

Abstract
Six-state amino acid recoding strategies are commonly applied to combat the effects of compositional heterogeneity and substitution saturation in phylogenetic analyses. While these methods have been endorsed from a theoretical perspective, their performance has never been extensively tested. Here, we test the effectiveness of six-state recoding approaches by comparing the performance of analyses on recoded and non-recoded data sets that have been simulated under gradients of compositional heterogeneity or saturation. In our simulation analyses, non-recoding approaches consistently outperform six-state recoding approaches. Our results suggest that six-state recoding strategies are not effective in the face of high saturation. Furthermore, while recoding strategies do buffer the effects of compositional heterogeneity, the loss of information that accompanies six-state recoding outweighs its benefits. In addition, we evaluate recoding schemes with 9, 12, 15, and 18 states and show that these consistently outperform six-state recoding. Our analyses of other recoding schemes suggest that under conditions of very high compositional heterogeneity, it may be advantageous to apply recoding using more than six states, but we caution that applying any recoding should include sufficient justification. Our results have important implications for the more than 90 published papers that have incorporated six-state recoding, many of which have significant bearing on relationships across the tree of life. [Compositional heterogeneity; Dayhoff 6-state recoding; S&R 6-state recoding; six-state amino acid recoding; substitution saturation.]
Compositional heterogeneity and substitution saturation are major challenges to phylogenetic inference. Compositional heterogeneity stems from the tendency of genes or organisms to have unequal proportions of amino acids (Collins et al. 1994; Foster and Hickey 1999). These unequal amino acid frequencies are caused by mutational and selective pressures acting at the nucleotide level (Singer and Hickey 2000; Knight et al. 2001), as well as differences in translational efficiency (Akashi and Eyre-Walker 1998). The combination of evolutionary and biological processes results in different amino acid compositions across taxa on the tree. Consequently, challenges to phylogenetic analyses arise when distantly related taxa share sequence similarities due to homoplasy (convergence), rather than descent from a common ancestor (Foster and Hickey 1999; Tarrío et al. 2001).
Similarly, phylogenetic reconstruction artifacts emerge under substitution saturation of amino acids. Substitution saturation occurs when there have been multiple amino acid substitutions at the same site washing out the evolutionary signal (Ho and Jermiin 2004). Like compositional heterogeneity, sequence saturation can lead to long branch attraction, driving unrelated taxa to group together in a clade due to homoplasy (Felsenstein 1978; Lawrence et al. 2019).
There is a large body of research on the conditions for state aggregation (or lumpability) in modeling character data such as DNA or amino acids (Kemeny and Snell 1976; Courtois 1977). Based on this foundation, matrix recoding has been proposed as a solution for both compositional heterogeneity and substitution saturation (Blanquart and Lartillot 2006; Susko and Roger 2007). Under matrix recoding methods, nucleotides or amino acids are lumped into groups based on function (Blanquart and Lartillot 2006). For example, under the RY nucleotide recoding strategy, purines (i.e., A and G) are coded with the character R and pyrimidines (i.e., T and C) are coded with the character Y (Woese et al. 1991; Phillips et al. 2001). In this recoding scenario, only transversion events are meaningful in a phylogenetic analysis. A similar recoding strategy has been implemented for amino acids, the most well-known being Dayhoff 6-state recoding. In Dayhoff 6-state recoding, chemically related amino acids that frequently replace each other are pooled together into six groups based on similar substitution scores in the Dayhoff (or PAM250) matrix (Dayhoff et al. 1978): AGPST, DENQ, HKR, ILMV, FWY, and C (Embley et al. 2003a; Hrdy et al. 2004). Thus, only amino acid changes between categories, and not within categories, are considered substitutions. Since the introduction of Dayhoff 6-state recoding, several other six-state amino acid recoding strategies based around other scoring matrices have been developed. For example, S&R 6-state recoding (Susko and Roger 2007; Feuda et al. 2017) is based on the JTT matrix (Jones et al. 1992) and KGB 6-state recoding (Kosiol et al. 2004; Feuda et al. 2017) is based on the WAG matrix (Williams et al. 2011).
Authors have increasingly been applying six-state recoding to phylogenetic analyses. To date, there are at least 91 phylogenetic studies that have implemented six-state amino acid recoding strategies, with the highest number of studies published in 2019 (Table 1). Several of these studies have proposed controversial topologies based on results from recoded matrices with deep implications across the tree of life (e.g., Rodríguez-Ezpeleta and Embley 2012; Feuda et al. 2017; Laumer et al. 2018; Puttick et al. 2018; Marlétaz et al. 2019). For example, the relationships of non-bilaterian animals have a major influence on how we understand the origin and evolution of key animal innovations (e.g., true epithelia, the gut, neural and muscle cell types), and recent papers using six-state recoding have major implications on how these relationships are viewed (Feuda et al. 2017; Laumer et al. 2018). While amino acid recoding has been considered from a theoretical perspective (Davidson et al. 2002; Embley et al. 2003a; Hrdy et al. 2004), and there have been comparisons between different recoding strategies (Susko and Roger 2007), there has not been extensive empirical testing of the widely applied six-state recoding approaches. Historically, simulation has been an effective strategy for empirically testing the performance of phylogenetic approaches (Kuhner and Felsenstein 1994; Swofford et al. 2001; Zwickl et al. 2002; Kubatko and Degnan 2007; Huang and Knowles 2016). In this study, we simulate data sets with a gradient of either compositional heterogeneity or saturation and compare the performance of maximum-likelihood analyses on six-state recoded data sets to the same analyses on non-recoded data sets. We also run a subset of these analyses using 9-, 12-, 15-, and 18-state recoding schemes and compare these results to those achieved with six-state recoded and non-recoded matrices.
Table 1.
Publications that use six-state amino acid recoding
Note: Asterisk indicates the publication included recoding approaches in a main figure to test if this strategy was appropriate.
Materials and Methods
Reproducibility and Transparency Statement
Custom scripts, command lines, and data used in these analyses are available in GitHub (https://github.com/josephryan/Hernandez_Ryan_2021_Recoding), Dryad (https://doi.org/10.5061/dryad.5mkkwh757) and Zenodo (https://zenodo.org/record/4660589). To maximize transparency and minimize confirmation bias, all analyses were pre-planned using phylotocol (DeBiasse and Ryan 2019) and pre-registered using the Center for Open Science’s pre-registration platform (https://osf.io/smj6k/ and https://osf.io/6ubgj/). Prior to the initial submission of this manuscript, we made four changes to the original plan outlined in our phylotocol. Details of changes and all versions of our phylotocol are available in our GitHub repository (see Section 5 “Amendment History” in the phylotocol). Briefly, our changes included (1) adding tests of compositional heterogeneity to our original plan to test saturation, (2) incorporating P4 after realizing that Seq-Gen was not well suited for testing compositional heterogeneity, (3) adding deep splits evaluation criteria, and (4) adding statistical tests and testing alternative Dayhoff strategies. The latest version of our phylotocol includes all of these changes along with the additional analyses we made in response to reviews of our manuscript by three reviewers (prior to running new analyses).
Overview of Empirical Data Sets Employed
The following methods can be divided into two main analyses: compositional heterogeneity and saturation. Both analyses employ empirical data from the following papers: Chang et al. (2015) hereafter “Chang,” and Feuda et al. (2017) hereafter “Feuda.” The topologies from Chang and Feuda are based on the same data set which is made up of 51,940 amino acid positions from 78 taxa representing a wide range of animals and 9 non-animal outgroups. Feuda extensively applied six-state amino acid recoding to this data set in a reanalysis of the Chang study, which did not use recoding.
For the compositional heterogeneity analysis, we use several hypothetical 20-taxon symmetrical trees which consist of four clades (named clade-A, clade-B, clade-C, and clade-D) made up of five taxa each (Fig. 1a), and apply global parameters estimated from the Chang data set. For the saturation analysis, we use the topologies reported in Chang and Feuda. More details on these analyses are provided below.
Figure 1.

Six-state recoding approaches produce more incorrect trees under various levels of compositional heterogeneity. a) Trees used for simulations. The value in the name of the tree (e.g., 0.008 in Tree 0.008) denotes the length in substitutions per site of the stem branches of clade-A and clade-B, and stem branches of clade-C and clade-D (highlighted in orange and with arrows). b) Percentage of 1000 trees that did not reconstruct a monophyletic group of taxa from clade-A and clade-B and monophyletic group of taxa from clade-C and clade-D.
