

Abstract
Invasive fungal infections have escalated from a rare curiosity to a major cause of human mortality around the globe. This is in part due to a scarcity in the number of antifungal drugs available to combat mycotic disease, making the discovery of novel bioactive compounds and determining their mode of action of utmost importance. The development and application of chemical genomic assays using the model yeast Saccharomyces cerevisiae has provided powerful methods to identify the mechanism of action of diverse molecules in a living cell. Furthermore, complementary assays are continually being developed in fungal pathogens, most notably Candida albicans and Cryptococcus neoformans, to elucidate compound mechanism of action directly in the pathogen of interest. Collectively, the suite of chemical genetic assays that have been developed in multiple fungal species enables the identification of candidate drug target genes, as well as genes involved in buffering drug target pathways, and genes involved in general cellular responses to small molecules. In this review, we examine current yeast chemical genomic assays and highlight how such resources provide powerful tools that can be utilized to bolster the antifungal pipeline.
Keywords: chemical genomics, fungal pathogens, Candida, Cryptococcus, antifungal, Saccharomyces cerevisiae
Introduction
Fungal diseases are ubiquitous in nature, threatening biodiversity and world food supplies, as well as accounting for billions of human infections each year in both developing and developed nations.1 Over the past several decades, advances in modern medicine that rely heavily on the use of immunosuppressive drugs, including chemotherapy and transplantation surgery, as well as infections with HIV, have dramatically increased the number of individuals with compromised immunity. While most individuals suffering from mycotic infection experience relatively benign superficial symptoms, immunocompromised individuals are highly susceptible to invasive infections that ravage crucial tissues and organs.1,2 These infections have notoriously poor clinical outcomes, where mortality rates often exceed 50% even with therapeutic intervention.1 Furthermore, invasive fungal infections incur the majority of the $7.2 billion in fungal-associated direct medical costs in the United States alone, despite comprising fewer than 1% of relevant hospital visits.3 These considerable health and economic burdens are likely gross underestimates due to inadequate diagnostics and an absence of mycological surveillance.1 In conjunction with lax antifungal stewardship that applies unnecessary selective pressure on fungal populations, it is not surprising that we are bearing witness to ever-increasing levels of drug resistance across established and emerging fungal pathogens.4–7
Over 90% of fungal-related mortality is attributable to opportunistic Candida, Cryptococcus, and Aspergillus species.1 Candida albicans is a commensal member of the human microbiota, but is also a primary causative agent of life-threatening bloodstream infections.8,9 In addition, the unprecedented, global rise of non-albicans Candida species with problematic levels of intrinsic and acquired drug resistance is deeply concerning. Since its sudden global emergence in 2009, Candida auris has been implicated in numerous nosocomial outbreaks, with 93% of C. auris clinical isolates exhibiting resistance to the most widely deployed antifungal class, the azoles, and 4% of strains being recalcitrant to all available antifungal classes.6,10 Similarly, high levels of antifungal resistance have been documented for Candida glabrata, which currently represents the second most commonly isolated Candida species in the United States and Europe.11,12 Cryptococcosis often manifests as devastating central nervous system infections in immunocompromised patients and has associated mortality rates approaching 70% in endemic regions.13 Cryptococcus neoformans and Cryptococcus gattii are the predominant culprits, with C. gatti also capable of causing disease in immuno-competent hosts.14,15 Lastly, Aspergillus fumigatus has a universal environmental presence and is responsible for over 200,000 reported invasive infections annually with burgeoning azole resistance reported.1,16
Currently, there are only three major classes of clinically used antifungal drugs to treat systemic infection, which interfere with just two central fungal processes (Fig. 1).11,17 Azoles are the most widely deployed antifungals owing to their broad-spectrum activity, favorable safety profile, and oral bioavailability.18,19 They target lanosterol 14-α-demethylase, diverting ergosterol biosynthesis toward the production of a toxic sterol intermediate that exerts a severe cell membrane stress, blocking further cell growth and division (Fig. 1B).18,19 Unfortunately, their fungistatic action and widespread, prophylactic use in both medicine and agriculture poses strong directional selection pressure for the evolution of resistance.4,7,16 Polyenes, such as amphotericin B, directly bind and remove ergosterol from the cell membrane acting as a sterol sponge (Fig. 1B).20 Although acquired resistance in clinically relevant contexts remains rare,4,7,21 polyenes are instead associated with undesirable host nephrotoxicity. Thus, amphotericin B is usually deployed as a last resort therapy against cryptococcal meningitis22 and multidrug-resistant Candida infections.23 Finally, the echinocandins are the only novel drug class approved for clinical application in the past two decades.11 They noncompetitively inhibit 1,3-β-d-glucan synthase, depriving the fungal cell wall of an integral polymeric component, leading to the induction of cell wall stress and eventually cellular death (Fig. 1B).4,7
Figure 1.
