

Abstract
Hypertrophic cardiomyopathy (HCM) is a disease of the myocardium caused by mutations in sarcomeric proteins with mechanical roles, such as the molecular motor myosin. Around half of the HCM-causing genetic variants target contraction modulator cardiac myosin-binding protein C (cMyBP-C), although the underlying pathogenic mechanisms remain unclear since many of these mutations cause no alterations in protein structure and stability. As an alternative pathomechanism, here we have examined whether pathogenic mutations perturb the nanomechanics of cMyBP-C, which would compromise its modulatory mechanical tethers across sliding actomyosin filaments. Using single-molecule atomic force spectroscopy, we have quantified mechanical folding and unfolding transitions in cMyBP-C domains targeted by HCM mutations that do not induce RNA splicing alterations or protein thermodynamic destabilization. Our results show that domains containing mutation R495W are mechanically weaker than wild-type at forces below 40 pN and that R502Q mutant domains fold faster than wild-type. None of these alterations are found in control, nonpathogenic variants, suggesting that nanomechanical phenotypes induced by pathogenic cMyBP-C mutations contribute to HCM development. We propose that mutation-induced nanomechanical alterations may be common in mechanical proteins involved in human pathologies.
Keywords: hypertrophic cardiomyopathy, protein mechanics, cMyBP-C, single-molecule, AFM, contraction
Graphical Abstract

Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac muscle disease, affecting up to 1 in 200 individuals.2,3 Macroscopically, HCM is characterized by thickened left ventricular walls and reduced size of the left ventricular chamber, while at the tissue level, HCM myocardium typically shows interstitial fibrosis and fiber disarray (Figure 1a). These structural changes occur alongside functional defects such as diastolic dysfunction, which can lead to the most severe consequences of the disease including heart failure and sudden cardiac death.4–6 Despite encouraging advances,7 currently there are no therapies to revert nor prevent HCM pathogenesis, and clinical management relies on long-term palliative treatments and surgical procedures.4,5
Figure 1.
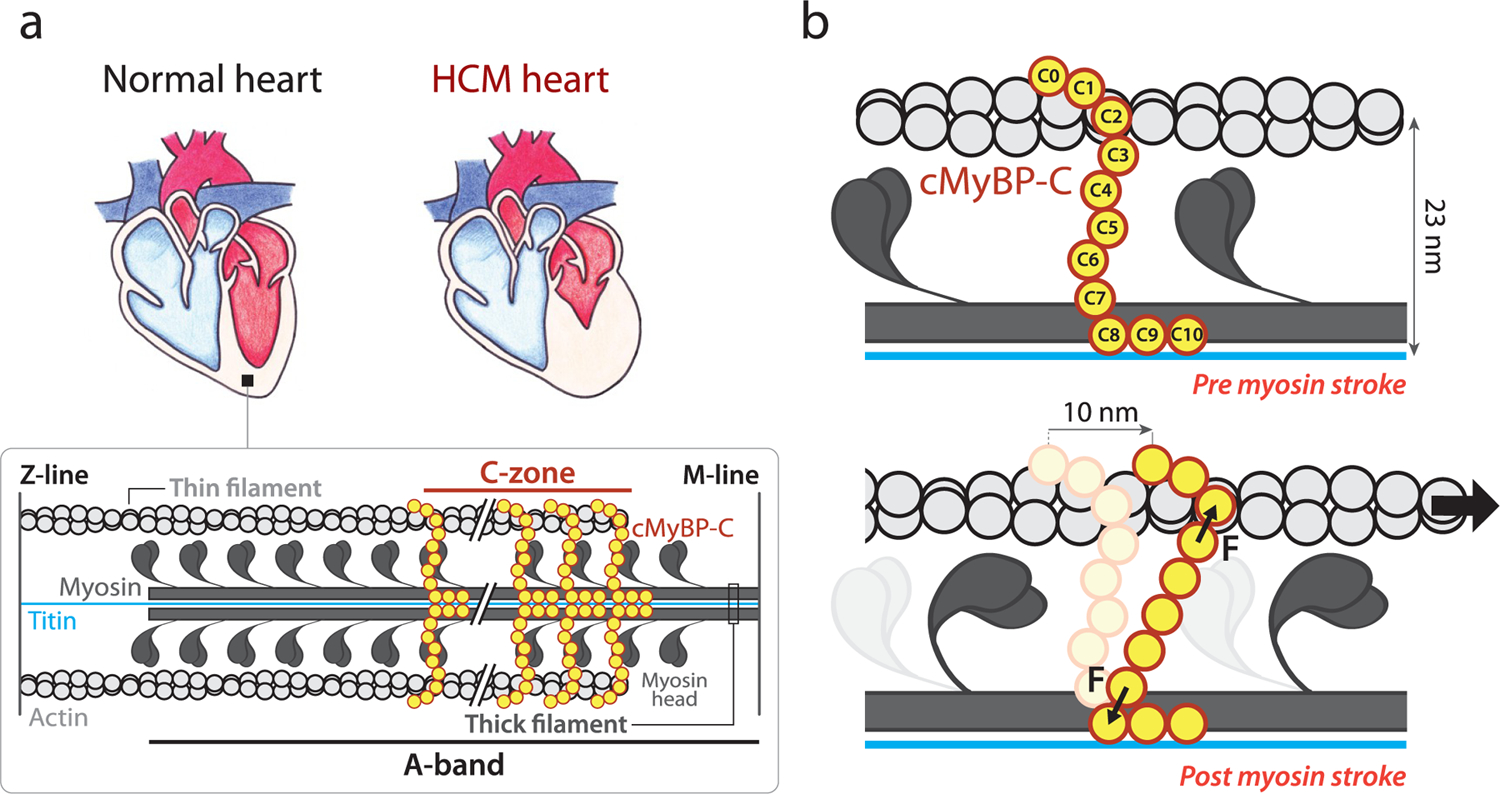
Overview of the mechanical role of cMyBP-C in the sarcomere. (a) Comparison of a healthy heart and an HCM counterpart, which shows thicker left ventricular walls and reduced left ventricle volume. Inset: Schematics of the sarcomere, whose contraction relies on actin-based thin filaments that glide over myosin-containing thick filaments thanks to myosin power strokes. cMyBP-C (in yellow) is located in the C-zone, a part of the A-band of the sarcomere. The M-line and the Z-line structures, which arrange filaments supporting sarcomere organization, are also shown.1 (b) cMyBP-C tethers are subject to mechanical force during a 10 nm myosin power stroke. Interfilament distance is indicated.
The majority of HCM cases are caused by autosomal dominant mutations targeting mechanical proteins of the sarcomere, the basic contractile unit of cardiomyocytes (Figure 1a).5,8,9 In sarcomeres, myosin heads use the energy coming from ATP hydrolysis to extend from the myosin backbone in the thick filaments, establish cross-bridges with the neighboring actin thin filaments, and generate ∼10 nm power strokes that propel the thin filaments past the thick ones, leading to muscle contraction (Figure 1b).4,8,10–12 Cardiac myosin-binding protein C (cMyBP-C), a well-known modulator of sarcomere activity, is a multidomain protein located in the C-zone of the sarcomere, grouped in nine regularly spaced transverse stripes that are 43 nm apart from each other.13 cMyBP-C’s C-terminal C8–C10 domains run axially along the thick filament, anchoring cMyBP-C to the myosin backbone and titin. The central and N-terminal domains of the protein extend radially from the thick filament toward the thin filament located ∼23 nm away (Figure 1).13–19
The regulation of contraction by cMyBP-C occurs through complex and modulatable, but incompletely understood, mechanisms involving interactions with partner proteins in the sarcomere.15,16,20 For instance, the N-terminal region of cMyBP-C has been shown to activate thin filaments and inhibit thick filaments.21 Indeed, recent experiments suggest that cMyBP-C promotes a low-activity, super-relaxed state (SRX) of myosin.22 In addition, the ability of cMyBP-C to tether thin and thick filaments can result in further mechanical modulation. Since the lifetime of the interaction between cMyBP-C and actin filaments is on the order of 20–300 ms,23 i.e., at least one order of magnitude longer than myosin power strokes,24 contraction is expected to induce substantial mechanical strain on cMyBP-C that can contribute to the slowing of actomyosin motion (Figure 1b).25,26 Hence, how cMyBP-C tethers respond to a mechanical load27–29 can be fundamental for its modulatory role on sarcomere power generation.
MYH7 and MYBPC3, which encode β-myosin heavy chain and cMyBP-C, respectively, are the most frequently mutated genes in HCM, accounting for 80% of cases.15,16,18 The vast majority of pathogenic variants in MYBPC3 result in cMyBP-C truncations that induce HCM via reduction in total cMyBP-C content (protein haploinsufficiency).15,26,31–36 However, other rare MYBPC3 pathogenic mutations only cause single amino acid substitutions that result in full-length, mutant cMyBP-C proteins that can incorporate into sarcomeres to the same levels as the wild-type (WT) protein.33,37 This includes the most frequent mutation in HCM (p.R502W).38 The molecular deficits, or phenotypes, of these missense mutations remain largely unexplored. Some of them have been proposed to disrupt cMyBP-C interaction with actomyosin filaments39–41 or to induce extensive protein destabilization;42,43 however, many HCM-causing missense mutants appear to operate via alternative, unidentified mechanisms.44 Prompted by the ability of cMyBP-C to establish mechanical tethers that modulate sarcomere contraction (Figure 1b), we hypothesized that HCM-causing mutations may perturb the nanomechanics of cMyBP-C, leading to altered sarcomere activity. Here, we have used single-molecule force spectroscopy by atomic force microscopy (AFM) to test this hypothesis.45 We have found that HCM-causing mutations can affect the mechanical stability and folding rate of the targeted domains, raising the possibility that alteration of cMyBP-C nanomechanics contributes to HCM pathogenesis.
RESULTS AND DISCUSSION
Selection of Pathogenic and Nonpathogenic cMyBP-C Variants.
Following strict assignment of pathogenicity based on clinical and epidemiological data,44 we selected four pathogenic missense mutations (p.R495Q, p.R495W, p.R502Q, and p.R502W) and two nonpathogenic variants (p.G507R and p.A522T) targeting cMyBP-C. The variants are located in exon 17 of the MYBPC3 gene, which together with exon 16 codifies for the C3 domain of cMyBP-C (Supplementary Text S1; Figure 2).46 Many HCM missense mutations cluster in C3,37,44 but no protein interactors have been described for this domain, as expected from its location far from the anchoring points of cMyBP-C to actomyosin filaments.47,48 Hence, mutations targeting C3 are arguably not predicted to affect cMyBP-C interactions, which is in agreement with the normal sarcomere localization of missense mutant cMyBP-C proteins in HCM myocardium.33,37 In addition, the high-resolution structure of C3 is known, which can help interpret the effects of the mutations (Figure 2).30 We first verified that the pathogenic mutations do not induce RNA splicing defects or extensive protein structural destabilization, two classical protein haploinsufficiency drivers linked to pathogenicity in 45% of cMyBP-C missense mutations.44
Figure 2.
