

Summary:
As a general rule, successful antineoplastic treatments induce an antitumor immune response, even if they were initially designed to target cancer cell–autonomous pathways. In this issue of Blood Cancer Discovery, Gulla and colleagues reveal that the proteasome inhibitor bortezomib induces immunogenic stress and death in multiple myeloma cells, thus explaining its therapeutic efficacy.
See related article by Gulla et al., p. 468.
Until recently, malignant disease has been viewed as a purely cell-autonomous disease mediated by the progressive accumulation of genetic and epigenetic alterations in premalignant and malignant cells that override cell-intrinsic tumor-suppressive mechanisms, including senescence and apoptosis. Nowadays, this view has been challenged, and it has become increasingly clear that malignant disease is also the result of inefficient immunosurveillance, meaning that tumors can only develop after their recognition and after elimination by the immune system has failed. Beyond academic considerations on the etiology of neoplastic disease, this concept has major practical implications. Indeed, the recent approvals of immune-checkpoint blockers (mostly targeting PD-1 and PD-L1) across a wide spectrum of previously intractable malignant diseases illustrates the possibility to transiently reactivate failed immunosurveillance to obtain tangible benefits in a substantial (though admittedly insufficient) fraction of patients with cancer.
The cell-autonomous vision of cancer has profoundly influenced the way in which cancer drugs have been conceived and developed. After the widespread implementation of chemotherapeutic cytotoxicants that indiscriminately kill proliferating cells of any kind, explaining their major side effects, the idea arose to develop “targeted” or “personalized” anticancer agents as part of a novel “precision medicine.” Thus, drugs should target the oncogene to which a cancer cell is “addicted,” for example, an activating mutation in an oncogenic kinase, thereby acting on malignant but not normal cells. Alternatively, drugs should target “non-oncogene addiction” (NOA), the phenomenon where tumor cells present stress phenotypes that render them vulnerable to specific agents. One such drug that targets NOA is bortezomib, an inhibitor of the 26S proteasome used for the treatment of multiple myeloma (MM). Now, in the current issue of Blood Cancer Discovery, Gulla and colleagues (1) report that bortezomib has the capacity to induce potent anticancer immune responses.
Although anticancer drugs, be they nonspecific chemotherapeutics or targeted agents, were supposed to mediate their effects by direct cytostatic or cytotoxic effects on malignant cells, it turned out that their long-term effects in patients relied on the induction of antitumor immune responses. Thus, several major chemotherapeutic agents, including anthracyclines and oxaliplatin, induced “immunogenic cell death” (ICD), meaning that they stress and kill cancer cells in such a way that they become recognizable to the immune system (2). Accordingly, accumulating evidence supports the idea that anticancer immune response elicited by chemotherapy determines patient prognosis (2). Ironically, imatinib, the first drug developed to target an oncogenic tyrosine kinase, BCR–ABL [the ABL kinase constitutively activated by a chromosomal translocation, in Philadelphia chromosome–positive chronic B-cell leukemias (CLL, chronic lymphocytic leukemia)] turned out to owe its long-term success against CLL [and later against gastrointestinal stroma tumors (GIST), which depend on another oncogenic kinase, KIT, also inhibited by imatinib] to immune responses as well (3). In this case, cell-autonomous effects on CLL or GIST cells increase their susceptibility to immune attack by cytotoxic T cells (CTL) and natural killer cells coupled to a reduction of immunosuppressive cell types (such as regulatory T cells and myeloid-derived suppressor cells). Moreover, imatinib inhibits nonmutated tyrosine kinases in immune cells, in particular in dendritic cells (DC), increasing their immunostimulatory action (3). Thus, targeted therapies acting on oncogenic kinases may have immunologic off-target effects that explain their therapeutic success.
Bortezomib has been suspected of having immunostimulatory effects since the discovery that it induces the expression of heat shock protein 90 on MM cells, favoring their recognition by DCs (4). The translocation of intracellular proteins to the cell surface is indeed a hallmark of ICD. In particular, the endoplasmic reticulum (ER) chaperone calreticuline (CALR) translocates from the ER lumen to the plasma membrane surface, where it acts as an “eat-me” signal to favor the phagocytosis of dying cells by DCs. CALR exposure has been observed in response to a wide range of ICD-inducing chemotherapeutics (2) and some targeted agents such as crizotinib, a tyrosine kinase inhibitor (5). Gulla and colleagues now report that bortezomib induces classic, CALR-dependent ICD while providing molecular and translational insights into the therapeutic relevance of bortezomib-induced ICD (1).