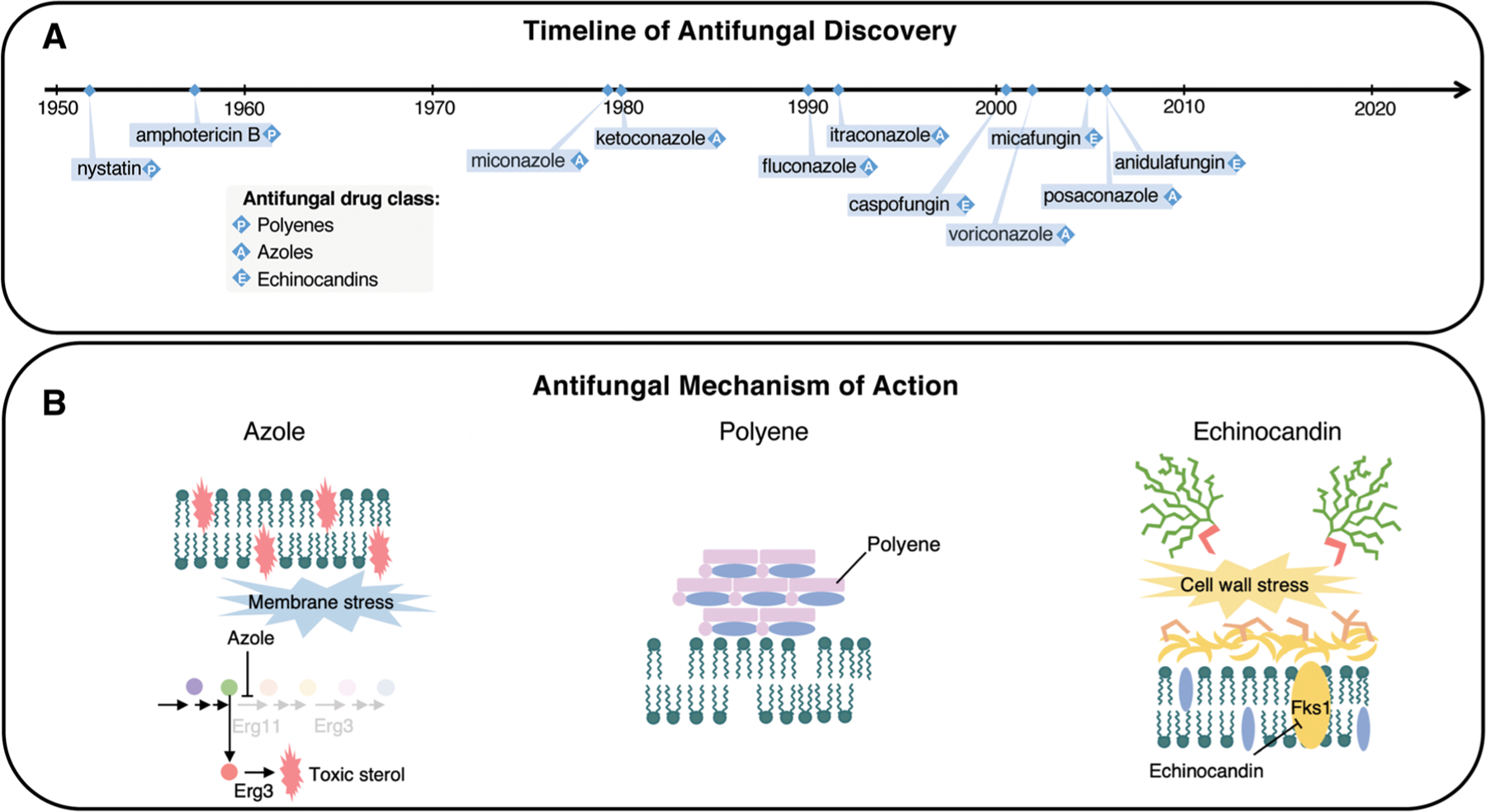
Timeline of antifungal discovery, and antifungal drug mechanisms of action. (A) The clinical introduction of antifungal agents belonging to the three major clinically used classes: polyenes, azoles, and echinocandins. (B) The mechanisms of action of antifungal drugs. Azoles exert fungistatic activity by inhibiting lanosterol 14-α-demethylase (encoded by ERG11), which leads to a block in ergosterol synthesis and the accumulation of toxic sterol intermediates, produced by Erg3 (left panel). Polyenes act as a fungicidal “sterol sponge” by forming extra-membranous aggregates that extract ergosterol from lipid bilayers (middle panel). Fungal cell walls are composed of (1,3)-β-d-glucan covalently linked to (1,6)-β-d-glucan, as well as chitin and mannan. Echinocandins prevent the synthesis of (1,3)-β-d-glucan by inhibiting the (1,3)-β-d-glucan synthase (encoded by FKS1 in C. albicans and by both FKS1 and FKS2 in S. cerevisiae); this results in a loss of cell wall integrity (right panel). Adapted, with permission, from Ref 134.