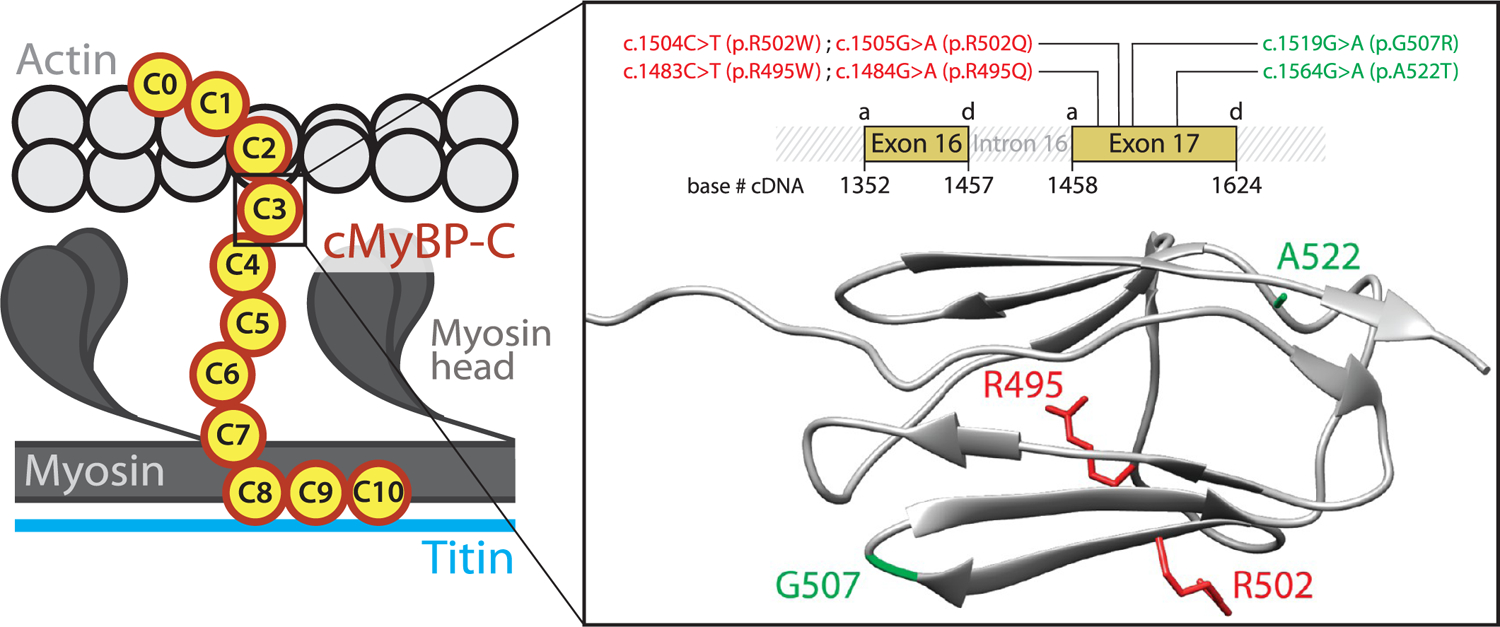
cMyBP-C variants tested in this report. The variants target the C3 central domain of cMyBP-C. Inset: The variants, which induce single nucleotide substitutions in MYBPC3 exon 17, are presented using both cDNA and protein nomenclatures. Variants are colored according to their pathogenicity (red: pathogenic mutations; green: nonpathogenic variants). MYBPC3 exons 16 and 17 code for the C3 domain, and the position of their acceptor (a) and donor (d) splicing sites in the cDNA sequence is indicated. The ribbon diagram presents the immunoglobulin (Ig)-like fold of the C3 domain, in which several β-strands arrange in a Greek key β-sandwich (pdb code 2mq0).30 The side chains of the residues targeted by the variants are highlighted.
Preservation of normal RNA processing in p.R495Q and p.R502W mutations has been observed before using human myocardial biopsies.33,50 In the case of mutation p.R502Q, no RNA splicing alteration has been detected using the leukocyte fraction of human blood samples44 and mini-gene constructs,51 two more readily available biological sources that provide results in excellent agreement with those obtained using myocardial samples.44 Using the former method, we studied RNA splicing of mutant p.R495W and also sought additional validation that p.R495Q and p.R502W do not induce RNA splicing alteration. We amplified by RT-PCR the region between exons 15 and 21 of MYBPC3 mRNA and observed that amplification of WT and mutant samples results in ∼700 bp bands, as expected (Supplementary Figure S1a; Supplementary Text S1). Preservation of canonical RNA splicing in the mutants was further confirmed by Sanger sequencing, which allows the detection of the variants in heterozygosis and the visualization of the correct 16/17 and 17/18 exon–exon boundaries (Supplementary Figure S1b).
To examine mutant protein stability, we did far-UV circular dichroism (CD) experiments using recombinant domains (Supplementary File S1; Supplementary Figure S2a). In agreement with previous reports, we found that WT and mutant domains have highly similar CD spectra showing a minimum at 215 nm, typical of β-structure-containing proteins (Supplementary Figure S3a).30,44,52 The only substantial difference was found in the spectrum of R502W, which displays a lower CD signal at 230 nm (Supplementary Figure S3b). Since the high-resolution structures of WT and R502W C3 are very similar,30 we interpret this change as originating from the absorption of the extra tryptophan in the mutant.53 To explore if mutations induce structural destabilization, we examined the stability of the mutant domains at increasing temperatures by tracking the CD signal at 215 nm (Supplementary Figure S4a). The temperature at the midpoint of the denaturing transition, or melting temperature (Tm), informs about the thermal stability of the domain. All mutations retain close-to-WT thermal stability (Supplementary File S1; Supplementary Figure S4b). The maximum drop in Tm was 5.3 °C for mutant R502Q; however, this limited decrease in thermal stability can also be found in nonpathogenic missense variants targeting C3 and therefore cannot explain the pathogenicity of the mutation.44
To confirm our observation that mutations do not induce extensive protein thermodynamic destabilization, we aimed at determining equilibrium thermodynamic parameters. However, thermal denaturation of C3 variants was largely irreversible, as indicated by non-native CD spectra obtained after ramping down the temperature to 25 °C (Supplementary Figure S3). Hence, to estimate the free energy (ΔG) of unfolding, we undertook urea-induced denaturation experiments, in which reversibility of protein unfolding transitions is generally favored.54 At 25 °C, urea concentrations over 4 M induced evident and largely reversible changes in the CD spectra of WT and mutant C3 domains (Supplementary Figures S5, S6a). We estimated ΔG of unfolding from the analysis of CD signals at 217 nm at increasing urea concentrations (Supplementary Figure S6b–h). Taking into account that the unfolding transition for some domains was not 100% reversible, we consider measured ΔG values as apparent (ΔGapp). ΔΔGapp (ΔGapp(WT) – ΔGapp(mutant)) at 25 °C was close to zero for all mutant domains, providing further evidence that pathogenic mutations do not cause destabilization of C3 (Supplementary Table S1; Supplementary Figure S6i,j). We obtained similar results at 37 °C, although limited spectral differences between folded and unfolded states at this temperature challenge accurate estimation of ΔGapp (Supplementary Figures S7, S8). In summary, none of the pathogenic variants studied in this report induce extensive protein destabilization, as determined using both thermal and chemical denaturation assays. These results are in agreement with largely preserved half-life and sarcomere localization of mutants R495Q and R502W in primary cardiomyocytes.37
Mechanical Stability of Mutant cMyBP-C Domains.
The results in the previous section show that the selected pathogenic MYBPC3 missense mutations preserve RNA splicing and protein stability, and thus it is unlikely that they cause HCM through classical protein haploinsufficiency. Most probably, these mutants are incorporated into the sarcomere but fail to provide proper functionality.33,37,55 Since cMyBP-C tethers are subject to mechanical force in the sarcomere (Figure 1b), we used AFM to examine whether mutations alter the mechanical stability of the C3 domain (Figure 3). We first produced polyproteins consisting of eight repetitions of the WT or the mutant C3 domains (Supplementary File S1; Supplementary Figure S2b). Using AFM, these polyproteins were subject to 40 pN/s increasing pulling force while monitoring their length in a buffer solution at physiological pH (7.2) and ionic strength (150 mM NaCl). Mechanical unfolding of a C3 domain within the polyprotein results in the extension of the polypeptide by 24–25 nm. The presence of multiple such unfolding steps fingerprints successful single-polyprotein recordings (Figure 3a).49 We measured the force at which mechanical unfolding occurs in the 40 pN/s force ramp for hundreds of WT and mutant domains (Supplementary File S2) and built distributions of unfolding forces (Figures 3b, 4a; Supplementary Figure S9). We found that the mean unfolding force (⟨Fu⟩) of the WT C3 domain is 90.6 pN, in agreement with previous measurements on cMyBP-C multidomain constructs.27,29 All pathogenic and nonpathogenic variants induce slight mechanical destabilization under our experimental conditions, as indicated by lower mean unfolding forces (Figure 3c; Table 1). However, this destabilization may not hold under physiological force regimes, as elaborated below.
Figure 3.
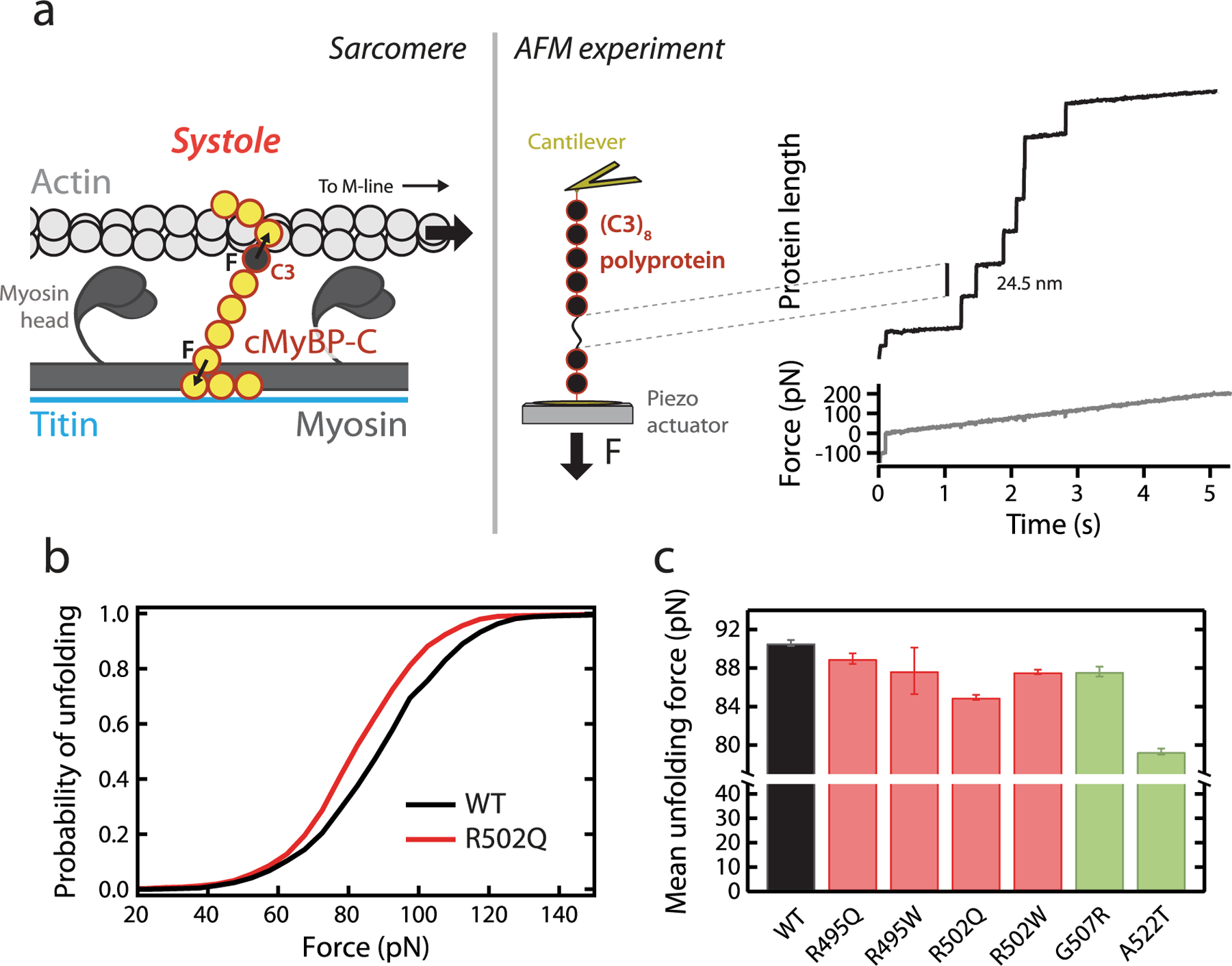
Characterization of the mechanical stability of WT and mutant C3 domains by single-molecule force spectroscopy by AFM. (a) Left: cMyBP-C tethers experience end-to-end mechanical force during the contraction of actomyosin filaments in systole. The position of the C3 domain within cMyBP-C is indicated. Right: The mechanical properties of a (C3)8 polyprotein are measured using single-molecule AFM. In these experiments, a single polyprotein is tethered between a cantilever and a moving piezo actuator, and its length is recorded while a linear increase in force is applied. Unfolding events are detected as step increases in the length of 24–25 nm.49 (b) Cumulative probability of unfolding with force for WT (n = 1033 unfolding events) and R502Q domains (n = 1254 unfolding events) during a 40 pN/s force ramp. (c) Mean unfolding forces in force-ramp experiments, as obtained from Gaussian fits to distributions of unfolding forces (see also Table 1). Error bars correspond to 83% confidence intervals. Bars are colored according to the pathogenic status of the mutation (pathogenic, red; nonpathogenic, green).