Testing Six-State Recoding Performance on Compositional Heterogeneity
We used the script comphet.pl (available in our GitHub repository) to simulate amino acid data in P4 (Foster 2004) on four hypothetical 20-taxon balanced trees (Fig. 1a). We chose P4 because it specializes in simulating data in which amino acid (or nucleotide) composition varies across the tree. Using the amino acid rates of substitution estimated from the Chang data set, we simulated sequences that were 1000 amino acids in length under the GTR model. To introduce compositional heterogeneity, we used a balanced tree and generated one set of amino acid frequencies for clade-A and clade-C and a different set of frequencies for clade-B and clade-D. We paired amino acids by starting with the order of the 20 amino acids commonly used as input to standard phylogenetic programs (i.e., A, R, N, D, C, Q, E, G, H, I, L, K, M, F, P, S, T, W, Y, V), divided them in half (i.e., [A-I] and [L-V]), and paired the two groups (i.e., (A, L), (R, K), (N, M) (D, F), (F, P), (Q, S), (E, T), (G, W), (H, Y), (I, V)). For clade-B and clade-D, we used the amino acid frequencies estimated from the Chang data set (Supplementary Table S1 available on dryad at https://doi.org/10.5061/dryad.5mkkwh757). For clade-A and clade-C, we added 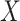 to the amino acid in each of the 10 frequency pairs that had the lowest frequency in the Chang data set and subtracted
to the amino acid in each of the 10 frequency pairs that had the lowest frequency in the Chang data set and subtracted 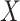 from the other, where
from the other, where 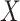 is the inflation parameter (i.e., 0.1, 0.5, 0.9) multiplied by the lowest frequency of the pair.
is the inflation parameter (i.e., 0.1, 0.5, 0.9) multiplied by the lowest frequency of the pair.
For example, the Chang frequencies for the amino acids 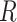 and
and 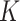 are 0.063 and 0.080, respectively. These frequencies were used for clade-B and clade-D without adjustment. To determine the increment value
are 0.063 and 0.080, respectively. These frequencies were used for clade-B and clade-D without adjustment. To determine the increment value 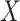 under the inflation parameter 0.1, we multiplied the frequency of
under the inflation parameter 0.1, we multiplied the frequency of 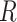 , which is the lowest of the pair, by 0.1 (
, which is the lowest of the pair, by 0.1 (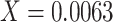 ). We then added
). We then added 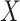 to the Chang frequency of
to the Chang frequency of 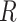 (
(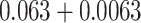 ) and subtracted
) and subtracted 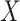 from the Chang frequency of
from the Chang frequency of 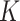 (
(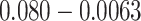 ). We rounded these values to three decimal places (because P4 requires frequencies to add up to 1 and the sum of non-rounded frequencies was often slightly above or below 1) for a final set of frequencies of
). We rounded these values to three decimal places (because P4 requires frequencies to add up to 1 and the sum of non-rounded frequencies was often slightly above or below 1) for a final set of frequencies of 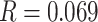 and
and 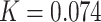 . See pseudocode in the Supplementary material available on dryad or the CompHet.pm module in our GitHub repository for the code used to implement this strategy. See Supplementary materials available on dryad for comparisons of results using 1000 random pairing strategies that show that the paring strategy described in the previous paragraph does not bias the results in favor of non-recoding.
. See pseudocode in the Supplementary material available on dryad or the CompHet.pm module in our GitHub repository for the code used to implement this strategy. See Supplementary materials available on dryad for comparisons of results using 1000 random pairing strategies that show that the paring strategy described in the previous paragraph does not bias the results in favor of non-recoding.
We recoded each simulated data set with both Dayhoff 6-state recoding and S&R 6-state recoding (Supplementary Table S2 available on dryad), and then reconstructed maximum-likelihood trees of these recoded data sets using the GTR multi-state model and of the non-recoded data sets using the Dayhoff and JTT models in RAxML (Stamatakis 2014). We calculated Robinson–Foulds distances (Robinson and Foulds 1981) between each of the resultant 48,000 phylogenies and the trees used for simulation using TOPD/FMTS (Puigbo et al. 2007). We also scored trees based on deep splits, a custom metric (see the is_mono.pl script in the GitHub repository) that evaluates the monophyly of the clade that includes clade-A and clade-B (this evaluation, by definition, also includes the monophyly of the clade that includes clade-C and clade-D). The rationale for this metric rather than Robinson–Foulds distances is that it focused on errors that were most likely due to convergent amino acid compositions (i.e., the pulling together of compositionally homogeneous but unrelated clades or tips). We evaluated deep split accuracy for each combination of model, recoding type (including no recoding), and level of applied compositional heterogeneity (i.e., inflation parameter). We performed chi-squared tests to compare the number of incorrect trees between non-recoding and recoding approaches. To correct for multiple chi-squared testing, we applied the Bonferroni correction at which 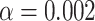 .
.
Testing Six-State Recoding Performance on Saturation
We used Seq-Gen (Rambaut and Grass 1997) to simulate the evolution of amino acids on the Chang and Feuda trees (incorporating both topology and branch-length estimates). We chose Seq-Gen because it has a branch length scaling factor parameter that allows for straight-forward introduction of saturation into simulations. We confirmed that increasing the branch length scaling factor parameter in Seq-Gen linearly increased levels of saturation (Supplementary Fig. S1 available on dryad) using the script seq-gen_saturation_test.pl (available in the accompanying GitHub repository). Next, we performed 1000 simulations per combination of tree (Chang and Feuda), branch length scaling factor parameter (1–20), and model of amino acid substitution (either Dayhoff or JTT) for a total of 80,000 data sets. We simulated an additional 1000 data sets on the Chang topology for a subset of branch length scaling factor parameters (1, 5, 10, 15, 20) under the GTR model using the amino acid rates of substitution, amino acid frequencies (up to three decimal places as in our P4 analysis), and gamma rate heterogeneity estimated from the Chang data set with maximum-likelihood (see shell script run_seqgen_estimated_model.sh in our GitHub repository for detailed parameters), bringing the grand total to 85,000 data sets. Each data set included 1000 amino acid columns.
For simulations performed on the Chang tree, we increased the branch length scaling factor parameter from 1 to 20 in increments of 1. The Feuda tree was produced from recoding the Chang data set (Feuda et al. 2017), and because trees produced from recoded data have substantially fewer substitutions and therefore shorter branch lengths, we multiplied each branch length on the Feuda tree by 2.6 (based on our calculation that the sum of branch lengths in the recoded tree was 2.6 times shorter than the sum of branch lengths in the non-recoded Chang tree).
We performed maximum-likelihood analyses with RAxML for each set of sequences produced from simulations over the Chang and Feuda topologies. For the data sets simulated with Dayhoff and JTT substitution models, we reconstructed trees using the generating model, the six-state recoding scheme derived from that model, and for a subset of branch length scaling factor parameters (1, 5, 10, 15, 20) we also reconstructed trees using LG, a sub-optimal model in this context, as it was not the model used for the simulations. For the data sets simulated with the GTR substitution model, we generated trees using Dayhoff and Dayhoff 6-state recoding. We produced 180,000 phylogenies in total to test saturation. To test the performance of each recoding (or non-recoding) scheme, we calculated Robinson–Foulds distances between the topology used for simulation (i.e., Chang or Feuda) and the reconstructed trees generated from simulated sequences using TOPD/FMTS. We used a 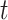 -test to determine if there were significant differences in Robinson–Foulds distances between recoded and non-recoded data sets for each branch length scaling factor. To correct for multiple
-test to determine if there were significant differences in Robinson–Foulds distances between recoded and non-recoded data sets for each branch length scaling factor. To correct for multiple 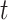 -tests, we applied the Bonferroni correction at which
-tests, we applied the Bonferroni correction at which 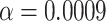 .
.
Testing Alternative Recoding Strategies on Compositional Heterogeneity
To test the effect of the number of states on recoding, we developed alternative Dayhoff 9-, 12-, 15-, and 18-state recoding strategies. The first step in these analyses was to determine the optimal amino acid binning strategy for each number of tested states. Since the number of possible bins for each state is finite, ideally, we would use an exhaustive algorithm to identify the binning scheme that maximizes the sum of intra-bin substitution scores originating from the log odds matrix for PAM 250 (Dayhoff et al. 1978). Unfortunately, as pointed out by Susko and Roger (2007), the number of possible bins is very large (e.g., there are roughly 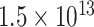 choices of bins under an eight-state recoding strategy) and an exhaustive algorithm is computationally intractable. Instead, we calculated the sum of intra-bin scores using the PAM 250 log odds matrix (see score.pl in our GitHub repository) for several binning schemes that incorporated subsets of the Dayhoff 6-state recoding bins and chose the best-scoring binning strategies from this set (Supplementary Table S3 available on dryad). We also compared our best binning strategies to those proposed in Susko and Roger (2007) using the PAM 250 log odds matrix to calculate intra-bin substitution scores, and in all cases, the scores we generated were higher, except for one which had an equal score (not entirely surprising given that the Susko and Roger bins were optimized for JTT recoding).
choices of bins under an eight-state recoding strategy) and an exhaustive algorithm is computationally intractable. Instead, we calculated the sum of intra-bin scores using the PAM 250 log odds matrix (see score.pl in our GitHub repository) for several binning schemes that incorporated subsets of the Dayhoff 6-state recoding bins and chose the best-scoring binning strategies from this set (Supplementary Table S3 available on dryad). We also compared our best binning strategies to those proposed in Susko and Roger (2007) using the PAM 250 log odds matrix to calculate intra-bin substitution scores, and in all cases, the scores we generated were higher, except for one which had an equal score (not entirely surprising given that the Susko and Roger bins were optimized for JTT recoding).