Gulla and colleagues found that bortezomib induces CALR exposure to the surface of human or mouse MM cells and that CALR is required for the phagocytosis of bortezomib-treated MM cells by monocyte-derived DCs (1). Coculture of bortezomib-treated human MM cells, DCs, and T lymphocytes led to an increase in the frequency of CD4+ effector memory (EM) cells, CD8+ EM cells, and CD8+ T EM cells reexpressing CD45RA (TEMRA) that was not seen when bortezombib-treated MM cells were removed from the coculture or when bortezomib alone was added to cocultures of DCs and T cells. When bortezomib-killed murine MM cells were injected into syngeneic immunocompetent mice, they induced an immune response that prevented the growth of MM cells injected 1 week later. This vaccination effect was reduced when the Calr gene was inactivated in MM cells. Moreover, established MMs only responded to bortezomib treatment in vivo when established in immunocompetent rather than in immunodeficient mice. Although knockout of Calr in MM cells did not affect their apoptotic response to bortezomib in vitro, it did abolish the efficacy of bortezomib against MM tumors developing in immunocompetent mice. These results demonstrate that a CALR-dependent immune response is required for optimal therapeutic efficacy of bortezomib against MM. Driven by this consideration, Gulla and colleagues determined the effects of bortezomib on the transcriptome of MM tumors evolving in immunocompetent mice. In CALR-expressing (but not in CALR-deficient) MM, bortezomib induced 90 immune-related genes that composed the “ICD signature.” High expression of the human orthologs of the mouse genes found in this signature correlated with clinical outcome of MM patients treated with bortezomib-based regimens (1).
Of note, among these 90 genes, 57 could be classified as interferon-stimulated genes, and bortezomib was able to activate a type-1 interferon response in MM cells in vitro, meaning that genes encoding interferon alpha1 (IFNA1), interferon beta1 (IFNB1), and chemokine (C-X-C motif) ligand 9 (CXCL9, a ligand acting on the receptor CXCR3 on T cells to attract them into the tumor bed) were upregulated (1). This response apparently was decisive for the bortezomib effects against MM because a neutralizing antibody specific for type-1 interferon receptor 1 (IFNAR1) interfered with tumor growth reduction by bortezomib in immunocompetent mice. Moreover, in mice bearing histocompatible MMs treated with bortezomib, IFNAR1 blockade prevented the upregulation of CXCL9, and low CXCL9 mRNA expression correlated with poor overall survival in MM patients (1). In sum, it appears that bortezomib can induce a type-1 interferon response, a phenomenon that has previously been linked to ICD and has been dubbed as “viral mimicry” (6). Thus, like other ICD-inducing drugs, bortezomib stresses and kills MM cells in a way that mimics viral infection to the degree that high levels of type-1 interferons are induced.
Gulla and colleagues then characterized the molecular and cellular mechanisms that explain how a 26S proteasome inhibitor can cause several hallmarks of ICD including CALR exposure and a type-1 interferon response (1).
In the context of ICD, CALR exposure is usually preceded by the induction of the integrated stress response, consisting of the phosphorylation of eukaryotic initiation factor 2α (eIF2α) by the ER stress kinase eIF2α kinase-3 (EIF2AK3, best known as PERK; ref. 2). Indeed, this pathway was activated in bortezomib-treated MM cells (1). In response to chemotherapeutic ICD inducers, this phosphorylation event occurs downstream of a general inhibition of DNA-to-RNA transcription (7). High-dose bortezomib reduces global RNA synthesis in osteosarcoma cells (7), suggesting that this might also occur in MM cells (Fig. 1). However, this conjecture remains to be explored.
Figure 1.