Figure 4.
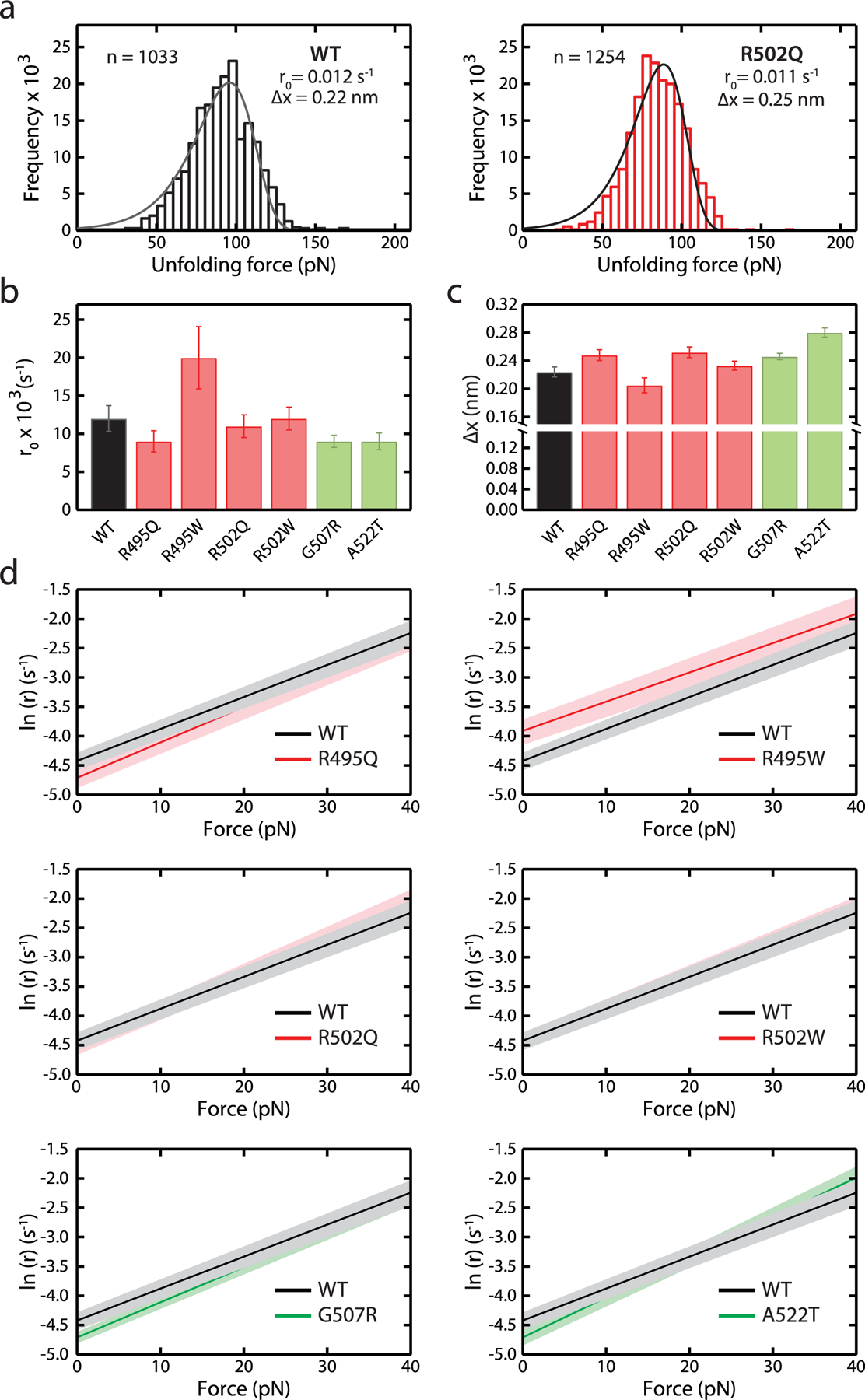
Characterization of r0 and Δx parameters of WT and mutant C3 domains according to Bell’s model. (a) Distributions of unfolding forces obtained for WT and R502Q domains in a 40 pN/s force ramp. Distributions were fit to Bell’s model56 (solid lines), and the resulting rate of unfolding at zero force, r0, and distance to the transition state, Δx, are indicated. (b, c) r0 and Δx values for WT and mutant C3 domains (see also Supplementary Figure S9 and Table 1). Error bars correspond to 83% confidence intervals. Bars are colored according to the pathogenic status of the mutation (pathogenic, red; nonpathogenic, green). (d) Force dependency of rates of unfolding, r, according to Bell’s model (red: pathogenic, green: nonpathogenic). 83% confidence intervals are indicated as shaded areas.
Table 1.
Nanomechanical Properties of WT and Mutant C3 Domainsa
| variant | pathogenic variant | ⟨Fu⟩ (pN) | r0 (s−1) | Δx (nm) | rf (s−1) |
|---|---|---|---|---|---|
| R495Q | yes | 89.0 ± 0.6 | 0.009 ± 0.001 | 0.25 ± 0.01 | 0.28 ± 0.14 |
| R495W | yes | 87.7 ± 2.4 | 0.020 ± 0.004 | 0.21 ± 0.01 | 0.11 ± 0.03 |
| R502Q | yes | 85.0 ± 0.3 | 0.011 ± 0.002 | 0.25 ± 0.01 | 2.1 ± 0.8 |
| R502W | yes | 87.6 ± 0.2 | 0.012 ± 0.002 | 0.23 ± 0.01 | 0.08 ± 0.02 |
| G507R | no | 87.6 ± 0.5 | 0.009 ± 0.001 | 0.25 ± 0.01 | 0.17 ± 0.10 |
| A522T | no | 79.3 ± 0.3 | 0.009 ± 0.001 | 0.28 ± 0.01 | 0.14 ± 0.04 |
| WT | 90.6 ± 0.3 | 0.012 ± 0.002 | 0.22 ± 0.01 | 0.16 ± 0.06 |
Errors are 83% confidence intervals from the fits used to calculate the mechanical parameters. If these intervals do not overlap, differences are considered statistically significant (see Methods). ⟨Fu⟩, mean unfolding force; r0, unfolding rate at zero force; Δx, distance to the transition state; rf, folding rate.
Experimental ⟨Fu⟩ values are a consequence of the underlying free energy landscapes and the specific pulling conditions. Extrapolation of AFM data to alternative ranges of forces can be achieved using models that consider how the energy landscape is shaped by the applied mechanical force. We have done so by fitting our data to Bell’s model (Figure 4a; Supplementary Figure S9).56,57 According to this model, the rate of mechanical unfolding (r) is dependent on force (F) according to
| (1) |
where r0 is the rate of unfolding at zero force, Δx is the distance to the transition state of the mechanical unfolding reaction, kb is the Boltzmann constant, and T is the absolute temperature. Fits show that both pathogenic and nonpathogenic mutations can affect r0 and/or Δx, therefore altering the mechanical behavior of C3 domains in a force-dependent manner (Figure 4b,c; Table 1). Using the parameters obtained from the fits, we estimated mechanical unfolding rates at low forces, which are challenging to probe experimentally using AFM but can be relevant in the context of cMyBP-C function in the sarcomere. This analysis showed that R495W domains unfold significantly faster than WT counter-parts at forces below 40 pN, whereas the rest of the mutants behave very similarly to WT, including control, nonpathogenic variants (Figure 4d).
In addition to the higher unfolding rate at low forces, the mechanical behavior of R495W was also characterized by an unfolding force distribution showing a tail at high forces (Supplementary Figure S9). The reasons for this behavior, which has been observed before in metalloproteins,59,60 may result from alternative unfolding pathways in the mutant. To investigate the molecular origin of the peculiar mechanical properties of mutant R495W, we ran atomistic molecular dynamics (MD) simulations. In Supplementary Figure S10a we show force extension plots for three independent runs for WT and R495W C3 domains. In all curves we found a similar force peak at ∼700 pN. As expected, the rupture forces in the simulations were notably higher than experimental values due to the many orders of magnitude higher loading rates in the computational unfolding.61 In most of the trajectories we also found an earlier event resulting in a lower force peak. These rupture processes correspond to the breaking of the mechanical clamp characteristic of the Ig-like fold (Supplementary Figure S10b).62,63 Specifically, the hydrogen bonds formed between the A and B/G β-strands are broken first, resulting in the low force peak, and those between the A′ and G strands come second and are the main determinants of the mechanical strength of the domain observed experimentally.62 We show snapshots taken from one WT trajectory illustrating the different steps in the mechanical unfolding of WT C3 in Supplementary Figure S10c. The events captured by this trajectory resemble observations for both the WT and the mutant domain in all replicates. Hence, although we detect a tendency to lower unfolding forces in R495W (WT: 729 ± 54 pN; R495W: 619 ± 29 pN; errors are standard error of the mean (SEM), p = 0.15; two tailed unpaired t test), no noticeable changes in the mechanical unfolding pathways of WT and R495W were observed. We could detect no interaction between residue 495 and the mechanical clamp either, so taking into account all evidence from the simulations, we consider highly unlikely that the R495W mutation causes major changes in the mechanical unfolding mechanism of C3. An alternative possibility is that the mechanical changes induced by the mutation are mediated by subtle allosteric rearrangements. This type of coupling may be challenging to capture in MD simulations, due to the weak cooperativity of classical force fields.64
Mechanical Folding in Missense Mutants of cMyBP-C.
To determine the ability of the different C3 domains to refold following mechanical unfolding, we did unfolding–quench–probe experiments (Figure 5a).65 In these experiments, proteins are first pulled in a force ramp to high forces (unfolding pulse). Then, force is relaxed to 0 pN, at which domains can regain the folded state (quench pulse). Finally, in the probe pulse, the protein is pulled back to high forces. Unfolding steps in the probe pulse report on domains that refolded during the quench pulse. To obtain folding fractions, the number of unfolding events in the probe and unfolding pulses are compared. Folding rates (rf) were estimated by measuring folding fractions at different quench times. Compared to WT, R502Q shows a 13× increased rf. The remaining pathogenic mutations and nonpathogenic variants do not cause significant changes in folding rate (Figure 5b,c; Supplementary Figure S11; Table 1).
Figure 5.
