell sample to the microfluidics channel, which eliminate the degradation of cells and mRNA molecules. After developing the two-electroporation device, we examined how initial mRNA concentration, corresponding CAR expression in T-cells, and the cytotoxic lifespan of the manufactured CAR T-cells are inter-related. Finally, a compartmental model of the T-cell cytoplasm was developed to theoretically study the translation of mRNA into CAR molecules. Results from theoretical study was used to understand the experimental results and to make predictions about the characteristics of mRNA-based CAR T-cells.
Experimental/Methods Section
Interdigitated Electrodes
Microscale interdigitated electrodes were manufactured on glass substrates. Traditional micro-fabrication methods such as photolithography, metal evaporation, lift-off, and dicing were utilized. Detailed protocols were discussed elsewhere [18].
Electroporation Device
First, microelectrodes with appropriate dimensions were manufactured. Detailed discussion about the dimensions of the electrodes will be discussed in the Results and Discussion. Then Epoxy enclosures (approximate height 1 mm) with open top were produced to concentrate the cell sample over the electrode array. No microfluidics pumps or syringes were used in the experiments.
Finite Element Modeling
(a). Design electrodes
COMSOL Multiphysics 5.3a (Burlington, MA) was used to design electrodes. To setup COMSOL simulations, briefly, interdigitated electrodes were drawn to scale using AutoCAD (Autodesk, San Rafael, CA) software, imported into COMSOL, and analyzed with the AC/DC electric current (EC) module. The frequency domain was used in simulations to incorporate the varying frequency studies. In all the analyses, buffer properties were set for 0.01x PBS (conductivity (σ) = 0.03 S/m, relative dielectric constant (εr) = 72). The electric potential, electric field, and electric field gradients were calculated and utilized to design the width of the electrodes and the spacing between individual electrodes.
(b). Transmembrane potential
AC/DC module of COMSOL were developed to calculate the induced transmembrane potentials of T-cells. We used the shell model technique for this calculation, which allowed us to mimic the cell membrane. A cell membrane thickness of 0.5 nm was used in all calculations. Electric conductivity (σ) of 1.1 S/m and dielectric constant value (εr) of 71 for the T-cell cytoplasm were used. σ and εr values for cell membrane were 5×10−8 S/m and 13.55, respectively. An initial/resting transmembrane voltage of −70 mV was utilized across T-cell membranes.
T-cell Isolation, Culturing K-562, and SUP-B15 Cells
These procedures were carried out according to previously published protocols [18].
Culturing Jurkat Cells
The Jurkat cell line was received from Dr. Dorsam’s group at North Dakota State University. Jurkat cells were cultured using a growth medium containing 89% RPMI 1640, 10% Fetal Bovine Serum (FBS), and 1% Penicillin/Streptomycin in an incubator (5% CO2 and 37°C). Cells were passaged every two days during their log phase (at between 80%–90% confluence) and used in experiments. To prepare cell samples for experiments, first cell sample was spun down at 500xg for 5 min. Then the cell sample was suspended in the electroporation buffer.
Preparation of the Electroporation Buffer
The electroporation buffer was 0.01x PBS with a conductivity of σ = 0.03 S/m, supplemented with sucrose to obtain an osmolarity of 180mOsm/L. To make the buffer, one part (by volume) of RNAse free 10x PBS (pH 7.4; Invitrogen, Carlsbad, CA) was mixed with 999 parts of RNAse free DI water (Invitrogen). Then, the osmolarity of the solution was adjusted to 180 mOsm/L using RNAse free D(+) Sucrose.
Electroporation Experiments
Upon isolation of T-cells from whole blood, they were suspended in StemCells®Easysep buffer (StemCell technologies, Cambridge, MA). T-cells were then spun down at 1200 rpm and re-suspended in electroporation buffer with a cell density of 1×106 cells/mL. mRNA molecules that produce EGFP were purchased (Biolegend, San Diego, CA) and CAR molecules against the CD19 antigen molecules on the ALL cells were commercially manufactured (G&P Biosciences, Santa Clara, CA). The fourth generation of CAR design was used to synthesis mRNA as it demonstrated complete remission for two-thirds of leukemia patients in the initial clinical trials. About 100 μL of the cell sample was added to the electrode, and experiments were conducted (patterning and electroporation) with the following conditions:
P-DC: Cell patterned with AC (sinusoidal) voltage (1 Vpp and 100 kHz, and 1 Vpp and 10 kHz) and transfected with 3 (0.5 ms per pulse) DC pulses of 9 Vp.
P-AC: Cell patterned with AC (sinusoidal) voltage (1 Vpp and 100 kHz, and 1 Vpp and 10 kHz) and transfected with sinusoidal voltage (8 Vpp and 100 Hz) for 150 ms.
NP- DC: Transfected with 3 DC pulses (0.5 ms per pulse) of 9 Vp and no-cell patterns were produced.
NP-AC: Transfected with sinusoidal voltage (8 Vpp and 100 Hz) for 150 ms and no cell patterns were produced.
Analysis of CAR (targeting CD19 antigen) expression
mRNA concentrations of 50, 25, and 5 μg/mL with approximately 1×105 T-cells for each experiment were used in experiments. For each mRNA concentration, after patterning and electroporation experiments, about 3.9 mL of cell culture medium was added to the sample and divided into 10 equal volumes, pipetted to the 24-well plate (Falcon® 24-well Clear Flat Bottom, Corning, NY) and incubated at 37°C and 5% CO2. The cell culture medium was composed of Iscove’s Modified Dulbecco’s Medium (IMDM) with 20% Fetal Bovine Serum (FBS; ATCC, Manassas, VA) by volume and 0.5% IL2 (by volume; Miltenyi Biotec, Cambridge, MA). To evaluate the CD19 expression, a sample from a well was collected, mixed with 100 nM of Biotin-SP (long spacer) AffiniPure F(ab’)2 Fragment Goat Anti-Mouse IgG (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) and incubated on ice (4°C) for 25 minutes. Then the cells were washed with PBS, and 200 nM of FITC-Streptavidin (Biolegend, San Diego, CA) was added, and the solution was mixed thoroughly. Next, the sample was incubated at room temperature for 5 minutes and then the cells were washed with PBS, and 100 nM of propidium iodide (PI; excitation: 493 nm, emission 636 nm; Thermofisher, Waltham, MA) was added to analyze cell viability. Flow cytometry (fluorescence intensity vs. cell number) was performed, and the average fluorescence intensity and the cell viability of each sample were analyzed.
Cytotoxicity Analysis
T-cells (~1×105) were electroporated (methods described above) with 50 μg/mL of mRNA. After electroporation, about 3.9 mL of medium (as described in the CAR analysis) was added and pipetted to 10 wells (equal volumes) in a 24 well plate. Then, 100 μL of the sample containing about 25000 SUP B-15 cells was added to each well, and the cell mixture was incubated at 37°C with 5% CO2 (T cell: SUP B-15 ratio = 4:1). The cells were taken out of the wells at specific time points, and each cell quantity was counted using flow cytometry. We used the forward-scattering and side-scattering feature of flow cytometry to count the SUP B-15 cell and T-cell populations. A similar procedure was used to analyze the cytotoxicity toward K-562 cells.
Mathematical modeling
A compartmental model developed for the externally injected (by electroporation) mRNA to CAR (targeting CD19) translation in the cytoplasm of a T-cell.
Definitions
R(t) = Externally injected mRNA concentration in a T-cell at time t
f(t) = Concentration of mRNA molecules in the cytoplasm of a T-cell injected by electroporation
kd1 = Rate of mRNA degradation in the cytoplasm of a T-cell
kd2 = Rate of CAR degradation in a T-cell
kd3 = Rate of CAR synthesis
p(t) = Concentration of CAR in a T-cell at time t
R0 = Initial concentration (t=0) of mRNA in the cytoplasm
Assumptions
Fundamental Equations
| (1) |
| (2) |
| (3) |
| (4) |
Solutions
| (5) |
| (6) |
Values
Results and Discussion
The electroporation devices we used in experiments had interdigitated microelectrodes manufactured on glass substrates. Interdigitated microelectrodes are parallel electrode pairs that produce largely uniform electric fields, but near the electrodes, there can be some non-uniformity of electric fields or electric field gradient (∇(E2)). Large number of biomedical assays, including cell separation, molecular concentration, and detection employed the dielectrophoretic (DEP) force produced using interdigitated electrodes [19–20]. The magnitude of the DEP force is proportional to ∇(E2). The DEP force is a valuable tool to develop biological assays because it can easily be produced using the microelectrodes, and the direction of the force is tunable to be toward the electrodes (attractive DEP force) or away from the electrodes (repulsive DEP force). For example, Gupta et al. used DEP force for the separation of circulating tumor cells from blood samples, while Velmanickam et al. demonstrated the efficient trapping of micro-RNA molecules on interdigitated electrodes [19–20]. A recent study by Guo and Zhu has demonstrated the successful use of DEP force to produce single-cell patterns on electrodes [21]. We used DEP force to pattern T-cells as single files. Ideally, as outlined in previous paragraphs, for controlled and tunable transfection of T-cells with mRNA, it is required to have one single file T-cell pattern per electrode pair. Moreover, as the middle of each electrode pair has uniform electric field, T-cell pattern needed to produce in the middle of the electrode pair.
We studied the possibility of using DEP force to produce one single-file T-cell pattern per electrode pair. Using the reported values for dielectric properties of cells such as the cytoplasmic conductivity and dielectric constant, and applied potential value of about 1Vpp, we estimated the DEP force to be about 1 nN on T-cells. We have found that at least 1 nN DEP force is required to manipulate T-cells and produce single file T-cell patterns. Inglis et al. have used smaller magnetophoretic forces (~10 pN) to separate leukocytes from whole blood cells. Moreover, the magnetophoretic force was used to deflect the leukocytes from the main flow channel to a side-channel [22]. In comparison, we found that stronger forces (nN range) are needed to hold cells between electrodes and produce T-cell patterns. Prior to experiments, we directly pipetted the cell sample that was suspended in the electroporation buffer on the electrodes. Subsequently, we studied the frequency dependent DEP force on T-cells by manually changing the frequency from 1 kHz to 1 MHz and the magnitude of the potential from .1–5 Vpp. We manually observed (using bright field or fluorescence microscope) the attraction (attractive DEP force) or repulsion (repulsive DEP force) of T-cells by the DEP force. We found that at about 50 kHz, DEP force changes from repulsive to attractive. Strongest attractive and repulsive DEP forces were produced at 100 kHz and 10 kHz, respectively. After examining the frequency dependent DEP forces of T-cells, we used a combination of attractive and repulsive DEP forces to produce T-cells patterns. First, attractive DEP force (produced at 100 kHz) was used to pull the cells toward electrodes, and then repulsive DEP force (produced at 10 kHz) was used to push cells away from electrodes to the middle region between the electrodes. It was found that the use of attractive and repulsive forces was an efficient way to produce T-cell patterns with almost all T-cells in the sample. Repulsive DEP force alone could also be used but it was difficult to produce single file T-cells patterns with almost all T-cells in the sample. The T-cell viability after exposing to DEP forces was over 95%. The viability analysis was performed using commercially available Live/Dead assay kits (Life technologies, Carlsbad, CA). T-cell patterning using attractive and repulsive DEP forces takes about 3 min to complete.
