

Abstract
As a conserved molecular chaperone, heat shock protein 90 (Hsp90) maintains the stability and homeostasis of oncoproteins and helps cancer cells survive. DNA-dependent protein kinase catalytic subunit (DNA-PKcs) plays a pivotal role in the non-homologous end-joining pathway (NHEJ) for DNA double-strand breaks (DSBs) repair. Tumor cells contain higher levels of DNA-PKcs to survive by the hostile tumor micro-environment and various anti-tumor therapies. Here, we showed that increased levels of Hsp90α, Hsp90β, and DNA-PKcs correlated with a poor overall survival in hepatocellular carcinoma (HCC). We revealed that Hsp90 N-terminal domain and C-terminal domain have different effects on DNA-PKcs protein and mRNA levels. The stability of DNA-PKcs depended on Hsp90α N-terminal nucleotide binding domain (NBD). Transcription factor SP1 regulates the transcription of PRKDC (gene name of DNA-PKcs) and is a client protein of Hsp90. Inhibition of Hsp90 N-terminal by STA9090 decreased the location of Hsp90α in nucleus, Hsp90α-SP1 interaction, SP1 level and the binding of Hsp90α/SP1 at the proximal promoter region of PRKDC. Since hyperthermia induces DSBs with increases level of DNA-PKcs, combined STA9090 treatment with hyperthermia effectively delayed the tumor growth and significantly decreased DNA-PKcs levels in xenografts model. Consistently, inhibition of Hsp90 increased the number of heat shock-induced γ-H2AX foci and delayed the repair of DSBs. Altogether, our results suggest that Hsp90 inhibitor STA9090 decreases DNA-PKcs protein stability and PRKDC mRNA level, which provide a theoretical basis for the promising combination therapy of hyperthermia and Hsp90 inhibitor in HCC.
Keywords: Hepatocellular carcinoma, Hsp90 inhibition, Hyperthermia, DNA-PKcs, DNA damage repair
Introduction
Hepatocellular carcinoma (HCC) is the most common form of liver cancer as it accounts for almost 90% of the cases and the incidence is estimated to reach over one million cases by 2025 (1). However, more efficacious treatments still need to be developed to decrease HCC mortality (2). Heat shock protein 90 (Hsp90) and its client proteins are overexpressed in cancer, Hsp90α (inducible form) and Hsp90β (constitutive form) are two isoforms of Hsp90 in humans (3). Recent studies have shown that more than 400 client proteins, rely on Hsp90 proteins for the folding and persistence in an active form (4). Hsp90 has been considered as a strict cytosolic molecular chaperone, but Hsp90 in the nucleus also plays vital roles. Hence, Hsp90 has been explored as a therapeutic target of cancer treatment for decades. Unexpectedly, the adverse effects, such as structural instability and hepatotoxicity, have been reported with early Hsp90 inhibitors (5, 6), STA9090 (Ganetespib) is the only Hsp90 inhibitor, which halted in the clinical trial phase III (7). Therefore, there is an urgent need to overcome these limitations for the development of therapies based on Hsp90 inhibitors (8, 9).
Heat stress (HS) or hyperthermia is an adjuvant therapeutic modality to inhibit the growth of cancer cells (10). High temperatures (40°C~46°C) render the tumor cells more sensitive to the effects of radiation and chemotherapy (11). Clinical trials have shown the benefits of using hyperthermia combined with chemotherapy to treat patients with a wide range of cancers, including HCC (12). It has been proposed that hyperthermia induces DNA damage, cell cycle arrest and apoptosis in cancer cells (13). HS can activate DNA-PKcs as a DNA damage response (DDR) (14). DNA-PKcs actively participates in the DSB repair by regulating the non-homologous end-joining (NHEJ) (15). Recently, the expression and function of DNA-PKcs have been studied in various human cancers, and DNA-PKcs has been involved in cancer initiation, progression, and apoptosis resistance. Therefore, DNA-PKcs represents a novel and critical target for the development of anticancer therapy (16, 17). Earlier studies showed that DNA-PKcs was a client of Hsp90 (http://www.picard.ch/downloads/Hsp90facts.pdf) (18), Hsp90 inhibition can selectively inhibit tumor cell proliferation by decreasing the levels of key DSB repair proteins (19). Our previous research also showed that hyperthermia enhanced the efficacy of the Hsp90 inhibitor 17-DMAG by impairing the G2/M transition, the combination of hyperthermia with 17-DMAG represents a useful and potential therapeutic strategy for HCC (20), but the effects of different Hsp90 inhibitors on DNA-PKcs and the detailed mechanism remains unclear.
Materials and Methods
Human tissue
HCC and adjacent non-tumoral tissues were collected in 95 patients at Taizhou Hospital (Zhejiang, China). The patient study was approved by the ethics committee. This study has been conducted in accordance with ethical standards and according to the Declaration of Helsinki and the national and international guidelines. All patients provided a written informed consent for the use of the surgical samples. None of the patients had been treated with chemotherapy or radiotherapy before the ablation of the liver tumor. The 95 cases were followed up for 7 years. The detailed clinical case information and records of 95 patients are listed in Supplementary Table1.
Plasmids
The HA-tag fusion of Hsp90α full-length, the N-terminal nucleotide binding domain (NBD)-middle domain (MD), the MD, the MD-C-terminal dimerization domain (CTD), the C-terminal MEEVD motif of Hsp90α, and FLAG-Hsp90α are kind gift from Matthias Mayer (ZMBH, Heidelberg University, Germany) (21).
Cell culture and reagents
HepG2 (SCSP-510), Huh7 (SCSP-526), and L02 (GNHu 6) were purchased from the Cell Bank, Chinese Academy of Medical Sciences (Shanghai, China). MHCC97H were obtained from the Liver Cancer Institute, Zhongshan Hospital, Fudan University, Shanghai, China. HepG2-Luc were constructed by Cloud-Clone Corp (Wuhan, China). All cell lines were authenticated, and cells were thawed upon arrival, expanded, and stored in a liquid nitrogen tank. The absence of mycoplasma cell contamination was confirmed using MycAwayTM-Color One-Step Mycoplasma Detection Kit (40611ES, Yeasen, China). All the cell lines were cultured in Dulbecco’s modified Eagle’s medium (Gibco, USA) supplemented with 10% fetal bovine serum (Gibco, USA) at 37°C, 5% CO2. Ganetespib (STA9090) (22) (S1159), Novobiocin (NB) (23) (S2492), NVP-BEP800 (24) (S1498), MG132 (S2619), and cycloheximide (CHX) (S7418) were purchased from SelleckChem (USA).
Cell Transfection
Hsp90AA1 sgRNA sequences and the siHsp90α and siHsp90β sequences were synthesized by iGeneBio (Guangzhou, China). The transfection of CRISPR-Cas9 plasmids for Hsp90α knockout, siHsp90α and siHsp90β were carried out with LIPO3000 (L3000015, Life Technologies, USA). According to the protocol, after 48 h, cells were collected, and incubated in the selection medium containing 800 μg/mL G418 (G001, MDBio, Inc, China) for 2–3 weeks. The successfully Hsp90AA1 knockout clones were confirmed by western blotting (21).
Western blotting
Western blot analysis was performed as described previously (21). We used the following antibodies: Hsp90α (6251422, Enzo, USA), Hsp90α (8165s, CST, USA), Hsp90β (5087s, CST, USA), DNA-PKcs (SC-390849, Santa Cruz, USA), γ-H2AX (9718s, CST, USA), HA (H3663, Sigma, USA), FLAG (C:KM8002, Sungene Biotech Co, China), SP1 (9389S, CST, USA) and β-actin (RM2001, Ray Antibody, China). The secondary antibodies (Donkey anti-Mouse/Rabbit/Rat IRDye 680/800) were from Abcam (USA). Western blots were scanned using Li-COR Odyssey, and band intensities were analyzed by ImageJ v1.8.0.
Immunohistochemistry (IHC)
Immunohistochemistry (IHC) was performed on paraffin sections of HCC patient tissues according to the standard LSAB protocol (BOSTER, China). We used the primary antibodies against Hsp90α (1:10000) (6251422, Enzo, USA), Hsp90β (1:2000) (5087s, CST, USA), DNA-PKcs (1:100) (SC-390849, Santa Cruz, USA), Isotype-matched IgG was used as negative controls. The IHC staining was graded by two independent pathologists who had no knowledge about the patient history. The staining intensity was scored as negative, weak positive, moderate positive, strong positive using 0, 1, 2, and 3 as scores, respectively (21).
Quantitative Real-time PCR (RT-qPCR) and reverse transcription-PCR (RT-PCR)
Total RNA from L02, Huh7, HepG2, and MHCC97H cells was isolated with the TRIzol reagent (9109, Takara, Japan) according to the manufacturer’s instructions. Then 1 μg RNA was reverse transcribed into cDNA using PrimeScript™ RT reagent kit (CRR047A, Takara, Japan). RT-qPCR and RT-PCR was performed using SYBR Premix Ex Taq and Premix Taq (Takara, Japan) and Premix Taq (Takara, Japan), respectively. RT-qPCR was subsequently done using a Roche LightCycler 480 PCR Detection System (Roche, Switzerland). The sequences of the primers used were listed in Supplementary Table 2. The cycling conditions were as follows: 1 cycle of 95°C for 5 min, 40 cycles of 95 for 1 min, 60°C for 30 s, and 72°C for 30 s. The melting curve obtained at the end of the RT-qPCR was used to determine the specificity of the reaction. The relative mRNA expression levels were calculated using the 2−ΔΔCt method. For the RT-PCR, the reaction was performed on a GeneAMP 9600 system with the same conditions. Subsequently, the PCR products were separated using electrophoresis on an agarose gel, and the quantification was performed using ImageJ v1.8.0. All mRNA expression levels were normalized to β-actin expression.
