

Abstract
Background:
To collect preliminary data on the effects of mexiletine on cortical and axonal hyperexcitability in sporadic amyotrophic lateral sclerosis (ALS) in a phase 2 double-blind randomized controlled trial.
Methods:
Twenty ALS subjects were randomized to placebo and mexiletine 300 or 600 mg daily for 4 wk and assessed by transcranial magnetic stimulation and axonal excitability studies. The primary endpoint was change in resting motor threshold (RMT).
Results:
RMT was unchanged with 4 wk of mexiletine (combined active therapies) as compared to placebo, which showed a significant increase (P = .039). Reductions of motor evoked potential (MEP) amplitude (P = .013) and accommodation half-time (P = .002), secondary outcome measures of cortical and axonal excitability, respectively, were also evident at 4 wk on mexiletine.
Conclusions:
The relative stabilization of RMT in the treated subjects was unexpected and could be attributed to unaccounted sources of error or chance. However, a possible alternative cause is neuromodulation preventing an increase. The change in MEP amplitude and accommodation half-time supports the reduction of cortical and axonal hyperexcitability with mexiletine.
Keywords: amyotrophic lateral sclerosis, axonal excitability, outcome research, randomized controlled clinical trial, transcranial magnetic stimulation
1 ∣. INTRODUCTION
Cortical and axonal hyperexcitability have been previously demonstrated in patients with both familial amyotrophic lateral sclerosis (ALS) and sporadic ALS (SALS) using transcranial magnetic stimulation (TMS)1-5 and threshold tracking nerve conduction studies (TTNCS).5,6 Such hyperexcitability, also shown in vitro and in a murine model for ALS, has been hypothesized to play a role in its pathogenesis. Cortical hyperexcitability has been demonstrated in ALS to be heralded by reduction of short interval intracortical inhibition (SICI)7,8 measured by TMS and associated with an adverse prognosis.7-15 In contrast, changes in resting motor threshold (RMT) have been more variable in ALS, with some studies suggesting a reduction of RMT in early disease and an increased threshold in later disease stages.7,12 In conjunction with cortical hyperexcitability, increased strength duration time constant (SDTC) and abnormalities of threshold electrotonus have also been reported in ALS, indicating axonal hyperexcitability, and have been associated with clinical features of ALS, including cramps, fasciculations, and split hand syndrome, as well as shorter survival.16-20
Mexiletine, a cardiac antiarrhythmic agent and use-dependent sodium channel blocker, has been shown to inhibit neuronal hyperexcitability both in vitro21 and in vivo.22 Mexiletine has also been found recently to be safe and largely tolerable in ALS subjects and to reduce the frequency and severity of muscle cramps,23 considered to be a manifestation of axonal hyperexcitability of peripheral motor nerves in this disease. The study failed to show significant effects on markers of disease progression but was not powered to do so.23 To assess the potential for mexiletine as a biotherapeutic in ALS, we performed an additional randomized placebo-controlled multicenter study using mexiletine to determine its effects on pharmacodynamic markers of cortical and axonal excitability using single and dual pulse TMS and TTNCS, respectively.
2 ∣. METHODS
2.1 ∣. Standard protocol approvals, registrations, and patient consents
The study was performed at 10 member sites of the Northeast ALS clinical trials consortium from February 2017 to September 2018. The institutional review board of each participating site approved the study protocol and all amendments. Written informed consent was obtained at the screening visit. The study was registered on clinicaltrials.gov (NCT02781454). The full protocol can be accessed as an online supplemental file.
2.2 ∣. General study design
Subjects with SALS were randomized in a 1:1:1 ratio to receive for 4 wk either (a) 600 mg/day of mexiletine, (b) 300 mg/day of mexiletine, or (c) placebo divided into two doses daily. We decided upon two dosages of the medication, 300 and 600 mg/day, based on the previous mexiletine study which demonstrated some tolerability concerns (mostly severe nausea) among subjects treated with 900 mg/day. The study drug and matching placebo was manufactured by the University of Iowa research pharmacy and distributed by the Clinical Materials Services Unit of the University of Rochester. Subjects were assigned to treatment groups using a computer-generated permuted-block randomization schedule, stratified by site, by a coordination center (Massachusetts General Hospital).
2.3 ∣. Subject selection criteria
Eligible subjects were 18 to 80 y old with possible, laboratory-supported probable, probable, or definite SALS as defined by revised El Escorial criteria,24 slow vital capacity (SVC) of ≥50% of predicted value, and disease duration ≤60 mo from symptom onset. Given the interest in testing effects of mexiletine on cortical and axonal hyperexcitability, subjects had to have a resting motor threshold of less than or equal to 83% of maximum stimulator output by TMS and a median compound muscle action potential (CMAP) of at least 1.5 mV by TTNCS. Subjects were excluded if they had a history of previous myocardial infarction, cardiomyopathy, or cardiac arrhythmia, previous use of mexiletine, implantation of a diaphragmatic pacer ≤60 days prior to the baseline study visit, or use of another investigation medication ≤30 days prior to the baseline study visit. They had to be on a stable dosage of riluzole and edaravone and any medication used to treat muscle cramps for ≥60 days or have been off these medications ≥30 days prior to randomization.
2.4 ∣. Study procedures
Screening procedures for all ALS subjects included obtaining informed consent, collection of demographic information, vital signs, medication review, and neurophysiological testing (TMS and TTNCS). Procedures also included obtaining the ALS diagnostic history, performing a physical and neurological examination, electrocardiography, hand-held dynamometry (HHD), laboratory safety panel (blood chemistry, complete blood count, serum pregnancy test for women of child-bearing potential, and urinalysis), administration of the revised ALS Functional Rating Scale (ALSFRS-R) questionnaire, and seated slow vital capacity (SVC).
Eligible subjects were assessed at a baseline visit performed within 21 days of the screening visit, at which time the study medication was initiated, a week 4 visit when the study medication was discontinued, and at a week 8 visit. Subjects were contacted by telephone at week 1. Procedures at all in-person visits included recording of vital signs, electrocardiography, assessment of adverse events (AEs), neurophysiological testing, and assessment of muscle cramps and fasciculations. A muscle cramp was defined as a sustained painful muscle contraction lasting seconds to minutes as distinguished from a fasciculation, which was defined as an involuntary muscle twitch. At baseline, subjects were asked to estimate the frequency of muscle cramps they had experienced in the previous 7 and 30 days as well as the intensity of associated pain on a visual analog scale from 0 to 10 during those time periods. Subjects were also asked to recall the average duration (hours/day) of fasciculations experienced during the previous 14 days, whether they occurred with exertion, rest, or both, and the degree to which the fasciculations interfered with their daily activities (none, minimal, or moderate) as well as any interference with sleep.
Two-day training workshops were held for all site neurophysiologists for TMS at the Berenson-Allen Center for Noninvasive Brain Stimulation at Beth Israel Deaconess Medical Center, Boston, and for TTNCS at the Neuromuscular Disorders clinic at Beth Israel Deaconess Medical Center, Boston. Prior to performing testing on ALS subjects, neurophysiologists at all sites had to perform TMS and TTNCS on the same healthy volunteer over a minimum of 3 separate days. These studies were reviewed remotely by blinded TMS and TTNCS evaluators (Drs. Courtney McIlduff and Steve Vucic, respectively) to ensure intra-operator reliability. All TMS was performed with a figure-8 coil using either a BiStim device (MagStim, Whitland South West Wales, UK) using Signal software (Cambridge, UK) or a MagVenture system (Alpharetta, GA, USA). RMT was assessed by single pulse TMS performed over the motor cortex with recording over the abductor pollicis brevis (APB) and first dorsal interosseous (FDI) muscles (FDI as control) using an AD Instruments Power Lab T26 amplifier and Labchart software. The side of stimulation was determined based on assessment of strength of the APB muscles bilaterally using manual muscle testing (MMT), with the left motor cortex stimulated and the right hand tested if neither muscle demonstrated weakness (5/5 on the Medical Research Council scale). Otherwise, testing was performed on an affected but only moderately weak muscle (≥3/5), stimulating from the contralateral motor cortex.