We compared the binning schemes that scored the highest for each recoding strategy (Table 2) against the Dayhoff and Dayhoff 6-state recoded matrices by testing their performance under reasonably high levels of compositional heterogeneity. We recoded the data that we simulated for the compositional heterogeneity analysis [data simulated with inflation parameter 0.5 using the hypothetical tree 0.002 (Fig. 1a)] using our Dayhoff 9-, 12-, 15-, and 18-state recoding strategies and reconstructed maximum-likelihood trees in RAxML. As in the main compositional heterogeneity analysis outlined above, we calculated deep splits scores (using the script is_mono.pl), to test the monophyly of the clade that included clade-A and clade-B and the clade that included clade-C and clade-D. We also performed a chi-squared test to compare the number of incorrect trees produced under Dayhoff-18 recoding (see Results for rationale) to those produced under non-recoding. To correct for multiple chi-squared testing, we applied the Bonferroni correction at which 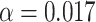 .
.
Table 2.
Best scoring binning schemes optimized on the Dayhoff matrix
| Dayhoff recoding | Binning scheme |
|---|---|
| 9-state | DEHNQ ILMV FY AST KR G P C W |
| 12-state | DEQ MLIV FY KHR G A P S T N W C |
| 15-state | DEQ ML IV FY G A P S T N K H R W C |
| 18-state | ML FY I V G A P S T D E Q N H K R W C |
Results
The Efficacy of Six-State Recoding Under a Compositional Heterogeneity Gradient
We simulated data with various levels of compositional heterogeneity by setting the amino acid frequencies of two non-sister five-taxon clades (e.g., clade-A and clade-C in Fig. 1a) to be highly divergent to the amino acid frequencies of the other two non-sister major clades (e.g., clade-B and clade-D in Fig. 1a) on a balanced 20-taxon tree. We adjusted the level of compositional heterogeneity by increasing the frequency differences of each amino acid between the two sets of frequencies by a factor that we call the inflation parameter. We adjusted the impact of introduced compositional heterogeneity by varying the length of the stem branches leading to those four clades (Fig. 1a). We tested the impact of sequence length by generating alignments of length 1000, 2000, 3000, 4000, and 5000 amino acids (see Supplementary material available on dryad for methods). For each simulated data set, we generated maximum-likelihood trees using recoding and non-recoding approaches. We scored these trees based on Robinson–Foulds distances from the true tree, as well as on whether a tree recovered the two major 10-taxon clades (i.e., a clade containing all clade-A and clade-B taxa and a clade containing all clade-C and clade-D taxa).
For each tree, we simulated 10,000,000 data sets with no introduced compositional heterogeneity (i.e., inflation parameter set to 0) to generate a null distribution of comp-het indices, to which we compared the compositionally heterogeneous data sets. We reconstructed trees on data simulated over hypothetical tree 0.002 for the first 1000 out of these 10,000,000 data sets. In our phylogenetic analyses of these 1000 data sets lacking compositional heterogeneity, recoded data sets performed consistently worse than non-recoded data sets (Supplementary Fig. S2 available on dryad).
Analyses of non-recoded data sets consistently produced trees that were more accurate than those produced on recoded data sets using both our deep splits metric and Robinson–Foulds distances. Despite changes to the stem branch length on the tree and level of compositional heterogeneity implemented by the inflation parameter, non-recoding methods produced more accurate trees (Fig. 1b; Supplementary Fig. S3 available on dryad). While the performance of the recoding approaches diminished at a slower rate than non-recoding approaches (Fig. 1b) under increasing compositional heterogeneity, non-recoding performed significantly better than recoding in all cases tested, except under the highest level of compositional heterogeneity and shortest stem branch (Supplementary Tables S4 and S5 available on dryad). We explored how data size impacted the performance of recoding methods in combination with compositional heterogeneity (details of these analyses are described in Supplementary material available on dryad). As sequence length increased, phylogenetic analyses of non-recoded data sets outperformed analyses of recoded data sets, except under the highest level of compositional heterogeneity (Supplementary Fig. S4 available on dryad; i.e., inflation parameter 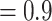 ). Additionally, we explored the effect of tree shape and compositional heterogeneity on recoding methods (analyses described in Supplementary material available on dryad). These results were consistent in that non-recoding methods outperformed recoding under all levels of compositional heterogeneity tested (Supplementary Fig. S6 available on dryad).
). Additionally, we explored the effect of tree shape and compositional heterogeneity on recoding methods (analyses described in Supplementary material available on dryad). These results were consistent in that non-recoding methods outperformed recoding under all levels of compositional heterogeneity tested (Supplementary Fig. S6 available on dryad).
To gauge how our simulated data compared to real data in terms of the levels of compositional heterogeneity, we scored real and simulated data sets using the average relative compositional frequency variability (RCFV) score (Kück and Struck 2014). Higher RCFV scores indicate greater variability in amino acid composition across a data set. We found that the level of compositional heterogeneity (as measured by RCFV) in data sets simulated with the inflation parameter set to 0.9 was substantially higher than the majority of real data sets. The median RCFV score was 0.088 for all data sets simulated under the inflation parameter of 0.9, while the median RCFV score for data from papers in Table 1 was 0.036 (Supplementary Fig. S5 available on dryad). We reason that our simulated data sets therefore are substantially compositionally heterogeneous since these published data sets, many of which used compositional heterogeneity as justification for the application of recoding, are likely enriched for compositional heterogeneity.
The Efficacy of Six-State Recoding Under a Saturation Gradient
We simulated data sets on the Chang and Feuda trees under the Dayhoff and JTT models with increasing levels of saturation. Under all tested levels of saturation, phylogenetic reconstructions using the Dayhoff and LG models on non-recoded data matrices that were simulated under the Dayhoff model produced trees with fewer errors on average (as measured by Robinson-Foulds distances from the true tree) than those that used the Dayhoff 6-state recoded matrix (Fig. 2a). The results were similar for data simulated under the JTT model, where trees reconstructed with the JTT and LG models on non-recoded data matrices contained fewer errors on average across all tested levels of saturation compared to reconstructions with the S&R 6-state recoded matrix (Fig. 2b). The results were consistent regardless of which tree (i.e., Chang or Feuda) was used for data simulations (Supplementary Fig. S7 available on dryad). As saturation increased, the performance of recoding approaches decreased at a faster rate than non-recoding approaches (Supplementary Fig. S7 available on dryad). 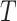 -tests performed for each branch length scaling factor parameter showed that Robinson–Foulds distances were significantly higher for recoded data sets compared to non-recoded data sets (P-value < 2.2e-16).
-tests performed for each branch length scaling factor parameter showed that Robinson–Foulds distances were significantly higher for recoded data sets compared to non-recoded data sets (P-value < 2.2e-16).
Figure 2.

Six-state recoding approaches produce more errors under increasing levels of saturation. Robinson–Foulds distances were calculated for 1000 runs for each branch length scaling factor parameter. All data were simulated on the Chang tree. a) Data sets simulated under the Dayhoff model. b) Data sets simulated under the JTT model. c) Data sets simulated under the GTR model using the amino acid rates of substitution, amino acid frequencies, and gamma rate heterogeneity estimated from the Chang data set.
We also simulated data under the GTR model using the amino acid rates of substitution, amino acid frequencies, and gamma rate heterogeneity parameters estimated from the Chang data set. Phylogenetic analyses of data simulated under GTR resulted in fewer errors on average when reconstructed with non-recoded Dayhoff matrices compared to reconstructions with the Dayhoff 6-state recoded matrices (Fig. 2c). 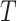 -tests carried out for each branch length scaling factor parameter indicated that recoded approaches performed significantly worse than non-recoded approaches (P-value < 2.2e–16).