Immunofluorescence microscopy
Cells were fixed in 4% paraformaldehyde, washed with PBS, permeabilized with −20°C precooled methanol. The immunodetection was performed by incubating the cells with anti-Hsp90α (1:1000) (6251422, Enzo, USA), anti-Hsp90β (1:500) (5087s, CST, USA), anti-DNA-PKcs (1:200) (SC-390849, Santa Cruz, USA), anti-HA (1:100) (H3663, Sigma, USA), anti-SP1 (1:100) (GTX110593, GeneTex, USA) and anti-γ-H2AX (1:200) (9718s, CST, USA) at 4°C overnight and subsequently with Alexa Fluor 594 anti-Rat (1:100) (A-21209, Life Technologies, USA), Alexa Fluor 488 anti-mouse (1:100) (A-11001, Life Technologies, USA), Alexa Fluor 488 anti-Rabbit (1:100) (A31628, Life Technologies, USA), and Alexa Fluor 555 anti-Rabbit (1:100) (A31572, Life Technologies, USA) antibodies at room temperature for 2 h. The nuclei of cells were stained with DAPI for 5 min. Images were taken on an Olympus FV1000 Confocal Laser Scanning Microscope (Tokyo, Japan).
In vivo experiments
Five-week-old male BALB/c nude mice were obtained from the Southern Medical University Laboratory Animal Centre. All procedures in this animal study were approved by the Institutional Animal Care and Use Committee of Southern Medical University (Approval code L2020029) and were carried out according to the principles of the NIH Guide for the Care and Use of Laboratory Animals. We injected 5 × 106 HepG2-Luc cells in 100 μl DMEM subcutaneously into the flanks of the mice. After 10 days, the mice were divided into four groups according to the treatment. STA9090 stock solution was diluted in 10% DMSO with 20% Cremophor RH (MB2572, Sigma, USA). The final formulation of 5.5 mg/ml STA9090 contained 10% DMSO, 18% Cremophor RH, 3.6% dextrose (158968, Sigma, USA), 68.4% ddH2O. Mice in the STA9090 group and combination group exposed to STA9090 and hyperthermia were injected intraperitoneally with STA9090 at a dose of 12.5 mg/kg three times a week for 10 days. Mice in the control and hyperthermia groups were injected intraperitoneally with the final formulation without STA9090. Two hours after injection, mice in the hyperthermia and the combination groups were transferred into a 42°C thermostatic cabinet and kept there for 2 h. The weights of the mice and the longest and shortest diameters of the xenograft tumors were measured before each treatment. After 10 days, all mice were sacrificed, and the xenograft tumors were isolated. The tumor volume was calculated according to the following formula: longest diameter × (shortest diameter)2 × 0.5.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were conducted using the published protocol (25). Ultrasonic systems (Scientz JY88-Ⅱ, China) was optimized to produce DNA segments ranging from 150 to 500 bp as determined by agarose gel electrophoresis. Hsp90α or SP1 interacted DNA fragments were precipitated by incubating the cell extract with specific primary Hsp90α antibody (1:200) (6251422, Enzo, USA) or SP1 antibody (1:100) (9389S, CST, USA) overnight. The complexes were captured using Protein A/G-Sepharose beads (IP10–10ML, Millipore, MA, USA). The primers of Hsp90α/SP1 ChIP-PCR used were listed: Forward: 5’–GAGTCCACCTGGCGAGT-3’, Reverse: 5’- CGCCGCTGTGATTGG-3’. The DNA samples of ChIP and input were analyzed by PCR and separate in a 2% agarose gel staining with EB.
Statistical analysis
The overall survival rate was estimated using the Kaplan-Meier method, and patients were divided into high versus low expression group based on the median value for each gene. Statistical p values were analyzed using a two-tailed Student t test. The data were represented as mean ± standard deviation (SD) from three independent experiments. The differences between groups were analyzed by SPSS software 21.0, n represents number of samples analyzed. P < 0.05 was considered as statistically significant.
Results
High levels of Hsp90α, Hsp90β, and DNA-PKcs in HCC
We collected proteomics data from Fuchu He et al. (26) on the Chinese Human Proteome Project (CNHPP) website (http://liver.cnhpp.ncpsb.org/). We compared the protein expression profiles of Hsp90α, Hsp90β, and DNA-PKcs in patient tumors and adjacent tissues. Hsp90α, Hsp90β, and DNA-PKcs proteins were all up-regulated in tumoral tissues (Fig. 1A). The proteomic data showed a positive correlation between the protein levels of Hsp90α and DNA-PKcs, and between the protein amounts of Hsp90β and DNA-PKcs in patients with HCC (Fig. 1C). The mRNA expression profiles of Hsp90AA1, Hsp90AB1, and PRKDC in HCC samples were obtained from The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/). The mRNA levels of Hsp90AA1, Hsp90AB1, and PRKDC were higher in HCC (Fig. 1B), and a significant mRNA expression correlation between Hsp90AA1 and PRKDC and between Hsp90AB1 and PRKDC (Fig. 1D). In addition, protein (Fig. 1E) and mRNA (Fig. 1F–G) levels of Hsp90α, Hsp90β, and DNA-PKcs increased in hepatoma cell lines (Huh7, HepG2, and MHCC97H) as compared with those in the non-malignant liver cell line L02. These findings indicate that DNA- PKcs amounts increase in parallel with the Hsp90α and Hsp90β levels in HCC.
Figure 1.
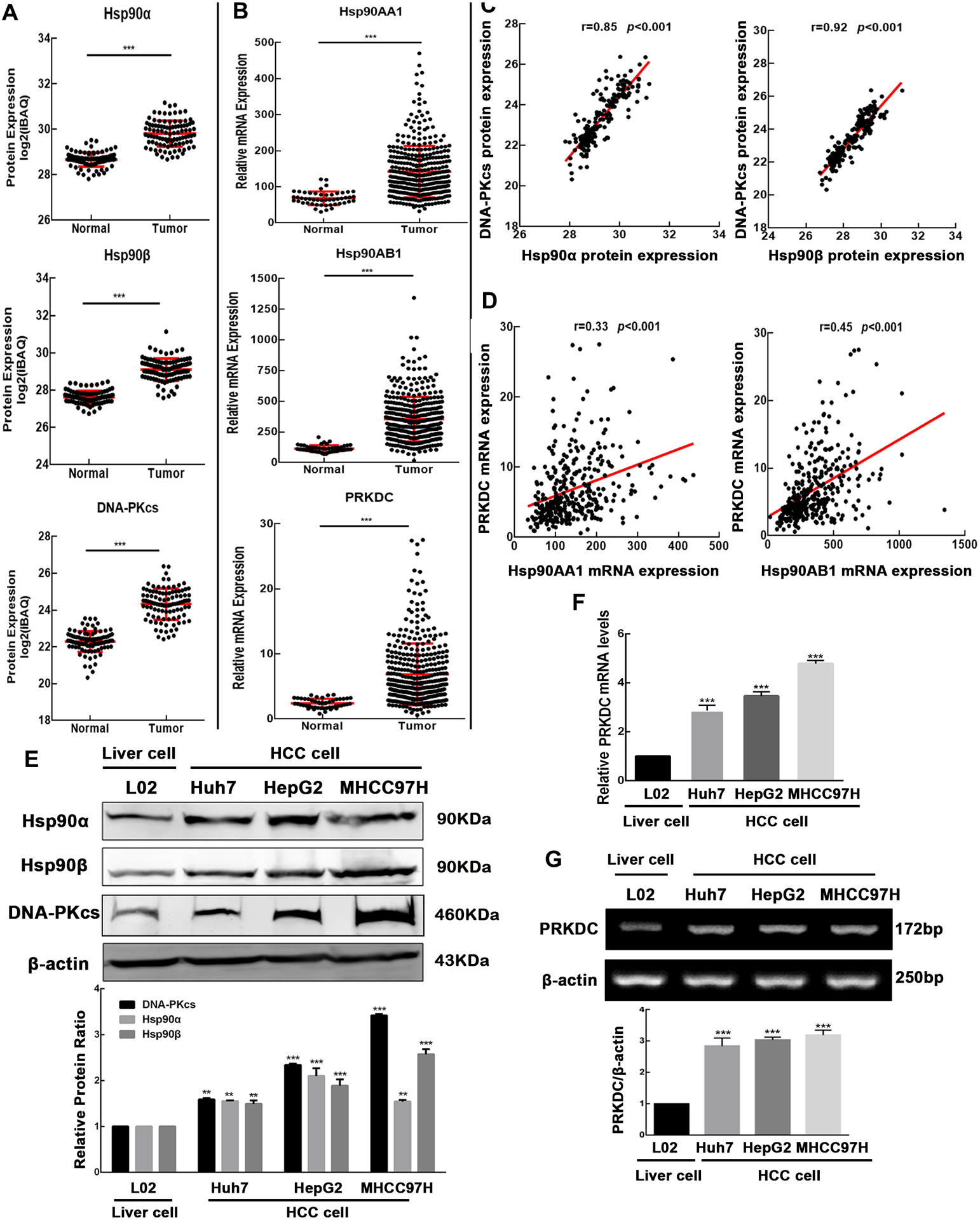
Up-regulation of Hsp90α, Hsp90β, DNA-PKcs levels in HCC. We analyzed data obtained from Fuchu He et al. on the CNHPP data portal. (A) Protein levels of Hsp90α, Hsp90β and DNA-PKcs increased and (C) a positive correlation between the Hsp90α and DNA-PKcs, Hsp90β and DNA-PKcs. Quantification of mRNA in tumors and adjacent tissue was obtained from TCGA. (B) Increase mRNA levels of Hsp90AA1, Hsp90AB1, PRKDC and (D) a positive correlation between the Hsp90AA1 and PRKDC, Hsp90AB1 and PRKDC were obtained in HCC. (E) Higher Hsp90α, Hsp90β and DNA-PKcs levels were measured in HCC cell lines Huh7, HepG2, and MHCC97H than in liver cell line L02 cells using Western blotting. RT-qPCR (F) and RT-PCR (G) analyses showed increased levels of the mRNA. Band intensities were quantified by ImageJ v1.8.0, and values represent mean ± SD, n = 3, **p < 0.01, ***p < 0.001.