The stagnant discovery of novel antifungal compounds over the past several decades can be attributed to a variety of factors. First and foremost, the number of fungal-specific cellular pathways that can be therapeutically targeted is restricted due to the eukaryotic similarities between fungi and humans, compared with more evolutionarily distant microbes, such as bacteria.18,19 In addition, an estimated 80% of molecules with published fungicidal activity are not pursued due to inherent unsuitable properties, namely promiscuous functionality, as well as a lack of whole-cell bioactivity due to difficulties with permeating across the fungal cell wall and membrane.18,24 Poor target specificity and lack of whole-cell bioactivity also contribute to high attrition rates during pre-clinical development. Finally, the scientific complexities of antifungal development are exacerbated by a chronic dearth of research initiatives and resource investment. Yet, despite these challenges, scientific advancements have offered great hope for the discovery and development of novel antifungal agents. Phenotype-based identification of bioactive molecules, which prioritizes potent activity within the context of intact fungal cells, can now be coupled with a vast array of functional genomics resources to enable subsequent target deconvolution.25–28 In this review, we explore current chemical-genomic approaches developed for Saccharomyces cerevisiae, as well as those resources available in clinically important fungal pathogens, emphasizing their potential to revitalize antifungal drug discovery.
The power of yeast genetics
Since its genome sequence was assembled almost 25 years ago,29 S. cerevisiae has prevailed as the de facto eukaryotic model system, guiding our understanding of the eukaryotic cell and serving as an invaluable resource of genomic tools and reagents. Systematic genetic and phenotypic analyses in this species were enabled by a consortium of scientific laboratories that developed a comprehensive collection of deletion mutants, where each of the ~6000 yeast open reading frames was replaced with a kanMX dominant, drug-resistance marker.30 This strategy also encompassed the inclusion of strain-specific molecular sequences: two unique 20 base-pair oligonucleotides that are flanked by universal priming sequences. These strain-specific barcodes surround each kanMX drug-resistance marker and serve as a unique molecular identifier for each strain in the S. cerevisiae genome.31 This systematic endeavor defined the set of ~1000 S. cerevisiae genes essential for viability in laboratory growth conditions and generated a set of ~5000 viable haploid deletion mutants.30,32 Notably, the essential gene set is not only contingent on standard growth conditions but also strain background, which has implications for drug development and how researchers might select potential drug targets. Importantly, this strategy motivated several other laboratories to develop additional genome-wide tools to enable proteomic studies, such as the yeast comprehensive temperature-sensitive mutant collection,33,34 the yeast titratable-promoter collection of essential genes,35,36 the tandem affinity purification (TAP-tagged) collection,37 the GFP (green fluorescent protein) collection,38 and genome-scale two-hybrid resources.39 More recently, complementary strain collections were constructed in which subsets of the ~1000 essential yeast genes were individually altered to produce conditional or hypomorphic alleles with the potential to be assayed at a semipermissive state, enabling systematic analysis of essential genes.40 Collectively, these mutant collections have enabled the development and application of high-throughput methodologies that have revealed incredible complexities inherent of eukaryotic biological systems.