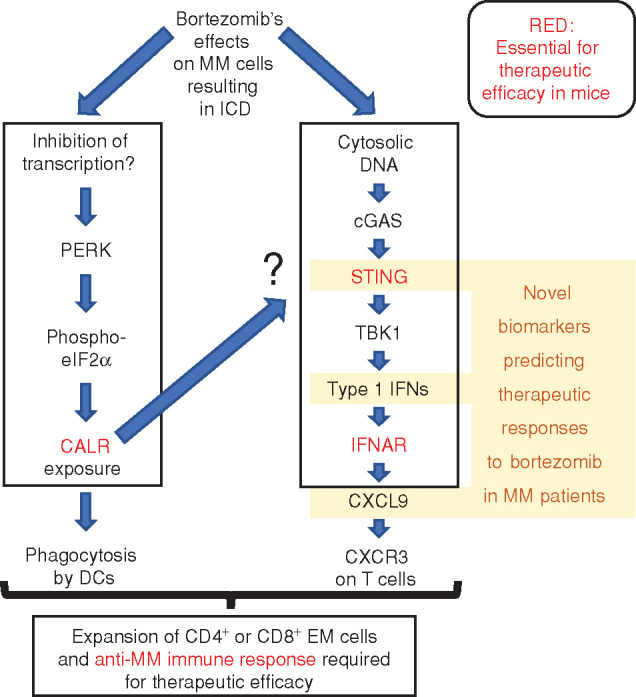
Hypothetical cascade of molecular, cellular, and immunologic effects of bortezomib. In MM cells, bortezomib induces CALR exposure, as well as a type-1 interferon (IFN) response. Several of the signaling elements involved in the cascade are shown. Note that knockout of the CALR gene abolishes the induction of a type-1 IFN response. However, the level at which this latter response is abolished is elusive, as indicated by the question mark. ICD then results in an anti-MM immune response that is required for bortezomib to be efficient in vivo. Several elements of the cascade have prognostic impact on patients with MM treated with bortezomib. Molecules and processes that are required for optimal bortezomib effects in mice (CALR, STING, IFNAR, immune response) are marked in red. For details, see the main text.
Bortezomib induced type-1 interferon responses by an increase in cytosolic DNA in the form of micronuclei, accumulation of the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), activation of transmembrane protein 173 (TMEM173, best known as STING), and activating phosphorylation of TANK-binding kinase 1 (TBK1; ref. 1). Knockout of STING abolished this latter event; prevented the induction of IFNA1, IFNB1, and CXCL9 gene transcription by bortezomib; and abolished the capacity of bortezomib-treated human MM cells to stimulate CD4+ EM and CD8+ EM cells in cocultured immune cells (Fig. 1). Nonetheless, the STING pathway appeared to be suboptimally activated in response to bortezomib, meaning that a synthetic STING agonist, ADUS-100, could trigger a stronger TBK1 phosphorylation in vitro. When combined with bortezomib, ADUS-100 also improved therapeutic responses in a mouse MM model, as it increased the infiltration of the tumors by T lymphocytes. Such synergistic effects were lost upon knockout of STING in mouse MM cells or in immunodeficient mice (1). Of note, in MM patients, STING mRNA levels positively correlated with the expression of the 57 interferon-stimulated genes found in the ICD cluster, pleading for the clinical relevance of this pathway (1).
In response to cGAS activation, STING is known to translocate to the ER (8), suggesting a possible intersection with the CALR exposure pathway ignited at the ER. However, knockout of STING did not interfere with bortezomib-induced CALR exposure (1). Conversely, knockout of CALR prevented the bortezomib-mediated induction of interferon-stimulated genes in vivo, in the MM mouse model (1), suggesting that CALR is somehow required for the cytosolic DNA–cGAS–STING–TBK1–type-1 interferon pathway to be activated (Fig. 1). However, this conjecture requires further exploration by in vitro experimentation. In particular, the exact level at which CALR deficiency interrupts this pathway remains to be determined. Cancer cells can escape immunosurveillance by suppressing the CALR exposure pathway at multiple levels (9). Hence, it would not be surprising that MM cells developed such deficiencies as a result of immunoselection, in particular after relapse from bortezomib-based therapeutic regimens.
In summary, the work by Gulla and colleagues reveals that ICD accompanied by CALR exposure and viral mimicry is an important clinical feature of MM treatment by bortezomib (1), echoing recent results on another efficient anti-MM drug, belantamab mafodotin, which also acts as a potent ICD inducer (10). This anti-BCMA antibody conjugated to maleimidocaproyl monomethyl auristatin F was FDA approved in August 2020 for the treatment of relapsed or refractory MM. Logically, clinical studies evaluating the possibility of combining bortezomib or belantamab mafodotin with anti–PD-1 antibodies will be underway soon (ClinicalTrials.gov Identifiers NCT03848845 and NCT04258683). It will be interesting to see whether the combination of ICD inducers with other immunotherapies, including STING agonists, will further improve the clinical management of MM.