High protein levels of Hsp90α, Hsp90β, and DNA-PKcs predict poor overall survival of patients with HCC
The expression of Hsp90α, Hsp90β and DNA-PKcs in tumoral and adjacent tissues from the liver of 95 patients with HCC was investigated using IHC (Fig. 2A). It showed a significantly higher protein expression of Hsp90α, Hsp90β and DNA-PKcs in tumors compared with that in adjacent tissues (Fig. 2B). We investigated the influence of Hsp90α, Hsp90β and DNA-PKcs expression on the clinical progression of patients with HCC. Higher Hsp90α, Hsp90β and DNA-PKcs expression was associated with reduced overall survival in the 95 HCC patients (Fig. 2C). Furthermore, using the data from the TCGA database, we found that higher mRNA levels of Hsp90AA1, Hsp90AB1, and PRKDC correlated with a worse outcome in patients with HCC. These results are consistent with the HCCDB data (http://lifeome.net/database/hccdb/home.html) (Fig. S1).
Figure 2.
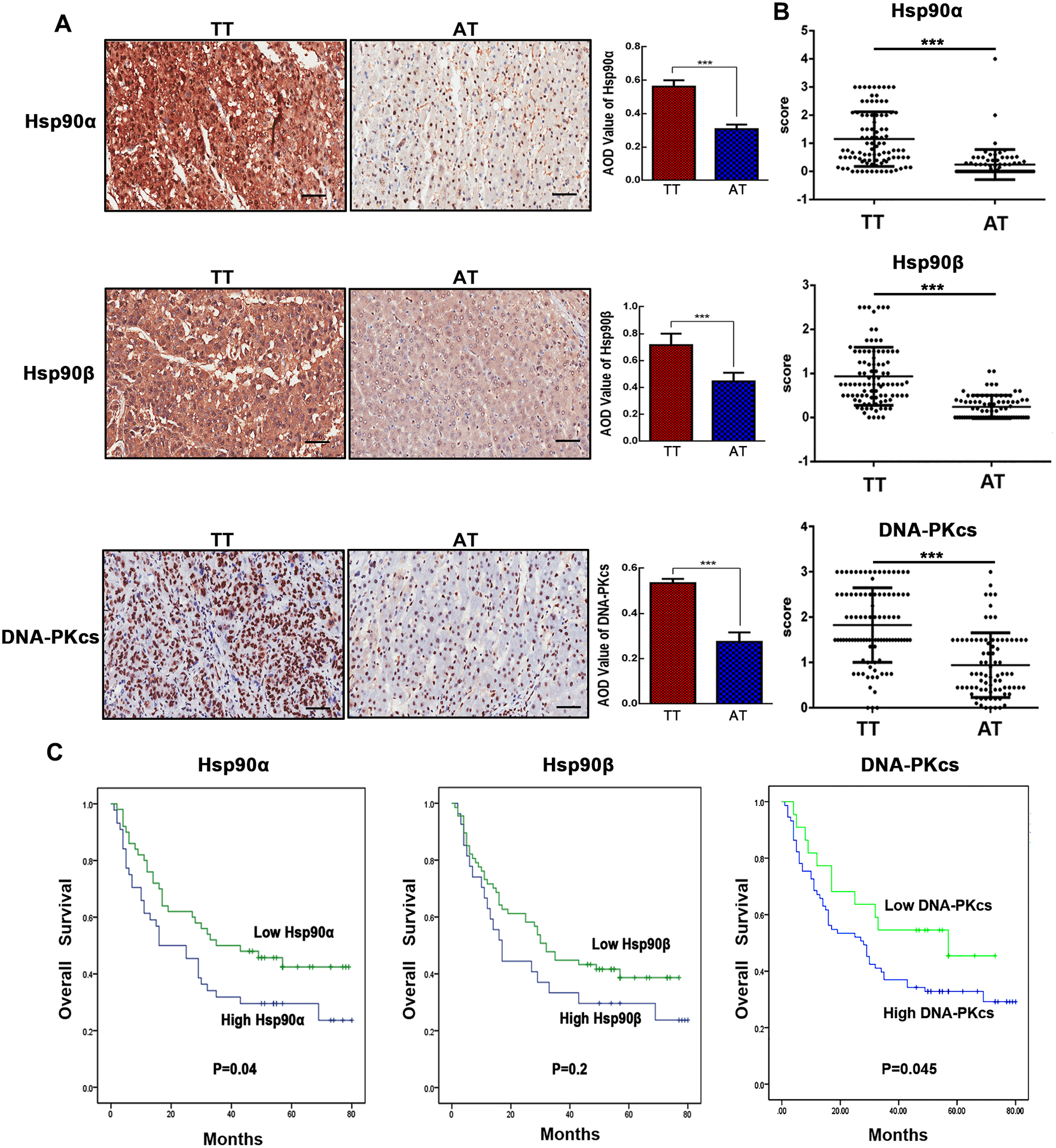
HCC tumor tissues exhibit higher levels of Hsp90α, Hsp90β and DNA-PKcs and predict poor overall survival of patients with HCC. (A) The protein expression of Hsp90α, Hsp90β and DNA-PKcs in tumor tissue (TT) or adjacent tissue (AT) of patients with HCC was detected using immunohistochemistry. Original magnification: 4X and 20X. Scale bar is 50 μm. The AOD (average optical density) value of Hsp90α, Hsp90β, and DNA-PKcs staining was quantified by Image J v1.8.0. (B) Tumor tissue had higher Hsp90α, Hsp90β, and DNA-PKcs expression by analyzing the final scores in TT and AT. (C) Kaplan-Meier survival analysis of patients with HCC based on Hsp90α, Hsp90β and DNA-PKcs protein levels. Low Hsp90α, Hsp90β, and DNA-PKcs: IHC score 0–1; High Hsp90α, Hsp90β and DNA-PKcs: IHC score 2–3. Values represent mean ± SD, n=95, ***p < 0.001.
The stability of DNA-PKcs depends on Hsp90α N-terminal nucleotide binding domain (NBD) in HCC cells
DNA-PKcs is one of Hsp90 client proteins (18), our data confirmed Hsp90α and Hsp90β could positively regulate DNA-PKcs level. DNA-PKcs was reduced under STA9090 (Fig. S2A–D), NVP-BEP800 (Fig. S3A–B), and Hsp90α, Hsp90β knockdown (Fig. S2E–F) (Fig. S3C), and Hsp90AA1 knock-out (KO) (Fig. S2G) treatment in HepG2 cells. Moreover, the transient transfection with a FLAG-Hsp90α-encoding plasmid rescued the expression of DNA-PKcs to wild-type levels in HepG2 KO cells (Fig. S2G), and the overexpression of Hsp90α and Hsp90β (Fig. S2H) increased DNA-PKcs levels, the proteasome inhibitor MG132 substantially reversed Hsp90α and Hsp90β knockdown-induced down-regulation of DNA-PKcs, demonstrating DNA-PKcs was degraded by ubiquitin-dependent pathway (Fig. S3D). Since the N-terminal nucleotide binding domain (NBD), or middle domain (MD), or C-terminal dimerization domain (CTD) of Hsp90α can interact with the client proteins respectively (27), to determine whether the Hsp90α domain(s) has a causal effect on DNA-PKcs stability, the level of DNA-PKcs was investigated in HepG2 and Huh7 cells treated with the NBD-binding inhibitor STA9090 and the CTD-binding inhibitor novobiocin (NB). STA9090 induced a clear decrease of DNA-PKcs, consistent with the reduction observed in Fig. S2A–D. In contrast, the NB increased DNA-PKcs expression level (Fig. 3A). Since different domain of Hsp90α alone can function as chaperones to interact with client proteins (28), to further prove this hypothesis that the Hsp90α domain(s) contributes to the level of DNA-PKcs, we transiently transfected the plasmids encoding HA-tagged Hsp90α full-length, or HA-tagged NBD-MD, MD, and MD-CTD domain of Hsp90α into HepG2 cells. DNA-PKcs protein amounts were significantly increased by Hsp90α NBD-MD and were reduced by Hsp90α MD-CTD (Fig. 3B). To elucidate the Hsp90α domain(s) interacts with DNA-PKcs and contributes to its stability, Hsp90α were immunoprecipitated from HepG2 cells treated with STA9090 or NB. Less Hsp90α was pulled down with DNA-PKcs antibody in STA9090 treated cells, while more Hsp90α associated with DNA-PKcs in NB group (Fig. 3C). Confocal microscopy results revealed Hsp90α was found throughout the cytoplasm and nucleus, and colocalized with DNA-PKcs in nuclei. After STA9090 treatment, the cytoplasmic Hsp90α level (Fig. S4A) and cytoplasmic STA9090 (Fig. S4B) increased dramatically, while less nuclear Hsp90α/STA9090 location and decreased Hsp90α-DNA-PKcs colocalization was observed. While treated with NB, HepG2 cells presented with more nucleus Hsp90α and increased Hsp90α-DNA-PKcs colocaliztion (Fig. S4B). To further confirm the effects of Hsp90α different domain on intracellular location and Hsp90α-DNA-PKcs interaction, we transiently transfected plasmids encoding NBD-MD, MD and MD-CTD constructs as HA fusions into HepG2 cells, the co-IP results revealed that DNA-PKcs associated with the NBD-MD construct of Hsp90α, but less with MD-CTD domain (Fig. 3D). The immunofluorescence images of HA and DNA-PKcs (Fig. S4C) also demonstrated the transfection of Hsp90α MD-CTD induced less nuclear Hsp90α/STA9090 location and decreased Hsp90α-DNA-PKcs colocalization, while the transfection of Hsp90α NBD-MD increased nucleus Hsp90α location and Hsp90α-DNA-PKcs interaction, consistent with the treatment of STA9090 and NB, respectively. To examine whether the decreased DNA-PKcs level was due to the degradation through the ubiquitin-proteasomal pathway after STA9090 treatment, we monitored the level of DNA-PKcs after STA9090 treatment in combination with the proteasome inhibitor MG132 or the protein synthesis inhibitor CHX. Our data showed that the DNA-PKcs levels increased in cells treated with STA9090 combined with MG132 compared with STA9090 alone (Fig. 3E). Consistently, the level of DNA-PKcs clearly decreased in cells treated with both STA9090 and CHX (Fig. 3F), suggested Hsp90 N-terminal domain inhibition promoted DNA-PKcs degradation. To determine whether NB treatment increased DNA-PKcs stability, HepG2 cells were treated with CHX and NB, the DNA-PKcs levels were increased in cells treated with NB plus CHX compared with CHX alone (Fig. 3G), demonstrating that NB promoted the stability of DNA-PKcs.