The functional redundancy and extensive buffering within the eukaryotic cell became evident once it was revealed that only ~17% of genes in the S. cerevisiae genome are essential under standard laboratory conditions.30 For decades prior, geneticists had investigated relationships between gene pairs, mapping out genetic interactions on a case-by-case basis. A genetic interaction between two genes is observed when a phenotype caused by a mutation in one gene depends on a mutation in another gene such that the combined effect deviates from expectation based on the individual effects, with synthetic lethality representing the extreme case of a negative genetic interaction (Fig. 2A and B).41–43 For example, one of the first examples of synthetic lethality was described with loss-of-function mutant alleles of both TUB1 and TUB3, both of which encode α-tubulin.44 With the completion of the yeast deletion collection, researchers began systematically mapping all genetic interactions in S. cerevisiae. To do so, synthetic genetic array (SGA) was developed as an automated method that combined arrays of gene deletion mutants with robotic manipulation for high-throughput construction of combinations of mutant alleles and identification of genetic interactions.45 In its first two applications, SGA methodology crossed at first eight followed by ~130 gene-specific query mutant strains to the ~5000 haploid deletion mutants resulting in complex networks of thousands of synthetic lethal or synthetic sick interactions.45,46 Recently, the combination of SGA with a genome-scale colony size-scoring methodology enabled the assessment of growth defects associated with approximately 18 million yeast gene deletion pairs. These large-scale studies measured the fitness of single and double mutants to identify nearly 550,000 negative genetic interactions and 350,000 positive genetic interactions, enabling the assembly of the first complete genetic interaction network for any organism.40,47
Figure 2.
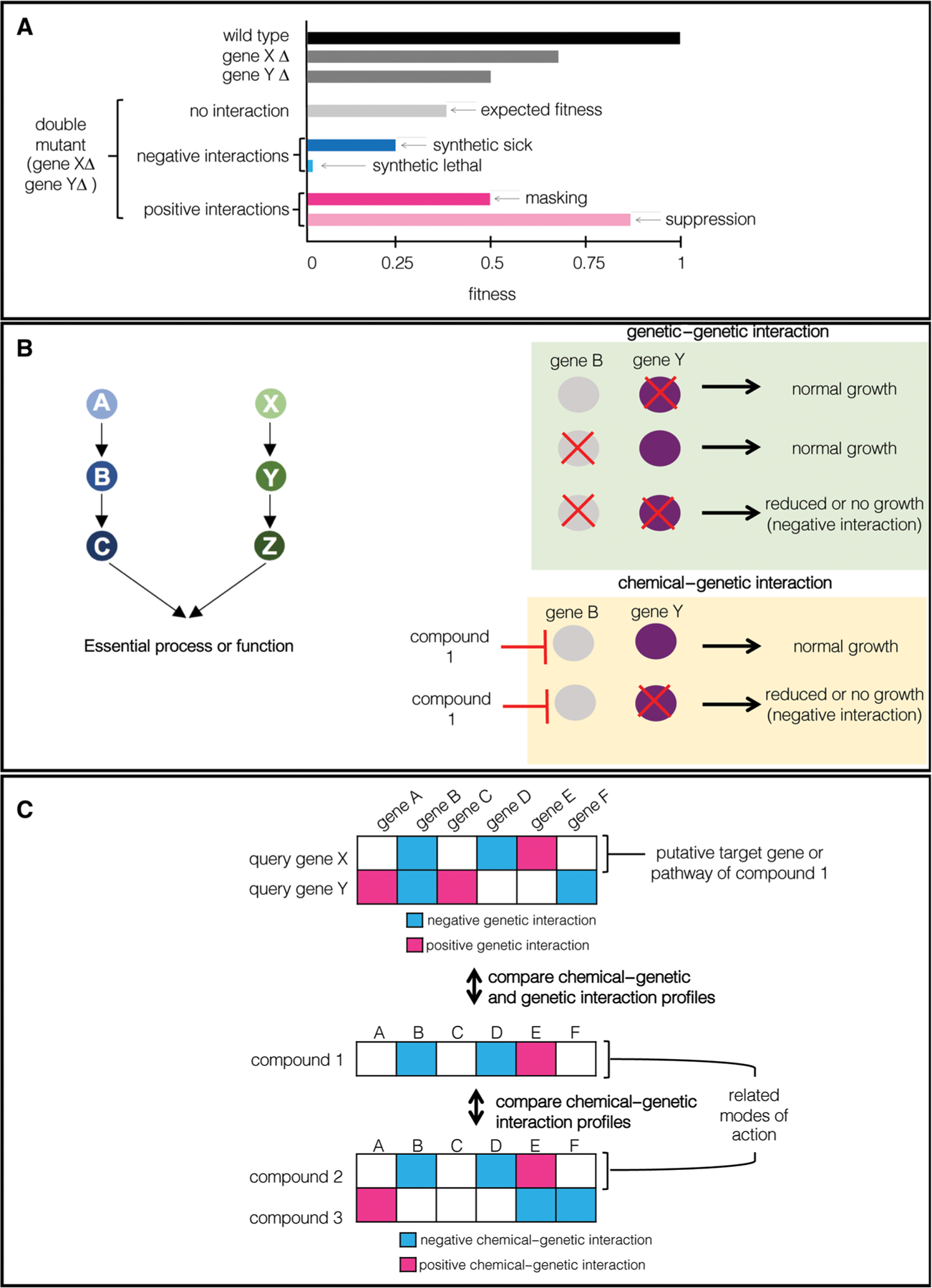
Application of genetic–genetic and chemical–genetic interactions for compound to target annotation. (A) A genetic interaction between two genes is observed when a phenotype caused by a mutation in one gene is exacerbated by a mutation in another gene. A negative genetic interaction occurs if the observed fitness of the double mutant is less than the double mutant fitness expected from a multiplicative model. A positive genetic interaction occurs if the observed fitness of the double mutant is greater than the double mutant fitness expected from a multiplicative model. (B) Mutations in genes residing in parallel pathways typically culminate in a negative genetic–genetic interaction (green box). Genetic inhibition of one pathway combined with pharmacological inhibition of a parallel pathway typically results in a negative chemical–genetic interaction (yellow box). (C) Comparing genetic–genetic and chemical–genetic interaction profiles can facilitate functional interpretation of chemical-genetic screening data for a given compound.