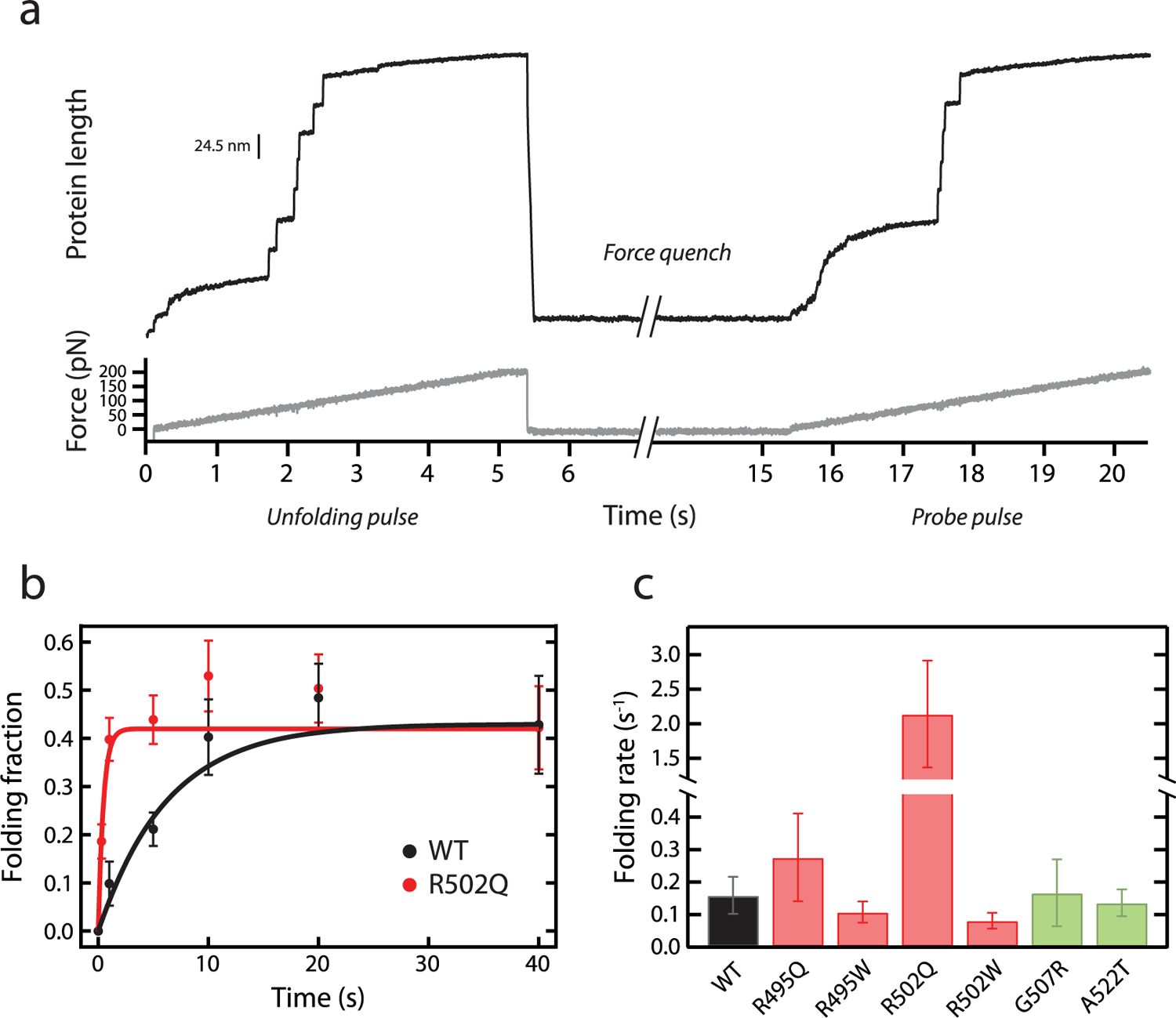
Characterization of mechanical folding of WT and mutant C3 domains. (a) Representative trace of mechanical refolding experiments by AFM. A single (C3)8 polyprotein is subject to an unfolding pulse; then force is quenched to 0 pN, and finally the protein is pulled again to high forces in a probe pulse. Folding fractions are calculated comparing the number of unfolding events in the probe and the unfolding pulses. In the example shown, 5 out of 7 domains refolded during the quench pulse. (b) Folding fractions of WT and R502Q C3 domains at different quench times. Lines are exponential fits to the data. Error bars are SEM estimated by bootstrapping58 (n ≥ 56 and n ≥ 86 unfolding events for all WT and R502Q data points, respectively). (c) Mechanical folding rates for WT C3 and its mutants, obtained from exponential fits to refolding data (see also Supplementary Figure S11 and Table 1). Error bars are 83% confidence intervals. Bars are colored according to the pathogenic status of the mutation (pathogenic, red; nonpathogenic, green).
cMyBP-C Nanomechanical Phenotypes in Context.
It has been proposed that HCM mutations lead to increased sarcomeric output and myocyte hypercontractility.22,66–68 Indeed, mavacamten, an inhibitor of myosin motor activity, is able to prevent development of HCM in mouse models69 and also shows beneficial effects in clinical trials.7 Many HCM-causing mutations in myosin affect directly the mechanochemical cycle of the protein, resulting in enhanced force generation.70,71 In addition, the proportion of low-activity SRX myosin heads is decreased in HCM myocytes devoid of cMyBP-C but is normalized upon treatment with mavacamten.22,72,73 The emerging unifying view is that, regardless of the specific mutation, HCM is primarily a mechanical disease at the molecular level; however, it remains unknown how missense mutations in the central region of cMyBP-C can lead to hypercontractility.74 In this work we have studied several pathogenic mutations targeting the central C3 domain of cMyBP-C, the region of the protein with the highest number of HCM-causing missense mutations.15,37,44 We verified that our variants do not induce classical protein haploinsufficiency drivers specifically associated with other HCM mutations (Supplementary Figures S1, S4).44 Hence, these variants trigger HCM by yet-to-identify molecular mechanisms.
The modulatory mechanisms of cMyBP-C on contraction are complex and far from being completely understood. While the C8–C10 C-terminal domains of cMyBP-C play a structural role providing strong anchorage to the thick filament, the C0–C2 N-terminal fragments can bind both actin filaments and myosin globular heads, resulting in sophisticated control of their activity through direct mechanical load (Figure 1b) or via conformational changes that are dependent on phosphorylation and calcium levels.16 In this highly intricate regulatory landscape, several possibilities can be envisioned by which altered cMyBP-C nanomechanics, as detected here for R495W and R502Q mutants (Figures 4d, 5c), can perturb sarcomere function.
Let us first consider a purely mechanical scenario. Current models on cMyBP-C modulation of sarcomere contraction state that the central region of the protein is subject to end-to-end mechanical force that results in a viscous drag slowing actomyosin motion (Figure 1b).16,23,25–27,75,76 This drag force depends on cMyBP-C stiffness. If we assume that five cMyBP-C domains bridge radially the thick and thin filaments,13 using the freely jointed chain (FJC) model of polymer elasticity,77 we can predict that the force experienced by cMyBP-C tethers increases by >250% during a 10 nm myosin power stroke (4.4 nm contour length, Lc, per domain, and 20 nm Kuhn length, Lk) (Figure 6).78 This force is expected to decrease to less than pre-power-stroke values if a cMyBP-C domain mechanically unfolds in the process (Lc = 0.4 nm per amino acid, 90 amino acid domain size, Lk = 1.32 nm for unfolded polypeptide regions).78 According to our AFM data, this low-force state is up to 66% more frequent in mutant R495W (Figure 4d), which is expected to reduce the average viscous load generated by mutant cMyBP-C. We speculate that the higher speed of folding detected for R502Q could alter other steps of the mechanochemical cycle of actomyosin filaments, for instance by generating more mechanical work during relaxation.79 It is important to stress that force estimates in Figure 6 are highly dependent on the specific geometry of cMyBP-C tethers and on the mechanical parameters considered. Although the model exemplifies how cMyBP-C viscous load can be reduced by mechanically labile domains, the exact forces experienced by cMyBP-C in the sarcomere, which so far remain unknown, may differ from the values shown in Figure 6.
Figure 6.
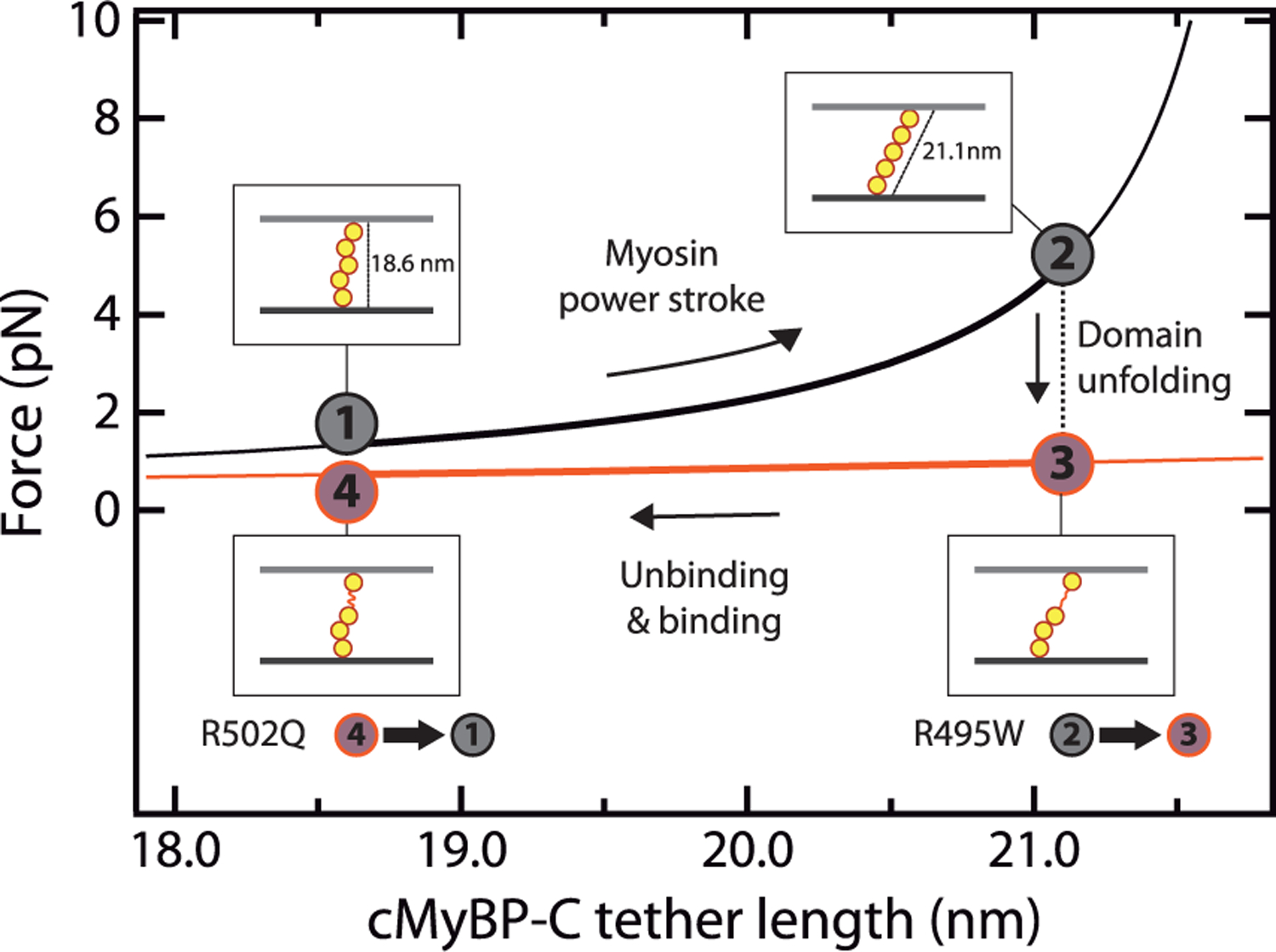
Model of mechanical modulation by cMyBP-C tethers and the influence of domain unfolding. FJC-estimated increase in force generated by a fully folded cMyBP-C tether (black) during a myosin power stroke. If one of the domains of cMyBP-C unfolds, force is reduced (orange). The model considers a radial distribution of domains C3–C7,13 and that anchoring C2 and C8 domains (not shown for simplicity) are located at the center of the thin and thick filaments, respectively, so that they contribute half their diameter to bridge the 23 nm interfilament space.19 The increase in cMyBP-C length during a myosin power stroke was estimated using the Pythagorean theorem considering a 10 nm power stroke.12 In the graph, we also considered that cMyBP-C can dissociate from actin sites at a slower rate than that of myosin power strokes. Mutation R495W increases the rate of mechanical unfolding of C3, whereas R502Q leads to faster C3 folding at low forces.
The interaction of N- and C-terminal domains of cMyBP-C with several sarcomeric proteins, resulting in modulation of their activity, is well documented.16 The possibility exists that the central domains of cMyBP-C transitorily participate in those binding reactions or have yet unidentified binding partners. Hence, as described in mutants targeting N- and C-terminal domains,40,80,81 mutations in central domains of cMyBP-C could also interfere with binding reactions either directly or allosterically. Indeed, we have proposed that hampered interaction of cMyBP-C with the mesa region of myosin can lead to sarcomere hypercontractility and HCM.68 In this regard, we have found that the C3 domain of cMyBP-C binds myosin sS1 with an affinity that is comparable to that of C0C2-sS1 interaction (Supplementary Figure S12, see Methods). This unexpected observation raises the possibility that mutations in C3 perturb the interaction between cMyBP-C and myosin, altering the number of myosin heads available for force generation as recently observed for myosin R403Q.82 Furthermore, we speculate that the mechanical unfolding of cMyBP-C central domains could result in the reinforcement or disarming of binding sites, similarly to other proteins under mechanical load such as titin and talin.65,83 In this scenario, altered mutant cMyBP-C nanomechanics would also cause perturbations of the interaction landscape of cMyBP-C.