-tests carried out for each branch length scaling factor parameter indicated that recoded approaches performed significantly worse than non-recoded approaches (P-value < 2.2e–16).
Furthermore, we tested the combined effects of sequence length and saturation on the performance of recoding strategies (see Supplementary material available on dryad for methods). Increases in sequence length minimized the impact of saturation and reduced errors in phylogenetic reconstruction for both recoding and non-recoding methods. However, non-recoding methods performed significantly better on all sequence lengths and levels of saturation, except for on the largest simulated data set with the lowest level of saturation where results from recoded and non-recoded analyses were equivocal (Supplementary Fig. S8 and Table S6 available on dryad).
The Effect of Alternative Recoding Strategies on Compositional Heterogeneity
We used the data simulated under inflation parameter 0.5 (mid-level of compositional heterogeneity) using the hypothetical tree 0.002 (short stem branches; Fig. 1a) from the main compositional heterogeneity analysis to test Dayhoff 9-, 12-, 15-, and 18-state recoding strategies and compared the performance of these methods to Dayhoff 6-state recoding and non-recoding. As in the main compositional heterogeneity analysis outlined above, trees were assessed by deep splits to determine if they recovered the two compositionally heterogeneous 10-taxon clades (i.e., a monophyletic group of clade-A and clade-B, and a monophyletic group of clade-C and clade-D). The percentage of trees that passed these criteria increased as the number of Dayhoff states increased with Dayhoff 18-state recoding outperforming all other strategies including the non-recoding approach (Fig. 3). Non-recoding outperformed all other recoding strategies except Dayhoff 12- and 15-state recoding under the highest level of compositional heterogeneity (inflation parameter 0.9; Fig. 3c). We performed a chi-squared test to determine if the differences in numbers of incorrect trees between analyses run with Dayhoff 18-state recoding and those run without recoding were significant. The difference was significant only under the highest level of compositional heterogeneity (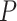 -values for inflation parameters 0.1, 0.5, and 0.9: 0.4314, 0.2183, and 6.622e-06, respectively).
-values for inflation parameters 0.1, 0.5, and 0.9: 0.4314, 0.2183, and 6.622e-06, respectively).
Figure 3.

Dayhoff 9-, 12-, 15- and 18-state recoding produce fewer incorrect trees than Dayhoff 6-state recoding under various levels of compositional heterogeneity. Trees were reconstructed by applying the non-recoded (NR) Dayhoff matrix or alternative Dayhoff recoding strategies (the number of states in the recoding strategy is indicated by digits). Incorrect trees did not include a monophyletic group of taxa from clade-A and clade-B and monophyletic group of taxa from clade-C and clade-D. The 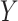 -axis refers to percentage out of 1000 trees.
-axis refers to percentage out of 1000 trees.
Discussion
The philosophy underlying recoding strategies in phylogenetics is that sacrificing some information is beneficial in cases where homoplasy is high, as is the case when there is substantial heterogeneity in nucleotide or amino acid composition or when data sets are highly saturated. Six-state amino acid recoding has been proposed as a strategy to improve phylogenetic reconstruction in the presence of compositional heterogeneity and saturation (Embley et al. 2003a; Hrdy et al. 2004; Martin et al. 2005). While there have been simulation analyses that compare different binning schemes (Susko and Roger 2007; Nesnidal et al. 2010), there are few if any studies that compare the accuracy of six-state recoding to non-recoding approaches. In this study, we used simulations under gradients of compositional heterogeneity and saturation to compare the performance of six-state amino acid recoding strategies. Remarkably, we found that non-recoding approaches outperformed six-state recoding approaches in all of our comparisons. Our results show that while six-state recoding seems to be less affected by increases in compositional heterogeneity, it does not overcome the penalty of information loss even under the highest levels of compositional heterogeneity (Fig. 1b). Furthermore, we found that six-state recoding performs poorly when applied to highly saturated data sets. As such, we conclude that the costs of information loss associated with the six-state recoding schemes are too great to justify applying these strategies.
We confirm that Dayhoff 6-state recoding is inappropriate for phylogenetic inference and our analyses with S&R 6-state recoding show that limitations extend beyond Dayhoff matrices, as six-states likely are too few for reliable phylogenetic analysis. It is possible that not all recoding strategies are inappropriate. Specifically, we found that our Dayhoff 9-, 12-, 15-, and 18-state recoding strategies performed better than the standard Dayhoff 6-state recoding approach for all tested levels of compositional heterogeneity (Fig. 3). Dayhoff 18-state recoding performed the best under all gradients of compositional heterogeneity and may comprise the optimum balance of minimizing compositional heterogeneity while maximizing information retention. However, we do not advocate blindly applying Dayhoff 18-state recoding, especially since significant improvement only occurs under the most extreme compositional heterogeneity setting (0.9), which we show is uncommon in real data sets based on RCFV scores (RCFV scores 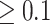 occurred in 6 out of 25 sampled publications; Supplementary Table S7 available on dryad). Nevertheless, conservative recoding approaches under very high levels of compositional heterogeneity may be justified provided that these approaches are properly tested.
occurred in 6 out of 25 sampled publications; Supplementary Table S7 available on dryad). Nevertheless, conservative recoding approaches under very high levels of compositional heterogeneity may be justified provided that these approaches are properly tested.
Applying a recoding method that is data set specific may be another tactic to handle compositional heterogeneity or saturation. Susko and Roger (2007) and Nesnidal et al. (2010) applied this strategy by testing several recoding binning schemes informed by their data sets of interest. Tailoring the level and/or type of recoding to the amount of compositional heterogeneity and saturation, perhaps on a column-by-column basis, may be a successful approach, but further testing using such a tailored method would be necessary. Since only a handful of studies have investigated different recoding schemes, it is clear that more analyses are required to gain an understanding of the impact of alternative recoding methods for compositionally heterogeneous and/or saturated data sets.
Implications
There are at least 91 publications that use six-state amino acid recoding, with 2019 seeing more than any year to date (Table 1). Many of these studies have proposed controversial topologies with profound implications across the tree of life including bacteria, archaea, unicellular eukaryotes, fungi, animals, and plants. We have shown that six-state recoding greatly reduces information content and therefore often results in suboptimal phylogenetic reconstructions. We suggest that these data sets should be reevaluated using criteria that assess the amount of compositional heterogeneity within data sets, and/or reanalyzed using non-recoding approaches unless extreme levels of compositional heterogeneity are evident. When applying recoding, it would be beneficial to determine the number of states in a recoding strategy based on the level of compositional heterogeneity using approaches that we have applied in this study. Nevertheless, we advocate caution when interpreting results stemming from analyses that have employed six-state recoding and contend that published analyses in which six-state recoding approaches substantially influenced the conclusions might need to be revisited.
Acknowledgments
We thank Melissa DeBiasse for providing comments on an earlier version of the manuscript and Gordon Burleigh, Christine Schnitzler, Marta Wayne, and Bryan Kolaczkowski for feedback on this project. The authors would like to express their thanks to David Swofford and Gavin Naylor for influential discussions at an early stage of the project. We also thank Michal Kowalewski for providing guidance on statistics employed and Luis Vargas for assistance in troubleshooting scripts. Lastly, we thank UF SACNAS for providing a supportive space for scientific growth and encouragement. The views expressed in this paper do not necessarily reflect the views of those acknowledged.
Supplementary Material
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.5mkkwh757.