Ideally, as discussed earlier, to minimize the shadowing effect outlined previously, it requires one single file T-cell pattern per pair of interdigitated electrodes. In addition, to expose every single-cell to tunable and uniform electric fields during electroporation, the single file cell pattern should be located between the microelectrodes. During the initial experiments, we have found that careful engineering of DEP force on the cells is required to produce desired cell patterns. For example, DEP force is directly proportional to ∇(E2). The magnitude of ∇(E2) depends on the geometrical parameters such as spacing between electrodes and width of the electrodes, and the magnitude of the applied potential. Moreover, the spacing between electrodes and width of the electrodes control the locations of highest and lowest ∇(E2), and the magnitude of the applied electric potential contributes to the magnitude of ∇(E2). To further understand how these variables contribute to produce desired cell patterns for electroporation experiments, we developed series of finite element simulations and studied impact of those variables. Figure 1 summarizes our findings. We have calculated ∇(E2) in the Figure 1(a, c, e) and Figure S1 (supplementary figure). Approximate locations of the electrodes are indicated by solid orange lines. The circles with broken white lines show the locations of cells prior to applying attractive DEP force. White arrows show the travel path of a cell when attractive DEP is applied. The red arrows show the potential travel path of a cell when a repulsive DEP is applied. The circles with broken yellow lines show the locations where cell patterns are produced. First, we studied the cell-pattern formation when spacing between electrodes is significantly greater than the diameter of the cells. Cell diameter of 5 μm and spacing between electrodes value of 10 μm were used in the calculation. Figure 1(a) shows the calculated ∇(E2) values for the applied potential of .1 Vpp. Positive DEP force attracts the cells towards the highest ∇(E2) or to the electrode edges [23]. When the DEP force changed from attractive to repulsive, the cells concentrate on the locations that have the lowest ∇(E2) [23]. Since the spacing between electrodes is significantly greater than the diameter of the cells, multiple single file cell patterns are produced. In addition, some cells can concentrate on the electrodes. The cells located on the electrodes may not electroporate to the same degree as the other cells located between electrodes. It is because cells located on the electrodes expose to low electric fields than other cells. Figure 1(b) shows an example of the cell patterns produced for the conditions discussed in Figure 1(a). We have used the electric potential of .1 Vpp and spacing between electrodes and the width of the electrodes are about 2–3 times of the cell diameter. Since there are conglomerate cell patterns are produced, as we explained earlier, these electrode dimensions and the potential are not suitable for electroporation experiments.
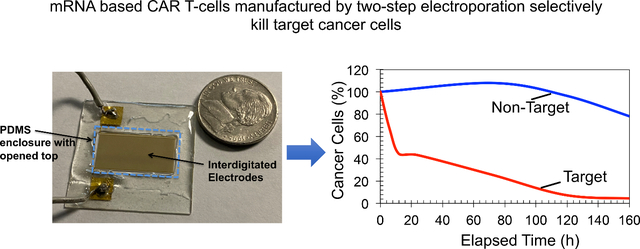
Figure 1:
Design of interdigitated electrodes for effective single-file T-cell patterning needed for two-step electroporation. (a and c) Calculated electric field gradients (∇E2) produced by high (Figure 1(c)) and low (Figure 1(a)) electric potentials, and incorrect electrode dimensions. No effective single-file cell patterns could be produced for these cases. (b and d) Fluorescence images of the cell patterns produced by the electrodes discussed in (a) and (c). (e) Calculated (∇E2) for properly designed electrode width and spacing between electrodes as well as correct electric potential values. Note that this electrode design and potential could produce only one single-file cell patterns between each electrode pair. (f) Fluorescence image of suitable cell patterns for two-step electroporation. The device design discussed in Figure 1(e) was used in experiments. (g) Picture of the patterned single-file primary T-cells using the design with proper dimensions of electrodes and electric potential values. Scale bars are 5 μm in figures 1(a, c, and e), 10 μm in figures 1(b, d and f) and 20 μm in figure 1(g). Cells were stained with Calcein for easy visualization.
Figure 1(c) shows the ∇(E2) produced by higher applied electric potential (5 Vpp) than the previous case (.1Vpp) and the spacing of the electrodes are approximately same size as the cell diameter and width of the electrodes (10 μm) are larger than the cell diameter (5 μm). For this electric potential, even with proper electrode dimensions, single file cell patterns are not useful for expected tunable and uniform electroporation as repulsive DEP forces push the cells away in the vertical direction from the electrodes. Figure 1(d) shows the cell patterns produced under the conditions discussed in Figure 1(c). Note that each cell could be different vertical position above the electrodes, and each cell exposes to different electric field values during electroporation, which is not suitable for the proposed tunable and uniform electroporation. Figure 1(e) illustrates ideal conditions needed to produce desired single-cell pattern between electrodes. We have used the electric potential of 1 Vpp and electrode dimensions (width and spacing between electrodes were about 5–7 μm) are approximately equal to the cell diameter (5 μm). Electric potential value and electrode dimensions are sufficient to produce the largest ∇(E2) and attractive DEP force near electrodes and smallest ∇(E2) or repulsive DEP force between electrodes. Figure 1(f) shows a picture of the single file cell patterns produced using the conditions discussed in Figure 1(e). In addition, we have also investigated another case, where an electric potential of 1 Vpp is applied, but the width of the electrodes (15 μm) is larger than the cell diameter (5 μm) and spacing between the electrodes are approximately same size as the cell diameter (Figure S1). As indicated in the Figure S1, when repulsive DEP force is applied, some cells could go over the electrodes, and other cells concentrate between electrodes. As described earlier, the cells located on the electrodes may not electroporate to the same degree as the other cells located between electrodes. Therefore, this design is also not suitable for producing the desired single-file cell pattern. We did not experimentally produce any cell patterns with these conditions. In the experiments shown in Figures 1(b, d, and f), we used cultured Jurkat cells, as they closely resemble T-cells. After designing electrodes and optimizing the electric potentials, we switched to using primary T-cells isolated from whole blood samples and performed T-cell patterning experiments (Figure 1(g)). The single file T-cell patterns illustrated in figure 1(g) contain a single file of T-cells between individual electrodes. Cell samples with 106 cells/mL were used in the cell patterns shown in Figure 1(g).
We found that, depending on the cell diameter, proper dimensions for spacing between individual electrodes, width of the electrodes, and the electric potential, is needed to produce desired single file cell patterns. Moreover, the spacing between electrodes and the width of the electrodes needed to be approximately 1.5 times the cell diameter. For example, an electrode spacing of 10 μm and an electrode width of 10 μm were needed to pattern T-cells (~7 μm) in the electrode array. Selection of magnitude of the electric potential is based on the cell diameter, cell-type and conductivity of the buffer [19–21]. Generally, we have found that, for smaller cells (e.g., T-cells with diameter ~7 μm) suspended in low conductivity buffer (e.g., 0.03 S/m), 1 Vpp potential is sufficient.
Once the cells are patterned as single files between electrodes, electroporation was used to transfect cells with mRNA molecules. DeBruin and Krassowska studied single-cell electroporation using an electromagnetic model and explained the fundamental theory behind the pore formation on cell membranes and its limits [24]. It was demonstrated that a threshold value of 1 V for the induced transmembrane potential is needed to initiate the electroporation [24]. Above 1 V, the resealing capability decreases, and subsequently, significant damages to the cell take place. This evidence demonstrates the importance of the induced transmembrane potentials. Moreover, nearly 1V is needed to electroporate cells without cell damage or harming cell viability. We calculated the induced transmembrane potentials on patterned cells (with DEP force) and randomly placed cells (no DEP force used).
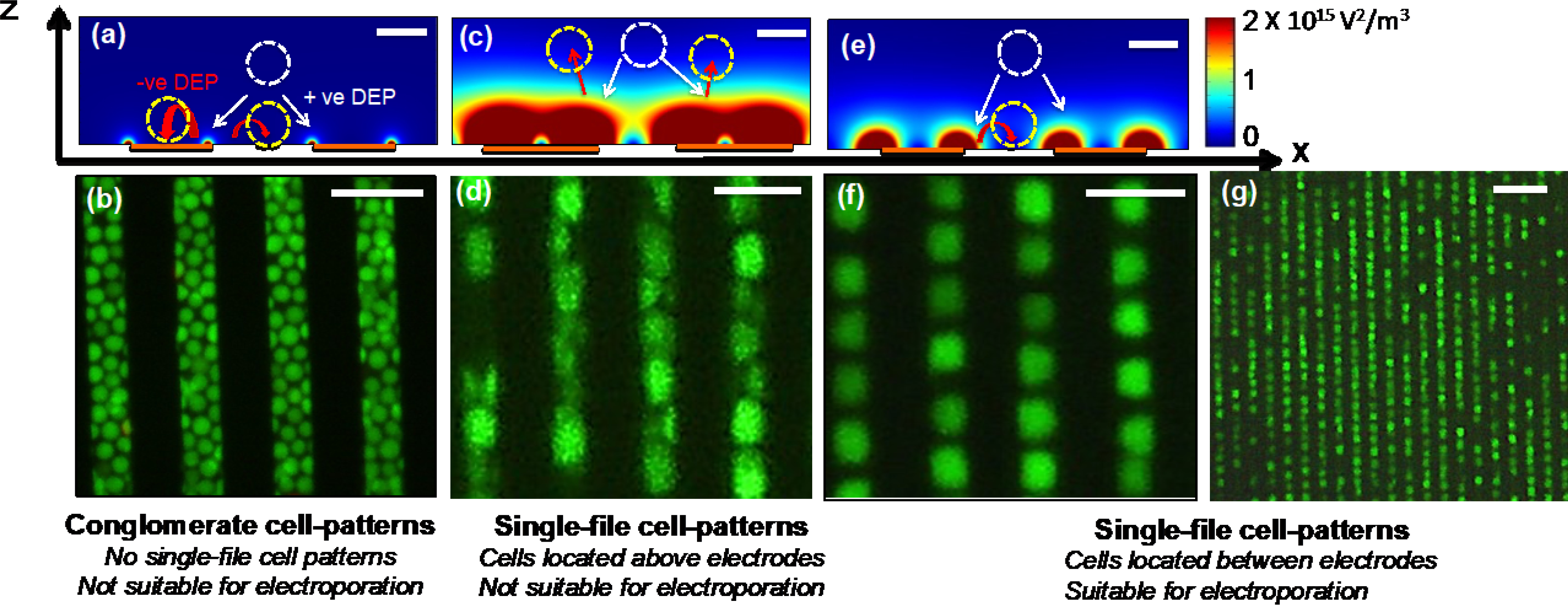
Figure 2(a) shows the isometric view of randomly placed cells, including the locations of each cell and the calculated induced transmembrane electric potential values. Note that some cells are placed on the electrodes (e.g., A, B, and C), and some other cells are located between electrodes (e.g., D and E). For further comparison, in the figure right below Figure 2(a), we calculated the induced transmembrane electric potential values of the randomly placed cells. Figures 2(b and c) illustrate cell locations and the calculated induced transmembrane electric potentials of two single file cell patterns. The figure right below figures 2(b and c) shows the induced transmembrane electric potentials of each cell. Next, we extracted the maximum values of the induced transmembrane electric potential values of each cell. Also, we extended our analysis and calculated the maximum induced transmembrane electric potential values of the cells that were placed in traditional cuvette-based electroporation devices that use macro electrodes (Figure 2(d)). The largest variation in the maximum induced transmembrane electric potential of 1.40±1.2 V was calculated for the macro electrode-based cuvette electroporation. 1V induced transmembrane potential with the smallest variation was calculated for patterned cells in microelectrodes (1±0.05 V), and randomly placed cells in the microelectrodes had an induced transmembrane potential of 1.05±0.2 V. By comparison of calculated induced membrane electric potential values of each case, patterned cells with micro-electrodes have almost identical induced transmembrane potential values. Therefore, all the patterned T-cells could uniformly transfect with mRNA. In addition, mRNA levels in T-cells could be varied by simply varying the external electric potential.