Figure 3.
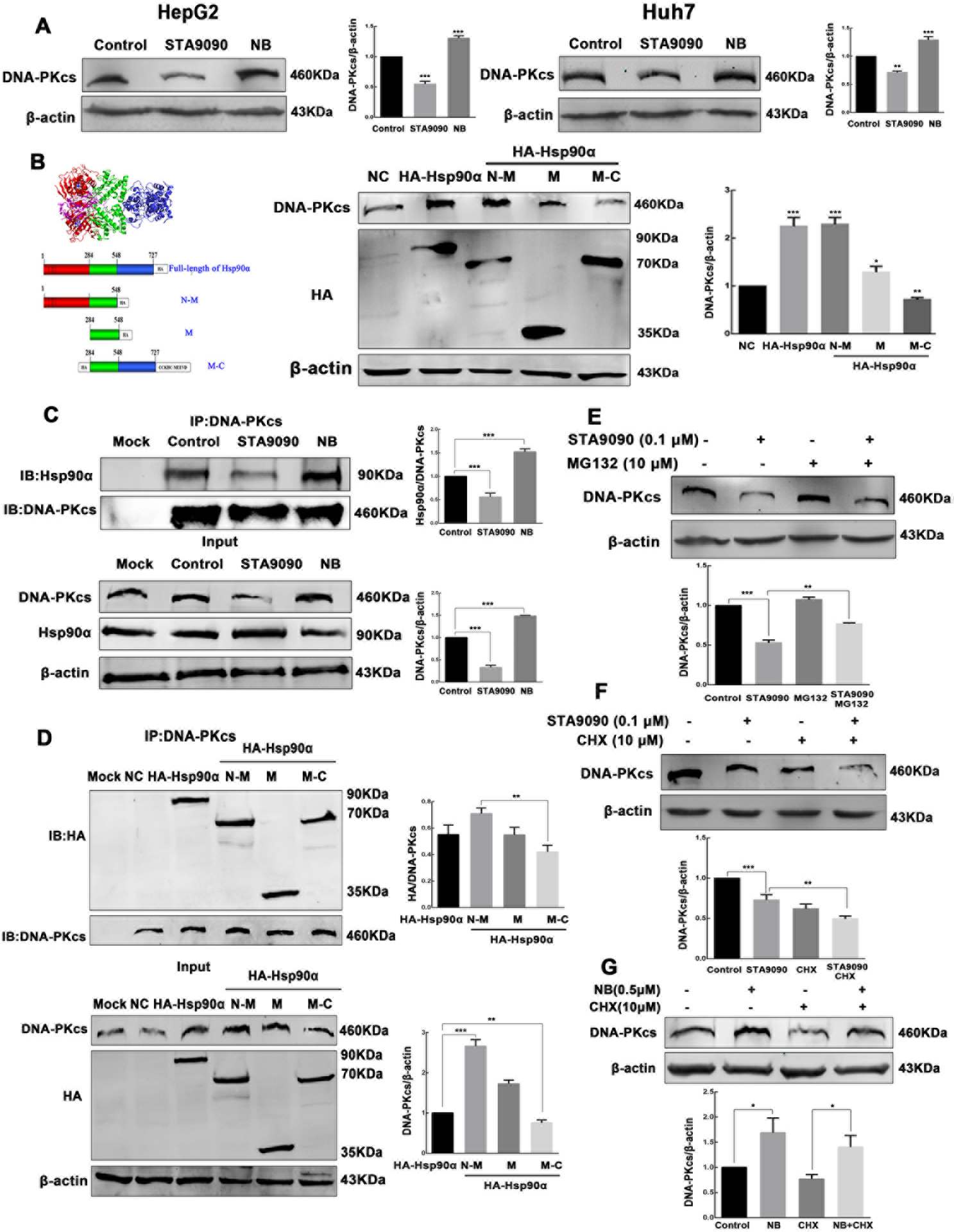
The stability of DNA-PKcs depends on the NBD of Hsp90α. (A) HepG2 and Huh7 cells were incubated with 0.1 μM STA9090 and 0.5 μM NB for 24 h as indicated. DNA-PKcs and β-actin levels were determined by Western blot. (B) HepG2 cells were transiently transfected with plasmids encoding HA-tagged Hsp90α full-length or the HA-tagged NBD-MD, MD, and MD-CTD domains. DNA-PKcs, HA, and β-actin levels were assayed by Western blot. (C) Immunoprecipitation of DNA-PKcs was performed with lysates of HepG2 cells after STA9090 and NB treatment. Hsp90α and DNA-PKcs levels were determined by Western blot. (D) HA-tagged Hsp90α full-length or the HA-tagged NBD-MD, MD, and MD-CTD constructs used for the interaction study. Immunoprecipitation was performed with anti‐DNA-PKcs. Membranes were probed with anti‐HA and anti‐DNA-PKcs antisera. (E) Cells treatment with STA9090 (0.1 μM for 24 h) and MG132 (10 μM for 6 h) increased DNA-PKcs levels compared with incubation with STA9090 alone. (F) STA9090 (0.1 μM for 24 h) combined with CHX (10 μM for 6 h) decreased DNA-PKcs amounts compared with STA9090 alone. (G) NB (0.5 μM for 24 h) combined with CHX (10 μM for 6 h) increased DNA-PKcs amounts compared with CHX alone. Band intensities were quantified by Image J v1.8.0, and values represent mean ± SD, n = 3, *p < 0.05. **p < 0.01, ***p < 0.001.
N-terminal Hsp90 inhibitor STA9090 reduces the binding of Hsp90α/SP1 to PRKDC promoter and decreases PRKDC transcription
Since the above-presented data indicated that the different effects between STA9090 and NB on DNA-PKcs protein levels, and different cytoplasmic/nucleus location of Hsp90α, we wondered whether STA9090 and NB have also different influences on DNA-PKcs mRNA transcription. In parallel with the STA9090 and NB treatment, we transiently transfected plasmids encoding HA-tagged Hsp90α full-length, or HA-tagged NBD-MD, MD, and MD-CTD of Hsp90α into HepG2 cells. The abundance of PRKDC mRNA significantly decreased upon STA9090 treatment and HA-tagged MD-CTD overexpression, but dramatically increased upon NB treatment and HA-tagged NBD-MD overexpression (Fig. 4A–B). It suggested that Hsp90 influenced the PRKDC transcription. To confirm this, the ChIP assay in HepG2 cells treated with STA9090 and NB was carried out to mimic the in vivo recruitment of Hsp90α to the promoter of PRKDC. The results revealed that the PRKDC promoter could be pulled down by anti-Hsp90α antibodies and confirming that STA9090 significantly decreased the binding of Hsp90α to PRKDC promoter, but NB treatment caused an increased binding (Fig. 4C). Recent reports have found that the function of Hsp90 in nuclear events can regulate transcription factors and the general transcription machinery, thus contributing to regulation of gene expression. As such, a probable mechanism driving the broad reach of Hsp90 in DNA binding events is the maintenance of transcription factors (29, 30). To check this possibility. We focused on 103 Hsp90 related transcription factors from the list of Hsp90 Interactor (PicardLab, https://www.picard.ch/) as candidates for PRKDC transcription. Besides, the 1500 bp fragment upstream of the transcriptional start site (TSS) of PRKDC was determined as promoter region. Sequences of PRKDC promoter region were download from USCS database (http://genome.ucsc.edu/). PROMO was performed to analyze the transcript factors binding at the promoter region of PRKDC. The result showed that 72 potential transcript factors binding in the promoter region of PRKDC. There were 10 candidate PRKDC transcription factors related to Hsp90 screened by Venn diagram (Fig.S5A). Next, Cistrome database (http://cistrome.org/) was used to obtain the ChIP-seq data of the 10 candidate transcription factors. Four ChIP-seq data (SP1, IRF2, ZBTB20 and NFRKB) were included and had significant binding signals in Cistrome database (Fig. S5B). The correlation of candidate transcription factors, Hsp90α and DNA-PKcs were analyzed using the data from CNHPP database. The results indicated that the positive correlation of both Hsp90α/SP1 and DNA-PKcs/SP1 were highly related (Fig. S5C). However, the relationship of Hsp90α/NFRKB and DNA-PKcs/NFRKB were negative correlation. Thus, we speculated that Hsp90α, as a transcriptional cofactor, participated in the process of SP1 transcribing PRKDC. Further, immunoprecipitation with anti-Hsp90α antibody followed by immunoblotting with anti-Hsp90α and anti-SP1 antibodies was performed in HepG2 cells treated with STA9090 or NB, as shown in Fig. 4D. In addition, the confocal microscopy observed the co-localization between the SP1 and Hsp90α (Fig. 4E). The result revealed that less colocalization of SP1 with Hsp90α in the nucleus after STA9090 treatment, however, both SP1 and Hsp90α were significantly colocalized under NB treatment. Besides, confocal immunofluorescence revealed that SP1 associated with the N-M construct of Hsp90α, less SP1 and M-C domain of Hsp90α were colocalized in the nucleus (Fig. S5D). SP1 is considered as a factor to determine the core activity of the promoter by direct interaction with other factors of the basal transcriptional machinery and by cooperation with several transcriptional activators (31–33). Therefore, we performed ChIP assay to examine Hsp90α/SP1 interaction to the PRKDC promoter. The results revealed that −564~−666 bp to TSS site of the PRKDC promoter could be pulled down by anti-Hsp90α or anti-SP1, and STA9090 significantly decreased the binding of Hsp90α/SP1 to PRKDC promoter, NB increased the binding (Fig. 4F). Taken together, these results indicated that Hsp90α interacted with SP1 in the nucleus, Hsp90α-SP1 interaction is important for the PRKDC transcription, and STA9090 decreased the binding of Hsp90α/SP1 to PRKDC promoter and reduced PRKDC transcription. Besides, the expression of the PRKDC was also reduced by Hsp90 inhibition under heat stress, immunofluorescence staining confirmed that STA9090 induced a decrease of Hsp90 in the nucleus, thereby might affect Hsp90 function inside nucleus (Fig. S6). Hyperthermia can induce DDR, and the increased level of PRKDC mRNA was confirmed by RT-qPCR and RT-PCR analyses, consistent with our hypothesis, the abundance of PRKDC mRNA significantly decreased upon STA9090 inhibitor treatment (Fig. 4G–H).