Funding
This work was supported by the National Science Foundation under Grant Number 1542597; and the Graduate Research Fellowship Program to A.M.H. Additional funding to A.M.H. was provided by the Florida Education Fund Mcknight Doctoral Fellowship Program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Andersson J.O., Hirt R.P., Foster P.G., Roger A.J.. 2006. Evolution of four gene families with patchy phylogenetic distributions: influx of genes into protist genomes. BMC Evol. Biol. 6:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi H., Eyre-Walker A.. 1998. Translational selection and molecular evolution. Curr. Opin. Genet. Dev. 8:688–693. [DOI] [PubMed] [Google Scholar]
- Aouad M., Taib N., Oudart A., Lecocq M., Gouy M., Brochier-Armanet C.. 2018. Extreme halophilic archaea derive from two distinct methanogen Class II lineages. Mol. Phylogenet. Evol. 127:46–54. [DOI] [PubMed] [Google Scholar]
- Ballesteros J.A., Santibáñez López C.E., Kováč L., Gavish-Regev E., Sharma P.P.. 2019. Ordered phylogenomic subsampling enables diagnosis of systematic errors in the placement of the enigmatic arachnid order Palpigradi. Proc. R. Soc. B Biol. Sci. 286:20192426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benavides L.R., Cosgrove J.G., Harvey M.S., Giribet G.. 2019. Phylogenomic interrogation resolves the backbone of the Pseudoscorpiones tree of life. Mol. Phylogenet. Evol. 139:106509. [DOI] [PubMed] [Google Scholar]
- Benavides L.R., Pinto-da-Rocha R., Giribet G.. 2021. The phylogeny and evolution of the flashiest of the armored harvestmen (Arachnida: Opiliones). Syst. Biol. syaa080. [DOI] [PubMed] [Google Scholar]
- Bennett G.M., Mao M.. 2018. Comparative genomics of a quadripartite symbiosis in a planthopper host reveals the origins and rearranged nutritional responsibilities of anciently diverged bacterial lineages. Environ. Microbiol. 20:4461–4472. [DOI] [PubMed] [Google Scholar]
- Blanquart S., Lartillot N.. 2006. A Bayesian compound stochastic process for modeling nonstationary and nonhomogeneous sequence evolution. Mol. Biol. Evol. 23:2058–2071. [DOI] [PubMed] [Google Scholar]
- Borowiec M.L., Lee E.K., Chiu J.C., Plachetzki D.C.. 2015. Extracting phylogenetic signal and accounting for bias in whole-genome data sets supports the Ctenophora as sister to remaining Metazoa. BMC Genomics. 16:987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochier-Armanet C., Forterre P., Gribaldo S.. 2011. Phylogeny and evolution of the Archaea: one hundred genomes later. Curr. Opin. Microbiol. 14:274–281. [DOI] [PubMed] [Google Scholar]
- Burki F., Okamoto N., Pombert J.-F., Keeling P.J.. 2012. The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proc. R. Soc. B Biol. Sci. 279:2246–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E.S., Neuhof M., Rubinstein N.D., Diamant A., Philippe H., Huchon D., Cartwright P.. 2015. Genomic insights into the evolutionary origin of Myxozoa within Cnidaria. Proc. Natl. Acad. Sci. USA. 112:14912–14917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S., Xian W., Fu Y., Marin B., Keller J., Wu T., Sun W., Li X., Xu Y., Zhang Y., Wittek S., Reder T., Günther G., Gontcharov A., Wang S., Li L., Liu X., Wang J., Yang H., Xu X., Delaux P.M., Melkonian B., Wong G.K.S., Melkonian M.. 2019. Genomes of subaerial zygnematophyceae provide insights into land plant evolution. Cell. 179: 1057–1067. [DOI] [PubMed] [Google Scholar]
- Collins T.M., Wimberger P.H., Naylor G.J.P.. 1994. Compositional bias, character-state bias, and character-state reconstruction using parsimony. Syst. Biol. 43:482–496. [Google Scholar]
- Courtois P.J. 1977. Decomposability: queueing and computer system applications. New York:Academic Press. [Google Scholar]
- Cox C.J., Foster P.G., Hirt R.P., Harris S.R., Embley T.M.. 2008. The archaebacterial origin of eukaryotes. Proc. Natl. Acad. Sci. USA. 105:20356–20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha T.J., Giribet G.. 2019. A congruent topology for deep gastropod relationships. Proc. R. Soc. B Biol. Sci. 286:20182776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson E.A., van der Giezen M., Horner D.S., Embley T.M., Howe C.J.. 2002. An [Fe] hydrogenase from the anaerobic hydrogenosome-containing fungus Neocallimastixfrontalis L2. Gene. 296:45–52. [DOI] [PubMed] [Google Scholar]
- Dayhoff M., Schwartz R., Orcutt B.. 1978. 22 A model of evolutionary change in proteins. In: Atlas of protein sequence and structure, Vol. 5. Silver Spring (MD): National Biomedical Research Foundation. p. 345–352. [Google Scholar]
- DeBiasse M.B., Ryan, J.F.. 2019. Phylotocol: promoting transparency and overcoming bias in phylogenetics. Syst. Biol. 68: 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derelle R., Lang B.F.. 2012. Rooting the eukaryotic tree with mitochondrial and bacterial proteins. Mol. Biol. Evol. 29:1277–1289. [DOI] [PubMed] [Google Scholar]
- Derelle R., Torruella G., Klimeš V., Brinkmann H., Kim E., Vlĉek È., Lang B.F., Eliáš M.. 2015. Bacterial proteins pinpoint a single eukaryotic root. Proc. Natl. Acad. Sci. USA. 112:E693–E699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delsuc F., Brinkmann H., Chourrout D., Philippe H.. 2006. Tunicates and not cephalochordates are the closest living relatives of vertebrates. Nature. 439:965–968. [DOI] [PubMed] [Google Scholar]
- Deschamps P., Moreira D.. 2009. Signal conflicts in the phylogeny of the primary photosynthetic eukaryotes. Mol. Biol. Evol. 26: 2745–2753. [DOI] [PubMed] [Google Scholar]
- Domman D., Horn M., Embley T.M., Williams T.A.. 2015. Plastid establishment did not require a chlamydial partner. Nat. Commun. 6:6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn C.W., Hejnol A., Matus D.Q., Pang K., Browne W.E., Smith S.A., Seaver E., Rouse G.W., Obst M., Edgecombe G.D., Sørensen M. V., Haddock S.H.D., Schmidt-Rhaesa A., Okusu A., Kristensen R.M., Wheeler W.C., Martindale M.Q., Giribet G.. 2008. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 452:745–749. [DOI] [PubMed] [Google Scholar]
- Eitel M., Francis W.R., Varoqueaux F., Daraspe J., Osigus H.-J., Krebs S., Vargas S., Blum H., Williams G.A., Schierwater B., Wörheide G.. 2018. Comparative genomics and the nature of placozoan species. PLOS Biol. 16:e2005359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embley M., van der Giezen M., Horner D.S., Dyal P.L., Foster P.. 2003a. Mitochondria and hydrogenosomes are two forms of the same fundamental organelle. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 358:191–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embley T.M., van der Giezen M., Horner D., Dyal P., Bell S., Foster P.. 2003b. Hydrogenosomes, mitochondria and early eukaryotic evolution. IUBMB Life (International Union Biochem. Mol. Biol. Life). 55:387–395. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 1978. Cases in which Parsimony or Compatibility Methods will be Positively Misleading. Syst Biol. 27:401–410. [Google Scholar]
- Feuda R., Dohrmann M., Pett W., Lartillot N., Wö G., Pisani D., De C.W.. 2017. Improved modeling of compositional heterogeneity supports sponges as sister to all other animals. Curr. Biol. 27:3864–3870. [DOI] [PubMed] [Google Scholar]
-
Fitzpatrick D.A., Creevey C.J., McInerney J.O..