Truncating mutations in cMyBP-C that cause HCM result in normal levels of mutant mRNA but no detectable truncated polypeptide in the myocardium, probably due to its degradation by cellular protein quality control systems, such as the ubiquitin/proteasome system (UPS). Beyond insufficient mechanical modulation in cMyBP-C-deficient myocardium, the exhaustion of these protein control systems can also contribute to the pathogenesis of HCM.84–87 Since mechanical destabilization results in increased protein unfolding, a well-known trigger of the UPS,88 it is possible that nanomechanical destabilization of cMyBP-C missense mutants can result in activation of protein quality control systems.
CONCLUSIONS
Our single-molecule experiments show that pathogenic missense mutations in cMyBP-C can induce mechanical destabilization and alteration in the mechanical folding properties of the targeted domain. We propose that these nanomechanical phenotypes, which are not found in nonpathogenic variants, can perturb the function of cMyBP-C in the sarcomere by several mechanisms, potentially contributing to HCM pathogenesis. Similar nanomechanical phenotypes may be also found in pathogenic mutations targeting other proteins with mechanical roles, such as titin,89 talin,90 filamin,91 lamin,92 and α-catenin.93 Future studies will investigate the prevalence of nanomechanical phenotypes in other cMyBP-C mutations, as well as their impact in low-force transitions that are now amenable for experimental observation94 and in the mechanical output of mutant actomyosin filaments.26
METHODS
Human Samples.
The procurement of human samples was achieved following the principles outlined in the Declaration of Helsinki according to a project approved by the Comitéde Ética de Investigación of Instituto de Salud Carlos III (PI 39_2017) and by the Ethics Committee of the Naples University Federico II “Carlo Romano” (Protocol number 157/13).
Selection of Mutants.
Nonpathogenic variants have allele frequencies that are incompatible with HCM prevalence.44 The four pathogenic mutations are enriched in HCM patients with respect to control population and have been found in multiple families cosegregating with the disease (criteria PS4 and PP1 of pathogenicity by the American College of Medical Genetics, respectively), as retrieved from the Health in Code (HIC)-Mutations database. HIC-Mutations encompasses information about >155 000 individuals from thousands of articles in the literature as well as from HIC clinical reports.
Analysis of RNA Splicing.
Leukocytary fractions from venous blood were treated with Trizol (Thermo Fisher Scientific) to extract total RNA. Then, cDNA was obtained by retro-transcription of total RNA with random primers and Superscript IV VILO Master Mix (Thermo Fisher Scientific). To amplify the region spanning MYBPC3 exons 15 through 21, we used primers MyBPC forward (5′-CAAGCGTACCCTGACCATCA-3′) and MyBPC reverse (5′-GGATCTTGGGAGGTTCCTGC-3′). To avoid amplification of genomic DNA, the annealing region of the reverse primer targets the exon 20/21 junction (Supplementary Text S1). The resulting PCR amplification products were purified using the Qiaquick PCR purification kit (Qiagen). Finally, the purified fragments were sequenced using the Sanger method. We consider that standard RNA processing is not altered when electropherograms allow the unambiguous reading of the expected WT MYBPC3 cDNA sequence at the junction between exons 16, 17, and 18. Since patients carry mutations in heterozygosis, a double peak corresponding to the WT and mutant base was detected at the variant position, as expected.
Protein Expression and Purification.
The production and purification of the (C3)8 polyproteins was done as reported before,49 except for the case of nonpathogenic variants, which were also expressed by overnight induction of BLR(DE3) E. coli cultures with 0.4 mM isopropyl β-D-1-thiogalactopyranoside, at 18 °C and 250 rpm agitation. Mutations were introduced by PCR. Polyprotein-coding mutant cDNAs were obtained by an iterative cloning strategy using BamHI, BglII, and KpnI restriction enzymes, as described.95,96 To produce monomeric C3 domains, the corresponding cDNA was cloned in a custom-modified pQE80L plasmid using BamHI and BglII restriction enzymes. Sanger sequencing was performed on all final expression plasmids. Monomers and mutant polyproteins were purified following the same protocol as for wild-type (C3)8,49 which includes two rounds of purification using nickel-based affinity and size-exclusion chromatographies. In this final step, elution was carried out in 10 mM Hepes, pH 7.2, 150 mM NaCl, and 1 mM EDTA for polyproteins, while monomers were generally recovered in 20 mM NaPi, pH 6.5, and 63.6 mM NaCl (CD buffer). SDS-PAGE analysis was carried out to evaluate the purification process and to identify the purest and highest concentrated fractions. Proteins were stored at 4 °C. For chemical denaturation, size-exclusion chromatography was done in 50 mM ammonium bicarbonate pH 7, and resulting samples were freeze-dried using a VirTis BenchTop K lyophilizer (40–100 mTorr, >12 h). To conduct chemical denaturation and renaturation experiments, proteins were reconstituted in CD buffer including appropriate urea concentrations. When needed, protein reconstitution was followed by concentration using Amicon-Ultra 0.5 mL centrifugal filter unit 3 K (Sigma) to reach suitable protein concentrations. Alternatively, protein samples were not freeze-dried but buffer-exchanged using Amicon-Ultra devices. The motor domain of human β-cardiac myosin expressed recombinantly as a single-headed short S1 construct (sS1) was used for binding experiments (see below). The sS1 construct with an eight-residue PDZ-binding affinity clamp motif at the C-terminus (sS1-AC) was coexpressed with FLAG-tagged human essential light chain in murine myoblast cells (C2C12) using adenoviral vectors. As described previously,97 sS1-AC was purified using FLAG affinity chromatography followed by anion-exchange chromatography. The cMyBP-C C0C2 fragment was purified as described before.68
Circular Dichroism.
CD experiments were conducted in a Jasco J-810 spectropolarimeter. To obtain CD spectra, protein fractions at 0.1–0.5 mg/mL were tested in CD buffer, and signal was recorded every 0.2 nm at a speed of 50 nm/min. Data were collected with standard sensitivity (100 mdeg). Four different scans were performed for each construct, which were later averaged to obtain the final spectra. The baseline contribution of the CD buffer was subtracted from all protein spectra (including those in the presence of urea, see below), which were finally normalized according to protein concentration. Protein concentrations were estimated from A280 measurements considering theoretical extinction coefficients according to ProtParam Tool (Supplementary File S1).98 To examine thermal stability, protein samples were heated at 30 °C/h from 25 to 85 °C using a Peltier temperature controller while recording CD signal at 215 nm (0.5 °C data pitch). The recorded CD signal changes as the protein denatures and unfolds, and the melting temperature (Tm) was calculated by performing a sigmoidal fitting to the denaturing curves using IGOR Pro (Wavemetrics). A 0.1 cm path-length cuvette was used in these thermal denaturation experiments, resulting in 370–400 V dynode voltage signals. Chemical denaturation was investigated by progressively adding urea to the protein samples to a final concentration of 5.8–6.5 M while recording CD spectra.54 With that aim, an 8 M urea stock was prepared in CD buffer. Urea solutions were used on the same day they were prepared or stored at −80 °C for no longer than 24 h. Due to urea addition, protein samples were diluted from 0.5–0.8 mg/mL to 0.1–0.2 mg/mL at the end of the titration. To accommodate this decrease in protein concentration, both 0.1 and 0.2 cm path-length cuvettes were used. To improve signal-to-noise ratio, normalized CD spectra were smoothed by means of a binomial algorithm using IGOR Pro (Wavemetrics). Chemical denaturation curves were obtained by plotting the CD signal at 217 nm at increasing urea concentrations since this wavelength shows the largest differences between folded and unfolded spectra. Denaturation curves were then fitted to eq 2, which considers that ΔG of unfolding is linearly dependent on the concentration of urea:99
| (2) |
where x is the urea concentration; Yn and Yu are the native and unfolded CD signals in the absence of urea, respectively; mn and mu consider the dependence of the CD signals with urea concentration for the native and unfolded states, respectively; m is the slope determining ΔG variation with urea concentration; R is the gas constant; and T is the absolute temperature. For simplicity, we considered mn and mu as zero, which is in agreement with the CD signal plateaus observed at low and high urea concentrations in most of the denaturing curves. Fitting of denaturation curves was done using IGOR Pro (Wavemetrics). To study the reversibility of the unfolding reaction, first we induced the denaturation of the protein samples in the presence of high urea concentrations of 5.8–6.4 M. From this starting point, CD buffer was progressively added so that the urea was diluted to a final concentration of around 0.7 M. Hence, protein concentration decreased during the titration from 0.6–0.8 mg/mL to 0.06–0.09 mg/mL. As in the denaturation experiments, 0.1 and 0.2 cm path-length cuvettes were used depending on protein concentration. To track the renaturation process, CD spectra were processed as explained above for the denaturation transitions.
Single-Molecule Atomic Force Spectroscopy.
Single-molecule AFM experiments were done in an AFS force-clamp spectrometer (Luigs & Neumann).45 1–20 μL of a 0.04–1.5 mg/mL solution of the purified polyprotein in 10 mM Hepes, pH 7.2, 150 mM NaCl, and 1 mM EDTA buffer were deposited onto a gold-coated coverslip (Luigs & Neumann). We used silicon nitride MLCT cantilevers (Bruker AFM Probes), with a reflective 60 nm gold coating on their back side. These cantilevers were calibrated using the thermal fluctuations method.100 Typical spring constants were in the range of 15–20 pN/nm. Single polyproteins were picked up by pressing the cantilever onto the gold surface at a contact force of 500–2000 pN for 0.8–2 s and subject to a 40 pN/s linear increase in force until their detachment. During this stretching, the length of the polyprotein is measured and unfolding events are detected as specific stepwise increases in extension. The mechanical unfolding of C3 domains results in steps of 24–25 nm; only traces containing two or more unfolding events and with detachment forces >175 pN were analyzed.49 Traces where the fingerprint was interrupted by unidentifiable events were discarded. Unfolding forces were recorded in at least three different experiments performed with different cantilevers. Only experiments with low laser interference (peak-to-peak height in baseline force–extension traces lower than 25 pN) were included in the analysis of unfolding forces.45 Mean unfolding forces were obtained from Gaussian fits to histograms of unfolding forces. Some of the unfolding data for WT C3 have also been included in a technical report.49 Distributions were also fit to Bell’s model of protein unfolding under a force ramp to get the value of the unfolding rate at zero force, r0, and the distance to the transition state, Δx.56 For folding experiments, we programmed a force regime consisting of two ramps separated by a quench pulse. To quantify folding rates, we calculated folding fractions at different quench times. The bootstrap method was used to estimate standard errors of the mean for the folding fractions.58 Time courses of folding were fit to an exponential function:
| (3) |
where Fr(t) is the folding fraction as a function of time, R is the maximum folding fraction, and rf is the folding rate. Fits considered that the folding reaction was complete at 40 s. Values of R < 1 in the folding of Ig domains are common and are due to misfolded states that are highly dependent on experimental conditions.65,101 Hence, we focused on folding rate, which is a more robust molecular parameter. Analysis was done in IGOR Pro (Wavemetrics).
Molecular Dynamics Simulations.
To explore the conformational dynamics of the mechanical unfolding of WT and R495W C3, we ran atomistic MD simulations in explicit solvent. In the case of WT, we used as starting point the first structural model of the experimental NMR structure of C3 (2mq0).30 Since the structure of the mutant is not available, we generated it from the WT structure using the swapaa command implemented in Chimera.102 For both systems we followed identical simulation protocols. First we put the protein at the center of a simulation box with 30 nm in its longest dimension and 6 nm length in the other two dimensions. The protein was then solvated with ∼35 000 water molecules and Na+ and Cl− ions were introduced to enforce electroneutrality at a ∼0.1 M ion concentration. The boxes were then energy minimized and equilibrated using a 100 ps NVT simulation with position restraints in the protein heavy atoms, followed by a 200 ps NPT run to equilibrate the density. Starting from the last snapshot of this simulation, we ran three independent pulling simulations with a 10−5 nm/ps pulling speed for the separation between the N- and C-termini in the longest box dimension and a harmonic force constant of 1000 kJ/(mol·nm2). The equations of motion were integrated using a leapfrog stochastic dynamics integrator using a time step of 2 fs. Electrostatics were calculated using the particle-mesh Ewald method103 and a real-space 1.2 nm cutoff. Simulations were performed using the CHARMM36m force field and the modified version of the TIP3P water model.104–106 All simulations were performed using the Gromacs software package (versions 2018 and 2020).107 For the analysis of the simulation runs, we combined the suite of Gromacs analysis programs with the MDtraj Python library.108
Binding Assays by Microscale Thermophoresis.