Figure 4.
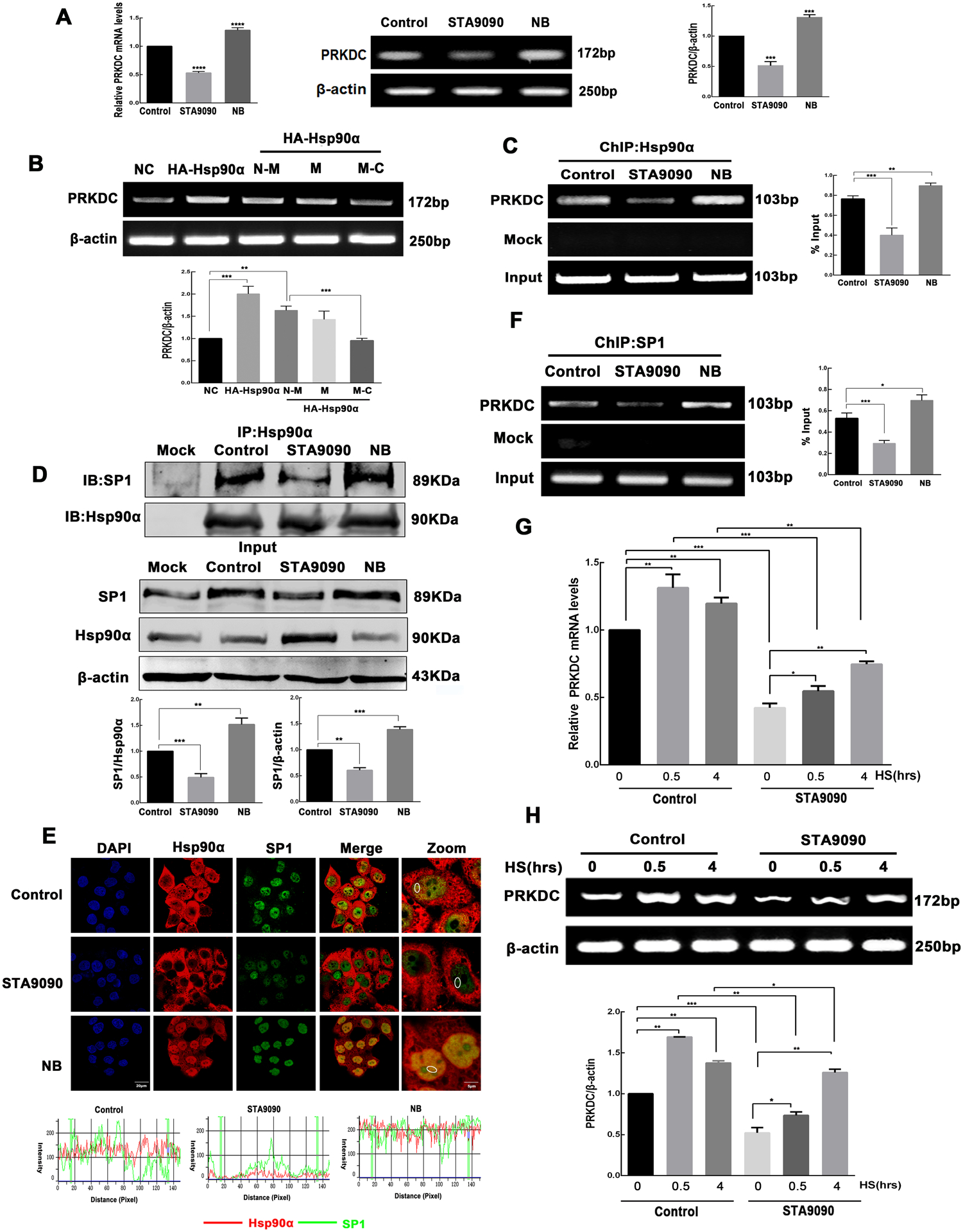
STA9090 reduces the binding of Hsp90α/SP1 to PRKDC promoter and decreases PRKDC transcription. (A) PRKDC mRNA levels were analyzed using RT-qPCR and RT-PCR in HepG2 cells incubated with 0.1 μM STA9090 and 0.5 μM NB for 24 h. β-actin was used as loading control. (B) HepG2 cells were transiently transfected with plasmids encoding HA-tagged Hsp90α full-length or the HA-tagged NBD-MD, MD, and MD-CTD domains. PRKDC and β-actin mRNA levels were analyzed using RT-PCR. (C) ChIP analysis of PRKDC in cells treated with STA9090 (0.1 μM) or NB (0.5 μM) for 24 h. Immunoprecipitation was performed using anti-Hsp90α antibodies followed by PCR analysis. (D) Immunoprecipitation of Hsp90α was performed with lysates of HepG2 cells after STA9090 and NB treatment. Hsp90α and SP1 levels were determined by western blotting. (E) Confocal immunofluorescence microscopy indicates the co-localization of Hsp90α and SP1 after STA9090 and NB treatment, the nucleus of the cells was stained with DAPI. Intensity plots of SP1 (green) and Hsp90α (red) of the representative circled areas of randomly selected cells for co-localization analysis. Scale bar is 20 μm. (F) ChIP analysis of PRKDC in cells treated with STA9090 (0.1 μM) or NB (0.5 μM) for 24 h. Immunoprecipitation was performed using anti-SP1 antibodies followed by PCR analysis. HepG2 cells were exposed to HS at 42°C for the indicated time (0, 0.5, and 4 h) in the presence or absence of STA9090 (0.1 μM) for 24 h. RT-qPCR (G) and RT-PCR (H) were performed to determine the PRKDC mRNA level. Band intensities were quantified by Image J v1.8.0, and values represent mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001.
Hyperthermia combined with STA9090 treatment inhibits HCC growth and decreases DNA-PKcs levels in vivo
Hyperthermia (20) sensitizing effects of Hsp90 inhibitor on HCC prompted us to investigate whether STA9090 could be an adjunct to hyperthermia. To this aim, we used HepG2-Luc cells for subcutaneous tumor xenografts in nude mice. Tumoral luminescence foci in animals treated with the combination of hyperthermia and STA9090 were much smaller than the foci observed in the control, HS, and STA-9090 groups (Fig. 5A). The average final tumor size was also significantly smaller in the combination group (Fig. 5B–C), consistent with our in vitro results and previously reported in vivo (20). The expression of DNA-PKcs in the combination group was significantly lower than that in the HS group as assessed using immunochemistry (Fig. 5D). Altogether, these results indicate that STA9090 sensitized HCC to hyperthermia in vivo. We hypothesized that this effect might be correlated with the levels of DNA damage repair protein DNA-PKcs.
Figure 5.