The mapping of genetic interactions in S. cerevisiae highlighted several critical observations with important implications for the development of novel antifungal therapies.45,47 While only ~1000 genes in S. cerevisiae are essential for viability,30 the identification of 550,000 negative genetic interactions between gene deletion pairs40,47 suggests the potential for combatting fungal infections may come from targeting multiple cellular nodes that together result in a lethal or sick phenotype (Fig. 2B).18,48 For example, in S. cerevisiae, FKS1 and its paralog FKS2 encode the biosynthetic enzyme for (1,3)-β-d-glucan synthesis and the molecular target of the echinocandins. While FKS1 and FKS2 are synthetic lethal, in keeping with echinocandin efficacy, FKS1 is also synthetic lethal with CHS3, a chitin synthase required for the synthesis of the cell wall component chitin,47 and inhibitors of chitin synthases, such as nikkomycin, are synergistic with caspofungin against numerous fungal pathogens.49,50 While this example highlights genetic interactions as determinants of the efficacy of compound combinations, the complexities of chemical action and genetic network density may preclude the prediction of synergism on a genome-wide scale.51 Thus, additional factors must be employed in order to identify effective antifungal combinations.
Chemical genomics in S. cerevisiae
The creation of the yeast deletion collection with strain-specific molecular barcodes enables the quantification of individual strains in a mixed population.31,52 Consequently, this approach has been widely used over the past two decades to investigate interactions between genes and small molecules.32,53–56 Such chemical-genetic assays are based on the concept that modifying the expression of a compound target (or the expression of other factors involved in a process that is targeted by the compound) alters the amount of compound required to effectively inhibit that target (see Figs. 3 and 4 below). This principle has been exploited at genome scale, allowing unbiased screening to determine a molecule’s mode-of-action in whole yeast cells. Benefits of the pooled chemogenomic format include the consumption of relatively little compound, which may be a limiting factor in terms of quantity and expense; the efficiency of processing pooled samples compared with profiling thousands of individual mutants; as well as the minimization of technical variation.26,27,52,57,58 To further enhance the utility of this approach, a set of donor strains, called Barcoders, was constructed that allows unique barcode sequences to be transferred to any S. cerevisiae strain collection in a rapid and cost-effective manner, enabling the application of parallel pooled approaches to a wide variety of complex bioassays.59
Figure 3.