The RMT was defined as the minimum stimulus intensity required for 50% of pulses to elicit a motor evoked potential (MEP) amplitude of at least 50 μV and was measured as a percentage of maximal stimulus output (MSO). Cortical inexcitability was demonstrated if no MEP could be distinguished with a maximal stimulus intensity of 100% upon multiple attempts. SICI was measured by dual pulse TMS with conditioned (80% of RMT) and test pulses (120% of RMT) at an interstimulus interval of 3 ms. Paired-pulse testing consisted of 98 pulses, divided into seven blocks of 14 pulses separated by pre-specified intervals between 5 and 7 s. Each block contained two single pulses with stimulation amplitude 80% of RMT, four single pulses with amplitude 120% of RMT, four paired pulses for SICI measurement (3 ms interstimulus interval), and four paired pulses for intracortical facilitation (ICF, 15 ms interstimulus interval) measurement. SICI and ICF were each defined as the ratio of the conditioned MEP amplitude to the unconditioned MEP amplitude. SICI, ICF, and MEP values were log-transformed prior to analysis given right skew of their distributions.
We report SICI−1 so that higher values reflect stronger inhibition. Input-output curves were generated by recording MEPs at varying levels of stimulation in a pseudo-randomized fashion using 3 blocks of 30 single pulses, with each block containing three pulses at stimulation levels 20% to 100% of MSO in 10% increments. The cortical silent period (CSP) was induced by having patients continuously activate the APB at approximately 30% of maximal voluntary contraction with a block of 10 single pulses delivered at 120% of the RMT. The CSP was recorded as the duration from the end of the MEP response to the resumption of electromyography activity. TMS raw data were analyzed blindly using custom MATLAB software (Mathworks, Natick, MA) by a single reviewer. TMS measurements were accepted for analysis if the overall study and individual measurements of RMT, input-output curves, and CSP were judged of good quality by the remote blinded evaluator, and RMT was ≤83% of MSO as required to perform the SICI stimulation protocol at 120% of RMT.
Axonal excitability of the median nerve was performed by stimulating with a Digitimer DS5 stimulator (Digitimer, UK) to stimulate and Viking electromyography machine for recording of motor responses. Median CMAPs were recorded by standard motor nerve conduction study at each site, with the side dictated by MMT as described for TMS. For both TMS and TTNCS, Natus electrodes were used. The cathode was placed 3 cm proximal to the distal wrist crease, and the anode was positioned 10 cm proximally along the median nerve and then 2 cm medially. Skin temperature was maintained above 32°C. Determinations were made of the SDTC, depolarizing threshold electrotonus 90-100 ms (TEd 90-100 ms), hyperpolarizing threshold electrotonus 90-100 ms (TEh 90-100 ms), accommodation half-time, and parameters of recovery cycle analysis including superexcitability and late subexcitability as well the peak CMAP amplitude using QTRACS software (Hugh Bostock, University College London) as previously described.25-28
2.5 ∣. Outcomes
The primary outcome was RMT due to its high within-subject reproducibility compared to other TMS parameters such as SICI.29 Secondary outcome measures included other parameters of TMS, specifically SICI, MEP amplitude obtained stimulating at 120% and 140% of the RMT and then normalized to the peak CMAP amplitude to account for decline in the latter as a reflection of lower motor neuron degeneration, and CSP, and the previously described measures of TTNCS, especially SDTC given that it is thought to reflect persistent inward sodium current.18 Additional secondary endpoints were changes in muscle cramp frequency and intensity on a 10 point visual analogue scale and fasciculations as a percentage of each day, as assessed by a daily diary tabulated weekly. Exploratory outcome measures included SVC, HHD of the APB, ALSFRS-R total score, and quality of life measured by the RAND-36 instrument.30
2.6 ∣. Sample size
Based on a report of a mean 4-wk change in RMT among 18 ALS patients of 6.2% and a SD of 5.1% (Dr. Steve Vucic, personal communication), it was estimated that a sample size of 60 subjects randomized 1:1:1 with up to 10% loss to follow-up would provide 80% power to detect an effect of a given dose of mexiletine on RMT if the increase in RMT relative to placebo was at least 5.3% based on a simple one-way ANOVA and two-sided alpha = 0.027 for each of the two active arms. This effect correlates to roughly 85% of the natural variation in RMT over 4 wk The same sample size would provide 80% power to detect a linear dose-response for a slope of 2.4% / 300 mg dose and an 80% probability of that the more effective mexiletine dosage would exhibit a greater increase in RMT in our sample, irrespective of compliance with assigned dose and thus reflecting variation in tolerance, if the difference in efficacy were at least 1.4%.
2.7 ∣. Statistical analysis
The effect of mexiletine on RMT was estimated from a modified intention-to-treat sample using a shared-baseline mixed model repeated-measures analysis with fixed effects of visit (four levels including the screening visit) and treatment x post-baseline visit interaction (2 visits x 3 treatment groups = 6 levels) and unstructured covariance among repeated measures (10 terms). Treatment-dependent differences in the 4-wk change in RMT and any sustained benefit at 8 wk were estimated using linear contrasts of the least-square means. The primary end point was the comparison of the average change in RMT from pre-treatment to week 4 for the two active groups vs. placebo, tested using a two-tailed Wald-test at alpha = 0.05. Secondary analyses tested for effects at each dosage separately and for a linear dose response. Correction for multiple comparisons was made for secondary analyses.
Equivalent models were used to analyze other pharmacodynamic markers obtained by TMS and NCS and clinical measures of progression. Measures with strongly right-skewed distributions and strictly positive values (SICI, ICF, MEP, SDTC, CMAP, and rheobase) were log-transformed for analysis and back-transformed for reporting. Frequency of muscle cramping was assessed using a similar generalized linear mixed model assuming that weekly counts followed a negative binomial distribution and including subject-specific random slopes with unstructured covariance of random intercepts and slopes. Pain from cramping and interference from fasciculations was analyzed in similar models assuming a normal distribution and identity link for pain and a multinomial distribution and cumulative logit link for interference from fasciculations.
Analyses of safety included frequency of AEs compared by negative binomial regression, proportion of subjects experiencing a given AE or serious adverse event (SAE) classified by MedDRA system organ class or preferred term by Fisher's exact test, occurrence of clinically significant clinical laboratory abnormalities by Fisher's exact, and trends in vital signs and ECG parameters by linear mixed models. Subjects were analyzed according to the treatment actually received. With a plan of 40 subjects exposed to mexiletine, the study would have an 80% probability of observing at least one instance of any safety outcome expected to occur in at least 4% of exposed patients, or 8% for events unique to a single dose. The study was estimated to have 80% power to detect treatment differences in event rates if the rates in the placebo arm were moderately frequent (20% to 50%) and mexiletine exposure increases the odds at least six-fold.
Analyses were performed using SAS (version 9.4, SAS Institute, Cary, NC). Recognizing the pilot nature of the study, both unadjusted comparison-wise two-tailed P-values to guide hypothesis generation and two-tailed P-values that adjust for one primary outcome, 12 primary and secondary outcomes; 32 primary, secondary, and exploratory pharmacodynamic outcomes; and 5 clinical outcomes for more formal hypothesis testing are reported.