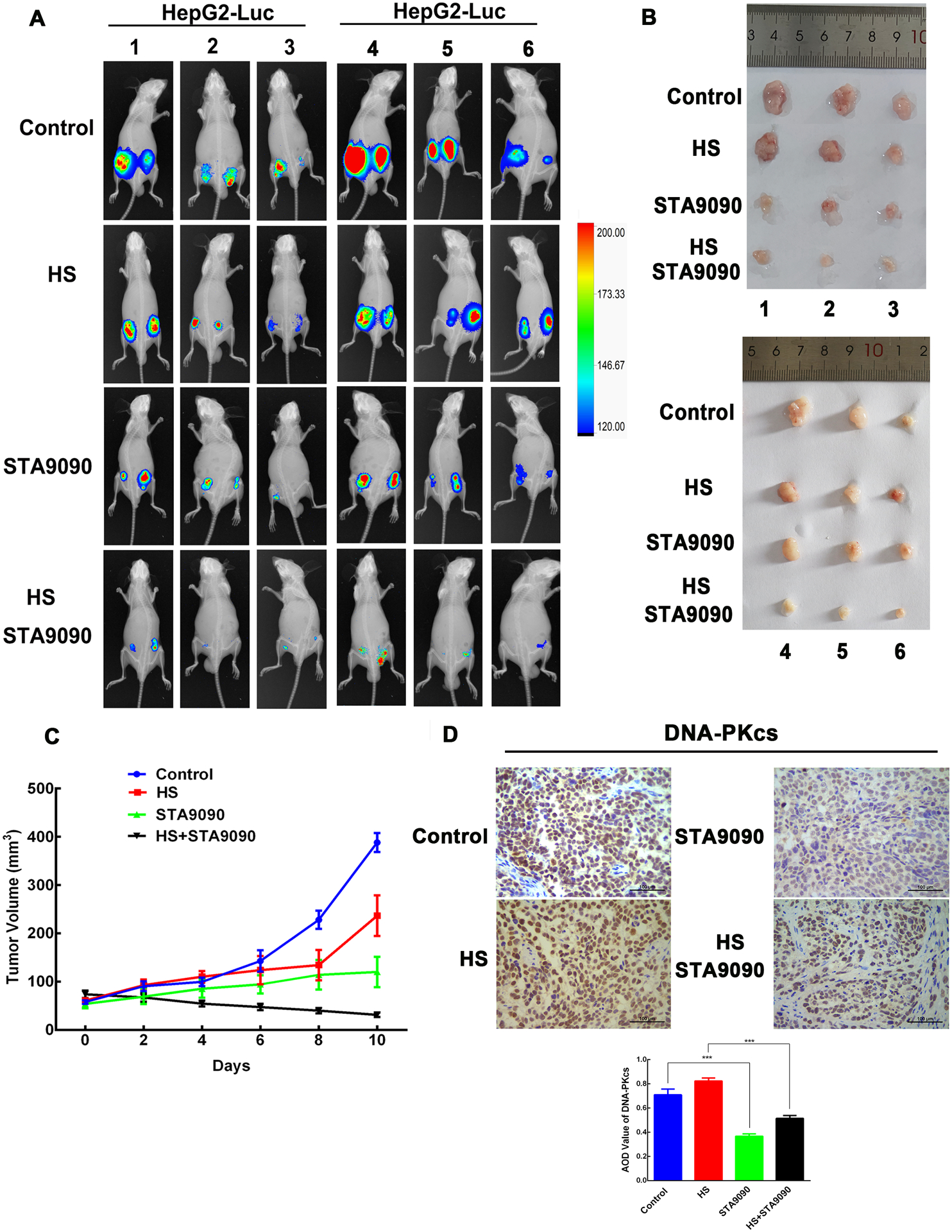
The combination of hyperthermia and STA9090 significantly reduced growth of xenograft tumors and decreased DNA-PKcs levels. HepG2-Luc cells (5 × 106 cells in 100 μl DMEM) were injected into both dorsal flanks of 4- to 5-week-old inbred BALB/c male nude mice. After 10 days, tumor-bearing mice were randomly divided into four groups as follows: Control (n=6 mice), HS (n=6 mice), STA9090 (n=6 mice), and the combination of STA9090 and HS (n=6 mice). Mice with xenograft tumors were kept for 2 h at 42°C and/or treated with STA9090 (12.5 mg/kg) three times a week for 10 days. (A) The luminescence was visualized using IVIS Lumina XRMS living imaging system. Appearance of mice tumors were shown. (B) Photographs of the tumors after 10 days of the HS and/or STA9090 treatment. (C) Growth curves of xenograft tumors in mice treated with STA9090 or HS alone or in combination (n = 6 mice). The combination of STA9090 and HS reduced tumor growth. (D) Staining of DNA-PKcs in tumors using immunohistochemistry. Values represent mean AOD ± SD of the tumors, n = 6, ***p < 0.001.
Inhibition of Hsp90 suppresses the increase of HS-induced DNA-PKcs and the repair of HS- induced DSB in HCC cells
HS induces γ-H2AX foci, which are indicator of DNA DSBs (34). In the NHEJ pathway. DNA-PKcs are recruited by Ku at the DSB, and DNA-PKcs regulates H2AX phosphorylation (35). We next examined the effects of Hsp90 inhibition on the expression of DNA-PKcs under HS. HepG2 cells were heated for 0, 0.5, and 4 h at 42°C in the absence or presence of STA9090, or of the siRNA targeting Hsp90α, or Hsp90β. We found that STA9090 decreased DNA-PKcs expression and suppressed the rise of DNA-PKcs levels after HS (Fig. 6A). Consistently, the DNA-PKcs levels also decreased in HepG2 cells transiently transfected with siHsp90α and siHsp90β that underwent HS as well (Fig. 6B). The changes of DNA-PKcs levels were further confirmed using confocal imaging in HepG2 cells (Fig. 6C–D). Next, we showed that more γ-H2AX foci, markers of severe DNA damage, were formed in response to the combination of STA9090 and HS (Fig. 6C). Similarly, the number of γ-H2AX foci increased significantly in treatment with siHsp90α or siHsp90β combined to HS by comparison with cells treated with siNC or undergoing HS alone (Fig. 6D).
Figure 6.
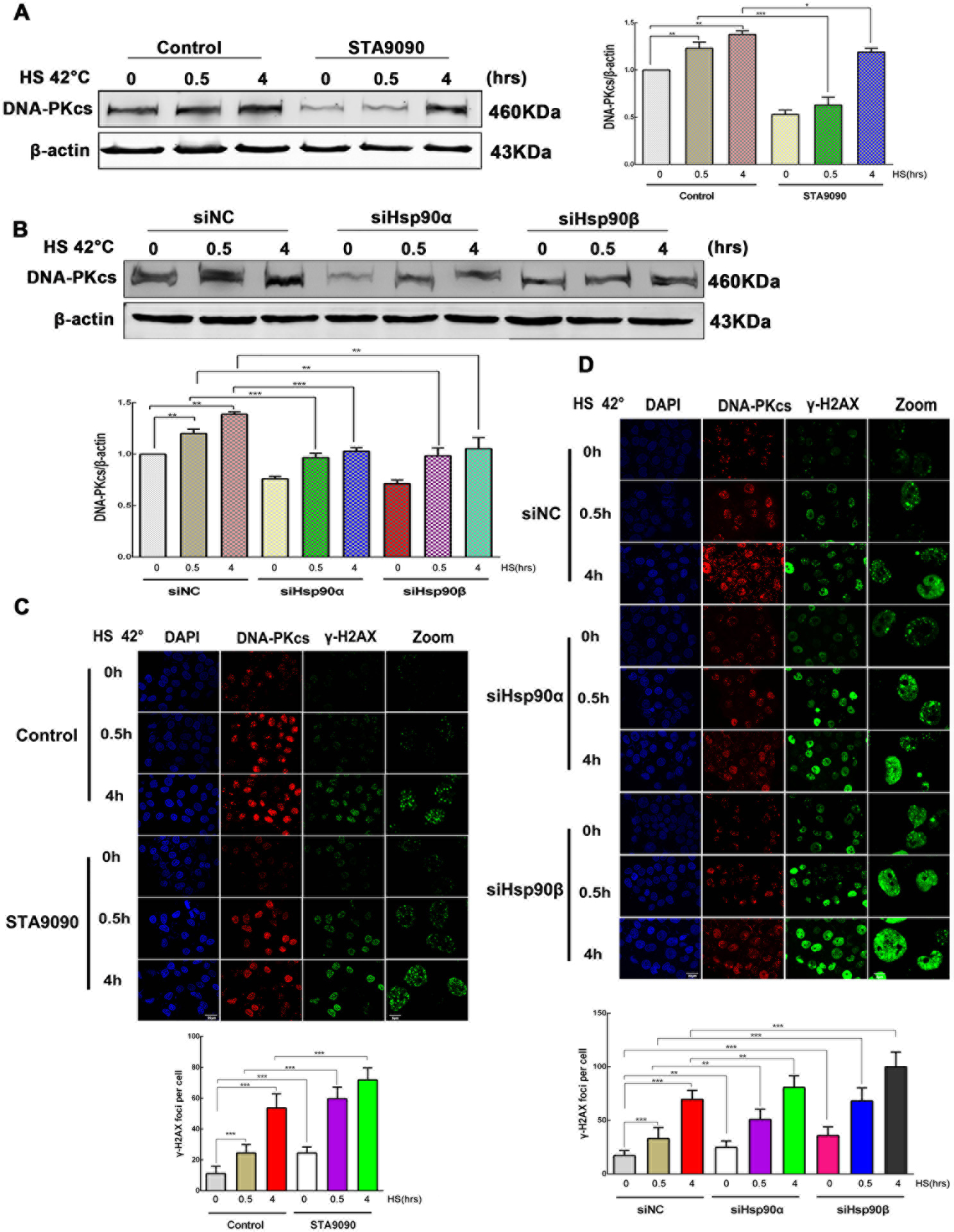
Effects of Hsp90 inhibition on HS-induced DNA-PKcs and γ-H2AX foci. (A) HepG2 cells were exposed to HS at 42°C for the indicated time (0, 0.5, and 4h) in the presence or absence of STA9090 (0.1μM). DNA-PKcs levels were detected using Western blot. (B) HepG2 cells were transiently transfected with siNC, siHsp90α, or siHsp90β for 48 h. Cells were then incubated for the specified times at 42°C. DNA-PKcs levels were analyzed using immunoblot. All data represent mean ±SD, n=3. *p < 0.05, **p < 0.01, ***p <0.001. (C) Immunofluorescence analysis of the effect of time-dependent HS (0, 0.5, and 4 h), combined or not with STA9090 (0.1μM) for 24 h, on DNA-PKcs and γ-H2AX foci. (D) Immunofluorescence analysis of the effect of time-dependent HS (0, 0.5, and 4 h), combined or not with siNC, siHsp90α, or siHsp90β for 48 h, on DNA-PKcs and γ-H2AX foci. Scale bar is 20 μm. For the quantification of γ-H2AX foci, 50 cells were counted. All data represent mean ±SD, n=50. **p < 0.01, ***p <0.001.
DNA damage was evaluated using the comet assay under neutral conditions. HepG2 cells were treated with STA9090 for 24 h (Fig. S7A–B), or transfected with siRNA targeting Hsp90α, or Hsp90β for 48 h (Fig. S7C–D) in combination or not with HS. In each case, the combination treatment generated a significantly higher number of DNA breaks than STA9090, siHsp90α, siHsp90β, or HS alone. This suggested that more severe DNA damage might correspond to a lower DNA repair capacity. These results indicate that Hsp90 inhibition suppresses the key NHEJ protein DNA-PKcs and reduces DSB repair in heat-stressed HCC cells. Therefore, Hsp90 inhibition might block DSB repair by the DNA-PKcs-mediated NHEJ pathway.