2006a. Genome phylogenies indicate a meaningful
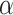 -proteobacterial phylogeny and support a grouping of the mitochondria with the Rickettsiales. Mol. Biol. Evol. 23:74–85. [DOI] [PubMed] [Google Scholar]
-proteobacterial phylogeny and support a grouping of the mitochondria with the Rickettsiales. Mol. Biol. Evol. 23:74–85. [DOI] [PubMed] [Google Scholar] - Fitzpatrick D.A., Logue M.E., Stajich J.E., Butler G.. 2006b. A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol. Biol. 6:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster P.G. 2004. Modeling compositional heterogeneity. Syst. Biol. 53:485–495. [DOI] [PubMed] [Google Scholar]
- Foster P.G., Cox C.J., Embley T.M.. 2009. The primary divisions of life: a phylogenomic approach employing composition-heterogeneous methods. Philos. Trans. R. Soc. B Biol. Sci. 364:2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster P.G., Hickey D.A.. 1999. Compositional bias may affect both DNA-based and protein-based phylogenetic reconstructions. J. Mol. Evol. 48:284–290. [DOI] [PubMed] [Google Scholar]
- Fu C.-J., Sheikh S., Miao W., Andersson S.G.E., Baldauf S.L.. 2014. Missing genes, multiple ORFs, and C-to-U type RNA editing in Acrasis kona (Heterolobosea, Excavata) mitochondrial DNA. Genome Biol. Evol. 6:2240–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haen K.M., Lang B.F., Pomponi S.A., Lavrov D. V.. 2007. Glass sponges and bilaterian animals share derived mitochondrial genomic features: a common ancestry or parallel evolution? Mol. Biol. Evol. 24:1518–1527. [DOI] [PubMed] [Google Scholar]
- He D., Sierra R., Pawlowski J., Baldauf S.L.. 2016. Reducing long-branch effects in multi-protein data uncovers a close relationship between Alveolata and Rhizaria. Mol. Phylogenet. Evol. 101:1–7. [DOI] [PubMed] [Google Scholar]
- Heinz E., Williams T.A., Nakjang S., Noël C.J., Swan D.C., Goldberg A. V., Harris S.R., Weinmaier T., Markert S., Becher D., Bernhardt J., Dagan T., Hacker C., Lucocq J.M., Schweder T., Rattei T., Hall N., Hirt R.P., Embley T.M.. 2012. The genome of the obligate intracellular parasite trachipleistophora hominis: new insights into microsporidian genome dynamics and reductive evolution. PLoS Pathog. 8:e1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hejnol A., Obst M., Stamatakis A., Ott M., Rouse G.W., Edgecombe G.D., Martinez P., Baguñà J., Bailly X., Jondelius U., Wiens M., Müller W.E.G., Seaver E., Wheeler W.C., Martindale M.Q., Giribet G., Dunn C.W.. 2009. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proc. R. Soc. London B Biol. Sci. 276:4261–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendy M.D., Penny D.. 1989. A Framework for the Quantitative Study of Evolutionary Trees. Syst. Zool. 38:297. [Google Scholar]
- Hill M.S., Hill A.L., Lopez J., Peterson K.J., Pomponi S., Diaz M.C., Thacker R.W., Adamska M., Boury-Esnault N., Cárdenas P., Chaves-Fonnegra A., Danka E., De Laine B.-O., Formica D., Hajdu E., Lobo-Hajdu G., Klontz S., Morrow C.C., Patel J., Picton B., Pisani D., Pohlmann D., Redmond N.E., Reed J., Richey S., Riesgo A., Rubin E., Russell Z., Rützler K., Sperling E.A., di Stefano M., Tarver J.E., Collins A.G.. 2013. Reconstruction of family-level phylogenetic relationships within demospongiae (porifera) using nuclear encoded housekeeping genes. PLoS One. 8:e50437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.Y.W., Jermiin L.S.. 2004. Tracing the decay of the historical signal in biological sequence data. Syst. Biol. 53:623–637. [DOI] [PubMed] [Google Scholar]
- Hrdy I., Hirt R.P., Dolezal P., Bardonová L., Foster P.G., Tachezy J., Martin Embley T.. 2004. Trichomonas hydrogenosomes contain the NADH dehydrogenase module of mitochondrial complex I. Nature. 432:618–622. [DOI] [PubMed] [Google Scholar]
- Huang H., Knowles L.L.. 2016. Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Syst. Biol. 65:357–365. [DOI] [PubMed] [Google Scholar]
- Jones D.T., Taylor W.R., Thornton J.M.. 1992. The rapid generation of mutation data matrices from protein sequences. Bioinformatics. 8:275–282. [DOI] [PubMed] [Google Scholar]
- Kayal E., Roure B., Philippe H., Collins A.G., Lavrov D. V.. 2013. Cnidarian phylogenetic relationships as revealed by mitogenomics. BMC Evol. Biol. 13:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemeny J.G., Snell J.L.. 1976. Finite Markov Chains. New York: Springer. [Google Scholar]
- Klinges J.G., Rosales S.M., McMinds R., Shaver E.C., Shantz A.A., Peters E.C., Eitel M., Wörheide G., Sharp K.H., Burkepile D.E., Silliman B.R., Vega Thurber R.L.. 2019. Phylogenetic, genomic, and biogeographic characterization of a novel and ubiquitous marine invertebrate-associated Rickettsiales parasite, Candidatus Aquarickettsia rohweri, gen. nov., sp. nov. ISME J. 13:2938–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R.D., Freeland S.J., Landweber L.F.. 2001. A simple model based on mutation and selection explains trends in codon and amino-acid usage and GC composition within and across genomes. Genome Biol. 2:research0010.1–0010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosiol C., Goldman N., Buttimore N.H.. 2004. A new criterion and method for amino acid classification. J. Theor. Biol. 228:97–106. [DOI] [PubMed] [Google Scholar]
- Kubatko L.S., Degnan J.H.. 2007. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56:17–24. [DOI] [PubMed] [Google Scholar]
- Kuhner M.K., Felsenstein J.. 1994. A simulation comparison of phylogeny algorithms under equal and unequal evolutionary rates. Mol. Biol. Evol. 11:459–468. [DOI] [PubMed] [Google Scholar]
- Lasek-Nesselquist E. 2012. A mitogenomic re-evaluation of the bdelloid phylogeny and relationships among the syndermata. PLoS One. 7:e43554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek-Nesselquist E., Gogarten J.P.. 2013. The effects of model choice and mitigating bias on the ribosomal tree of life. Mol. Phylogenet. Evol. 69:17–38. [DOI] [PubMed] [Google Scholar]
- Laumer C.E., Fernández R., Lemer S., Combosch D., Kocot K.M., Riesgo A., Andrade S.C.S., Sterrer W., Sørensen M. V., Giribet G.. 2019. Revisiting metazoan phylogeny with genomic sampling of all phyla. Proc. R. Soc. B Biol. Sci. 286:20190831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumer C.E., Gruber-Vodicka H., Hadfield M.G., Pearse V.B., Riesgo A., Marioni J.C., Giribet G.. 2018. Support for a clade of Placozoa and Cnidaria in genes with minimal compositional bias. Elife. 7:e36278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T.J., Amrine K.C.H., Swingley W.D., Ardell D.H.. 2019. TRNA functional signatures classify plastids as late-branching cyanobacteria. BMC Evol. Biol. 19:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leliaert F., Tronholm A., Lemieux C., Turmel M., DePriest M.S., Bhattacharya D., Karol K.G., Fredericq S., Zechman F.W., Lopez-Bautista J.M.. 2016. Chloroplast phylogenomic analyses reveal the deepest-branching lineage of the Chlorophyta, Palmophyllophyceae class. nov. Sci. Rep. 6:25367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemer S., Bieler R., Giribet G.. 2019. Resolving the relationships of clams and cockles: dense transcriptome sampling drastically improves the bivalve tree of life. Proc. R. Soc. B Biol. Sci. 286:20182684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux C., Otis C., Turmel M.. 2014. Chloroplast phylogenomic analysis resolves deep-level relationships within the green algal class Trebouxiophyceae. BMC Evol. Biol. 14:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Lemer S., Kirkendale L., Bieler R., Cavanaugh C., Giribet G.. 2020. Shedding light: a phylotranscriptomic perspective illuminates the origin of photosymbiosis in marine bivalves. BMC Evol. Biol. 20:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano-Fernandez J., Tanner A.R., Giacomelli M., Carton R., Vinther J., Edgecombe G.D., Pisani D.. 2019. Increasing species sampling in chelicerate genomic-scale datasets provides support for monophyly of Acari and Arachnida. Nat. Commun. 10:2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H. 2015. Evolutionary origin of a streamlined marine bacterioplankton lineage. ISME J. 9:1423–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H., Csüros M., Hughes A.L., Moran M.A.. 2013. Evolution of divergent life history strategies in marine alphaproteobacteria. MBio. 4:e00373–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H., Swan B.K., Stepanauskas R., Hughes A.L., Moran M.A.. 2014. Evolutionary analysis of a streamlined lineage of surface ocean Roseobacters. ISME J. 8:1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzano-Marín A., Coeur d’acier A., Clamens A.-L., Orvain C., Cruaud C., Barbe V., Jousselin E.. 2018. A freeloader? The highly eroded yet large genome of the serratia symbiotica symbiont of cinara strobi. Genome Biol. Evol. 10:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlétaz F., Peijnenburg K.T.C.A., Goto T., Satoh N., Rokhsar D.S.. 2019. A new spiralian phylogeny places the enigmatic arrow worms among gnathiferans. Curr. Biol. 29:312-318.e3. [DOI] [PubMed] [Google Scholar]
- Martin W., Deusch O., Stawski N., Grünheit N., Goremykin V.. 2005. Chloroplast genome phylogenetics: why we need independent approaches to plant molecular evolution. Trends Plant Sci. 10:203–209. [DOI] [PubMed] [Google Scholar]
- Masta S.E., Longhorn S.J., Boore J.L.. 2009. Arachnid relationships based on mitochondrial genomes: Asymmetric nucleotide and amino acid bias affects phylogenetic analyses. Mol. Phylogenet. Evol. 50:117–128. [DOI] [PubMed] [Google Scholar]
- Matsumoto T., Shinozaki F., Chikuni T., Yabuki A., Takishita K., Kawachi M., Nakayama T., Inouye I., Hashimoto T., Inagaki Y.. 2011. Green-colored plastids in the dinoflagellate genus lepidodinium are of core chlorophyte origin. Protist. 162:268–276. [DOI] [PubMed] [Google Scholar]
- Moore K.R., Magnabosco C., Momper L., Gold D.A., Bosak T., Fournier G.P.. 2019. An expanded ribosomal phylogeny of cyanobacteria supports a deep placement of plastids. Front. Microbiol. 10:1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan C.C., Foster P.G., Webb A.E., Pisani D., McInerney J.O., O’Connell M.J.. 2013. Heterogeneous models place the root of the placental mammal phylogeny. Mol. Biol. Evol. 30:2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroz L.L., Kocot K.M., Citarella M.R., Dosung S., Norekian T.P., Povolotskaya I.S., Grigorenko A.P., Dailey C., Berezikov E., Buckley K.M., Ptitsyn A., Reshetov D., Mukherjee K., Moroz T.P., Bobkova Y., Yu F., Kapitonov V. V., Jurka J., Bobkov Y. V., Swore J.J., Girardo D.O., Fodor A., Gusev F., Sanford R., Bruders R., Kittler E., Mills C.E., Rast J.P., Derelle R., Solovyev V. V., Kondrashov F.A., Swalla B.J., Sweedler J. V., Rogaev E.I., Halanych K.M., Kohn A.B.. 2014. The ctenophore genome and the evolutionary origins of neural systems. Nature. 510:109–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan Kutty S., Meusemann K., Bayless K.M., Marinho M.A.T., Pont A.C., Zhou X., Misof B., Wiegmann B.M., Yeates D., Cerretti P., Meier R., Pape T.. 2019. Phylogenomic analysis of Calyptratae: resolving the phylogenetic relationships within a major radiation of Diptera. Cladistics. 35:605-622. [DOI] [PubMed] [Google Scholar]
- Nesnidal M.P., Helmkampf M., Bruchhaus I., Hausdorf B.. 2010. Compositional heterogeneity and phylogenomic inference of metazoan relationships. Mol. Biol. Evol. 27:2095–2104. [DOI] [PubMed] [Google Scholar]
- Neumann J.S., Desalle R., Narechania A., Schierwater B., Tessler M.. 2020. Morphological Characters Can Strongly Influence Early Animal Relationships Inferred from Phylogenomic Data Sets. Syst. Biol. 0:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y., Kamikawa R., Hashimoto T., Inagaki Y.. 2012. Separate origins of group I introns in two mitochondrial genes of the Katablepharid Leucocryptos marina. PLoS One. 7:e37307. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
O’Halloran D.M., Fitzpatrick D.A., McCormack G.P., McInerney J.O., Burnell A.M..