Figure 2:
Calculated induced potential values of the patterned and randomly placed cells on interdigitated electrodes. The diameter of each T-cell was 7μm. (a) Isometric view shows the locations of cells with respect to electrodes of the randomly placed cells and their induced membrane potential values. The external electric potential was applied between interdigitated electrodes. The figure shows only a section of interdigitated electrodes. The figure right below (a) shows 2-D view of the calculated induced membrane potential values in (a). (b and c) Calculated induced potential values of patterned T-cells between electrodes. Moreover, (b) and (c) show two configurations of T-cell patterns. Figures right below show the 2-D view of the calculated induced membrane potentials. (d) Numerical values of calculated induced membrane potential values of patterned, randomly placed and cells placed on traditional cuvettes. Moreover, cuvettes represent the traditional macro electrode-based electroporation devices. (e) Calculated induced electric potential values of a cell placed in micro-and macro-electrodes. The red arrows indicate the direction of the electric field. The legend shows the conversion of colors to numerical values for (a), (b), (c) and (e).
To further understand the potential benefits of the electroporation of cells with micro- vs. macro-electrodes, we further extended our FEM simulation and calculated induced transmembrane electric potential values of cells placed between micro- and macro-electrodes (Figure 2(e)). Typically, dimensions of the macro electrodes (in cm) are significantly greater than the diameter of the cells (in μm), and microelectrodes and cells have dimensions in μm. Therefore, induced transmembrane potentials are produced locally in the cell membrane by the microelectrodes, while macro electrodes induce electric potentials in the entire half-spherical space of the cell (Figure 2(e)). In our earlier study, when we utilized electroporation with microelectrodes, we reported very high cell viability values (~100%) [35]. In addition, as the localized induced transmembrane potential of 1 V produced by microelectrodes does not cause significant irreversible cell damage, longer electroporation times could be used for the cells in microelectrodes. Longer electroporation times could be useful for transfecting cells with high or tunable levels of mRNA molecules. In contrast, macro-electrodes induce electric potentials with considerable variability, and therefore low cell viability and uneven (or low) transfection efficiency is expected.
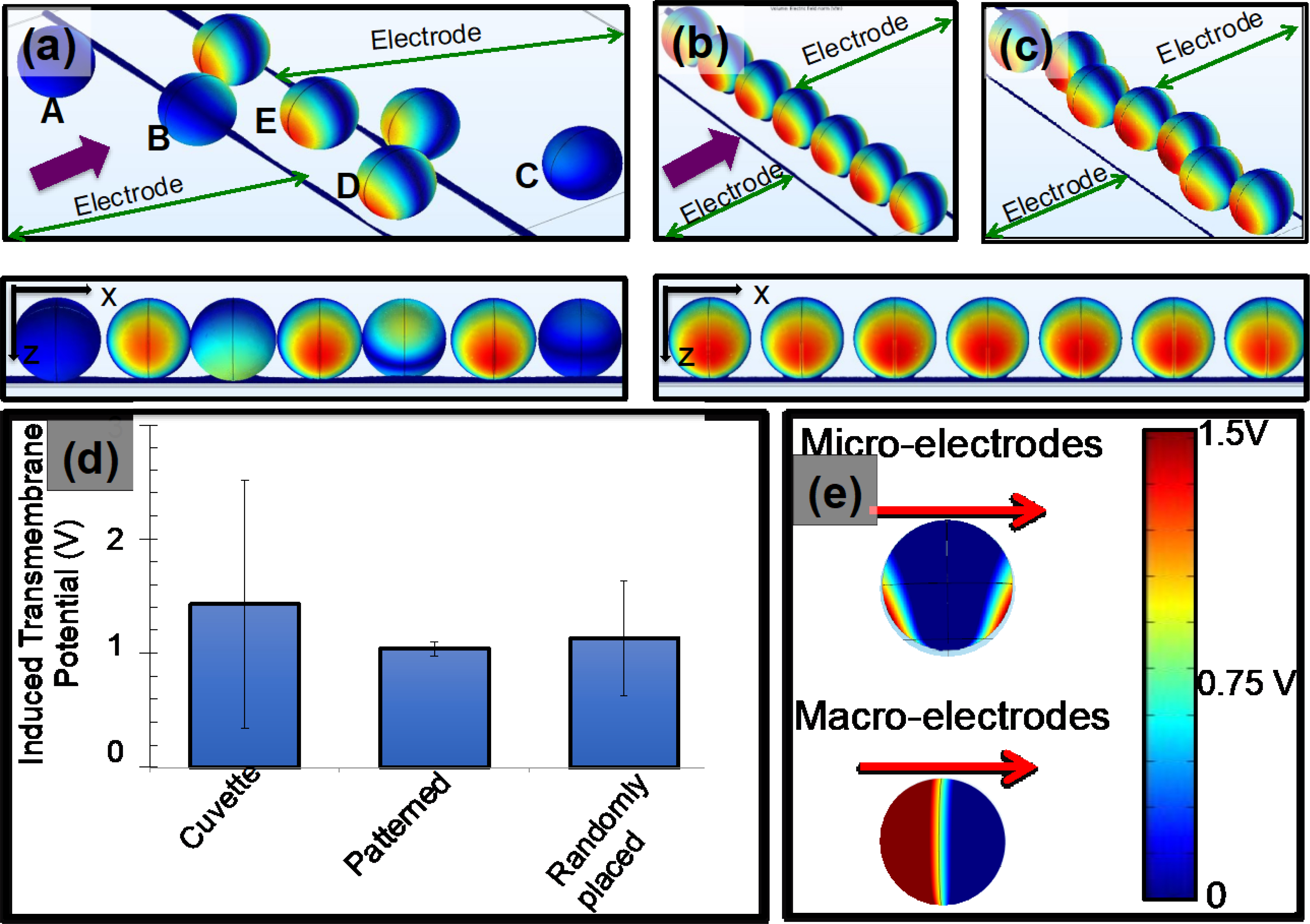
Several methods have been reported for electroporation, and the vast majority of these methods use DC pulses. Zhao at al. used DC electric fields produced by macro electrodes to electroporate T-cells and manufacture CAR T-cells [25]. AC electric fields with low frequencies (< 100 Hz) have also been used in electroporation [10]. Zhan et al. have demonstrated that low-frequency electric fields result in reduced damage to cells when compared with DC pulse electric fields [10]. Additionally, Chang et al. demonstrated that electric fields produced by combined AC potentials and DC pulses produce a sonication effect on molecules and improve the transport of molecules into cells [26]. We employed AC and DC electric fields to in the electroporation experiments. Figure 3 show the proposed two-step electroporation method. Figure 3(a) shows a picture of the device that we used for experiments. Figure 3(b) shows the fluorescence image (excitation: 493 nm, emission: 636 nm) of the patterned T-cells. Note there was no visible fluorescence was recorded after making cell patterns. Figure 3(c) illustrates the image of the cell sample after electroporation by DC pulses. T-cells were transfected with commercially available PI. Rectangle drawn with broken line shows a patterned and transfected single-file of T-cells. After transfection, cells were collected by pipetting and stained with Calcein molecules (Thermofisher) and re-patterned. The Calcein molecules enter into the live cells and produce strong florescence (excitation: 495 nm, emission: 515 nm). Figure 3(d) shows the image of the transfected and stained T-cells. T-cells with yellow shows the transfected viable cells. The rectangle drawn with broken line illustrates a single-file of T-cells. T-cell patterns shown in the Figures 3(c) and (d) were produced using 105 cells/mL.
Figure 3:
Steps of the CAR T-cell manufacturing using two-step electroporation. (a) Picture of a device used in experiments. In addition to interdigitated electrodes, we produced Epoxy enclosure with open top to avoid spill of cell sample beyond the electrodes. There were no infusion pumps to flow T-cell sample mixed with mRNA into the device. (b) Propidium iodide (PI) and Calcein molecules were used to demonstrate the transfection of T-cells in the device. Figure shows that fluorescence image of the patterned cells. (c) Fluorescence image of the cells after electroporation. T-cells were electroporated the PI molecules that are fluorescent. A rectangle with broken lines shows a single line of patterned T-cells. (d) After the electroporation with PI, cells were collected and stained with Calcein to evaluate the cell viability and pipetted back to the device, patterned and imaged. The images show a section of patterned cell sample. Yellow color cells indicate the stained cells with both PI and Calcein molecules. Rectangle with broken lines shows a single file of patterned cells. Scale bars in Figures 3(b, c and d) indicate 40 μm.