3 ∣. RESULTS
3.1 ∣. Subjects
The flow of subjects is shown in Figure 1. Between February 2017 and July 30, 2018 subjects were screened and 20 SALS subjects consented and enrolled from 10 centers (0-8 enrolled per site), with follow up completed by September 2018. The intended recruitment was 60 subjects but could not be reached even with a 1-y extension of the study. 20% of screened subjects were excluded due to cortical inexcitability. Of the 20 subjects randomized, all received the assigned study drug. One subject receiving placebo discontinued the study drug a few days prior to the week 4 visit due to inability to swallow the capsule but continued the study to its completion. Data from this subject is included in analyses. Baseline characteristics did not differ substantially among treatment groups for either clinical features (Table 1) or neurophysiological parameters (Table 2). Chance baseline differences in the distribution of El Escorial criteria and time since diagnosis are not expected to influence neurophysiologic outcomes, but this could not be evaluated within this study.
FIGURE 1.
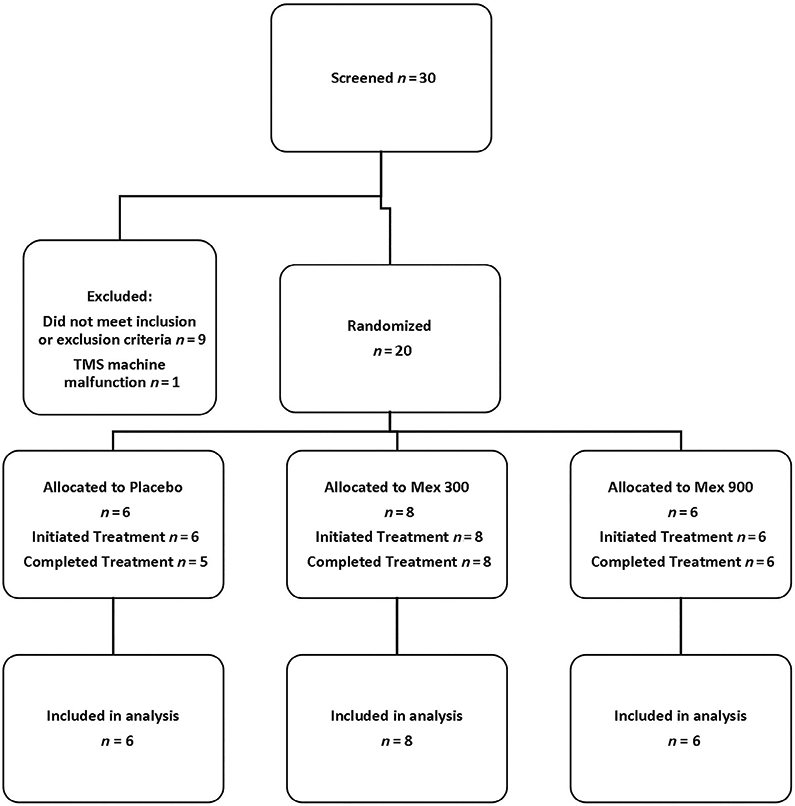
CONSORT diagram. Mex = mexiletine.; RMT = resting motor threshold.; CMAP = compound muscle action potential
TABLE 1.
Baseline characteristics of ALS subjects
| Treatment groups | |||
|---|---|---|---|
| Variable | Placebo n = 6 | Mx 300 mg n = 8 | Mx 600 mg n = 6 |
| Age (y) | 52.0 (11.3) | 58.5 (10.8) | 60.5 (14.1) |
| Men | 83.3% (5) | 62.5% (5) | 66.7% (4) |
| Race | |||
| Non-Hispanic | 100.0% (6) | 100.0% (8) | 83.3% (5) |
| Hispanic | 0.0% (0) | 0.0% (0) | 17.7% (1) |
| El Escorial criteria | |||
| Possible | 50.0% (3) | 12.5% (1) | 0.0% (0) |
| Probable laboratory | 33.3% (2) | 62.5% (5) | 83.3% (5) |
| Probable | 16.7% (1) | 25.0% (2) | 0.0% (0) |
| Definite | 0.0% (0) | 0.0% (0) | 16.7% (1) |
| Years since symptom onset | 1.8 (0.7) | 1.9 (0.9) | 2.1 (1.5) |
| Years since diagnosis | 0.7 (0.3) | 0.6 (0.5) | 1.2 (1.4) |
| Bulbar onset | 16.7% (1) | 12.5% (1) | 16.7% (1) |
| Taking riluzole | 83.3% (5) | 100.0% (8) | 66.7% (4) |
| ALSFRS-R total score | 41.3 (5.4) | 39.8 (4.2) | 38.7 (5.5) |
| SVC (max %-pred) | 92.4 (14.1) | 95.4 (14.8) | 87.9 (15.3) |
| BMI (kg/m2) | 25.5 (4.2) | 28.5 (7.3) | 28.3 (2.4) |
| Any cramps in previous 7 days (%) | 83.3% (5) | 87.5% (7) | 83.3% (5) |
| Cramps in previous 7 days | 10.2 (38.9) | 8.1 (7.0) | 12.8 (11.9) |
| Any cramps in previous 30 days (%) | 83.3% (5) | 100.0% (8) | 83.3% (5) |
| Cramps in previous 30 days | 42.3 (41.1) | 32.8 (30.2) | 53.7 (52.1) |
| Maximum cramp pain in previous 7 days | 2.0 (1.8) | 3.5 (2.1) | 2.5 (2.4) |
| Maximum cramp pain in previous 30 days | 2.3 (2.3) | 3.9 (1.6) | 2.8 (2.4) |
| Any fasciculations in previous 14 days | 100.0% (6) | 87.5% (7) | 83.3% (5) |
| Duration of fasciculations (h/day) | 5.7 (9.8) | 4.4 (8.8) | 3.6 (6.2) |
Note: Values are mean (SD) or % (N).
Abbreviations: ALSFRS-R, revised ALS function rating scale; BMI, body mass index; Mx, mexiletine; SVC, slow vital capacity.
TABLE 2.
Baseline neurophysiological parameters
| Treatment groups | |||
|---|---|---|---|
| Variable | Placebo n = 6 | Mx 300 mg n = 8 | Mx 600 mg n = 6 |
| RMT (%) | 54.5 (21.4) | 53.3 (19.5) | 55.8 (11.8) |
| SICI−1(mV/mV) | 1.03 (0.81) | 0.96 (0.53) | 0.71 (0.35) |
| MEP (120%) (mV) | 0.43 (0.44) | 0.73 (1.01) | 0.78 (0.79) |
| ICF (mV/mV) | 1.49 (0.94) | 1.32 (0.50) | 1.61 (1.41) |
| CSP (ms) | 47.8 (13.1) | 75.8 (61.4) | 93.9 (40.2) |
| MEP (150%) (mV) | 3.11 (2.21) | 2.27 (2.05) | 1.88 (1.86) |
| Peak CMAP (mV) | 5.53 (3.71) | 4.87 (2.16) | 7.20 (3.54) |
| SDTC (ms) | 0.53 (0.05) | 0.53 (0.12) | 0.51 (0.12) |
| Rheobase (mA) | 3.73 (1.45) | 3.11 (1.63) | 4.22 (3.51) |
| Accommodation half-time (ms) | 46.4 (6.72) | 42.4 (6.23) | 45.4 (3.03) |
| TEd (90-100) (%) | 45.0 (4.23) | 46.9 (6.32) | 45.0 (7.05) |
| TEh (90-100) (%) | −113(17.9) | −130 (20.4) | −122 (27.0) |
| Superexcitability (%) | −29 (8.55) | −30 (6.00) | −25 (11.5) |
| Subexcitability (%) | 13.6 (2.03) | 15.9 (7.69) | 12.1 (1.83) |
Note: Values are mean (SD).