Discussion
In this study, we showed that increased levels of Hsp90α, Hsp90β, and DNA-PKcs correlated with a poor overall survival in patients with HCC. We revealed that Hsp90 N-terminal domain and C-terminal domain have different effects on DNA-PKcs protein and mRNA levels. The stability of DNA-PKcs positively depended on Hsp90α N-terminal nucleotide binding domain. Inhibition of Hsp90 N-terminal decreased the location of Hsp90α in nucleus, Hsp90α-SP1 interaction, SP1 level and the interaction of Hsp90α/SP1 with the proximal promoter region of PRKDC.
The two chaperone sites of Hsp90 located in the N- and C-terminal fragments has been reviewed (27). It has been revealed the N- and C-terminal fragments are the independent molecular chaperone molecules (28). Interestingly, DNA-PKcs protein and mRNA levels were decreased or increased under STA9090 or NB treatment, which is consistent to transfection outcomes with the C- or N-terminal fragments of Hsp90α. Firstly, we found that STA9090 weakened the interaction between Hsp90 and DNA-PKcs, and degraded DNA-PKcs through the proteasome/ubiquitination pathway, presenting that the Hsp90α NBD is important for maintaining DNA-PKcs stability. Secondly, we proposed the participation of Hsp90 in regulating DNA-PKcs transcription. According to the previous studies, the C-terminal of Hsp90 is responsible for the cytoplasmic localization (36), nuclear Hsp90 could maintain the stability of the nuclear proteins, and regulate transcription factors, then contributing to gene expression (30, 37). Hence, the broad influence of Hsp90 on DNA binding factors might explain why Hsp90 has a different influence on PRKDC under STA9090 and NB treatment. Hsp90 in nuclear regulates the activity of transcription factors and the general transcription machinery (29, 30). A previous study reported that SP1 interacts with Hsp90α and SP1 is Hsp90 related transcription factors by binding to the GC-rich promoter region of the target genes (38). After the bioinformatics searching, we focus on the effect of Hsp90 inhibition on SP1, which is the transcription factor of PRKDC, and Hsp90 client protein. In the ChIP experiments, STA9090 decreased the binding of Hsp90α to the PRKDC promoter, while NB treatment increased the binding, and the same effect on SP1. Nevertheless, the main question addressed here is how the presence of Hsp90 induces PRKDC transcription. We believed the interaction between Hsp90α and SP1 are further used to meet a specialized need of PRKDC transcription in the nucleus. Numerous transcriptional cofactors cannot bind to DNA directly, but rather are recruited to the promoter through other transcription factors that directly bind to promoter, such as p300/CBP (39), and Hsp90 (40). And previous finding revealed that SP1 could recruit Hsp90α to the promoter region of the target genes (38). Our date found that while NB promoted the co-localization of SP1 with Hsp90α, STA9090 disassociated the interaction. Next, both anti- Hsp90α and anti-SP1 could pull down the DNA fragment from −564 to −666 bp of the TSS in the PRKDC promoter region, STA9090 prevented Hsp90α and SP1 from binding to the PRKDC promoter region, but NB promoted the bindings of Hsp90α and SP1. The schematic representation of that Hsp90 is involved in the SP1-dependent transcription of PRKDC under STA9090 and NB treatment shows in Fig.7.
Figure 7.
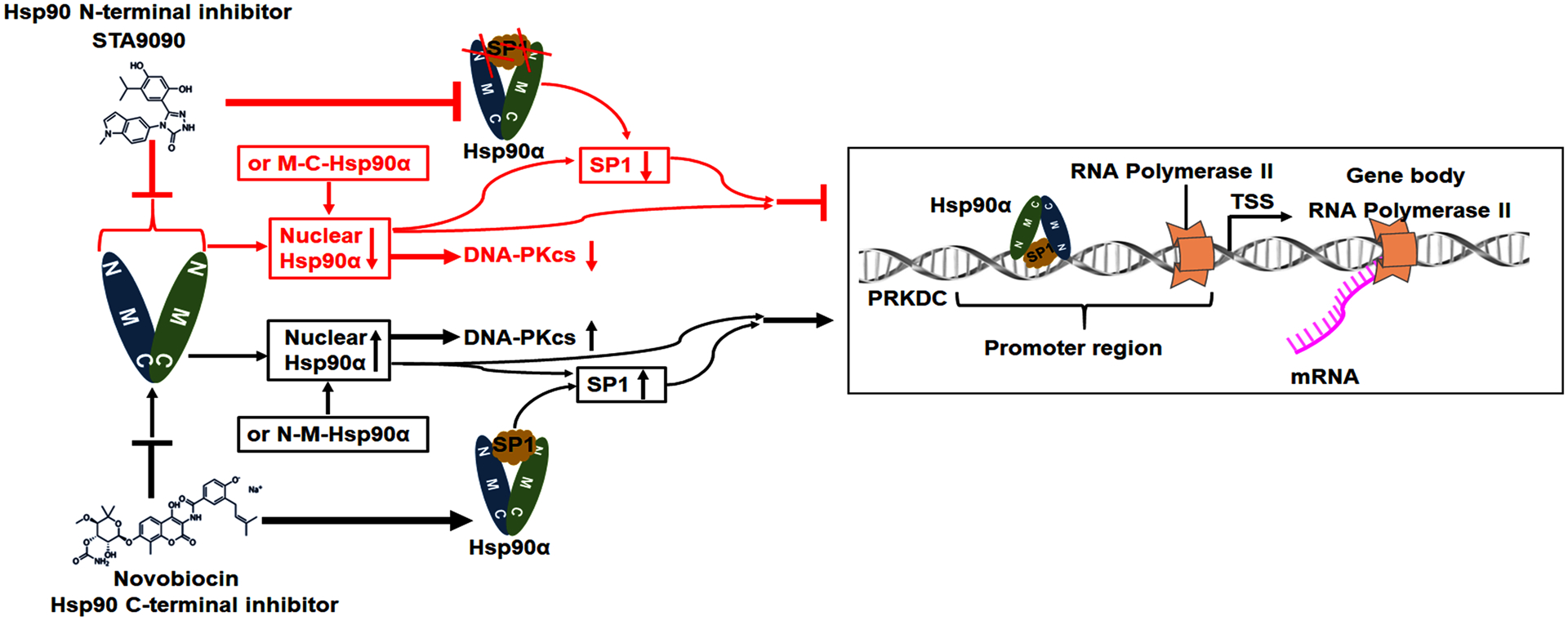
The schematic diagram illustrates that Hsp90 is involved in maintaining DNA-PKcs stability and the SP1-dependent transcription of PRKDC.
Hyperthermia is a DNA damage-based anticancer therapy. HS affects DNA structure and DNA-associated processes in cell cycle phase–dependent manner (41). As hyperthermia induces heat-shock response with heat-shock proteins (HSPs) and DDR with increased DNA-PKcs level, which both compromises its anticancer effect. Hsp90, one of the most abundant HPSs, is involved in various processes related to tumorigenesis (42). A study showed that HS activates YAP/TAZ to induce the heat shock transcriptome and suggested a potential combinational therapy (43). And Hsp90 regulates HDAC3-dependent gene transcription, 17AAG interferes with the nuclear translocation of HDAC3, and a possible connection between the nuclear HDAC3 and Hsp90 is suggested linking with DNA damage response (44). The combination with inhibitor and hyperthermia might be exploited to enhance the therapeutic effects alone. Indeed, clinical trial results based on Hsp90 inhibitors were unsatisfactory for decades (45). In this study, we focus on the effects of the combination of hyperthermia with Hsp90 inhibitor STA9090. We showed that the combination delayed HCC tumor growth and decreased major DSB repair player DNA-PKcs and increased DNA damage biomarker γ-H2AX foci, and a higher number of DNA breaks. We also found that down-regulation of Hsp90β produced more serious DSB than Hsp90α, suggesting a direct role for Hsp90β in DNA damage signaling, and inhibitors specific for Hsp90β might have stronger effects.
Taken together, our study demonstrated that Hsp90 inhibitor STA9090 suppressed DSB repair pathways by reducing DNA-PKcs protein stability and mRNA transcription. We revealed that the reason why Hsp90 N-terminal and C-terminal have different effects on DNA-PKcs and confirmed that Hsp90α regulated PRKDC transcription via SP1 in HepG2 cells. We presented Hsp90 in nuclear was essential for PRKDC transcription during DNA repair. Moreover, we provide evidence for combining hyperthermia with STA9090 as a promising therapeutic strategy. It remains to determine if the combination can overcome the disadvantages of using Hsp90 inhibitors or hyperthermia alone in clinical trials. It would also be interesting to clarify whether selectively targeting Hsp90-dependent transcription of PRKDC provides effective anticancer combination therapy in the future.
Supplementary Material
Acknowledgments
This work was supported by grants of the National Natural Science Foundation of China (NSFC) No. 81673216 to Xuemei Chen, No. 81741131 and 81671860 to Fei Zou. We thank Prof. Dr. Matthias P. Mayer (Zentrum fur Molekulare Biologie der University Heidelberg) for kindly providing the plasmid. We like to thank the kind gifts of STA9090 and STA9090-BODIPY from Weiwen Ying (OnTarget Pharmaceutical Consulting LLC). We are also greatly indebted to Dr. Hongyan Du from School of Laboratory Medicine and Biotechnology, Southern Medical University for sharing MHCC97H cell line.