We then examined the impact of T-cell patterning followed by electroporation on the transfection efficiency and cell viability. We calculated the transfection efficiency by quantifying the percentage of cells in sample that has measurable levels of protein expression (e.g., EGFP and CAR). These experiments were conducted on primary T-cells isolated from commercially available pooled whole blood samples of healthy individuals (Innovative Research, Novi, MI). For our initial experiments, we used commercially available mRNA molecules that produced enhanced green fluorescent protein (EGFP; TriLink Biotechnologies, San Diego, CA; excitation: 488 nm, emission: 509 nm). EGFP is a common target that has been used to quantify the transfection levels of T-cells in other studies [9, 25]. Figure 4(a) illustrates the transfection efficiency values for patterned and electroporated with AC and DC potentials, and randomly placed and electroporated with AC and DC potentials. We used commercially available flow cytometry (BD; Franklin Lakes, NJ) to measure the EGFP expression of the transfected cells. The transfection efficiency of the patterned and electroporated (AC electric fields) cells at 24h was about 50%, which is more than two times that of the randomly placed T-cells (23.8%). These values for patterned and electroporated with DC and randomly placed and electroporated with DC pulses were 50.8% and 26.2%, respectively. We have also found that transfection efficiency could be further increased by re-mixing T-cells, followed by re-patterning and electroporation (data not shown). The cell viability was analyzed after 24 h of electroporation. Commercially available live/dead assays (Millipore-Sigma, Burlington, MA) were used to measure the percentage of live cells. Patterned and electroporated cells with DC potential had the average cell viability values of 85.5%. Randomly placed and electroporated cells with DC potentials had the average cell viability values of 70.6%. Similarly, patterned and electroporated with AC potentials and randomly placed and electroporated with AC potentials had average cell viability values of 82.6% and 74.5%, respectively. As stated earlier, the difference in viability of patterned vs. randomly placed cell could be attributed to the induced membrane potentials produced by these methods. The transfection efficiency and cell viability values of two-step electroporation of primary T-cells is better than the traditional DC electroporation methods and comparable with flow through electroporation devices [9, 16].
Figure 4:
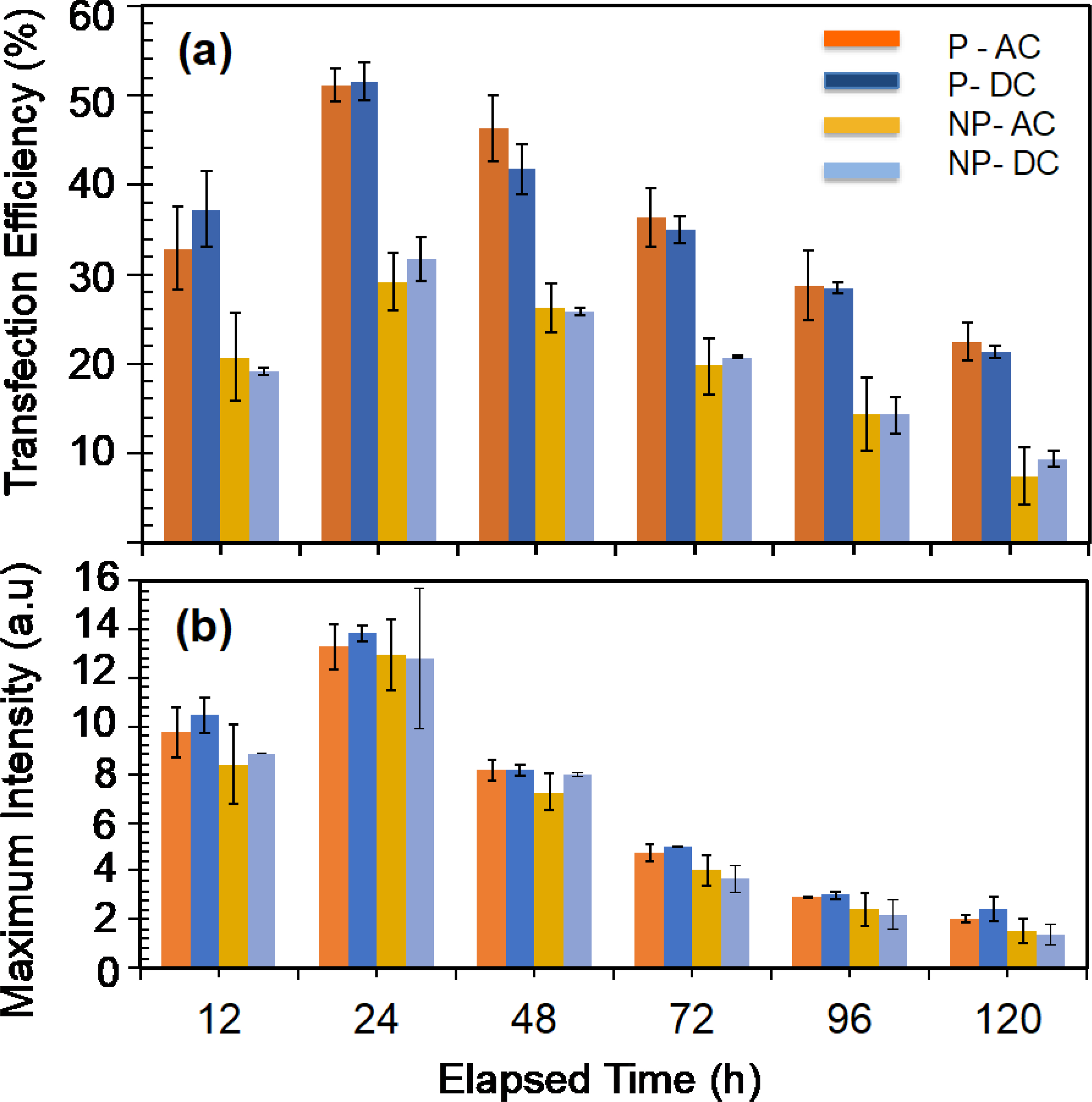
Transfection of T-cells with enhanced green fluorescence protein (EGFP) mRNA molecules and subsequent EGFP protein expression. (a) Variation of transfection efficiency (number of cells expressing detectable EGFP molecules) with time for patterned (denoted as P in the figure legend) and electroporated with AC and DC electric fields, and randomly placed (denoted as NP in the figure legend) and electroporated with AC and DC electric fields. (b) Variation of maximum fluorescence intensity with time for electroporated T-cells with AC and DC electric fields. Each electroporation experiment was repeated for patterned and randomly placed T-cells.
Regardless of the electroporation method or where T-cells were located (patterned or randomly placed), almost all the samples produced their highest EGFP expression after 24 h of electroporation (figure 4(a)). This shows that T-cells will take about 24 h to translate the majority of mRNA to EGFP. After the 24 h time point, the transfection efficiency was decreased. This decrease in EGFP expression could result from cell division or degradation of EGFP molecules. As the transfected cell divides, daughter cells may take up mRNA molecules from mother cells, and therefore a gradual decrease in the expression is expected. However, the division of primary T-cells is a complex process, and T-cell division under standard culture conditions may not be very effective. Vera et al. demonstrated that the expansion of T-cells requires rigorous culture conditions, and inefficient gas exchange, lack of nutrients, and waste accumulation could limit the T-cell expansion [27]. Therefore, we assumed that the number of T-cells is constant during the evaluation period.
To further understand the time-dependent degradation of GFP expression, we examined the maximum fluorescence intensity, which provides information about the maximum number of EGFP molecules expressed at any time point (Figure 4(b)). Patterned and electroporated T-cells produced the highest maximum fluorescence intensity values peaking at 24 h. We assumed that maximum fluorescence intensity could be proportional to the number of mRNA molecules injected into those cells at t=0. Therefore, by comparison, patterned and electroporated cells could have a higher quantity of mRNA molecules than randomly placed and electroporated cells. The maximum EGFP intensity drastically decreases for all 4 cases after the 24 h time point. Maximum EGFP expression at any given time after 24 h is also proportional to the maximum EGFP intensity at 24 h. By combining the two observations, we could conclude that the initial quantity of mRNA present in T-cells determine the maximum value of EGFP and the lifespan of the EGFP expression. The lifespan of EGFP is defined as the period (after transfection at t=0) at which measurable (by flow cytometry) EGFP is present. This observation is in agreement with the findings by Barret at al., which demonstrated that initial mRNA quantity in T-cells could dictate the lifespan of CAR molecules [28]. We then conducted CAR T-cell manufacturing experiments using the mRNA molecules that produce CARs that recognize the CD19 antigen on ALL cells. We selected the CD19 and ALL cells because CAR T-cell therapy is currently approved for treating ALL. Since the patterned and electroporated T-cells with AC and DC electric fields produced identical cell viability and transfection efficiency (Figure 4(a)), we used AC electric fields alone for the rest of the study. First, we examined how transfected T-cells with mRNA produced CAR molecules. Figure 5(a) shows the time-dependent CAR expression (average expression per cell) in T-cells for different concentrations of mRNA. We only varied the mRNA concentration between experiments and kept all other experimental parameters (such as electroporation time and frequency of the electric field) unchanged.