The possibility of a direct interaction between myosin and C3 was investigated using microscale thermophoresis (MST) measurements, a technique that readily captures the interaction between cMyBP-C and myosin sS1.68 C3 protein was prepared in a buffer containing 10 mM Tris-HCl (pH 7.5), 4 mM MgCl2, 1 mM EDTA, 1 mM DTT, and 50 mM KAc. C3 was fluorescently labeled via its His-tag using a commercially available His-tag labeling kit following the manufacturer’s protocol (NanoTemper Technologies). A detailed method of determination of binding affinity of two proteins using MST has been previously described.68 Briefly, unlabeled sS1-AC was titrated into a fixed concentration of fluorescently labeled C3 protein (84 nM). The experiment was carried out in a buffer containing 10 mM Tris-HCl at pH 7.5, 4 mM MgCl2, 1 mM EDTA, 1 mM DTT, 50 mM KAc, 1 mM ATP, and 0.05% Tween-20. Sixteen different concentrations of sS1-AC were prepared by serial dilution and mixed with fluorescently labeled C3 to generate a full binding isotherm. The mixture was incubated in the dark for 30 min at 23 °C. Samples were then loaded into NT.115 premium-treated capillaries (NanoTemper Technologies) and mounted in a Monolith NT.115 apparatus (NanoTemper Technologies) for data acquisition. C3 fluorescence was measured using a red LED (excitation 605–645 nm; emission 680–685 nm) at 45% excitation power; an IR laser was used at 60% power. All data were acquired at 23 °C. Data analysis was carried out with NT Affinity Analysis software (NanoTemper Technologies), which derives binding isotherms from the raw fluorescence traces. Following the same protocol, C0C2-sS1 binding isotherms were also set up for reference. Two independent measurements from two different preparations of C0C2, C3, and sS1 proteins were carried out.
Statistics.
Unless indicated otherwise, statistical significance of the differences in parameters between WT and mutant domains were inferred from 83% confidence intervals, which were estimated using IGOR Pro (Supplementary File S1, Table 1). No overlapping intervals suggest that the null hypothesis can be rejected with p < 0.05.109,110
Supplementary Material
ACKNOWLEDGMENTS
J.A.C. acknowledges funding from the Ministerio de Ciencia e Innovación (MCIN) through grants BIO2014–54768-P, BIO2017–83640-P (AEI/FEDER, UE), EIN2019–102966, RYC-2014–16604, and BFU2017–90692-REDT, the European Research Area Network on Cardiovascular Diseases (ERA-CVD/ISCIII, MINOTAUR, AC16/00045), and the Comunidad de Madrid (consortium Tec4Bio-CM, S2018/NMT-4443, FEDER). This work was supported by NIH grants RM1 GM33289 and HL117138 to J.A.S.; a Stanford Dean’s Postdoctoral Fellowship to D.P. and N.N.; and a Stanford Maternal and Child Health Research Institute (MCHRI) Postdoctoral Fellowship (1220552–140-DHPEU) to N.N. Financial support to D.D.S. comes from Eusko Jaurlaritza (Basque Government) through the project IT1254–19, and grants RYC-2016–19590 and PGC2018–099321-B-I00 from the MCIN (FEDER). The CNIC is supported by the Instituto de Salud Carlos III (ISCIII), MCIN, and the Pro CNIC Foundation and was a Severo Ochoa Center of Excellence (SEV-2015–0505). We acknowledge funding from ISCIII to the Centro de Investigación Biomédica en Red (CIBERCV), CB16/11/00425. C.S.C. is the recipient of an FPI-SO predoctoral fellowship, BES-2016–076638. M.R.P. was the recipient of a Ph.D. fellowship from the Italian Ministry of Education, Universities and Research (MIUR). C.P.L. was a recipient of a CNIC Master Fellowship. We thank N. Vicente for excellent technical support (through grant PEJ16/MED/TL-1593 from Consejería de Educación, Juventud y Deporte de la Comunidad de Madrid and the European Social Fund). We thank the Spectroscopy and Nuclear Magnetic Resonance Core Unit at CNIO for access to CD instrumentation and discussion about protein binding assays. We thank A. Thompson and S. Day for their insights. We thank all members of the Molecular Mechanics of the Cardiovascular System team for helpful discussions and the contribution of five anonymous reviewers.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.1c02242.
Supplementary text (genomic region where mutations of interest are located), supplementary figures (results of RNA splicing and protein stability experiments, extended force spectroscopy data, results of molecular dynamics simulations and binding isotherms), a supplementary table (thermodynamic parameters for protein unfolding), and supplementary references (PDF)
Data of mutant proteins (XLSX)
Raw AFM data (XLSX)
The authors declare the following competing financial interest(s): L.M. is a share-holder of Health in Code. J.A.S. is a Co-founder of, consults for, and owns stock in Cytokinetics, Inc.
A version of the manuscript has been submitted to a pre-print server.111
Contributor Information
Carmen Suay-Corredera, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain.
Maria Rosaria Pricolo, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain; Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Universitàdi Napoli Federico II, 80131 Naples, Italy.
Diana Velázquez-Carreras, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain.
Divya Pathak, Department of Biochemistry, Stanford University School of Medicine, Stanford, California 94305, United States; Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, California 94305, United States.
Neha Nandwani, Department of Biochemistry, Stanford University School of Medicine, Stanford, California 94305, United States; Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, California 94305, United States.
Carolina Pimenta-Lopes, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain.
David Sánchez-Ortiz, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain.
Iñigo Urrutia-Irazabal, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain.
Silvia Vilches, Heart Failure and Inherited Cardiac Diseases Unit, Department of Cardiology, Hospital Universitario Puerta de Hierro, 28222 Madrid, Spain; European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart, 28222 Madrid, Spain.
Fernando Dominguez, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain; Heart Failure and Inherited Cardiac Diseases Unit, Department of Cardiology, Hospital Universitario Puerta de Hierro, 28222 Madrid, Spain; European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart, 28222 Madrid, Spain; Centro de Investigación Biomédica en Red en Enfermedades Cardiovasculares (CIBERCV), 28029 Madrid, Spain.
Giulia Frisso, Dipartimento di Medicina Molecolare e Biotecnologie Mediche, Universitàdi Napoli Federico II, 80131 Naples, Italy; CEINGE Biotecnologie Avanzate, scarl, 80145 Naples, Italy.
Lorenzo Monserrat, Health in Code, 15006 A Coruña, Spain.
Pablo García-Pavía, Heart Failure and Inherited Cardiac Diseases Unit, Department of Cardiology, Hospital Universitario Puerta de Hierro, 28222 Madrid, Spain; European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart, 28222 Madrid, Spain; Centro de Investigación Biomédica en Red en Enfermedades Cardiovasculares (CIBERCV), 28029 Madrid, Spain; Universidad Francisco de Vitoria (UFV), 28223 Madrid, Spain.
David de Sancho, Polimero eta Material Aurreratuak: Fisika, Kimika eta Teknologia, Kimika Fakultatea, Euskal Herriko Unibertsitatea UPV/EHU, 20018 Donostia-San Sebastián, Spain; Donostia International Physics Center (DIPC), 20018 Donostia-San Sebastián, Spain.
James A. Spudich, Department of Biochemistry, Stanford University School of Medicine, Stanford, California 94305, United States; Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, California 94305, United States
Kathleen M. Ruppel, Department of Biochemistry, Stanford University School of Medicine, Stanford, California 94305, United States; Stanford Cardiovascular Institute, Stanford University School of Medicine, Stanford, California 94305, United States
Elías Herrero-Galán, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain.
Jorge Alegre-Cebollada, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain.
REFERENCES
- (1).Gautel M; Djinovic-Carugo K The Sarcomeric Cytoskeleton: From Molecules to Motion. J. Exp. Biol 2016, 219, 135–145. [DOI] [PubMed] [Google Scholar]
- (2).Braunwald E Hypertrophic Cardiomyopathy. In Hypertrophic Cardiomyopathy: Foreword by Bernard Gersh and Historical Context by Eugene Braunwald; Springer-Verlag: London, 2015; pp 1–8. [Google Scholar]
- (3).Semsarian C; Ingles J; Maron MS; Maron BJ New Perspectives on the Prevalence of Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol 2015, 65, 1249–1254. [DOI] [PubMed] [Google Scholar]
- (4).Teekakirikul P; Zhu W; Huang HC; Fung E Hypertrophic Cardiomyopathy: An Overview of Genetics and Management. Biomolecules 2019, 9, 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Roma-Rodrigues C; Fernandes AR Genetics of Hypertrophic Cardiomyopathy: Advances and Pitfalls in Molecular Diagnosis and Therapy. Appl. Clin. Genet 2014, 7, 195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Alcalai R; Seidman JG; Seidman CE Genetic Basis of Hypertrophic Cardiomyopathy: From Bench to the Clinics. J. Cardiovasc Electrophysiol 2007, 19, 104–110. [DOI] [PubMed] [Google Scholar]
- (7).Olivotto I; Oreziak A; Barriales-Villa R; Abraham TP; Masri A; Garcia-Pavia P; Saberi S; Lakdawala NK; Wheeler MT; Owens A; Kubanek M; Wojakowski W; Jensen MK; Gimeno-Blanes J; Afshar K; Myers J; Hegde SM; Solomon SD; Sehnert AJ; Zhang D; et al. Mavacamten for Treatment of Symptomatic Obstructive Hypertrophic Cardiomyopathy (Explorer-HCM): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2020, 396, 759–769. [DOI] [PubMed] [Google Scholar]
- (8).Akhtar M; Elliott P The Genetics of Hypertrophic Cardiomyopathy. Glob Cardiol Sci. Pract 2018, 2018, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Marston SB How Do Mutations in Contractile Proteins Cause the Primary Familial Cardiomyopathies? J. Cardiovasc Transl Res 2011, 4, 245–255. [DOI] [PubMed] [Google Scholar]
- (10).Squire JM Muscle Contraction: Sliding Filament History, Sarcomere Dynamics and the Two Huxleys. Glob Cardiol Sci. Pract 2016, 2016, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Krans JL The Sliding Filament Theory of Muscle Contraction. Nat. Educ 2010, 3, 66. [Google Scholar]
- (12).Spudich JA The Myosin Swinging Cross-Bridge Model. Nat. Rev. Mol. Cell Biol 2001, 2, 387–392. [DOI] [PubMed] [Google Scholar]
- (13).Lee K; Harris SP; Sadayappan S; Craig R Orientation of Myosin Binding Protein C in the Cardiac Muscle Sarcomere Determined by Domain-Specific Immuno-EM. J. Mol. Biol 2015, 427, 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pfuhl M; Gautel M Structure, Interactions and Function of the N-Terminus of Cardiac Myosin Binding Protein C (MyBP-C): Who Does What, with What, and to Whom? J. Muscle Res. Cell Motil 2012, 33, 83–94. [DOI] [PubMed] [Google Scholar]
- (15).Harris SP; Lyons RG; Bezold KL In the Thick of It: HCM-Causing Mutations in Myosin Binding Proteins of the Thick Filament. Circ. Res 2011, 108, 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Heling L; Geeves MA; Kad NM MyBP-C: One Protein to Govern Them All. J. Muscle Res. Cell Motil 2020, 41, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Flashman E; Redwood C; Moolman-Smook J; Watkins H Cardiac Myosin Binding Protein C: Its Role in Physiology and Disease. Circ. Res 2004, 94, 1279–1289. [DOI] [PubMed] [Google Scholar]