Abbreviations: CMAP, compound muscle action potential; ICF, intracortical facilitation; MEP, maximal evoked potential; Mx, mexiletine; RMT, resting motor threshold; SDTC, strength-duration time constant; SICI, short interval intracortical inhibition; TEd (90-100), depolarizing threshold electrotonus at 90-100 ms; TEh (90-100), hyperpolarizing threshold electrotonus at 90-100 ms.
3.2 ∣. Safety and tolerability
AEs by MedDRA system organ class are reported in Table 3. There were no significant differences among AEs comparing placebo with either dose of mexiletine. One SAE was reported, a deep vein thrombosis in a subject on 600 mg/day mexiletine. Compared to placebo, no significant differences were noted in laboratory safety studies, ECG, or vital signs. The study medications were well tolerated at both doses. Of the 20 subjects who initiated study drug, 1 subject on placebo did not complete the study as they had difficulty swallowing the capsule.
TABLE 3.
Treatment-emergent serious adverse events and adverse events by MedDRA system organ class and overall
| Placebo |
Mx 300 mg |
Mx 600 mg |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Serious adverse events | #E | #S | %S | #E | #S | %S | #E | #S | %S |
| Vascular disorders | 0 | 0 | 0% | 0 | 0 | 0% | 1 | 1 | 17% |
| Deep venous thrombosis | 0 | 0 | 0% | 0 | 0 | 0% | 1 | 1 | 17% |
| Overall | 0 | 0 | 0% | 0 | 0 | 0% | 1 | 1 | 17% |
| Placebo |
Mx 300 mg |
Mx 600 mg |
|||||||
| Adverse events | #E | #S | %S | #E | #S | %S | #E | #S | %S |
| Blood and lymphatic system disorders | 1 | 1 | 17% | 0 | 0 | 0% | 0 | 0 | 0% |
| Ear and labyrinth disorders | 0 | 0 | 0% | 0 | 0 | 0% | 1 | 1 | 17% |
| Eye disorders | 1 | 1 | 17% | 0 | 0 | 0% | 0 | 0 | 0% |
| Gastrointestinal disorders | 3 | 2 | 33% | 3 | 3 | 38% | 7 | 3 | 50% |
| General disorders and administration site conditions | 0 | 0 | 0% | 2 | 2 | 25% | 2 | 2 | 33% |
| Infections and infestations | 0 | 0 | 0% | 0 | 0 | 0% | 1 | 1 | 17% |
| Injury, poisoning and procedural complications | 0 | 0 | 0% | 0 | 0 | 0% | 2 | 2 | 33% |
| Investigations | 2 | 2 | 9% | 12 | 6 | 26% | 5 | 3 | 16% |
| Musculoskeletal and connective tissue disorders | 0 | 0 | 0% | 2 | 1 | 13% | 2 | 1 | 17% |
| Nervous system disorders | 2 | 2 | 33% | 3 | 3 | 38% | 5 | 2 | 33% |
| Renal and urinary disorders | 0 | 0 | 0% | 0 | 0 | 0% | 1 | 1 | 17% |
| Respiratory, thoracic and Mediastinal disorders | 0 | 0 | 0% | 2 | 1 | 13% | 0 | 0 | 0% |
| Skin and subcutaneous tissue disorders | 8 | 2 | 33% | 6 | 2 | 25% | 3 | 1 | 17% |
| Vascular disorders | 0 | 0 | 0% | 0 | 0 | 0% | 1 | 1 | 17% |
| Overall | 19 | 5 | 83% | 18 | 5 | 63% | 25 | 5 | 83% |
Note: #E, number of reported events; #S and %S, number and percentage of subjects experiencing at least once instance of a given type of adverse event, respectively. All comparisons were non-significant by Fisher's exact test.
3.3 ∣. Outcome measures
The changes in clinical markers of progression are summarized in Table 4. Cramp intensity was 1.3 units lower than placebo at weeks 3 and 4 for subjects on both 300 and 600 mg/day (P = .044 for the combined estimate). Effects on cramp frequency were not significant either among all subjects or among those who reported at least 10 or more cramps over the 30 days prior to baseline, but the point estimates suggested a large effect at the 600 mg/day dose (relative reductions of 56% among all subjects and 47% among those with 10+ cramps at baseline). There was no correlation between frequency of muscle cramps (mean number per week) or duration of fasciculations (percentage of days) and parameters of axonal excitability. Effects on fasciculations were not estimable due to limited variation among subjects. There were no significant differences in the rates of decline for ALSFRS-R, SVC, RAND-36, or HHD comparing placebo with mexiletine.
TABLE 4.
Neurophysiology & Markers of progression
| Change from baseline to week 4 (mean change or ratio*) |
Active treatment (300 and 600 mg combined) versus placebo from baseline to week 4 |
|||||
|---|---|---|---|---|---|---|
| Placebo | Mx 300 mg | Mx 600 mg | Estimated value | Unadj P value | Adj P value | |
| TMS parameters | ||||||
| RMT (%) | 4.746 (0.155,9.337) | −2.430 (−7.311,2.451) | 0.805 (−3.832,5.441) | −5.559 (−10.80,−0.315) | .039 | .039 |
| SICI−1 (mV/mV)* | 0.961 (0.634,1.459) | 0.655 (0.437,0.983) | 0.886 (0.608,1.291) | 0.792 (0.484,1.296) | .33 | >.99 |
| ICF (mV/mV)* | 0.915 (0.665,1.259) | 1.046 (0.772,1.417) | 0.941 (0.704,1.256) | 1.084 (0.744,1.581) | .66 | >.99 |
| MEP (120%)/peak CMAP (mV/mV)* | 1.308 (0.695,2.462) | 0.589 (0.315,1.102) | 0.490 (0.249,0.965) | 0.411 (0.208,0.810) | .013 | .16 |
| MEP (140%)/peak CMAP (mV/mV)* | 0.693 (0.340,1.412) | 0.678 (0.281,1.633) | 1.115 (0.502,2.475) | 1.254 (0.501,3.142) | .61 | >.99 |
| CSP (ms) | 4.037 (−30.84,38.91) | 3.937 (−31.75,39.62) | 4.823 (−29.76,39.41) | 0.343 (−41.36,42.04) | .99 | >.99 |
| TTNCS parameters | ||||||
| Peak CMAP (mV)* | 0.940 (0.706,1.252) | 1.217 (0.924,1.602) | 0.917 (0.657,1.279) | 1.123 (0.792,1.593) | .49 | >.99 |
| STDC (ms)* | 0.935 (0.837,1.045) | 0.895 (0.802,0.998) | 0.966 (0.848,1.099) | 0.994 (0.876,1.126) | .91 | >.99 |
| Rheobase (mA)* | 1.303 (0.987,1.721) | 1.187 (0.910,1.547) | 0.886 (0.646,1.215) | 0.787 (0.571,1.084) | .13 | >.99 |
| TEd (90-100) (%) | 0.103 (−5.771,5.978) | 0.957 (−4.855,6.768) | −0.258 (−6.997,6.482) | 0.246 (−6.709,7.202) | .94 | >.99 |
| TEh (90-100) (%) | −4.219 (−15.00,6.562) | −10.184 (−21.57,1.204) | 2.230 (−10.52,14.98) | 0.242 (−12.64,13.13) | .97 | >.99 |
| Accommodation. Half-time (ms) | 5.442 (0.977,9.907) | −2.619 (−7.075,1.837) | −1.243 (−5.980,3.494) | −7.373 (−11.52,−3.226) | .002 | .049 |
| Latency (ms) | −0.025 (−0.461,0.411) | 0.018 (−0.402,0.438) | −0.165 (−0.682,0.351) | −0.049 (−0.595,0.498) | .85 | >.99 |
| Superexcitability (%) | −0.724 (−4.975,3.526) | 2.234 (−2.129,6.597) | 1.511 (−3.391,6.413) | 2.597 (−2.772,7.966) | .32 | >.99 |
| Subexcitability (%) | 0.448 (−3.001,3.897) | −2.563 (−5.706,0.581) | −0.575 (−4.178,3.027) | −2.017 (−5.767,1.733) | .27 | >.99 |
| Clinical measures | ||||||
| ALSFRS-R | −0.827 (−2.141,0.487) | −1.414 (−2.554,−0.274) | −1.038 (−2.352,0.276) | −0.399 (−1.966,1.169) | .60 | >.99 |
| SVC (% predicted) | −0.991 (−5.450,3.467) | −0.532 (−4.425,3.361) | −6.918 (−11.37,−2.461) | −2.734 (−7.938,2.471) | .29 | >.99 |
| HHD (kg) | 0.566 (−1.185,2.316) | 0.763 (−0.753,2.279) | −0.649 (−2.400,1.101) | −0.509 (−2.606,1.589) | .62 | >.99 |
| Cramp number (weeks 3-4)* | 2.055 (0.394,10.726) | 2.323 (0.590,9.141) | 0.912 (0.200,4.162) | 0.708 (0.111,4.535) | .71 | >.99 |
| Cramp intensity (weeks 3-4)* | 2.861 (1.778,3.945) | 1.537 (0.487,2.586) | 1.597 (0.700,2.494) | −1.294 (−2.555,−0.034) | .044 | >.99 |
Note: Values are model estimate (95% CI). P value <.05 in bold.