Figure 5:
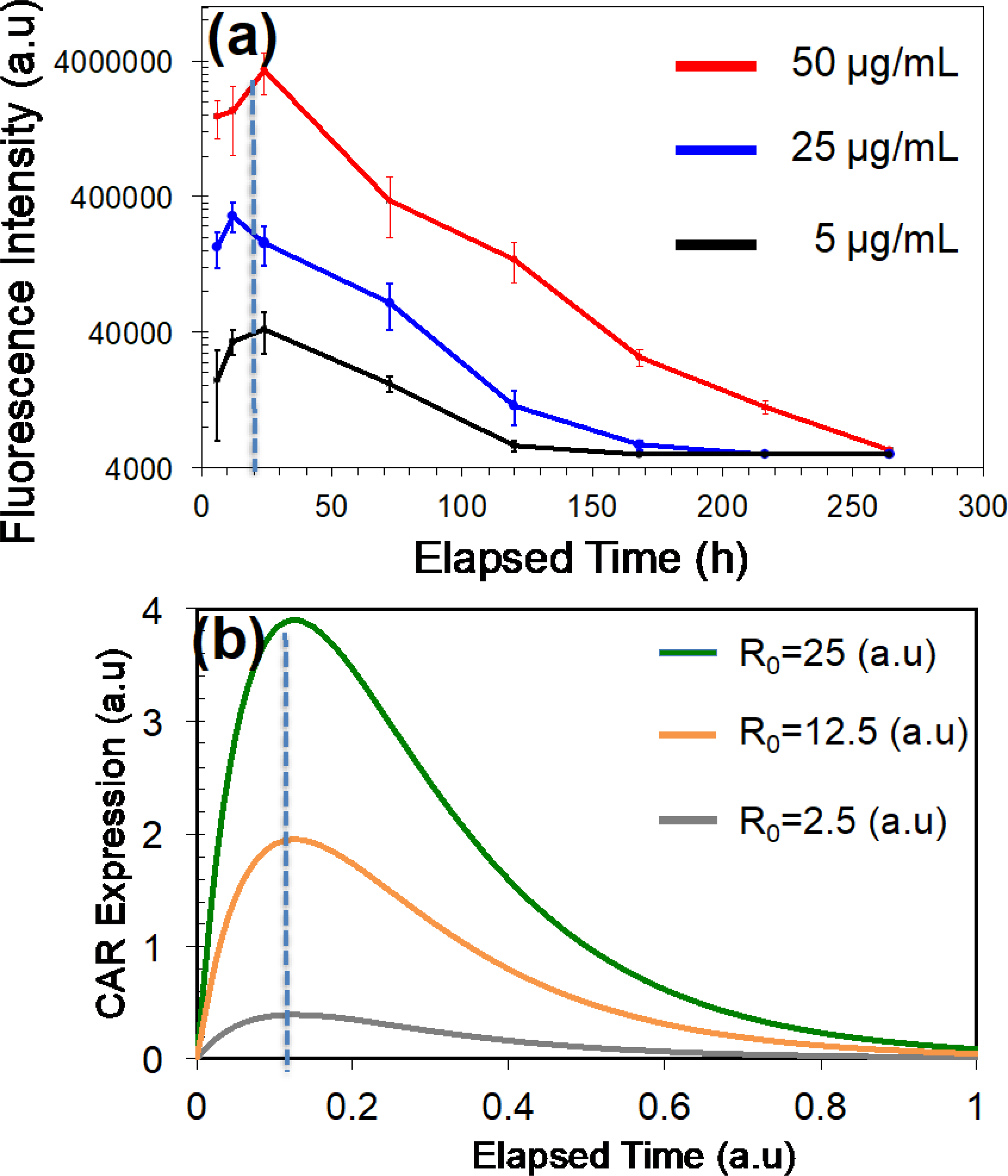
Variation of CAR (targeting CD19 antigen) with time for T-cells that are patterned and transfected (AC electric fields) with mRNA molecules. (a) Experimentally measured temporal variation of CAR with different mRNA concentrations in the buffer. (b) Calculated CAR expression variation with time. Calculation was performed using the expression for CAR expression obtained by solving equations derived for the compartmental model of a T-cell cytoplasm.
The CAR expression peaked earlier than that for EGFP molecules (24 h for CD19 and 48 h for EGFP), which shows that peaking time is dependent on the mRNA type. Also, Zhao at al. have reported similar results for GFP when electroporated T-cells with mRNA produced a maximum expression of GFP molecules within 24 h after electroporation [25]. According to Figure 5(a), the magnitude of the peak value of CAR is dependent on the mRNA concentration in the buffer. Therefore, magnitude of peak CAR expression proportional to the initial mRNAs levels in the T-cells at t=0. This data also demonstrates that lifespan of CAR expression in T-cells is proportional to the peak expression value at 24 h. The time point at which peaking of CAR occurs is independent of the mRNA concentration. Furthermore, CAR expression lasted over 10 days for the 50 μg/mL mRNA concentration. The duration of CAR expression in T-cells manufactured by our method is longer (over 10 days) than other reported studies by Birkholtz et al. (5 days), Zhao et al. (4 days), Pohl-Guimarães et al. (7 days), and Biossel et al. (2 days) [9, 25, 29 and 30].
Our findings on EGFP and CAR expressions are valuable information for broadly understanding the translation of mRNA to protein expression. Moreover, in the context of CAR T-cells, a lower decay rate is desirable for CAR molecules because sustained CAR expression could be beneficial for producing longer cytotoxicity. Furthermore, our findings could also be useful for manufacturing CAR T-cells with pre-determined lifespans of cytotoxicity. For example, one can produce highly active CAR T-cells (T-cells with very high CAR expression) or moderately active CAR T-cells (T-cells with low CAR expression) for desired time periods by changing the initial mRNA molarity in the electroporation buffer. In addition to mRNA molarity in the buffer, changing other experimental variables such as electroporation time will also be used to vary the transfection levels of mRNA in T-cells. To further improve our understanding about the CAR expression with time, we developed a compartment modelling approach (Figure 6).
Figure 6:
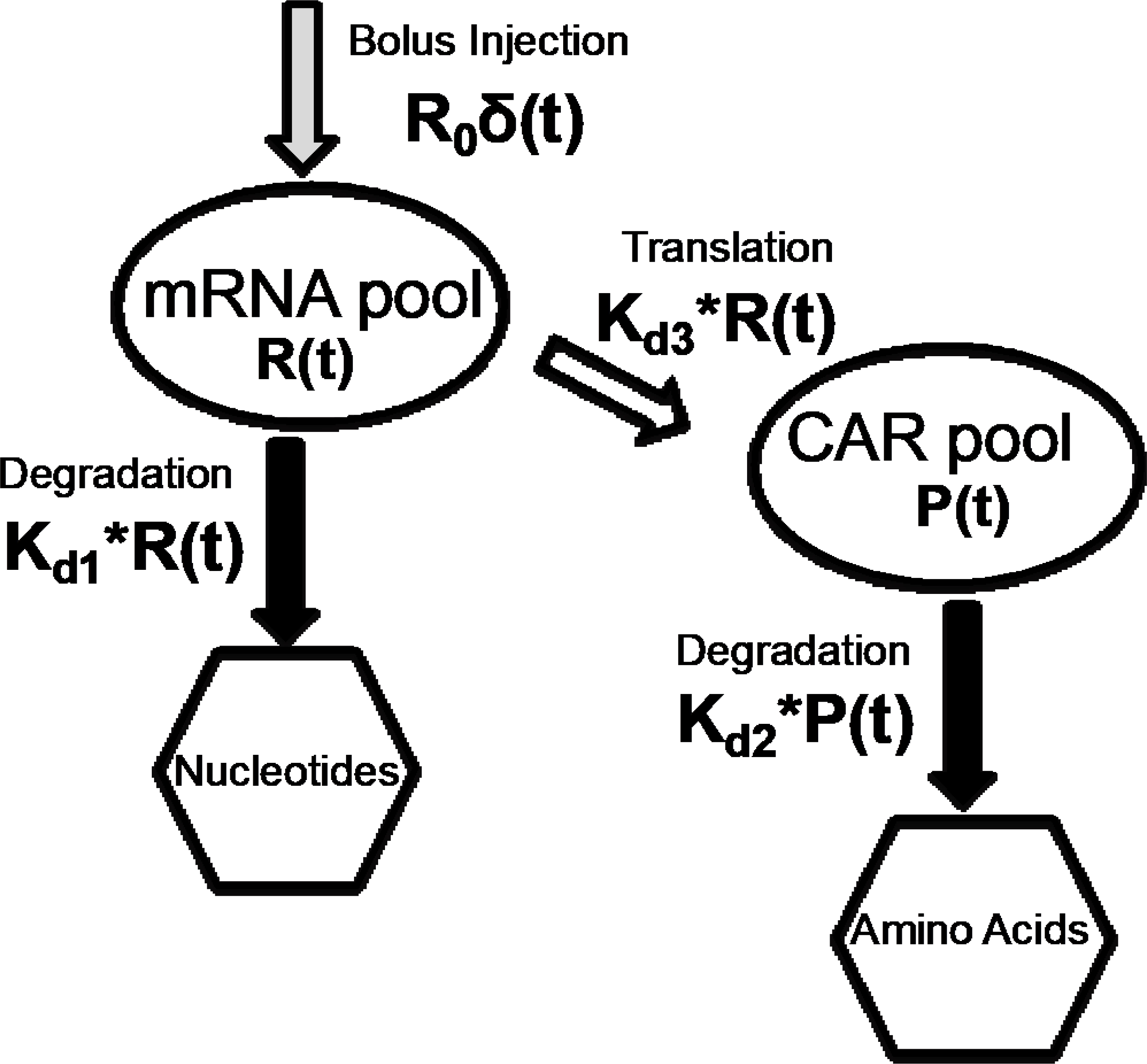
The compartmental model of the T-cell cytoplasm developed to study the expected CAR expression. The transfection of mRNA by electroporation was assumed as bolus input to the compartmental model. Basic equations were derived using time dependent mRNA degradation and translation as well as temporal variation of CAR degradation.
Hargrove et al. developed a compartmental modeling approach to understand the fundamental relationship between mRNA and protein in cells [31]. The model demonstrated the mathematical relation between protein expression and mRNA transcription, degradation, translation and protein degradation in the natural cellular environment. This model is inaccurate for externally injected mRNA molecules by electroporation because the latter process may occur independent of cell regulatory networks (e.g., no mRNA synthesis by the cell). Notably, there is a critical gap in the knowledge about the translation of externally injected mRNA into protein molecules. Figure 6 illustrates the compartmental model we developed to study the time-dependent CAR expression from externally injected mRNA by electroporation. The model utilized the mRNA degradation, protein degradation, and translation of mRNA to protein as variables. Schematic diagram in Figure 6 illustrates how mRNA molecules translate to CAR molecules in T-cells. We assumed that there was no CAR expression at t=0, and mRNA was injected as a bolus input (Eq. 3) at t=0 by electroporation. This assumption based on the fact that the electroporation time (in ms) was significantly smaller than the CAR expression time (in days). Two fundamental equations (Eqs. 2 and 4) were derived by assuming conservation of molecules (Eq. 1) in the cytoplasm of a T-cell. Eqs. 2 and 4 were solved analytically and results were used to derive analytical expressions for the time-dependent variation of mRNA (Eq. 5) and CD19 (Eq. 6).
We used the study published by Hargrove at al. to obtain numerical values for kd1, kd2, and kd3 [31]. However, degradation of CAR without target cells could be significantly less, and therefore we assumed a value of 1 day−1 for the rate of degradation. First, we studied how the initial mRNA level modulates the lifespan of the CAR expression or time-dependent expression of CAR in T-cells (Figure 5(b)). We calculated the CAR expression for the initial mRNA levels (R0) of 1, 10, and 25 (a.u). The data show that the lifespan of the CAR is proportional to the initial mRNA levels (R0). Additionally, the maximum level of CAR expression (or peak value of CAR expression) is also proportional to the initial mRNA levels. Surprisingly, the half-life of CAR expression does not change with R0. The half-life is defined as the time taking to achieve 50% reduction of the maximum expression of CAR. The model and the experimental data predict that the time point at which CAR peaking is independent of the mRNA concentration. These conclusions from the mathematical model agree with the experimental observations for CAR that target CD 19 antigen. By comparing experimental data with mathematical modeling, we can conclude that CAR expression, peak value for CAR, and the lifespan of CAR expression could be modulated by varying the initial mRNA concentration in T-cells.