- (18).Carrier L; Mearini G; Stathopoulou K; Cuello F Cardiac Myosin-Binding Protein C (MYBPC3) in Cardiac Pathophysiology. Gene 2015, 573, 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Irving TC; Konhilas J; Perry D; Fischetti R; de Tombe PP Myofilament Lattice Spacing As a Function of Sarcomere Length in Isolated Rat Myocardium. Am. J. Physiol Heart Circ Physiol 2000, 279, H2568–H2573. [DOI] [PubMed] [Google Scholar]
- (20).Previs MJ; Michalek AJ; Warshaw DM Molecular Modulation of Actomyosin Function by Cardiac Myosin-Binding Protein C. Pfluegers Arch 2014, 466, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kampourakis T; Yan Z; Gautel M; Sun YB; Irving M Myosin Binding Protein-C Activates Thin Filaments and Inhibits Thick Filaments in Heart Muscle Cells. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 18763–18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Toepfer CN; Wakimoto H; Garfinkel AC; McDonough B; Liao D; Jiang J; Tai AC; Gorham JM; Lunde IG; Lun M; Lynch IV TL; McNamara JW; Sadayappan S; Redwood CS; Watkins HC; Seidman JG; Seidman CE Hypertrophic Cardiomyopathy Mutations in MYBPC3 Dysregulate Myosin. Sci. Transl. Med 2019, 11, No. eaat1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Weith A; Sadayappan S; Gulick J; Previs MJ; Vanburen P; Robbins J; Warshaw DM Unique Single Molecule Binding of Cardiac Myosin Binding Protein-C to Actin and Phosphorylation-Dependent Inhibition of Actomyosin Motility Requires 17 Amino Acids of the Motif Domain. J. Mol. Cell. Cardiol 2012, 52, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Woody MS; Winkelmann DA; Capitanio M; Ostap EM; Goldman YE Single Molecule Mechanics Resolves the Earliest Events in Force Generation by Cardiac Myosin. eLife 2019, 8, No. e49266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Previs MJ; Beck Previs S; Gulick J; Robbins J; Warshaw DM Molecular Mechanics of Cardiac Myosin-Binding Protein C in Native Thick Filaments. Science 2012, 337, 1215–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).O’Leary TS; Snyder J; Sadayappan S; Day SM; Previs MJ MYBPC3 Truncation Mutations Enhance Actomyosin Contractile Mechanics in Human Hypertrophic Cardiomyopathy. J. Mol. Cell. Cardiol 2019, 127, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Karsai A; Kellermayer MS; Harris SP Mechanical Unfolding of Cardiac Myosin Binding Protein-C by Atomic Force Microscopy. Biophys. J 2011, 101, 1968–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Karsai A; Kellermayer MS; Harris SP Cross-Species Mechanical Fingerprinting of Cardiac Myosin Binding Protein-C. Biophys. J 2013, 104, 2465–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Michalek AJ; Howarth JW; Gulick J; Previs MJ; Robbins J; Rosevear PR; Warshaw DM Phosphorylation Modulates the Mechanical Stability of the Cardiac Myosin-Binding Protein C Motif. Biophys. J 2013, 104, 442–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang XL; De S; McIntosh LP; Paetzel M Structural Characterization of the C3 Domain of Cardiac Myosin Binding Protein C and Its Hypertrophic Cardiomyopathy-Related R502W Mutant. Biochemistry 2014, 53, 5332–5342. [DOI] [PubMed] [Google Scholar]
- (31).Marston S; Copeland O; Jacques A; Livesey K; Tsang V; McKenna WJ; Jalilzadeh S; Carballo S; Redwood C; Watkins H Evidence from Human Myectomy Samples That MYBPC3 Mutations Cause Hypertrophic Cardiomyopathy through Haploinsufficiency. Circ. Res 2009, 105, 219–222. [DOI] [PubMed] [Google Scholar]
- (32).van Dijk SJ; Dooijes D; dos Remedios C; Michels M; Lamers JM; Winegrad S; Schlossarek S; Carrier L; ten Cate FJ; Stienen GJ; van der Velden J Cardiac Myosin-Binding Protein C Mutations and Hypertrophic Cardiomyopathy: Haploinsufficiency, Deranged Phosphorylation, and Cardiomyocyte Dysfunction. Circulation 2009, 119, 1473–1483. [DOI] [PubMed] [Google Scholar]
- (33).Helms AS; Davis FM; Coleman D; Bartolone SN; Glazier AA; Pagani F; Yob JM; Sadayappan S; Pedersen E; Lyons R; Westfall MV; Jones R; Russell MW; Day SM Sarcomere Mutation-Specific Expression Patterns in Human Hypertrophic Cardiomyopathy. Circ.: Cardiovasc. Genet 2014, 7, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Barefield D; Kumar M; Gorham J; Seidman JG; Seidman CE; de Tombe PP; Sadayappan S Haploinsufficiency of MYBPC3 Exacerbates the Development of Hypertrophic Cardiomyopathy in Heterozygous Mice. J. Mol. Cell. Cardiol 2015, 79, 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Parbhudayal RY; Garra AR; Gotte MJW; Michels M; Pei J; Harakalova M; Asselbergs FW; van Rossum AC; van der Velden J; Kuster DWD Variable Cardiac Myosin Binding Protein-C Expression in the Myofilaments Due to MYBPC3 Mutations in Hypertrophic Cardiomyopathy. J. Mol. Cell. Cardiol 2018, 123, 59–63. [DOI] [PubMed] [Google Scholar]
- (36).Schuldt M; Pei J; Harakalova M; Dorsch LM; Schlossarek S; Mokry M; Knol JC; Pham TV; Schelfhorst T; Piersma SR; dos Remedios C; Dalinghaus M; Michels M; Asselbergs FW; Moutin M-J; Carrier L; Jiménez CR; van der Velden J; Kuster DWD Proteomic and Functional Studies Reveal Detyrosinated Tubulin As Treatment Target in Sarcomere Mutation-Induced Hypertrophic Cardiomyopathy. Circ.: Heart Failure 2021, 14, No. e007022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Helms AS; Thompson AD; Glazier AA; Hafeez N; Kabani S; Rodríguez J; Yob JM; Woolcock H; Mazzarotto F; Lakdawala NK; Wittekind SG; Pereira AC; Jacoby DL; Colan SD; Ashley EA; Saberi S; Ware JS; Ingles J; Semsarian C; Michels M; et al. Spatial and Functional Distribution of MYBPC3 Pathogenic Variants and Clinical Outcomes in Patients with Hypertrophic Cardiomyopathy. Circ: Genomic Precis Med 2020, 13, 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Walsh R; Thomson KL; Ware JS; Funke BH; Woodley J; McGuire KJ; Mazzarotto F; Blair E; Seller A; Taylor JC; Minikel EV; Exome Aggregation Consortium; MacArthur DG; Farrall M; Cook SA; Watkins H Reassessment of Mendelian Gene Pathogenicity Using 7,855 Cardiomyopathy Cases and 60,706 Reference Samples. Genet. Med 2017, 19, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ababou A; Gautel M; Pfuhl M Dissecting the N-Terminal Myosin Binding Site of Human Cardiac Myosin-Binding Protein C. Structure and Myosin Binding of Domain C2. J. Biol. Chem 2007, 282, 9204–9215. [DOI] [PubMed] [Google Scholar]
- (40).Ababou A; Rostkova E; Mistry S; Le Masurier C; Gautel M; Pfuhl M Myosin Binding Protein C Positioned to Play a Key Role in Regulation of Muscle Contraction: Structure and Interactions of Domain C1. J. Mol. Biol 2008, 384, 615–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).de Lange WJ; Grimes AC; Hegge LF; Spring AM; Brost TM; Ralphe JC E258K HCM-Causing Mutation in Cardiac MyBP-C Reduces Contractile Force and Accelerates Twitch Kinetics by Disrupting the cMyBP-C and Myosin S2 Interaction. J. Gen. Physiol 2013, 142, 241–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Pricolo MR; Herrero-Galan E; Mazzaccara C; Losi MA; Alegre-Cebollada J; Frisso G Protein Thermodynamic Destabilization in the Assessment of Pathogenicity of a Variant of Uncertain Significance in Cardiac Myosin Binding Protein C. J. Cardiovasc Transl Res 2020, 13, 867–877. [DOI] [PubMed] [Google Scholar]
- (43).Smelter DF; de Lange WJ; Cai W; Ge Y; Ralphe JC The HCM-Linked W792R Mutation in Cardiac Myosin-Binding Protein C Reduces C6 FnIII Domain Stability. Am. J. Physiol Heart Circ Physiol 2018, 314, H1179–H1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Suay-Corredera C; Pricolo MR; Herrero-Galan E; Velazquez-Carreras D; Sanchez-Ortiz D; Garcia-Giustiniani D; Delgado J; Galano-Frutos JJ; Garcia-Cebollada H; Vilches S; Dominguez F; Sabater Molina M; Barriales-Villa R; Frisso G; Sancho J; Serrano L; Garcia-Pavia P; Monserrat L; Alegre-Cebollada J Protein Haploinsufficiency Drivers Identify MYBPC3 Mutations That Cause Hypertrophic Cardiomyopathy 2020, 2020.05.04.20087726. medRxiv. 10.1101/2020.05.04.20087726v1 (accessed May 16, 2021). [DOI] [PMC free article] [PubMed]
- (45).Popa I; Kosuri P; Alegre-Cebollada J; Garcia-Manyes S; Fernandez JM Force Dependency of Biochemical Reactions Measured by Single-Molecule Force-Clamp Spectroscopy. Nat. Protoc 2013, 8, 1261–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Carrier L; Bonne G; Bahrend E; Yu B; Richard P; Niel F; Hainque B; Cruaud C; Gary F; Labeit S; Bouhour JB; Dubourg O; Desnos M; Hagege AA; Trent RJ; Komajda M; Fiszman M; Schwartz K Organization and Sequence of Human Cardiac Myosin Binding Protein C Gene (MYBPC3) and Identification of Mutations Predicted to Produce Truncated Proteins in Familial Hypertrophic Cardiomyopathy. Circ. Res 1997, 80, 427–434. [PubMed] [Google Scholar]
- (47).Sadayappan S; de Tombe PP Cardiac Myosin Binding Protein-C: Redefining Its Structure and Function. Biophys. Rev 2012, 4, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Ackermann MA; Kontrogianni-Konstantopoulos A Myosin Binding Protein-C: A Regulator of Actomyosin Interaction in Striated Muscle. J. Biomed. Biotechnol 2011, 2011, 636403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pimenta-Lopes C; Suay-Corredera C; Velázquez-Carreras D; Sánchez-Ortiz D; Alegre-Cebollada J Concurrent Atomic Force Spectroscopy. Commun. Phys 2019, 2, 91. [Google Scholar]
- (50).Marston S; Copeland O; Gehmlich K; Schlossarek S; Carrier L How Do MYBPC3 Mutations Cause Hypertrophic Cardiomyopathy? J. Muscle Res. Cell Motil 2012, 33, 75–80. [DOI] [PubMed] [Google Scholar]
- (51).Ito K; Patel PN; Gorham JM; McDonough B; DePalma SR; Adler EE; Lam L; MacRae CA; Mohiuddin SM; Fatkin D; Seidman CE; Seidman JG Identification of Pathogenic Gene Mutations in LMNA and MYBPC3 That Alter RNA Splicing. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 7689–7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Greenfield NJ Using Circular Dichroism Spectra to Estimate Protein Secondary Structure. Nat. Protoc 2006, 1, 2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kelly SM; Price NC The Use of Circular Dichroism in the Investigation of Protein Structure and Function. Curr. Protein Pept. Sci 2000, 1, 349–384. [DOI] [PubMed] [Google Scholar]
- (54).Walters J; Milam SL; Clark AC Practical Approaches to Protein Folding and Assembly: Spectroscopic Strategies in Thermodynamics and Kinetics. Methods Enzymol 2009, 455, 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Watkins H; Ashrafian H; Redwood C Inherited Cardiomyopathies. N. Engl. J. Med 2011, 364, 1643–1656. [DOI] [PubMed] [Google Scholar]
- (56).Schlierf M; Li H; Fernandez JM The Unfolding Kinetics of Ubiquitin Captured with Single-Molecule Force-Clamp Techniques. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 7299–7304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bell GI Models for the Specific Adhesion of Cells to Cells. Science 1978, 200, 618–627. [DOI] [PubMed] [Google Scholar]
- (58).Kosuri P; Alegre-Cebollada J; Feng J; Kaplan A; Ingles-Prieto A; Badilla CL; Stockwell BR; Sanchez-Ruiz JM; Holmgren A; Fernandez JM Protein Folding Drives Disulfide Formation. Cell 2012, 151, 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zheng P; Li H Highly Covalent Ferric–Thiolate Bonds Exhibit Surprisingly Low Mechanical Stability. J. Am. Chem. Soc 2011, 133, 6791–6798. [DOI] [PubMed] [Google Scholar]
- (60).Lei H; Guo Y; Hu X; Hu C; Hu X; Li H Reversible Unfolding and Folding of the Metalloprotein Ferredoxin Revealed by Single-Molecule Atomic Force Microscopy. J. Am. Chem. Soc 2017, 139, 1538–1544. [DOI] [PubMed] [Google Scholar]