Abbreviations: ALSFRS-R, ALS functional rating scale, revised; CI, confidence interval; CSP, cortical silent period; HHD, hand-held dynamometry; ICF, intracortical facilitation; motor evoked potential measured at 120% or 140% of RMT normalized to the peak compound muscle action potential amplitude = MEP (120% or 140%) / Peak CMAP; RMT, resting motor threshold; SICI, short interval intracortical inhibition; STDC, strength duration time constant; SVC, slow vital capacity; TEd (90-100), depolarizing threshold electrotonus at 90-100 ms; TEh (90-100), hyperpolarizing threshold electrotonus at 90-100 ms.
The change in neurophysiological parameters are shown in Figure 1 (cortical excitability) and Figure 2 (axonal excitability) and summarized in Table 4. RMT was unchanged from baseline to 4 wk with mexiletine (combined active therapies) as compared to placebo, which showed a significant increase (P = .039). The power for detecting a change in RMT under the originally assumed variance estimate and treatment difference with only 20 rather than 60 participants was 49%. Dose-dependent reduction of the MEP amplitude at 120% of the RMT normalized to the peak CMAP was evident at 4 wk, although the association was not significant after correction for multiple comparisons (P = .16). There were no other significant differences in the TMS parameters.
FIGURE 2.
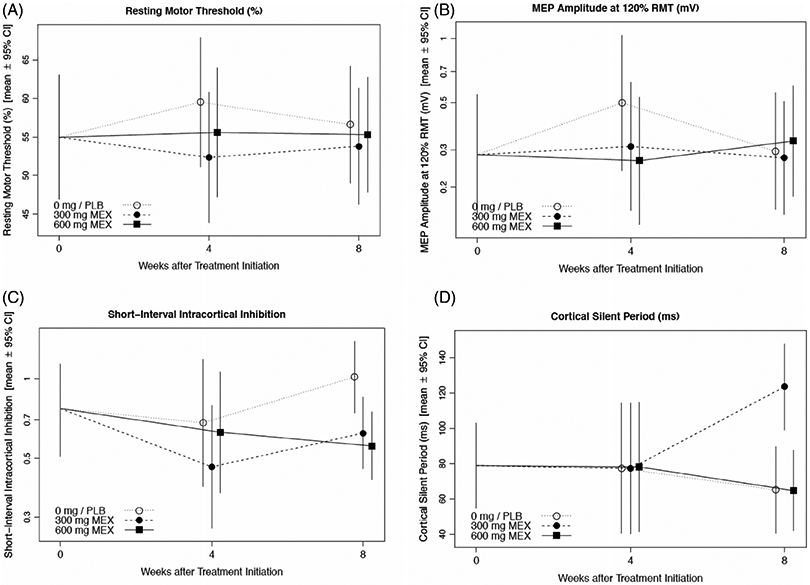
Mexiletine treatment effects on cortical excitability. Plots show treatment- and visit-specific estimates with 95% confidence bounds from a shared baseline, mixed model repeated measures analysis for: Resting motor threshold (A), MEP amplitude at 120% of RMT/ peak CMAP (B), short-interval intracortical inhibition (C), and cortical silent period (D)
Accommodation halftime was significantly reduced over 4 wk with mexiletine (adjusted P = .048).There were no other significant differences in the axonal excitability parameters (Figure 3).
FIGURE 3.
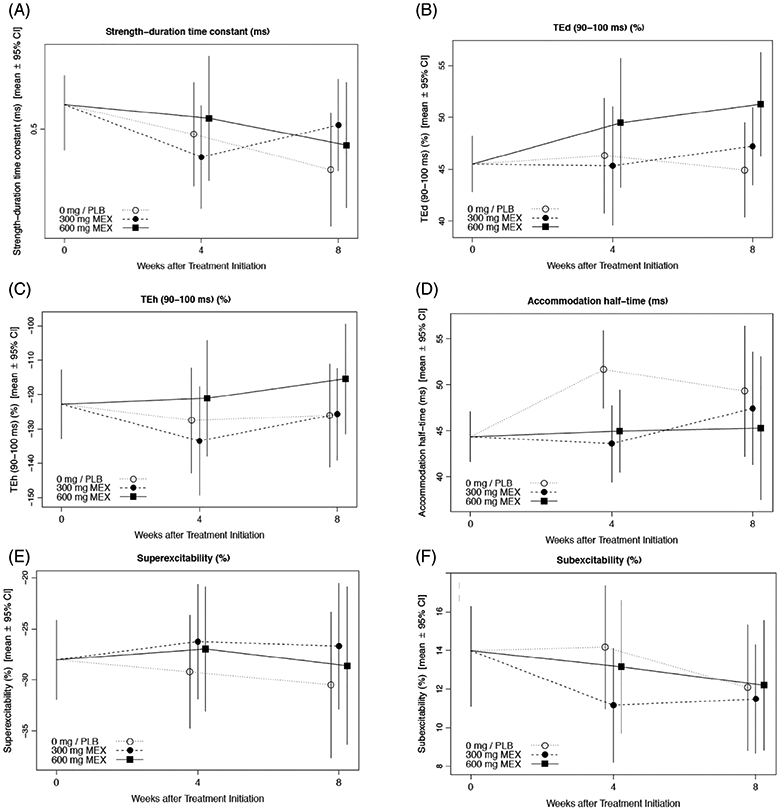
Mexiletine treatment effects on axonal excitability of peripheral motor nerves. Plots show treatment- and visit-specific estimates with 95% confidence bounds from a shared baseline, mixed model repeated measures analysis for: Strength-duration time constant (A), depolarizing threshold electrotonus, 90-100 ms (B), hyperpolarizing threshold electrotonus, 90-100 ms (C), accommodation half-time (D), superexcitability (E), and subexcitability (F)
4 ∣. DISCUSSION
This pilot study demonstrated that mexiletine was safe and generally tolerable at both 300 mg and 600 mg per day. There were no significant differences in the number of AEs seen with either dose of study medication compared to placebo. The only SAE seen, deep venous thrombosis in a subject on 600 mg a day that required hospitalization, is unlikely to be attributed to mexiletine which is not known to be prothrombotic. The single subject on placebo who discontinued study medication prematurely did so only due to difficulty swallowing the capsule which could not be opened and swallowed due to concerns about unblinding.