To further understand the characteristics of CAR T-cells manufactured in the two-step electroporation process, we examined the time-dependent variation of CAR expression when target cancer cells were present. We used the commercially available ALL cells (SUP-B15 cell line) that express the CD19 antigens as target cells. Briefly, we electroporated T-cells with mRNA, suspended both the electroporated T-cells and SUP-B15 cells (T-cell: SUP-B15 ratio = 4:1), and co-cultured in an incubator (37°C; 5% CO2). We did not change any parameters in the protocol that we used to manufacture CAR T-cells in Figure 5(a). Figure 7(a) shows the normalized temporal variation of average CAR per T-cell with and without ALL. By comparison, when SUB-B15 cells are present, the time taken for CAR expression to peak occurred earlier than for the case without SUP-B15 cells (8 h vs. 22 h; Figure 7(a)). Valitutti et al. demonstrated experimental evidence showing a rapid downregulation of triggered T-cell receptors after the T-cell activation by specific antigens, which is similar to the conjugation of CAR to antigens in SUP-B15. We used the developed mathematical model to predict the CAR expression in the presence of SUP-B15. Following the experimental evidence by Valitutti et al., we systematically increased the numerical values of 𝑘𝑘𝑑𝑑2 from 1 to 11 and calculated the CAR expression (Figure 7(b)) [32]. Our calculation shows that the peak value of CD19 appears earlier when target cells are present, which is in agreement with our experimental data. Moreover, by comparison of the time values (both experimental- and modeling-based), it can be concluded that CAR expression degrades about 10 times faster when the target cells are present.
Figure 7:
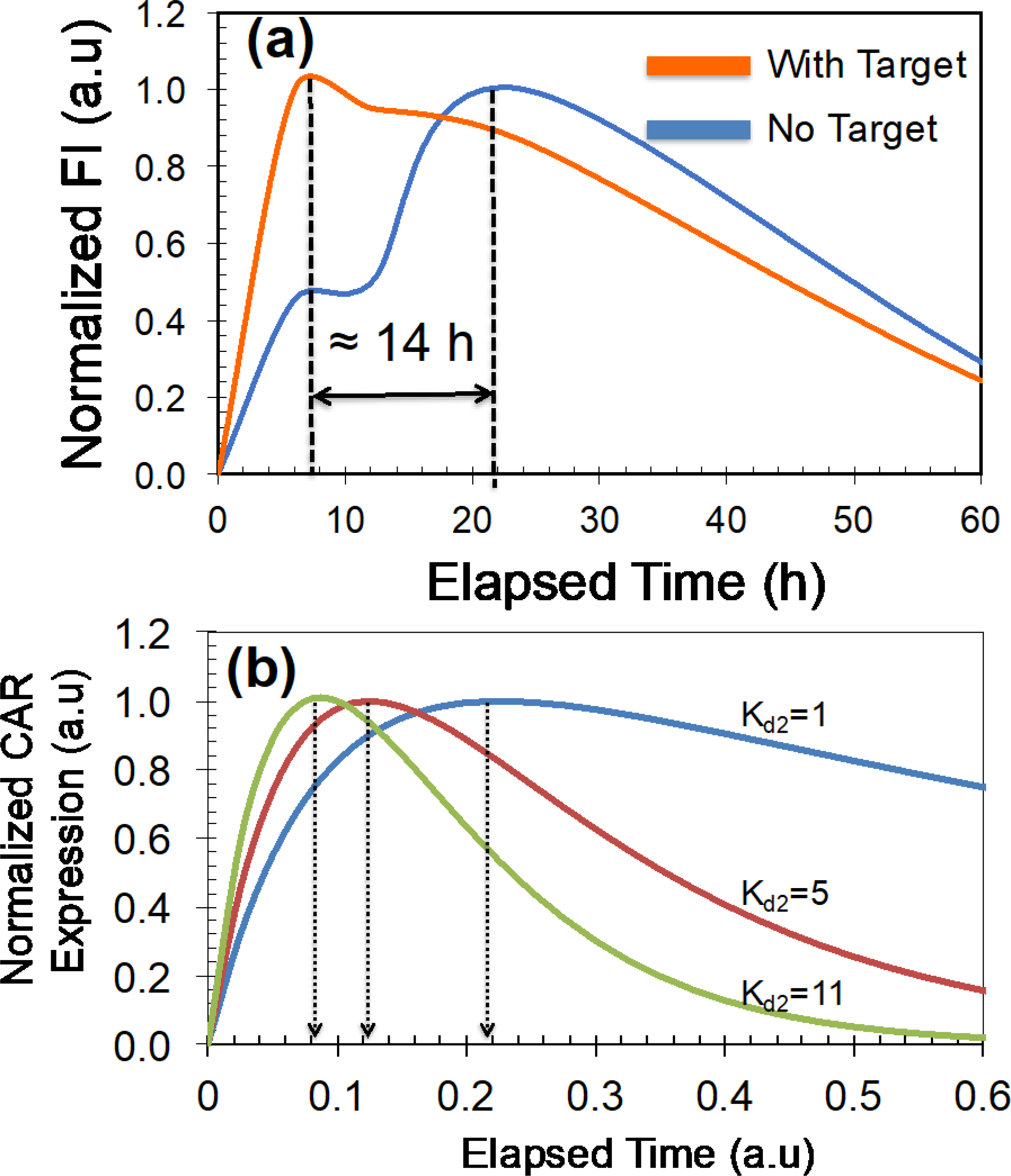
Temporal variation of CAR (targeting CD19 antigen) expression with and without target antigen molecules. Note that commercially available SUP-B15 cells expressing CD19 antigen on their cell surfaces were used in experiments. Transfection (AC electric fields) of T-cells with mRNA (50 μg/mL) was performed after cell patterning. We have normalized the CAR expressions to their peak values for comparison. (a) Experimentally measured CAR expression with time. (b) Calculated variation of CAR with time for kd2 values.
Next, we evaluated the manufactured CAR T-cells’ ability to kill or be cytotoxic towards the target cancer cells. Previous studies have demonstrated that mRNA-based CAR T-cells were able to kill cancer cells [9]. Birkholtz et al. demonstrated that mRNA-based CAR T-cells manufactured to target breast cancer cells through ErbB2 and CEA receptors have successfully lysed ErbB2+ and CEA+ tumor cells [9]. In addition, a similar study has reported that mRNA-based CAR T-cells exhibited cytolytic activities that are similar to viral-based CAR T-cells [9]. Furthermore, Birkholtz et al. demonstrated that the percentage lysis of cancer cells (target cells) is dependent on the ratio of target cells to CAR T-cells [9]. Moreover, the higher the target: effector ratio, the higher the cell lysis percentage reported. Biossel et al. have used flow cytometry to study the time dependent cytotoxicity produced by CAR T-cells [29]. Moreover, flow cytometry was used quantity the target cells and effector cells at each time point [29].
In the next set of experiments, we studied the cytotoxicity of manufactured CAR T-cells on cancer cells. As explained earlier, we manufactured CAR T-cells that recognize the CD19 antigens. We used the commercially available SUP-B15 cell line, which is ALL. Biossel et al., have also used SUP-B15 cells as the target cells for evaluating the cytotoxicity produced by recognition of CD19 antigen in ALLs [29]. Briefly, we co-cultured CAR T-cells and SUP-B15 cells (SUP-B15: CAR T-cells=1:4) in a standard 96-well plate. The number of CAR T-cells and SUP-B15 cells at each time point was quantified with flow cytometry. Figure 8 illustrates the variation of SUP-B15 cell quantity with time for electroporated (both patterned and randomly placed) cells. The number of CAR T-cells was almost constant during the evaluation period (data not shown). Data presented in Figure 8 show that CAR T-cells manufactured by patterning and electroporation kill cells faster than the cells electroporated without patterning. This may be because patterned and electroporated cells could have higher CAR molecules than randomly placed and electroporated cells. The presence of a large number of CAR molecules enhances the probability of finding the CD19 antigen molecules on ALLs. Birkholtz et al. demonstrated that a target: effector cell ratio of 1:2 produced only 2% cell lysis at 24 h when CAR T-cells manufactured with a commercially available electroporation system were used [9]. In comparison, CAR T-cells manufactured by patterned and electroporated cells produced about 60% cell lysis at 24 h. Cells electroporated without patterning had about 25% cell lysis at 24 h. These cell lysis percentages reflected the T-cell transfection by macro- vs. microelectrodes and patterned vs. randomly placed cells. Negative control experiments were performed by electroporating patterned T-cells without mRNA. We found that the SUP-B15 cell population increased by approximately 200% during the 7-day evaluation period (data not shown).