2006. The molecular phylogeny of a nematode-specific clade of heterotrimeric G-protein
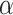 -subunit genes. J. Mol. Evol. 63:87–94. [DOI] [PubMed] [Google Scholar]
-subunit genes. J. Mol. Evol. 63:87–94. [DOI] [PubMed] [Google Scholar] - Ometto L., Cestaro A., Ramasamy S., Grassi A., Revadi S., Siozios S., Moretto M., Fontana P., Varotto C., Pisani D., Dekker T., Wrobel N., Viola R., Pertot I., Cavalieri D., Blaxter M., Anfora G., Rota-Stabelli O.. 2013. Linking genomics and ecology to investigate the complex evolution of an invasive Drosophila pest. Genome Biol. Evol. 5:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero-Bravo A., Goffredi S., Sabree Z.L.. 2018. Cladogenesis and genomic streamlining in extracellular endosymbionts of tropical stink bugs. Genome Biol. Evol. 10:680–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A., Braun E.L.. 2020. Phylogenetic analyses of sites in different protein structural environments result in distinct placements of the metazoan root. Biology. 9:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey L.W., Grant J., Tekle Y.I., Lasek-Nesselquist E., Morrison H.G., Sogin M.L., Patterson D.J., Katz L.A.. 2010. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst. Biol. 59:518–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean C., Deschamps P., López-García P., Moreira D., Brochier-Armanet C.. 2015. Extending the conserved phylogenetic core of archaea disentangles the evolution of the third domain of life. Mol. Biol. Evol. 32:1242–1254. [DOI] [PubMed] [Google Scholar]
- Pett W., Adamski M., Adamska M., Francis W.R., Eitel M., Pisani D., Wörheide G.. 2019. The role of homology and orthology in the phylogenomic analysis of metazoan gene content. Mol. Biol. Evol. 36:643–649. [DOI] [PubMed] [Google Scholar]
- Philip G.K., Creevey C.J., McInerney J.O.. 2005. The Opisthokonta and the Ecdysozoa may not be clades: stronger support for the grouping of plant and animal than for animal and fungi and stronger support for the Coelomata than Ecdysozoa. Mol. Biol. Evol. 22: 1175–1184. [DOI] [PubMed] [Google Scholar]
- Philippe H., Brinkmann H., Copley R.R., Moroz L.L., Nakano H., Poustka A.J., Wallberg A., Peterson K.J., Telford M.J.. 2011. Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature. 470:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H., Derelle R., Lopez P., Pick K., Borchiellini C., Boury-Esnault N., Vacelet J., Renard E., Houliston E., Quéinnec E., Da Silva C., Wincker P., Le Guyader H., Leys S., Jackson D.J., Schreiber F., Erpenbeck D., Morgenstern B., Wörheide G., Manuel M.. 2009. Phylogenomics revives traditional views on deep animal relationships. Curr. Biol. 19:706–712. [DOI] [PubMed] [Google Scholar]
- Philippe H., Poustka A.J., Chiodin M., Hoff K.J., Dessimoz C., Tomiczek B., Schiffer P.H., Müller S., Domman D., Horn M., Kuhl H., Timmermann B., Satoh N., Hikosaka-Katayama T., Nakano H., Rowe M.L., Elphick M.R., Thomas-Chollier M., Hankeln T., Mertes F., Wallberg A., Rast J.P., Copley R.R., Martinez P., Telford M.J.. 2019. Mitigating anticipated effects of systematic errors supports sister-group relationship between Xenacoelomorpha and Ambulacraria. Curr. Biol. 29:1818–1826. [DOI] [PubMed] [Google Scholar]
- Phillips M.J., Lin Y.-H., Harrison G., Penny D.. 2001. Mitochondrial genomes of a bandicoot and a brushtail possum confirm the monophyly of australidelphian marsupials. Proc. R. Soc. Lond. Ser. B Biol. Sci. 268:1533–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick K.S., Philippe H., Schreiber F., Erpenbeck D., Jackson D.J., Wrede P., Wiens M., Alié A., Morgenstern B., Manuel M., Wörheide G.. 2010. Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Mol. Biol. Evol. 27:1983–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani D., Pett W., Dohrmann M., Feuda R., Rota-Stabelli O., Philippe H., Lartillot N., Wörheide G.. 2015. Genomic data do not support comb jellies as the sister group to all other animals. Proc. Natl. Acad. Sci. USA. 112:15402–15407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons J., Ribera I., Bertranpetit J., Balke M.. 2010. Nucleotide substitution rates for the full set of mitochondrial protein-coding genes in Coleoptera. Mol. Phylogenet. Evol. 56:796–807. [DOI] [PubMed] [Google Scholar]
- Puigbo P., Garcia-Vallve S., McInerney J.O.. 2007. TOPD/FMTS: a new software to compare phylogenetic trees. Bioinformatics. 23:1556–1558. [DOI] [PubMed] [Google Scholar]
- Puttick M.N., Morris J.L., Williams T.A., Cox C.J., Edwards D., Kenrick P., Pressel S., Wellman C.H., Schneider H., Pisani D., Donoghue P.C.J.. 2018. The interrelationships of land plants and the nature of the ancestral embryophyte. Curr. Biol. 28:733–745. [DOI] [PubMed] [Google Scholar]
- Rambaut A., Grass N.C.. 1997. Seq-Gen: an application for the Monte Carlo simulation of DNA sequence evolution along phylogenetic trees. Bioinformatics. 13:235–238. [DOI] [PubMed] [Google Scholar]
- Raymann K., Forterre P., Brochier-Armanet C., Gribaldo S.. 2014. Global phylogenomic analysis disentangles the complex evolutionary history of DNA replication in Archaea. Genome Biol. Evol. 6:192–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D.F., Foulds L.R.. 1981. Comparison of phylogenetic trees. Math. Biosci. 53:131–147. [Google Scholar]
- Rodríguez-Ezpeleta N., Embley T.M.. 2012. The SAR11 group of alpha-proteobacteria is not related to the origin of mitochondria. PLoS One. 7:e30520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rota-Stabelli O., Lartillot N., Philippe H., Pisani D.. 2013. Serine codon-usage bias in deep phylogenomics: pancrustacean relationships as a case study. Syst. Biol. 62:121–133. [DOI] [PubMed] [Google Scholar]
- Ryan J.F., Pang K., Schnitzler C.E., Nguyen A.-D., Moreland R.T., Simmons D.K., Koch B.J., Francis W.R., Havlak P., Smith S.A., Putnam N.H., Haddock S.H.D., Dunn C.W., Wolfsberg T.G., Mullikin J.C., Martindale M.Q., Baxevanis A.D.. 2013. The genome of the ctenophore mnemiopsis leidyi and its implications for cell type evolution. Science. 342:1242592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwentner M., Combosch D.J., Pakes Nelson J., Giribet G.. 2017. A phylogenomic solution to the origin of insects by resolving crustacean-hexapod relationships. Curr. Biol. 27:1818–1824. [DOI] [PubMed] [Google Scholar]