Mexiletine (combined doses) demonstrated a significant reduction of cramp intensity compared to controls, corroborating the results of two recent randomized placebo-controlled studies.23,31 Treatment with mexiletine 600 mg/day led to a relative reduction of cramp frequency of more than 50%, though this was not statistically significant. Unlike the previous studies,23,31 no comparable effect was seen with 300 mg/day, possibly due to the small sample. There was no apparent effect on fasciculations. Studies have suggested that both cramps and fasciculations are manifestations of axonal hyperexcitability of peripheral motor nerves, but this report failed to show a correlation between parameters of axonal excitability and the rate of muscle cramps or the duration of fasciculations. Mexiletine also had no effect on clinical markers of progression, ALSFRS-R, SVC, HHD, and RAND-36, though the study was of short duration and insufficiently powered to detect such a change.
While this was a pilot study that was substantially underpowered, the results demonstrated limited, although significant, effects of mexiletine on the excitability of cortical motor neurons and peripheral motor nerve axons. Combined estimates revealed that mexiletine treatment in SALS subjects over 4 wk resulted in a relative stabilization of the RMT, the primary outcome measure, compared with placebo which showed an increase. RMT is thought to reflect the density of corticomotoneuronal projections as well as cortical excitability, in part reflective of increased persistent sodium current.32,33 Previous studies in ALS subjects have shown RMT to be increased,33,34 unchanged,35 or reduced.13,36 This variability is believed to reflect a number of clinical factors, with reduced RMT seen at earlier stages of disease, most notably in subjects with C9orf72 gene mutations suggesting an additional genetic contribution,36 and increased RMT and eventual cortical inexcitability seen with disease progression.12,35
Twenty percent of screened subjects in the current study exhibited cortical inexcitability, similar to that reported in previous studies.12,37,38 Cortical inexcitability has also been attributed to upper motor neuron degeneration.39 The relative stabilization of RMT by mexiletine compared to the patients on placebo, who demonstrated an increase of RMT, was not expected given the known inhibitory effects of the study medication on neuronal sodium channels. As it was unexpected, the stabilization of RMT with treatment may be attributed to chance, unforeseen technical considerations, or unknown medications influencing TMS. However, this finding could also suggest a neuromodulating effect of mexiletine in ALS on preventing an increase.12,35 Although sodium channel blockers have increased RMT in prior studies,40 the opposite effect in ALS could reflect a baseline cortical hyperexcitability in which opposing depolarization block could dominate the typical reduction in inward current.41,42 Such competing mechanisms could potentially result in opposite-appearing effects at different doses, as we observed. Indeed, such a mechanism could parallel the hyperpolarizing shift facilitating persistent sodium current activation by the anticonvulsant carbamazepine, a sodium channel blocker, in the setting of decreased sodium channel beta subunits.43 A more comprehensive longer and larger study would be required to corroborate the effect of mexiletine and substantiate this claim.
While no changes were noted for other secondary TMS endpoints including SICI, ICF, and CSP, mexiletine demonstrated a numerical dose-dependent reduction of the MEP amplitude at 120% of the RMT normalized to the peak CMAP (MEP [120%]/ peak CMAP), although the association was not significant after correction for multiple comparisons. Increases of MEP amplitudes have been shown in a number of studies in both SALS and familial ALS subjects, typically early in the disease.1,13,44,45 They are thought to be reflective of cortical hyperexcitability by alterations in neurotransmitter modulation.32 The physiological mechanisms that affect the MEP amplitude are believed to be independent of those that affect the RMT, which may explain the discrepant findings between the two measures in this study.32
Despite the lack of other findings by axonal excitability study, a significant decrease of accommodation half-time was shown, even after correcting for multiple comparisons. Accommodation half-time is a parameter derived from threshold electrotonus data and defined as the time to the midpoint between the peak threshold reduction and the average level at the end of the polarization.46 It is generally thought to be a measure of potassium channel gating, with activation of slow potassium channels leading to accommodation to depolarizing currents and reflected in a reduction of accommodation half-time.46 As mexiletine typically inhibits fast inactivated sodium channel, its effect on accommodation half-time is likely spurious in nature given also that there were no alterations in other measures of threshold electrotonus or recovery cycle analysis that reflect slow potassium channels. A recent phase 2 single blind randomized controlled trial investigating the effects of mexiletine in SALS subjects treated with either daily mexiletine 300 mg plus riluzole versus daily riluzole alone for 4 mo also failed to show an effect on parameters of axonal excitability, specifically, SDTC and latent addition.47 However, effects of mexiletine on accommodation half-time were not reported.
There were a number of limitations to this trial. Although efforts were made to enroll a diverse population of subjects, the recruited subjects were all Caucasian. Despite extending the duration of the study and increasing the number of sites, the trial was substantially under-enrolled and, thus, underpowered. There were many possible reasons for this, including clinical use of mexiletine to control cramps, competing enrollment into other ALS studies, concerns by potential subjects about possible discomfort in undergoing the neurophysiological procedures, logistical issues related to performing the lengthy procedures and transporting subject to multiple locations for the two neurophysiological tests at some sites, and a significant number of subjects who were ineligible based on the neurophysiological exclusion criteria. Necessitated by the challenges in recruitment, the disease duration inclusion was relaxed from less than 24 mo to less than 60 mo, which could have also had an effect on the markers of cortical hyperexcitability, thought to be more prominent earlier in the disease.35 Given the small number of patients and short duration, the study did not have power to detect a slowing of progression.
While the effect seen on RMT, the primary objective of the study, was the opposite of expected, possibly due to neuromodulation by mexiletine promoting stabilization, other neurophysiological parameters do provide evidence supporting a reduction of excitability by mexiletine of cortical motor neurons and peripheral motor nerve axons (as a surrogate marker of spinal motor neurons). Given these findings demonstrating target engagement, a longer and larger study using mexiletine may be warranted to determine more definitively its effects on disease progression.
ACKNOWLEDGMENTS
This study was supported by funding from the ALS Association and ALS Finding a Cure. None of the funding agencies were involved in the design and conduct of the study; the collection, management, analysis, and interpretation of the data; or the preparation, review, or approval of the manuscript. The authors thank the patients who participated in this study; the medical monitor, Nazem Atassi, MD (Massachusetts General Hospital), the members of the Steering Committee, Anthony Amato, MD (Brigham and Women's Hospital), Eric Macklin, PhD (Massachusetts General Hospital), and William David, MD, PhD (Massachusetts General Hospital), and Bernadette Gillick, PhD (University of Minnesota) for TMS technical support.