Figure 8:
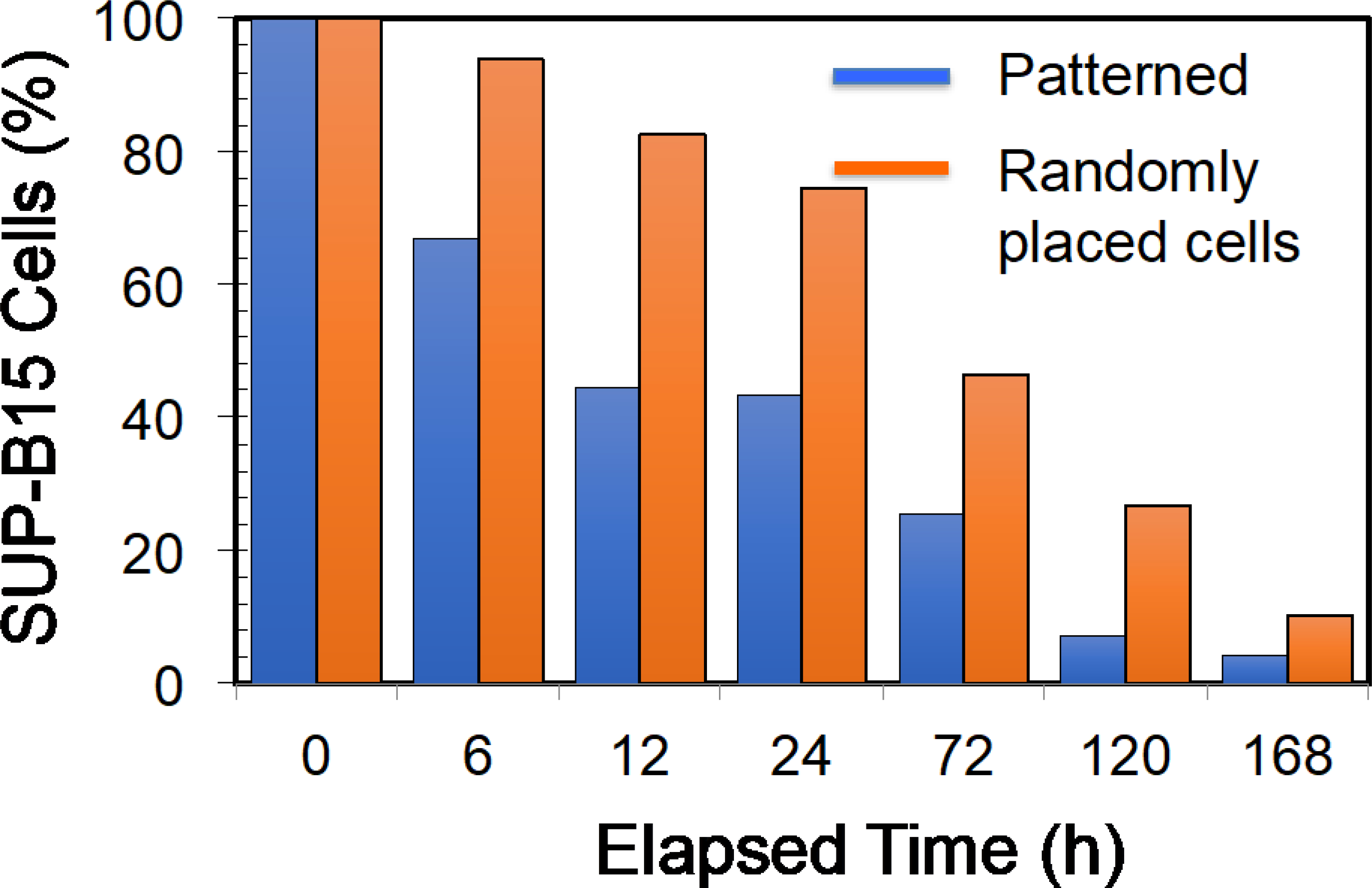
Variation of SUP-B15 cell number when co-cultured with CAR T-cells that recognizes the CD19 antigen on SUP-B15 cells. CAR T-cells were electroporated (AC electric fields) using 50 μg/mL mRNA in the electroporation buffer and co-cultured with SUP-B15 cells, which was started at t=0. Cell samples were electroporated after producing single file T-cell patterns or randomly placing T-cells in electrodes. Initially (t=0), percentage target cells were set to 100% for comparison of percentage changes with time.
We then investigated the cytotoxicity of CAR T-cells toward other leukemia cells (chronic myelogenous leukemia, K-562) that do not have any known expression of CD19 antigen molecules [28]. The CAR T-cell: K-562 ratio was 4:1. Figure 9 compares the variation of K-562 and SUP-B15 cells during the presence of CAR T-cells that express CD19. We found CAR T-cells manufactured by electroporation (both patterned and randomly placed) did not produce a significant reduction of K-562 cell numbers. We have also found that different reduction rates for SUP-B15. Moreover, in the first 20 h, about 60% of SUP-B15 cells were killed, and then only 38% of SUP-B15 cells were killed in the next 100 h, and finally, about 1% of SUP-B15 cells were killed in the last 25 h. As we discussed earlier, this could be representative of the probability of conjugating CAR molecules (T-cells) and CD19 antigen (ALL). As SUP-B15 reduces, the probability of conjugation could also reduce.
Figure 9:
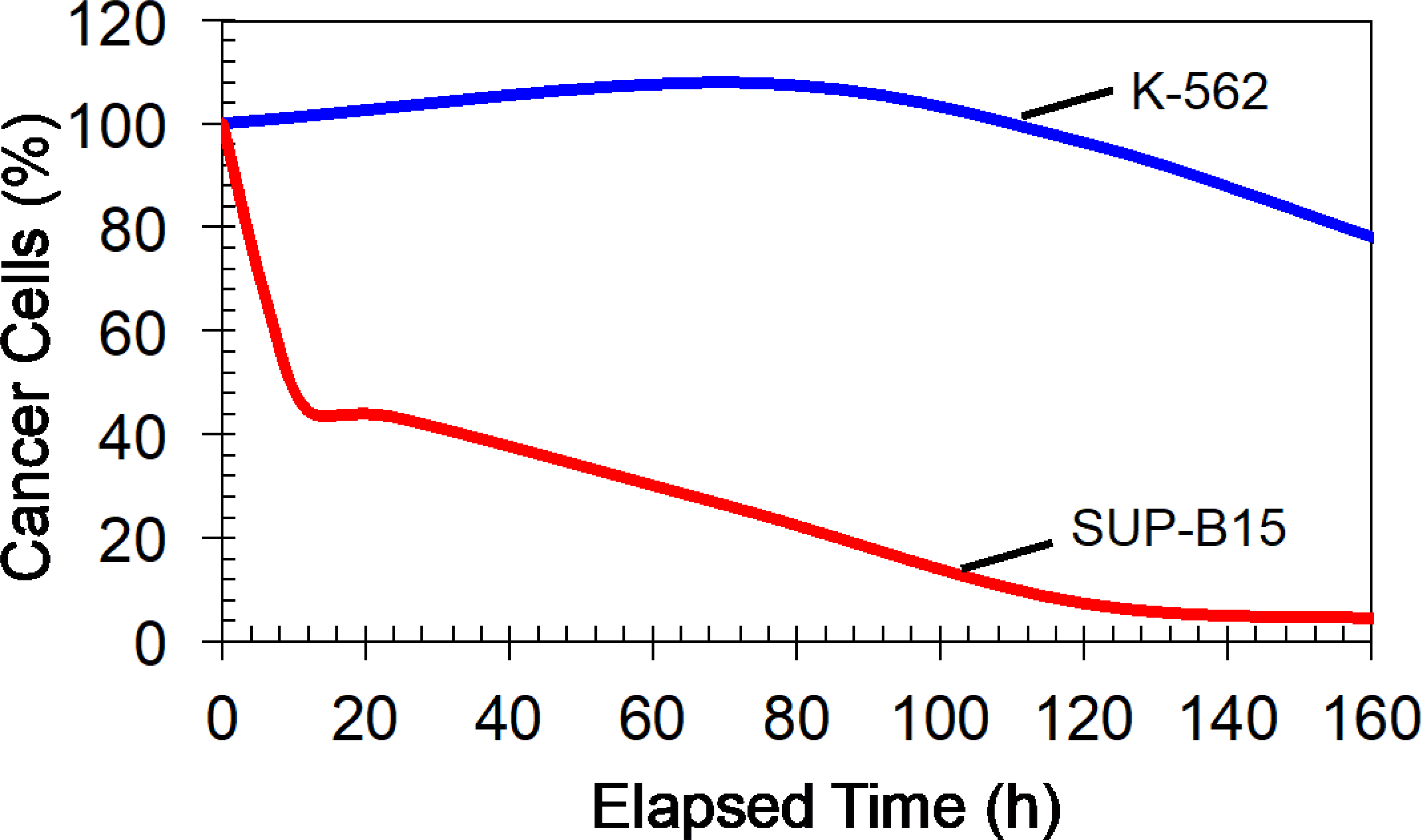
Demonstrate the selective cancer cells killing by CAR T-cells. CAR T-cells were manufactured by producing cell patterns and electroporated (AC electric fields) with mRNA molecules (50 μg/mL) that translated to CAR molecules that target CD 19 antigen molecules. CAR T-cells mixed with SUP-B15 (express CD 19 antigens) or K-562 cells (do not express CD19 antigens) and co-cultured (starting t=0).
Conclusions
We demonstrated that initial mRNA levels in T-cells modulate the peak value of target CAR expression, expression time, and lifespan of cytotoxicity. We have also found that half-life of CAR expression does not vary with initial mRNA content. In addition, two-step electroporation substantially increased the cell viability and transfection efficiency of the T-cells. Ideally, higher transfection efficiency is needed to produce a sufficient number of CAR T-cells from a small amount of a patient’s blood sample. Similarly, when there are CAR T-cells with long cytotoxic lifespans, a small number of CAR T-cells or a small amount of whole blood could be used for therapy. The cell viability after electroporation is also a critical factor because the removal of dead and dying cells is challenging. This is because there are no known T-cell surface antigens that we can utilize to purify dead or dying cells. The presence of non-viable cells in the CAR T-cell sample is very undesirable because these cells could produce serious side effects. To this end, our group recently demonstrated that dielectrophoretic cell separation could be utilized to purify T-cells in a label-free manner after electroporation [36].
We did not see a difference in maximum EGFP or CAR expression or lifespan of EGFP or CAR expression in T-cells manufactured with AC (100 Hz) and DC electric fields. However, studies have demonstrated that cell transfection rates from AC electroporation (in the kHz range) can be significantly higher than traditional DC electroporation [26]. The oscillating AC electric fields produce a mild sonication effect on molecules, which reduces molecular crowding near the pores and increases the transfection rate [26]. We also investigated the high-frequency (>1 kHz) AC electroporation and found that T-cells are difficult to electroporate at high frequencies. This could result from the low induced membrane potentials (< 1V) at high frequencies. Studies conducted by Chang et al. showed that they were able to electroporate and transfect red blood cells at higher frequencies (> 1 kHz) when high frequency electric fields were integrated with DC pulses [10]. Furthermore, it is highly unlikely that the low frequency electric fields (100 Hz) that we used in experiments could produce sonication effects on mRNA molecules. Generally, sonication effects are produced by either electrophoresis or dielectrophoresis. The electrophoresis is vanishingly small with AC electric fields. The dielectrophoresis produced in low frequency values (~100 Hz) for mRNA molecules is also very small because we found earlier that mRNA molecules require AC fields with 100 kHz or more to produce substantial dielectrophoresis effect [33].