- (61).Hummer G; Szabo A Kinetics from Nonequilibrium Single-Molecule Pulling Experiments. Biophys. J 2003, 85, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Marszalek PE; Lu H; Li H; Carrion-Vazquez M; Oberhauser AF; Schulten K; Fernandez JM Mechanical Unfolding Intermediates in Titin Modules. Nature 1999, 402, 100–103. [DOI] [PubMed] [Google Scholar]
- (63).Best RB; Fowler SB; Herrera JL; Steward A; Paci E; Clarke J Mechanical Unfolding of a Titin Ig Domain: Structure of Transition State Revealed by Combining Atomic Force Microscopy, Protein Engineering and Molecular Dynamics Simulations. J. Mol. Biol 2003, 330, 867–877. [DOI] [PubMed] [Google Scholar]
- (64).Best RB Atomistic Molecular Simulations of Protein Folding. Curr. Opin. Struct. Biol 2012, 22, 52–61. [DOI] [PubMed] [Google Scholar]
- (65).Alegre-Cebollada J; Kosuri P; Giganti D; Eckels E; Rivas-Pardo JA; Hamdani N; Warren CM; Solaro RJ; Linke WA; Fernandez JM S-Glutathionylation of Cryptic Cysteines Enhances Titin Elasticity by Blocking Protein Folding. Cell 2014, 156, 1235–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Davis J; Davis LC; Correll RN; Makarewich CA; Schwanekamp JA; Moussavi-Harami F; Wang D; York AJ; Wu H; Houser SR; Seidman CE; Seidman JG; Regnier M; Metzger JM; Wu JC; Molkentin JD A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016, 165, 1147–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Moss RL; Fitzsimons DP; Ralphe JC Cardiac MyBP-C Regulates the Rate and Force of Contraction in Mammalian Myocardium. Circ. Res 2015, 116, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Nag S; Trivedi DV; Sarkar SS; Adhikari AS; Sunitha MS; Sutton S; Ruppel KM; Spudich JA The Myosin Mesa and the Basis of Hypercontractility Caused by Hypertrophic Cardiomyopathy Mutations. Nat. Struct. Mol. Biol 2017, 24, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Green EM; Wakimoto H; Anderson RL; Evanchik MJ; Gorham JM; Harrison BC; Henze M; Kawas R; Oslob JD; Rodriguez HM; Song Y; Wan W; Leinwand LA; Spudich JA; McDowell RS; Seidman JG; Seidman CE A Small-Molecule Inhibitor of Sarcomere Contractility Suppresses Hypertrophic Cardiomyopathy in Mice. Science 2016, 351, 617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Adhikari AS; Kooiker KB; Sarkar SS; Liu C; Bernstein D; Spudich JA; Ruppel KM Early-Onset Hypertrophic Cardiomyopathy Mutations Significantly Increase the Velocity, Force, and Actin-Activated ATPase Activity of Human Beta-Cardiac Myosin. Cell Rep 2016, 17, 2857–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Adhikari AS; Trivedi DV; Sarkar SS; Song D; Kooiker KB; Bernstein D; Spudich JA; Ruppel KM β-Cardiac Myosin Hypertrophic Cardiomyopathy Mutations Release Sequestered Heads and Increase Enzymatic Activity. Nat. Commun 2019, 10, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).McNamara JW; Li A; Lal S; Bos JM; Harris SP; van der Velden J; Ackerman MJ; Cooke R; dos Remedios CG MYBPC3 Mutations Are Associated with a Reduced Super-Relaxed State in Patients with Hypertrophic Cardiomyopathy. PLoS One 2017, 12, No. e0180064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).McNamara JW; Singh RR; Sadayappan S Cardiac Myosin Binding Protein-C Phosphorylation Regulates the Super-Relaxed State of Myosin. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 11731–11736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Wijnker PJM; Friedrich FW; Dutsch A; Reischmann S; Eder A; Mannhardt I; Mearini G; Eschenhagen T; van der Velden J; Carrier L Comparison of the Effects of a Truncating and a Missense MYBPC3 Mutation on Contractile Parameters of Engineered Heart Tissue. J. Mol. Cell. Cardiol 2016, 97, 82–92. [DOI] [PubMed] [Google Scholar]
- (75).Luther PK; Winkler H; Taylor K; Zoghbi ME; Craig R; Padrón R; Squire JM; Liu J Direct Visualization of Myosin-Binding Protein C Bridging Myosin and Actin Filaments in Intact Muscle. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 11423–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Walcott S; Docken S; Harris SP Effects of Cardiac Myosin Binding Protein-C on Actin Motility Are Explained with a Drag-Activation-Competition Model. Biophys. J 2015, 108, 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Smith SB; Finzi L; Bustamante C Direct Mechanical Measurements of the Elasticity of Single DNA Molecules by Using Magnetic Beads. Science 1992, 258, 1122–1126. [DOI] [PubMed] [Google Scholar]
- (78).Li H; Linke WA; Oberhauser AF; Carrion-Vazquez M; Kerkvliet JG; Lu H; Marszalek PE; Fernandez JM Reverse Engineering of the Giant Muscle Protein Titin. Nature 2002, 418, 998–1002. [DOI] [PubMed] [Google Scholar]
- (79).Rivas-Pardo JA; Eckels EC; Popa I; Kosuri P; Linke WA; Fernandez JM Work Done by Titin Protein Folding Assists Muscle Contraction. Cell Rep 2016, 14, 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Mun JY; Kensler RW; Harris SP; Craig R The cMyBP-C HCM Variant L348P Enhances Thin Filament Activation through an Increased Shift in Tropomyosin Position. J. Mol. Cell. Cardiol 2016, 91, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Gruen M; Gautel M Mutations in β-Myosin S2 That Cause Familial Hypertrophic Cardiomyopathy (FHC) Abolish the Interaction with the Regulatory Domain of Myosin-Binding Protein-C. J. Mol. Biol 1999, 286, 933–949. [DOI] [PubMed] [Google Scholar]
- (82).Sarkar SS; Trivedi DV; Morck MM; Adhikari AS; Pasha SN; Ruppel KM; Spudich JA The Hypertrophic Cardiomyopathy Mutations R403Q and R663H Increase the Number of Myosin Heads Available to Interact with Actin. Sci. Adv 2020, 6, No. eaax0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).del Rio A; Perez-Jimenez R; Liu R; Roca-Cusachs P; Fernandez JM; Sheetz MP Stretching Single Talin Rod Molecules Activates Vinculin Binding. Science 2009, 323, 638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).van Dijk SJ; Bezold Kooiker K; Mazzalupo S; Yang Y; Kostyukova AS; Mustacich DJ; Hoye ER; Stern JA; Kittleson MD; Harris SP The A31P Missense Mutation in Cardiac Myosin Binding Protein C Alters Protein Structure but Does Not Cause Haploinsufficiency. Arch. Biochem. Biophys 2016, 601, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Schlossarek S; Carrier L The Ubiquitin-Proteasome System in Cardiomyopathies. Curr. Opin Cardiol 2011, 26, 190–195. [DOI] [PubMed] [Google Scholar]
- (86).Glazier AA; Hafeez N; Mellacheruvu D; Basrur V; Nesvizhskii AI; Lee LM; Shao H; Tang V; Yob JM; Gestwicki JE; Helms AS; Day SM HSC70 Is a Chaperone for Wild-Type and Mutant Cardiac Myosin Binding Protein C. JCI Insight 2018, 3, No. e99319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Wang L; Lai G; Chu G; Liang X; Zhao Y cMyBP-C Was Decreased via KLHL3-Mediated Proteasomal Degradation in Congenital Heart Diseases. Exp. Cell Res 2017, 355, 18–25. [DOI] [PubMed] [Google Scholar]
- (88).Chen B; Retzlaff M; Roos T; Frydman J Cellular Strategies of Protein Quality Control. Cold Spring Harbor Perspect. Biol 2011, 3, a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Anderson BR; Bogomolovas J; Labeit S; Granzier H Single Molecule Force Spectroscopy on Titin Implicates Immunoglobulin Domain Stability As a Cardiac Disease Mechanism. J. Biol. Chem 2013, 288, 5303–5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Haining AWM; von Essen M; Attwood SJ; Hytönen VP; del Río Hernández A All Subdomains of the Talin Rod Are Mechanically Vulnerable and May Contribute to Cellular Mechanosensing. ACS Nano 2016, 10, 6648–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Nakamura F; Stossel TP; Hartwig JH The Filamins. Cell Adhes Migr 2011, 5, 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Schreiber KH; Kennedy BK When Lamins Go Bad: Nuclear Structure and Disease. Cell 2013, 152, 1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Leckband DE; de Rooij J Cadherin Adhesion and Mechanotransduction. Annu. Rev. Cell Dev. Biol 2014, 30, 291–315. [DOI] [PubMed] [Google Scholar]
- (94).Popa I; Rivas-Pardo JA; Eckels EC; Echelman DJ; Badilla CL; Valle-Orero J; Fernandez JM A HaloTag Anchored Ruler for Week-Long Studies of Protein Dynamics. J. Am. Chem. Soc 2016, 138, 10546–10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Carrion-Vazquez M; Oberhauser AF; Fowler SB; Marszalek PE; Broedel SE; Clarke J; Fernandez JM Mechanical and Chemical Unfolding of a Single Protein: A Comparison. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 3694–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Alegre-Cebollada J; Badilla CL; Fernandez JM Isopeptide Bonds Block the Mechanical Extension of Pili in Pathogenic Streptococcus Pyogenes. J. Biol. Chem 2010, 285, 11235–11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Sommese RF; Sung J; Nag S; Sutton S; Deacon JC; Choe E; Leinwand LA; Ruppel K; Spudich JA Molecular Consequences of the R453C Hypertrophic Cardiomyopathy Mutation on Human Beta-Cardiac Myosin Motor Function. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 12607–12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Gasteiger E; Hoogland C; Gattiker A; Duvaud S; Wilkins MR; Appel RD; Bairoch A Protein Identification and Analysis Tools on the Expasy Server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, 2005; pp 571–607. [Google Scholar]
- (99).Santoro MM; Bolen DW Unfolding Free Energy Changes Determined by the Linear Extrapolation Method. 1. Unfolding of Phenylmethanesulfonyl.Alpha.-Chymotrypsin Using Different Denaturants. Biochemistry 1988, 27, 8063–8068. [DOI] [PubMed] [Google Scholar]
- (100).Hutter J; Bechhoefer J Calibration of Atomic-Force Microscope Tips. Rev. Sci. Instrum 1993, 64, 1868–1873. [Google Scholar]
- (101).Nunes JM; Mayer-Hartl M; Hartl FU; Muller DJ Action of the Hsp70 Chaperone System Observed with Single Proteins. Nat. Commun 2015, 6, 6307. [DOI] [PubMed] [Google Scholar]
- (102).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera-A Visualization System for Exploratory Research and Analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (103).Darden T; York D; Pedersen L Particle Mesh Ewald: An N·Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (104).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (106).MacKerell AD; Bashford D; Bellott M; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (107).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- (108).McGibbon RT; Beauchamp KA; Harrigan MP; Klein C; Swails JM; Hernández CX; Schwantes CR; Wang L-P; Lane TJ; Pande VS MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 2015, 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Payton ME; Greenstone MH; Schenker N Overlapping Confidence Intervals or Standard Error Intervals: What Do They Mean in Terms of Statistical Significance? J. Insect Sci 2003, 3, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Goldstein H; Michael JRH The Graphical Presentation of a Collection of Means. J. R Stat Soc. Ser. A Stat Soc 1995, 158, 175–177. [Google Scholar]
- (111).Suay-Corredera C; Pricolo MR; Velázquez-Carreras D; Pimenta-Lopes C; Sánchez-Ortiz D; Urrutia-Irazabal I; Vilches S; Domínguez F; Frisso G; Monserrat L; García-Pavía P; Herrero-Galán E; Alegre-Cebollada J Nanomechanical Phenotypes in cMyBP-C Mutants That Cause Hypertrophic Cardiomyopathy 2020, 2020.09.19.304618. bioRxiv. 10.1101/2020.09.19.304618v1 (accessed May 20, 2021). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.