Footnotes
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- 1.Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. NAT REV DIS PRIMERS 2021. 2021-01-21; 7(1):6. [DOI] [PubMed] [Google Scholar]
- 2.Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019. 2019-10-01; 16(10):589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blagg BS, Kerr TD. Hsp90 inhibitors: small molecules that transform the Hsp90 protein folding machinery into a catalyst for protein degradation. MED RES REV 2006. 2006-05-01; 26(3):310–38. [DOI] [PubMed] [Google Scholar]
- 4.Taipale M, Krykbaeva I, Koeva M, et al. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. CELL 2012. 2012-08-31; 150(5):987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta 2012. 2012-03-01; 1823(3):742–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoter A, El-Sabban ME, Naim HY. The HSP90 Family: Structure, Regulation, Function, and Implications in Health and Disease. INT J MOL SCI 2018. 2018-08-29; 19(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pillai RN, Fennell DA, Kovcin V, et al. Randomized Phase III Study of Ganetespib, a Heat Shock Protein 90 Inhibitor, With Docetaxel Versus Docetaxel in Advanced Non-Small-Cell Lung Cancer (GALAXY-2). J CLIN ONCOL 2020. 2020-02-20; 38(6):613–22. [DOI] [PubMed] [Google Scholar]
- 8.Moon SJ, Jeong BC, Kim HJ, et al. Bruceantin targets HSP90 to overcome resistance to hormone therapy in castration-resistant prostate cancer. THERANOSTICS 2021. 2021-01-20; 11(2):958–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Wang M, Lin Z, et al. Mild microwave ablation combined with HSP90 and TGFbeta1 inhibitors enhances the therapeutic effect on osteosarcoma. MOL MED REP 2020. 2020-08-01; 22(2):906–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arriortua OK, Garaio E, Herrero DLPB, et al. Antitumor magnetic hyperthermia induced by RGD-functionalized Fe3O4 nanoparticles, in an experimental model of colorectal liver metastases. Beilstein J Nanotechnol 2016. 2016-01-20; 7:1532–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jun HJ, Park SJ, Kang HJ, et al. The Survival Benefit of Combination Therapy With Mild Temperature Hyperthermia and an Herbal Prescription of Gun-Chil-Jung in 54 Cancer Patients Treated With Chemotherapy or Radiation Therapy: A Retrospective Study. INTEGR CANCER THER 2020. 2020-01-01; 19:1872145015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurwitz M, Stauffer P. Hyperthermia, radiation and chemotherapy: the role of heat in multidisciplinary cancer care. SEMIN ONCOL 2014. 2014-12-01; 41(6):714–29. [DOI] [PubMed] [Google Scholar]
- 13.Han SI, Duong HQ, Choi JE, et al. Hyperthermia switches glucose depletion-induced necrosis to apoptosis in A549 lung adenocarcinoma cells. INT J ONCOL 2008. 2008-04-01; 32(4):851–60. [PubMed] [Google Scholar]
- 14.Tomita M Involvement of DNA-PK and ATM in radiation- and heat-induced DNA damage recognition and apoptotic cell death. J RADIAT RES 2010. 2010-01-20; 51(5):493–501. [DOI] [PubMed] [Google Scholar]
- 15.Hnizda A, Blundell TL. Multicomponent assemblies in DNA-double-strand break repair by NHEJ. Curr Opin Struct Biol 2019. 2019-04-01; 55:154–60. [DOI] [PubMed] [Google Scholar]
- 16.Goodwin JF, Kothari V, Drake JM, et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. CANCER CELL 2015. 2015-07-13; 28(1):97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geng W, Tian D, Wang Q, et al. DNAPKcs inhibitor increases the sensitivity of gastric cancer cells to radiotherapy. ONCOL REP 2019. 2019-08-01; 42(2):561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehta RK, Pal S, Kondapi K, et al. Low-Dose Hsp90 Inhibitor Selectively Radiosensitizes HNSCC and Pancreatic Xenografts. CLIN CANCER RES 2020. 2020-10-01; 26(19):5246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. LANCET ONCOL 2013. 2013-08-01; 14(9):e358–69. [DOI] [PubMed] [Google Scholar]
- 20.Huang Z, Zhou X, He Y, et al. Hyperthermia enhances 17-DMAG efficacy in hepatocellular carcinoma cells with aggravated DNA damage and impaired G2/M transition. Sci Rep 2016. 2016-12-02; 6:38072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou X, Wen Y, Tian Y, et al. Heat Shock Protein 90alpha-Dependent B-Cell-2-Associated Transcription Factor 1 Promotes Hepatocellular Carcinoma Proliferation by Regulating MYC Proto-Oncogene c-MYC mRNA Stability. HEPATOLOGY 2019. 2019-04-01; 69(4):1564–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jhaveri K, Modi S. Ganetespib: research and clinical development. Onco Targets Ther 2015. 2015-01-20; 8:1849–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donnelly A, Blagg BS. Novobiocin and additional inhibitors of the Hsp90 C-terminal nucleotide-binding pocket. CURR MED CHEM 2008. 2008-01-20; 15(26):2702–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillan V, O’Neill K, Maitland K, Sverdrup FM, Devaney E. A repurposing strategy for Hsp90 inhibitors demonstrates their potency against filarial nematodes. PLoS Negl Trop Dis 2014. 2014-02-01; 8(2):e2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wen Y, Zhou X, Lu M, et al. Bclaf1 promotes angiogenesis by regulating HIF-1alpha transcription in hepatocellular carcinoma. ONCOGENE 2019. 2019-03-01; 38(11):1845–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Y, Sun A, Zhao Y, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. NATURE 2019. 2019-03-01; 567(7747):257–61. [DOI] [PubMed] [Google Scholar]
- 27.Mielczarek-Lewandowska A, Hartman ML, Czyz M. Inhibitors of HSP90 in melanoma. APOPTOSIS 2020. 2020-02-01; 25(1–2):12–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheibel T, Weikl T, Buchner J. Two chaperone sites in Hsp90 differing in substrate specificity and ATP dependence. Proc Natl Acad Sci U S A 1998. 1998-02-17; 95(4):1495–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calderwood SK, Neckers L. Hsp90 in Cancer: Transcriptional Roles in the Nucleus. ADV CANCER RES 2016. 2016-01-20; 129:89–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoveva A, Sawarkar R. Chromatin Immunoprecipitation (ChIP) of Heat Shock Protein 90 (Hsp90). Methods Mol Biol 2018. 2018-01-20; 1709:221–31. [DOI] [PubMed] [Google Scholar]
- 31.Taatjes DJ, Naar AM, Andel FR, Nogales E, Tjian R. Structure, function, and activator-induced conformations of the CRSP coactivator. SCIENCE 2002. 2002-02-08; 295(5557):1058–62. [DOI] [PubMed] [Google Scholar]
- 32.Sugawara T, Nomura E, Nakajima A, Sakuragi N. Characterization of binding between SF-1 and Sp1: predominant interaction of SF-1 with the N-terminal region of Sp1. J ENDOCRINOL INVEST 2004. 2004-02-01;27(2):133–41. [DOI] [PubMed] [Google Scholar]
- 33.Dunah AW, Jeong H, Griffin A, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. SCIENCE 2002. 2002-06-21; 296(5576):2238–43. [DOI] [PubMed] [Google Scholar]
- 34.Toivola DM, Strnad P, Habtezion A, Omary MB. Intermediate filaments take the heat as stress proteins. TRENDS CELL BIOL 2010. 2010-02-01; 20(2):79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.An J, Huang YC, Xu QZ, et al. DNA-PKcs plays a dominant role in the regulation of H2AX phosphorylation in response to DNA damage and cell cycle progression. BMC MOL BIOL 2010. 2010-03-06; 11:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Passinen S, Valkila J, Manninen T, Syvala H, Ylikomi T. The C-terminal half of Hsp90 is responsible for its cytoplasmic localization. Eur J Biochem 2001. 2001-10-01; 268(20):5337–42. [DOI] [PubMed] [Google Scholar]
- 37.Sawarkar R, Paro R. Hsp90@chromatin.nucleus: an emerging hub of a networker. TRENDS CELL BIOL 2013. 2013-04-01; 23(4):193–201. [DOI] [PubMed] [Google Scholar]
- 38.Hung JJ, Wu CY, Liao PC, Chang WC. Hsp90alpha recruited by Sp1 is important for transcription of 12(S)-lipoxygenase in A431 cells. J BIOL CHEM 2005. 2005-10-28; 280(43):36283–92. [DOI] [PubMed] [Google Scholar]
- 39.Xiao H, Hasegawa T, Isobe K. p300 collaborates with Sp1 and Sp3 in p21 (waf1/cip1) promoter activation induced by histone deacetylase inhibitor. J BIOL CHEM 2000. 2000-01-14; 275(2):1371–6. [DOI] [PubMed] [Google Scholar]
- 40.Echtenkamp FJ, Gvozdenov Z, Adkins NL, et al. Hsp90 and p23 Molecular Chaperones Control Chromatin Architecture by Maintaining the Functional Pool of the RSC Chromatin Remodeler. MOL CELL 2016. 2016-12-01; 64(5):888–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Velichko AK, Petrova NV, Kantidze OL, Razin SV. Dual effect of heat shock on DNA replication and genome integrity. MOL BIOL CELL 2012. 2012-09-01; 23(17):3450–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Staufer K, Stoeltzing O. Implication of heat shock protein 90 (HSP90) in tumor angiogenesis: a molecular target for anti-angiogenic therapy? Curr Cancer Drug Targets 2010. 2010-12-01; 10(8):890–7. [DOI] [PubMed] [Google Scholar]
- 43.Luo M, Meng Z, Moroishi T, et al. Heat stress activates YAP/TAZ to induce the heat shock transcriptome. NAT CELL BIOL 2020. 2020-12-01; 22(12):1447–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kotwal A, Amere SS. Hsp90 regulates HDAC3-dependent gene transcription while HDAC3 regulates the functions of Hsp90. CELL SIGNAL 2020. 2020-12-01; 76:109801. [DOI] [PubMed] [Google Scholar]
- 45.Gao Z, Garcia-Echeverria C, Jensen MR. Hsp90 inhibitors: clinical development and future opportunities in oncology therapy. Curr Opin Drug Discov Devel 2010. 2010-03-01; 13(2):193–202. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.