The CAR T-cells manufactured by our device expressed CARs that lasted for more than 10 days. From a therapeutic standpoint, longer cytotoxic lifespans are desired to avoid frequent infusions, as this drives up the cost and discomfort for the patient. However, Wang et al. argued that in real-world applications, when dealing with high tumor cell loads, a single infusion of CAR T-cells may not be sufficient [34]. Moreover, multiple doses of CAR T-cells over a lengthy period would be necessary. On the other hand, to address the personalized therapeutic needs, CAR T-cells with tunable cytotoxicity would be necessary and preferred. Our findings suggest that the initial CAR expression level in T-cells could dictate the lifespan of cytotoxicity. To increase the lifespan of cytotoxicity, one potential approach would be to increase the initial T-cell mRNA concentration. Although it is simple to increase the initial mRNA concentration in the electroporation buffer, it is counterproductive. This is because increased mRNA concentrations lead to molecular crowding near the pores and reduce transfection efficiency. Increasing the electroporation time or membrane potential will improve transfection efficiency, but these activities significantly reduce cell viability. As discussed earlier, our device/technique has produced high transfection rate and cell viability. Therefore, this device/technique could be used to further investigate the possibility of manufacturing CAR T-cells with significantly longer cytotoxic lifespans or even CAR T-cells with tunable lifespans.
Generally, about 106 CAR T-cells per kg of the patient is required for therapy [37]. Therefore, up to 80×106 CAR T-cell manufacturing capacity require to treat most of the individuals. Scaling up of the two-step electroporation is required to apply our device in treatments. Based on the current cell electroporation capacity of the current device, to accommodate the need of most individuals (up to 80 kg in weight), we estimate that scaled-up device could be a size of 6″ wafer. Additional research is needed to develop other essential capabilities in the clinical CAR T-cell manufacturing platform, such as automation and quality control methods. For example, recent developments in machine learning research have demonstrated the feasibility of developing quality control methods (such as the detection of cancer cells in the T-cells sample) and automation methods for biomanufacturing. Application of this technique beyond T-cells is also possible. However, depending on the cell diameter, a transfection device with proper dimensions of electrodes (e.g., spacing between electrodes and the width of the electrodes) is needed. Our future work will focus on developing this technique as a viable biomanufacturing platform for multiple cell-types.
Supplementary Material
Acknowledgement
DN acknowledges the financial support for National Institutes of Health (NIH) funded Dakota Cancer Collaborative on Translational Activity (DaCCoTA) program. BR acknowledges the graduate fellowship from biomedical engineering program at North Dakota State University. Authors thank the help of Greg Strommen and Jeffrey Erickson in manufacturing electrodes and ordering reagents needed for experiments. Authors would like to acknowledge the valuable suggestions to the manuscript provided by Dr. David Chang at Valley Hospital, Ridgewood, NJ.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1).Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M and Qu J, 2014. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science translational medicine, 6(224), pp.224ra25–224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Prasad V, 2018. Tisagenlecleucel—the first approved CAR-T-cell therapy: implications for payers and policy makers. Nature Reviews Clinical Oncology, 15(1), pp.11–12. [DOI] [PubMed] [Google Scholar]
- 3).Levine BL, Miskin J, Wonnacott K and Keir C, 2017. Global manufacturing of CAR T cell therapy. Molecular Therapy-Methods & Clinical Development, 4, pp.92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM and Rosenberg SA, 2010. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular Therapy, 18(4), pp.843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM and Kalos M, 2014. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer immunology research, 2(2), pp.112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Jordan CA, Neumann E and Sowers AE eds., Electroporation and electrofusion in cell biology. Springer Science & Business Media., 2013 [Google Scholar]
- 7).Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA and Morgan RA, 2006. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Molecular therapy, 13(1), pp.151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Van Tendeloo VF, Ponsaerts P, Lardon F, Nijs G, Lenjou M, Van Broeckhoven C, Van Bockstaele DR and Berneman ZN, 2001. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood, The Journal of the American Society of Hematology, 98(1), pp.49–56. [DOI] [PubMed] [Google Scholar]
- 9).Birkholz K, Hombach A, Krug C, Reuter S, Kershaw M, Kämpgen E, Schuler G, Abken H, Schaft N and Dörrie J, 2009. Transfer of mRNA encoding recombinant immunoreceptors reprograms CD4+ and CD8+ T cells for use in the adoptive immunotherapy of cancer. Gene therapy, 16(5), pp.596–604. [DOI] [PubMed] [Google Scholar]
- 10).Zhan Y, Cao Z, Bao N, Li J, Wang J, Geng T, Lin H and Lu C, 2012. Low-frequency ac electroporation shows strong frequency dependence and yields comparable transfection results to dc electroporation. Journal of controlled release, 160(3), pp.570–576. [DOI] [PubMed] [Google Scholar]
- 11).Krassowska W, & Filev PD (2007). Modeling electroporation in a single cell. Biophysical journal, 92(2), 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Pavlin M and Miklavčič D, 2008. Theoretical and experimental analysis of conductivity, ion diffusion and molecular transport during cell electroporation—relation between short-lived and long-lived pores. Bioelectrochemistry, 74(1), pp.38–46. [DOI] [PubMed] [Google Scholar]
- 13).Susil R, Šemrov D and Miklavčič D, 1998. Electric field-induced transmembrane potential depends on cell density and organizatio. Electro-and magnetobiology, 17(3), pp.391–399. [Google Scholar]
- 14).Mistani P, Guittet A, Poignard C and Gibou F, 2019. A parallel Voronoi-based approach for mesoscale simulations of cell aggregate electropermeabilization. Journal of Computational Physics, 380, pp.48–64. [Google Scholar]
- 15).Pavlin M, Pavselj N and Miklavcic D, 2002. Dependence of induced transmembrane potential on cell density, arrangement, and cell position inside a cell system. IEEE Transactions on Biomedical Engineering, 49(6), pp.605–612. [DOI] [PubMed] [Google Scholar]
- 16).Hsi P, Christianson RJ, Dubay RA, Lissandrello CA, Fiering J, Balestrini JL and Tandon V, 2019. Acoustophoretic rapid media exchange and continuous-flow electrotransfection of primary human T cells for applications in automated cellular therapy manufacturing. Lab on a Chip, 19(18), pp.2978–2992. [DOI] [PubMed] [Google Scholar]
- 17).Buie CR, Dominguez PAG, Ge Z and Moran JL, Massachusetts Institute of Technology, 2021. Microfluidic assay for rapid optimization of cell electroporation. U.S. Patent 10,947,526.
- 18).Ringwelski B, Jayasooriya V and Nawarathna D, 2020. Dielectrophoretic High Purity Isolation of Primary T-cells in Samples Contaminated with Leukemia Cells, for Biomanufacturing of Therapeutic CAR T-cells, 2020, Journal of Physics D: Applied Physics, 54(6), p.065402. [Google Scholar]
- 19).Gupta V, Jafferji I, Garza M, Melnikova VO, Hasegawa DK, Pethig R and Davis DW, 2012. ApoStream™, a new dielectrophoretic device for antibody independent isolation and recovery of viable cancer cells from blood. Biomicrofluidics, 6(2), p.024133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Velmanickam L, Bains M, Fondakowski M, Dorsam GP and Nawarathna D, 2018. iLluminate-miRNA: Paradigm for high-throughput, low-cost, and sensitive miRNA detection in serum samples at point-of-care. Journal of Physics D: Applied Physics, 52(5), p.055401. [Google Scholar]
- 21).Guo X and Zhu R, 2016. Controllable in-situ cell electroporation with cell positioning and impedance monitoring using microelectrode array. Scientific reports, 6(1), pp.1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Inglis DW, Riehn R, Austin RH and Sturm JC, 2004. Continuous microfluidic immunomagnetic cell separation. Applied Physics Letters, 85(21), pp.5093–5095. [Google Scholar]
- 23).Hamada R, Takayama H, Shonishi Y, Mao L, Nakano M and Suehiro J, 2013. A rapid bacteria detection technique utilizing impedance measurement combined with positive and negative dielectrophoresis. Sensors and Actuators B: Chemical, 181, pp.439–445. [Google Scholar]
- 24).DeBruin KA and Krassowska W, 1999. Modeling electroporation in a single cell. I. Effects of field strength and rest potential. Biophysical journal, 77(3), pp.1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA and Morgan RA, 2006. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Molecular therapy, 13(1), pp.151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Chang DC, 1989. Cell poration and cell fusion using an oscillating electric field. Biophysical journal, 56(4), pp.641–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Vera JF, Brenner LJ, Gerdemann U, Ngo MC, Sili U, Liu H, Wilson J, Dotti G, Heslop HE, Leen AM and Rooney CM, 2010. Accelerated production of antigen-specific T-cells for pre-clinical and clinical applications using Gas-permeable Rapid Expansion cultureware (G-Rex). Journal of immunotherapy (Hagerstown, Md.: 1997), 33(3), p.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH and Grupp SA, 2011. Treatment of advanced leukemia in mice with mRNA engineered T cells. Human gene therapy, 22(12), pp.1575–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Boissel L, Betancur M, Wels WS, Tuncer H and Klingemann H, 2009. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leukemia research, 33(9), pp.1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Pohl-Guimarães F, Yang C, Dyson KA, Wildes TJ, Drake J, Huang J, Flores C, Sayour EJ and Mitchell DA, 2019. RNA-modified T cells mediate effective delivery of immunomodulatory cytokines to brain tumors. Molecular Therapy, 27(4), pp.837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Hargrove JL and Schmidt FH, 1989. The role of mRNA and protein stability in gene expression. The FASEB Journal, 3(12), pp.2360–2370. [DOI] [PubMed] [Google Scholar]
- 32).Valitutti S, Müller S, Salio M and Lanzavecchia A, 1997. Degradation of T cell receptor (TCR)–CD3-ζ complexes after antigenic stimulation. The Journal of experimental medicine, 185(10), pp.1859–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Nawarathna D, Turan T and Wickramasinghe HK, 2009. Selective probing of mRNA expression levels within a living cell. Applied physics letters, 95(8), p.083117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Wang D, Starr R, Alizadeh D, Yang X, Forman SJ and Brown CE, 2019. In vitro tumor cell rechallenge for predictive evaluation of chimeric antigen receptor T cell antitumor function. JoVE (Journal of Visualized Experiments), (144), p.e59275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Nawarathna D, Unal K and Wickramasinghe HK, 2008. Localized electroporation and molecular delivery into single living cells by atomic force microscopy. Applied Physics Letters, 93(15), p.153111. [Google Scholar]
- 36).Jayasooriya VD and Nawarathna D, 2019. Label-free purification of viable human T-lymphocyte cells from a mixture of viable and non-viable cells after transfection by electroporation. Journal of Physics D: Applied Physics, 52(36), p.36LT01. [Google Scholar]
- 37).Hay KA and Turtle CJ, 2017. Chimeric antigen receptor (CAR) T cells: lessons learned from targeting of CD19 in B-cell malignancies. Drugs, 77(3), pp.237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.