- Schwentner M., Richter S., Rogers D.C., Giribet G.. 2018. Tetraconatan phylogeny with special focus on Malacostraca and Branchiopoda: highlighting the strength of taxon-specific matrices in phylogenomics. Proc. R. Soc. B Biol. Sci. 285:20181524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.-X., Todd Hittinger C., Rokas A.. 2017. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat. Ecol. Evol. 1:0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S., Clarke D.J., Lemmon A.R., Moriarty Lemmon E., Aitken A.L., Haddad S., Farrell B.D., Marvaldi A.E., Oberprieler R.G., McKenna D.D.. 2017. Phylogenomic data yield new and robust insights into the phylogeny and evolution of Weevils. Mol. Biol. Evol. 35:823–836. [DOI] [PubMed] [Google Scholar]
- Simion P., Philippe H., Baurain D., Jager M., Richter D.J., Di Franco A., Roure B., Satoh N., Quéinnec É., Ereskovsky A., Lapébie P., Corre E., Delsuc F., King N., Wörheide G., Manuel M.. 2017. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr. Biol. 27:958–967. [DOI] [PubMed] [Google Scholar]
- Singer G.A.C., Hickey D.A.. 2000. Nucleotide bias causes a genomewide bias in the amino acid composition of proteins. Mol. Biol. Evol. 17:1581–1588. [DOI] [PubMed] [Google Scholar]
- Song N., An S.-H., Yin X.-M., Zhao T., Wang X.-Y.. 2016. Insufficient resolving power of mitogenome data in deciphering deep phylogeny of Holometabola. J. Syst. Evol. 54:545–559. [Google Scholar]
- Sousa F., Foster P.G., Donoghue P.C.J., Schneider H., Cox C.J.. 2018. Nuclear protein phylogenies support the monophyly of the three bryophyte groups (Bryophyta Schimp.). New Phytol. 222: 565–575. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susko E., Roger A.J.. 2007. On reduced amino acid alphabets for phylogenetic inference. Mol. Biol. Evol. 24:2139–2150. [DOI] [PubMed] [Google Scholar]
- Swofford D.L., Waddell P.J., Huelsenbeck J.P., Foster P.G., Lewis P.O., Rogers J.S.. 2001. Bias in phylogenetic estimation and its relevance to the choice between parsimony and likelihood methods. Syst. Biol. 50:525–539. [PubMed] [Google Scholar]
- Szabó G., Schulz F., Toenshoff E.R., Volland J.-M., Finkel O.M., Belkin S., Horn M.. 2017. Convergent patterns in the evolution of mealybug symbioses involving different intrabacterial symbionts. ISME J. 11:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrío R., Rodríguez-Trelles F., Ayala F.J.. 2001. Shared nucleotide composition biases among species and their impact on phylogenetic reconstructions of the Drosophilidae. Mol. Biol. Evol. 18:1464–1473. [DOI] [PubMed] [Google Scholar]
- Telford M.J., Budd G.E., Philippe H.. 2015. Phylogenomic insights into animal evolution. Curr. Biol. 25:R876–R887. [DOI] [PubMed] [Google Scholar]
- Tikhonenkov D.V., Mikhailov K.V., Hehenberger E., Karpov S.A., Prokina K.I., Esaulov A.S., Belyakova O.I., Mazei Y.A., Mylnikov A.P., Aleoshin V.V., Keeling P.J.. 2020. New Lineage of Microbial Predators Adds Complexity to Reconstructing the Evolutionary Origin of Animals. Curr. Biol. 30:4500–4509. [DOI] [PubMed] [Google Scholar]
- Torruella G., Derelle R., Paps J., Lang B.F., Roger A.J., Shalchian-Tabrizi K., Ruiz-Trillo I.. 2011. Phylogenetic relationships within the Opisthokonta based on phylogenomic analyses of conserved single-copy protein domains. Mol. Biol. Evol. 29:531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uribe J.E., Irisarri I., Templado J., Zardoya R.. 2019. New patellogastropod mitogenomes help counteracting long-branch attraction in the deep phylogeny of gastropod mollusks. Mol. Phylogenet. Evol. 133:12–23. [DOI] [PubMed] [Google Scholar]
- Wang X., Lavrov D. V.. 2006. Mitochondrial genome of the Homoscleromorph Oscarella carmela (Porifera, Demospongiae) reveals unexpected complexity in the common ancestor of sponges and other animals. Mol. Biol. Evol. 24:363–373. [DOI] [PubMed] [Google Scholar]
- Wang Z., Wu M.. 2015. An integrated phylogenomic approach toward pinpointing the origin of mitochondria. Sci. Rep. 5:7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan N. V, Kocot K.M., Moroz L.L., Halanych K.M.. 2015. Error, signal, and the placement of Ctenophora sister to all other animals. Proc. Natl. Acad. Sci. USA. 112:5773–5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinheimer A.R., Aylward F.O.. 2020. A distinct lineage of Caudovirales that encodes a deeply branching multi-subunit RNA polymerase. Nat. Commun. 11:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T.A., Embley T.M., Heinz E.. 2011. Informational gene phylogenies do not support a fourth domain of life for nucleocytoplasmic large DNA viruses. PLoS One. 6:e21080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T.A., Szöllõsi G.J., Spang A., Foster P.G., Heaps S.E., Boussau B., Ettema T.J.G., Embley T.M.. 2017. Integrative modeling of gene and genome evolution roots the archaeal tree of life. Proc. Natl. Acad. Sci. USA. 114:E4602–E4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodniok S., Brinkmann H., Glöckner G., Heidel A.J., Philippe H., Melkonian M., Becker B.. 2011. Origin of land plants: do conjugating green algae hold the key? BMC Evol. Biol. 11:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woese C.R., Achenbach L., Rouviere P., Mandelco L.. 1991. Archaeal phylogeny: reexamination of the phylogenetic position of Archaeoglohus fulgidus in light of certain composition-induced artifacts. Syst. Appl. Microbiol. 14:364–371. [DOI] [PubMed] [Google Scholar]
- Wolfe J.M., Breinholt J.W., Crandall K.A., Lemmon A.R., Lemmon E.M., Timm L.E., Siddall M.E., Bracken-Grissom H.D.. 2019. A phylogenomic framework, evolutionary timeline and genomic resources for comparative studies of decapod crustaceans. Proc. R. Soc. B Biol. Sci. 286:20190079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L., Buenaventura E., Pape T., Narayanan Kutty S., Bayless K.M., Zhang D.. 2020. A phylotranscriptomic framework for flesh fly evolution (Diptera, Calyptratae, Sarcophagidae). Cladistics. 0:1–19. [DOI] [PubMed] [Google Scholar]
- Yoshida Y., Koutsovoulos G., Laetsch D.R., Stevens L., Kumar S., Horikawa D.D., Ishino K., Komine S., Kunieda T., Tomita M., Blaxter M., Arakawa K.. 2017. Comparative genomics of the tardigrades Hypsibius dujardini and Ramazzottius varieornatus. PLOS Biol. 15:e2002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Sun Y., Jiao N., Stepanauskas R., Luo H.. 2016. Ecological genomics of the uncultivated marine roseobacter lineage CHAB-I-5. Appl. Environ. Microbiol. 82:2100–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zverkov O.A., Mikhailov K. V., Isaev S. V., Rusin L.Y., Popova O. V., Logacheva M.D., Penin A.A., Moroz L.L., Panchin Y. V., Lyubetsky V.A., Aleoshin V. V.. 2019. Dicyemida and Orthonectida: two stories of body plan simplification. Front. Genet. 10:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwickl D.J., Hillis D.M.. 2002. Increased taxon sampling greatly reduces phylogenetic error. Syst. Biol. 51:588–598. [DOI] [PubMed] [Google Scholar]