Abbreviations:
- AE
adverse event
- ALS
amyotrophic lateral sclerosis
- ALSFRS-R
revised ALS Functional Rating Scale
- ANOVA
analysis of variance
- APB
abductor pollicis brevis
- CMAP
compound nerve action potential
- CSP
cortical silent period
- ECG
electrocardiogram
- FDI
first dorsal interosseous
- HHD
hand-held dynamometry
- ICF
intracortical facilitation
- MEP
motor evoked potential
- MMT
manual muscle testing
- MSO
maximal stimulus output
- RMT
resting motor threshold
- SAE
serious adverse event
- SALS
sporadic amyotrophic lateral sclerosis
- SDTC
strength duration time constant
- SICI
short interval intracortical inhibition
- SVC
slow vital capacity
- TEd 90-100 ms
depolarizing threshold electrotonus 90-100 ms
- TEh 90-100 ms
hyperpolarizing threshold electrotonus 90-100 ms
- TMS
transcranial magnetic stimulation
- TTNCS
threshold tracking nerve conduction studies
*. Mexiletine-2 ALS Study Group
Neurology Clinical Research Institute, Massachusetts General Hospital: Hong Yu, MPH (director, data management), Jennifer Yue (data management), Annette De Mattos (grants administration), Daniela Grasso (senior project manager), Lindsay Pothier (senior project manager), David Klements (project manager), Sara Vaughn (project manager), Melissa Ricker (assistant project manager), and Emily Engel (assistant project manager).
NEALS Outcomes and Monitoring Center, Barrow Neurological Institute: Meghan Hall (director of clinical monitoring), Ashley Sconzo (monitor), and Taylor Pitts (monitor).
University of Washington Medical Center: Laura Sissons-Ross (research coordinator) and Sharon Downing, RN (research coordinator).
Beth Israel Deaconess Medical Center: Hilda Gutierrez (research coordinator), Carmen Shin (research coordinator), and Peter Fried (TMS evaluator).
University of Michigan: Sean Meehan (co-investigator; current affiliation University of Waterloo, Waterloo, ON, Canada), Jayna Duell, RN (research coordinator); and Sangri Kim (research coordinator).
University of California, Irvine Medical Center: Veena Mathew, MSc (research coordinator) and Veronica Martin (research coordinator).
Barrow Neurological Institute: Nicole Turcotte (research coordinator), Gale Kittle (research coordinator), and Jacquelyn Nicolari, RN (research coordinator).
Medical University of South Carolina: Katrina Madden (research coordinator) and William Devries (TMS evaluator).
Columbia University: Natalia Leontovich (research coordinator) and Jessica Singleton (research coordinator).
University of Pittsburgh Medical Center: Danielle Rowlands, MSN (research coordinator).
Penn State Hershey Medical Center: Divpreet Kaur, MD (sub-investigator).
Augusta University: Brandy Quarles (research coordinator) and Kristy Bouchard (research coordinator).
Footnotes
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guideline.
DISCLOSURES OF CONFLICTS OF INTEREST
Dr. Weiss has consulted for Ra Pharma, Argenx, and Biogen and is a speaker for NuFactor. Dr. Macklin is a DSMB member for Shire Human Genetic Therapies and for Novartis, a Steering Committee member for a Biogen trial, and on the advisory board member for Cerevance, Intrance, and Inventram, and has received research support from Amylyx Pharmaceuticals, GlaxoSmithKline, and Mitsubishi Tanabe Pharma America. Professor Vucic reports receiving funding from the National Health & Medical Research Council Australia, honoraria from CSL Australia, and serving on scientific advisory boards for Biogen and ClENE. Dr. Wainger has received personal compensation from Amgen, Quralis, Apic Biosciences, Q-State Biosciences, Takeda, and is co-inventor on a patent for the use of potassium channel openers in neurodegenerative diseases. He is a New York Stem Cell Foundation–Robertson Investigator and has received research funding from the ALS Association, GlaxoSmithKline, AI Therapeutics, Target ALS, NIH, Revalesio, and Sanofi. Dr. Goutman is on the scientific advisory for Biogen and IFT Pharma. Dr. Goyal has received research support from Brainstorm Cell Therapeutics, Cytokinetics, Fulcrum, Kezar, Novartis, Octapharma, Orion, Orphazyme, served on Advisory Boards for Acceleron, Alexion, Argenx, Biogen, CSL Behring, Cytokinetics, MT Pharma, Novartis, Sanofi Genzyme, Sarepta, and is on the speaker's bureau for CSL. Dr. Rutkove has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Myolex, Biogen, Axcella Health, Merck. He has received personal compensation in an editorial capacity for Annals of Neurology and for serving on the Board of Directors of Myolex, Inc. He also holds stock and/or stock options in Myolex, Inc. Dr. Ladha reports receiving research support from the ALS Association, consulting for Biogen and Amylyx, and speaking for Biogen. Dr. Chen reports research support from the Muscular Dystrophy Association, ALS Association/ Barrow Neurological Institute, Flex Pharma, Inc., Healey ALS Center, NEALS, UCB, and consulting for Biogen. Dr. Harms has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Biogen, Sanofi, and Maze Therapeutics and received research support from Biogen. Dr. Brannagan has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Grifols, Ionis, Akcea, Alnylam, Shire, and CSL Behring and research support from Ionis, Alnylam, Viromed, Catalyst, Pharnext, Flexpharma, Novartis, and Grifols. Dr. Lacomis reports research support from Orion Pharma, Mitsubishi-Tanabe, and the ALS Association, and royalties from UpToDate. Dr. Wang has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Biogen and Argenx. He also holds stock and/or stock options in GWPH and Spark Therapeutics and has received research support from Fulcrum Therapeutic. Dr. Simmons has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Biohaven, Biogen, Cytokinetics. He has also received personal compensation in an editorial capacity for Muscle & Nerve and research support from Biogen, Biohaven, Cytokinetics, Genzyme, and Mallinckrodt. Dr. Rivner has consulted for and received research support from Alexion and Allergan, Research support from UCB, Mallinckrodt, Cytokinetics, Momenta, Shire Takeda, Orion, Biohaven, Catalyst, Grifols, Seikagaku, and RA pharmaceuticals. Dr. Shefner has received personal compensation from Cytokinetics, Avexis, Revalasio, Biohaven, Neurosense, MTPA, and Otsuka and for serving as neuromuscular section editor for UpToDate, and research support from Cytokinetics, Biogen, Brainstorm, MTPA, and Amylyx. Dr. Cudkowicz has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Takeda, Biogen, Cytokinetics, Sunovian, Immunitypharm, and Avexis. Dr. Atassi has received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Biogen, GSK, MT Pharma, and Neuropore Therapies. The remaining authors have no conflicts of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008;131:1540–1550. [DOI] [PubMed] [Google Scholar]
- 2.Mills K, Nithi K. Corticomotor threshold is reduced in early sporadic amyotrophic lateral sclerosis. Muscle Nerve. 1997;20:1137–1141. [DOI] [PubMed] [Google Scholar]
- 3.Ziemann U, Winter M, Reimers CD, Reimers K, Tergau F, Paulus W. Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis. Evidence from paired transcranial magnetic stimulation. Neurology. 1997;49:1292–1298. [DOI] [PubMed] [Google Scholar]
- 4.de Carvalho M, Evangelista T, Sales-Luís ML. The corticomotor threshold is not dependent on disease duration in amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:39–42. [DOI] [PubMed] [Google Scholar]
- 5.Bae JS, Simon NG, Menon P, Vucic S, Kiernan MC. The puzzling case of hyperexcitability in ALS. J Clin Neurol. 2013;9:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwai Y, Shibuya K, Misawa S, et al. Axonal dysfunction precedes motor neuronal death in amyotrophic lateral sclerosis. PLoS One. 2016;11(7):e0158596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vucic S, Kiernan MC. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain. 2006;129:2436–2446. [DOI] [PubMed] [Google Scholar]