Acknowledgments
L. Zitvogel and G. Kroemer are supported by the Ligue contre le Cancer (équipe labellisée), Agence National de la Recherche (ANR), Association pour la recherche sur le cancer (ARC), Association “Ruban Rose,” Cancéropôle Ile-de-France, Fondation pour la Recherche Médicale (FRM), the European Union Horizon 2020 Projects Crimson and Oncobiome, Fondation Carrefour, Institut National du Cancer (INCa), Inserm (HTE), Institut Universitaire de France, LeDucq Foundation, LabEx Immuno-Oncology (ANR-18-IDEX-0001), RHU Torino Lumière, Seerave Foundation, SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE), and SIRIC Cancer Research and Personalized Medicine (CARPEM). This study contributes to the IdEx Université de Paris ANR-18-IDEX-0001.
Footnotes
Blood Cancer Discov 2021;2:405–7
Authors' Disclosures
L. Zitvogel reports a patent for compounds and uses thereof to induce an immunogenic cancer cell death in a subject issued; a patent for compounds regulating calreticulin, KDEL receptor, and/or Erp-57 cell-surface exposure and uses thereof to evaluate the efficiency of a cancer treatment issued; a patent for inhibitors of protein phosphatase 1, GADD34, and protein phosphatase 1/GADD34 complex, preparation and uses thereof pending; a patent for kits and methods for detecting the ability to induce an immunogenic cancer cell death in a subject pending; and a patent for calreticulin for its use as a medication for the treatment of cancer in a mammal pending. G. Kroemer reports grants from Daiichi Sankyo, GlaxoSmithKline, Eleor, Kaleido, Lytix Pharma, PharmaMar, Samsara, Sanofi, Sotio, Vascage, and Vasculox and other support from Bristol Myers Squibb Foundation France, EverImmune, Samsara Therapeutics, and Therafast outside the submitted work, as well as a patent for compounds and uses thereof to induce an immunogenic cancer cell death in a subject issued, a patent for compounds regulating calreticulin, KDEL receptor, and/or Erp-57 cell-surface exposure and uses thereof to evaluate the efficiency of a cancer treatment issued, a patent for inhibitors of protein phosphatase 1, GADD34, and protein phosphatase 1/GADD34 complex, preparation and uses thereof pending, a patent for kits and methods for detecting the ability to induce an immunogenic cancer cell death in a subject pending, and a patent for calreticulin for its use as a medication for the treatment of cancer in a mammal pending.
References
- 1.Gulla A, Morelli E, Samur MK, Botta C, Hideshima T, Bianchi G, et al. Bortezomib induces anti–multiple myeloma immune response mediated by cGAS/STING pathway activation. Blood Cancer Discov 2021;2:468–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G.Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017;17:97–111. [DOI] [PubMed] [Google Scholar]
- 3.Zitvogel L, Rusakiewicz S, Routy B, Ayyoub M, Kroemer G.Immunological off-target effects of imatinib. Nat Rev Clin Oncol 2016;13:431–46. [DOI] [PubMed] [Google Scholar]
- 4.Spisek R, Charalambous A, Mazumder A, Vesole DH, Jagannath S, Dhodapkar MV.Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 2007;109:4839–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galluzzi L, Humeau J, Buque A, Zitvogel L, Kroemer G.Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol 2020;17:725–41. [DOI] [PubMed] [Google Scholar]
- 6.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med 2014;20:1301–9. [DOI] [PubMed] [Google Scholar]
- 7.Humeau J, Sauvat A, Cerrato G, Xie W, Loos F, Iannantuoni F, et al. Inhibition of transcription by dactinomycin reveals a new characteristic of immunogenic cell stress. EMBO Mol Med 2020;12:e11622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hopfner KP, Hornung V.Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 2020;21:501–21. [DOI] [PubMed] [Google Scholar]
- 9.Kroemer G, Zitvogel L.Subversion of calreticulin exposure as a strategy of immune escape. Cancer Cell 2021;39:449–51. [DOI] [PubMed] [Google Scholar]
- 10.Montes De Oca R, Bhattacharya S, Vitali N, Patel K, Kaczynski H, Shi HZ, et al. The anti-BCMA antibody-drug conjugate GSK2857916 drives immunogenic cell death and immune-mediated anti-tumor responses, and in combination with an OX40 agonist potentiatesin vivo activity. HemaSphere 2019;2:231. [Google Scholar]