- 8.Menon P, Kiernan MC, Vucic S. Cortical dysfunction underlies the development of the split-hand in amyotrophic lateral sclerosis. PLoS One. 2014;9(1):e87124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007;78:849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vucic S, Kiernan MC. Utility of transcranial magnetic stimulation in delineating amyotrophic lateral sclerosis pathophysiology. Handb Clin Neurol. 2013;116:561–575. [DOI] [PubMed] [Google Scholar]
- 11.Menon P, Geevasinga N, Yiannikas C, Howells J, Kiernan MC, Vucic S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: a prospective study. Lancet Neurol. 2015;14(5):478–484. [DOI] [PubMed] [Google Scholar]
- 12.Menon P, Higashihara M, van den Bos M, Geevasinga N, Kiernan MC, Vucic S. Cortical hyperexcitability evolves with disease progression in ALS. Ann Clin Transl Neurol. 2020;7(5):733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menon P, Kiernan MC, Vucic S. Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin Neurophysiol. 2015;126(4):803–809. [DOI] [PubMed] [Google Scholar]
- 14.Shibuya K, Park SB, Geevasinga N, et al. Motor cortical function determines prognosis in sporadic ALS. Neurology. 2016;87(5):513–520. [DOI] [PubMed] [Google Scholar]
- 15.Geevasinga N, Menon P, Howells J, Nicholson GA, Kiernan MC, Vucic S. Axonal ion channel dysfunction in c9orf72 familial amyotrophic lateral sclerosis. JAMA Neurol. 2015;72(1):49–57. [DOI] [PubMed] [Google Scholar]
- 16.Vucic S, Kiernan MC. Axonal excitability properties in amyotrophic lateral sclerosis. Clin Neurophysiol. 2006;117(7):1458–1466. [DOI] [PubMed] [Google Scholar]
- 17.Shibuya K, Misawa S, Nasu S, et al. Split hand syndrome in amyotrophic lateral sclerosis: different excitability changes in the thenar and hypothenar motor axons. J Neurol Neurosurg Psychiatry. 2013;84(9):969–972. [DOI] [PubMed] [Google Scholar]
- 18.Vucic S, Kiernan MC. Upregulation of persistent sodium conductances in familial ALS. J Neurol Neurosurg Psychiatry. 2010;81(2):222–227. [DOI] [PubMed] [Google Scholar]
- 19.Kanai K, Kuwabara S, Misawa S, et al. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain. 2006;129:953–962. [DOI] [PubMed] [Google Scholar]
- 20.Kanai K, Shibuya K, Sato Y, et al. Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(7):734–738. [DOI] [PubMed] [Google Scholar]
- 21.Fritz E, Izaurieta P, Weiss A, et al. Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J Neurophysiol. 2013;109(11):2803–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanai K, Kuwabara S, Arai K, Sung JY, Ogawara K, Hattori T. Muscle cramp in Machado-Joseph disease: altered motor axonal excitability properties and mexiletine treatment. Brain. 2003;126:965–973. [DOI] [PubMed] [Google Scholar]
- 23.Weiss MD, Macklin EA, Simmons Z, et al. Mexiletine ALS study group. A randomized trial of mexiletine in ALS: safety and effects on muscle cramps and progression. Neurology. 2016;86(16):1474–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on motor neuron diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 25.Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve. 1998;21:137–158. [DOI] [PubMed] [Google Scholar]
- 26.Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve. 2000;23:399–409. [DOI] [PubMed] [Google Scholar]
- 27.Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin Neurophysiol. 2001;112:1575–1585. [DOI] [PubMed] [Google Scholar]
- 28.Jankelowitz SK, Burke D. Axonal excitability in the forearm: normal data and differences along the median nerve. Clin Neurophysiol. 2009;120(1):167–173. [DOI] [PubMed] [Google Scholar]
- 29.Hermsen AM, Haag A, Duddek C, et al. Test-retest reliability of single and paired pulse transcranial magnetic stimulation parameters in healthy subjects. J Neurol Sci. 2016;362:209–216. [DOI] [PubMed] [Google Scholar]
- 30.Hays RD, Sherbourne CD, Mazel RM. The RAND 36-item health survey 1.0. Health Econ. 1993;2(3):217–227. [DOI] [PubMed] [Google Scholar]
- 31.Oskarsson B, Moore D, Mozaffar T, et al. Mexiletine for muscle cramps in amyotrophic lateral sclerosis: a randomized, double-blind crossover trial. Muscle Nerve. 2018;58(1):42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vucic S, Ziemann U, Eisen A, Hallett M, Kiernan MC. Transcranial magnetic stimulation and amyotrophic lateral sclerosis: pathophysiological insights. J Neurol Neurosurg Psychiatry. 2013;84(10):1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kiernan MC, Cikurel K, Bostock H. Effects of temperature on the excitability properties of human motor axons. Brain. 2001;124(Pt 4):816–825. [DOI] [PubMed] [Google Scholar]
- 34.Chervyakov AV, Bakulin IS, Savitskaya NG, et al. Navigated transcranial magnetic stimulation in amyotrophic lateral sclerosis. Muscle Nerve. 2015;51(1):125–131. [DOI] [PubMed] [Google Scholar]
- 35.Zanette G, Tamburin S, Manganotti P, Refatti N, Forgione A, Rizzuto N. Changes in motor cortex inhibition over time in patients with amyotrophic lateral sclerosis. J Neurol. 2002;249(12):1723–1728. [DOI] [PubMed] [Google Scholar]
- 36.Schanz O, Bageac D, Braun L, Traynor BJ, Lehky TJ, Floeter MK. Cortical hyperexcitability in patients with C9ORF72 mutations: relationship to phenotype. Muscle Nerve. 2016;54(2):264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huynh W, Dharmadasa T, Vucic S, Kiernan MC. Functional biomarkers for amyotrophic lateral sclerosis. Front Neurol. 2019;9:1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geevasinga N, Menon P, Sue CM, et al. Cortical excitability changes distinguish the motor neuron disease phenotypes from hereditary spastic paraplegia. Eur J Neurol. 2015;22(5):826–831. [DOI] [PubMed] [Google Scholar]
- 39.Pohl C, Block W, Träber F, et al. Proton magnetic resonance spectroscopy and transcranial magnetic stimulation for the detection of upper motor neuron degeneration in ALS patients. J Neurol Sci. 2001;190(1–2):21–27. [DOI] [PubMed] [Google Scholar]
- 40.Paulus W, Classen J, Cohen LG, et al. State of the art: pharmacologic effects on cortical excitability measures tested by transcranial magnetic stimulation. Brain Stimul. 2008;1(3):151–163. [DOI] [PubMed] [Google Scholar]
- 41.Wainger BJ, Kiskinis E, Mellin C, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Masson G, Przedborski S, Abbott LF. A computational model of motor neuron degeneration. Neuron. 2014;83(4):975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uebachs M, Opitz T, Royeck M, et al. Efficacy loss of the anticonvulsant carbamazepine in mice lacking sodium channel beta subunits via paradoxical effects on persistent sodium currents. J Neurosci. 2010;30(25):8489–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vucic S, Cheah BC, Yiannikas C, Kiernan MC. Cortical excitability distinguishes ALS from mimic disorders. Clin Neurophysiol. 2011;122(9):1860–1866. [DOI] [PubMed] [Google Scholar]
- 45.Geevasinga N, Menon P, Nicholson GA, et al. Cortical function in asymptomatic carriers and patients with C9orf72 amyotrophic lateral sclerosis. JAMA Neurol. 2015;72(11):1268–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howells J, Czesnik D, Trevillion L, Burke D. Excitability and the safety margin in human axons during hypothermia. J Physiol. 2013;591(12):3063–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shibuya K, Misawa S, Kimura H, et al. A single blind randomized controlled clinical trial of mexiletine in amyotrophic lateral sclerosis: efficacy and safety of sodium channel blocker phase II trial. Amyotrophic Lateral Scler Frontotemporal Degener. 2015;16(5–6):353–358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.