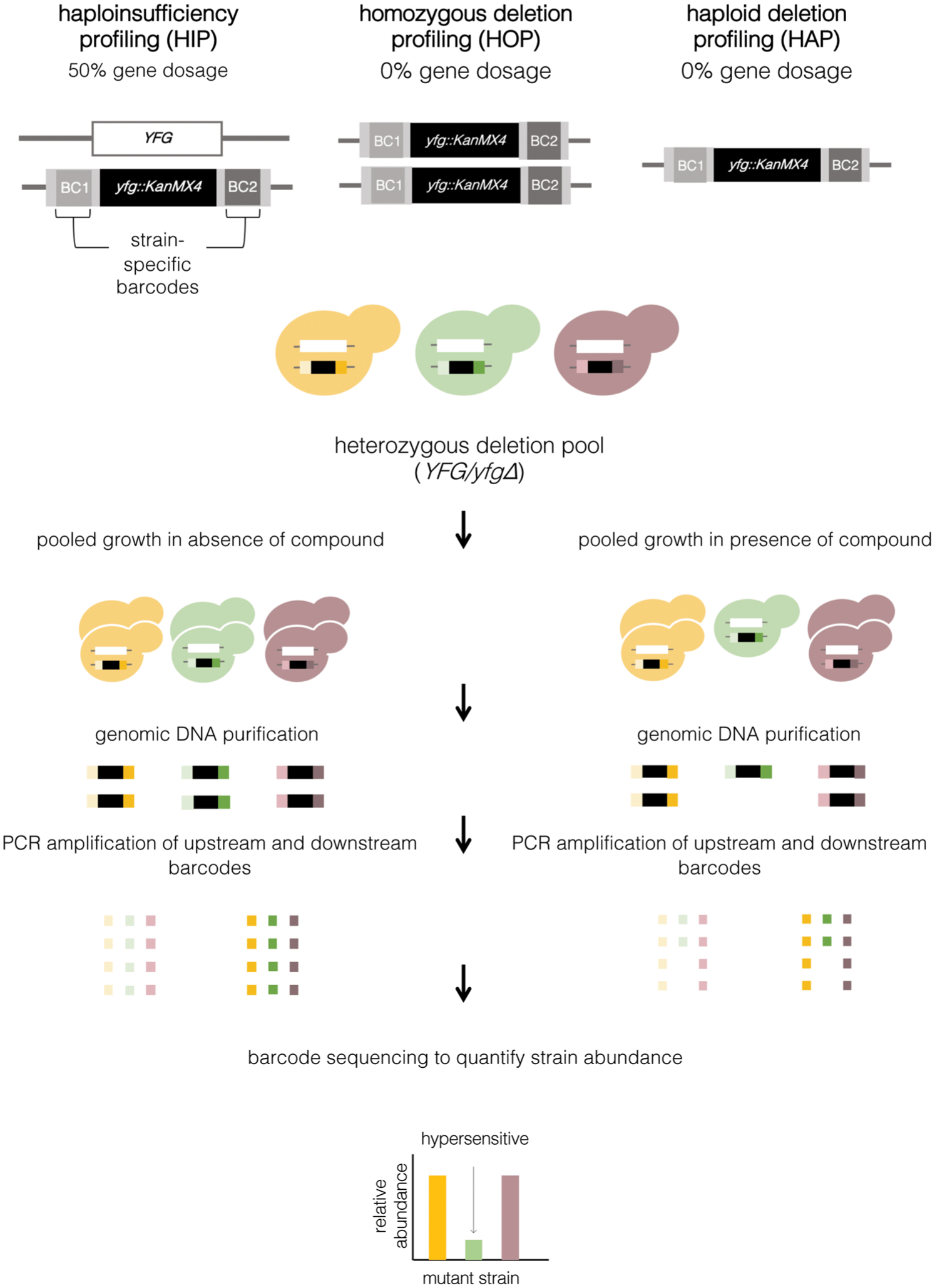
Reduced gene dosage chemical-genomic assays. Haploinsufficiency profiling (HIP), homozygous deletion profiling (HOP), and haploid deletion profiling (HAP) operate under a common principle. HIP employs heterozygous deletion mutants of essential or nonessential genes in a diploid for reduced gene dosage. HOP utilizes homozygous deletion mutants in a diploid parent and HAP utilizes haploid deletion mutants generated in a haploid parent to abolish the expression of nonessential genes. Individual strains within each genome-wide library are tagged with two unique DNA barcodes, upstream and downstream (BC1 and BC2), that permit simultaneous analysis within a single pool. Deletion libraries are grown competitively in the absence and presence of a compound of interest. Genomic DNA is isolated after a duration of pooled growth, and PCR amplification of strain-identifying barcodes is performed using universal primers for the upstream or downstream barcodes. High-throughput barcode sequencing and normalization to the untreated pool is used to quantify strain representation.
Figure 4.
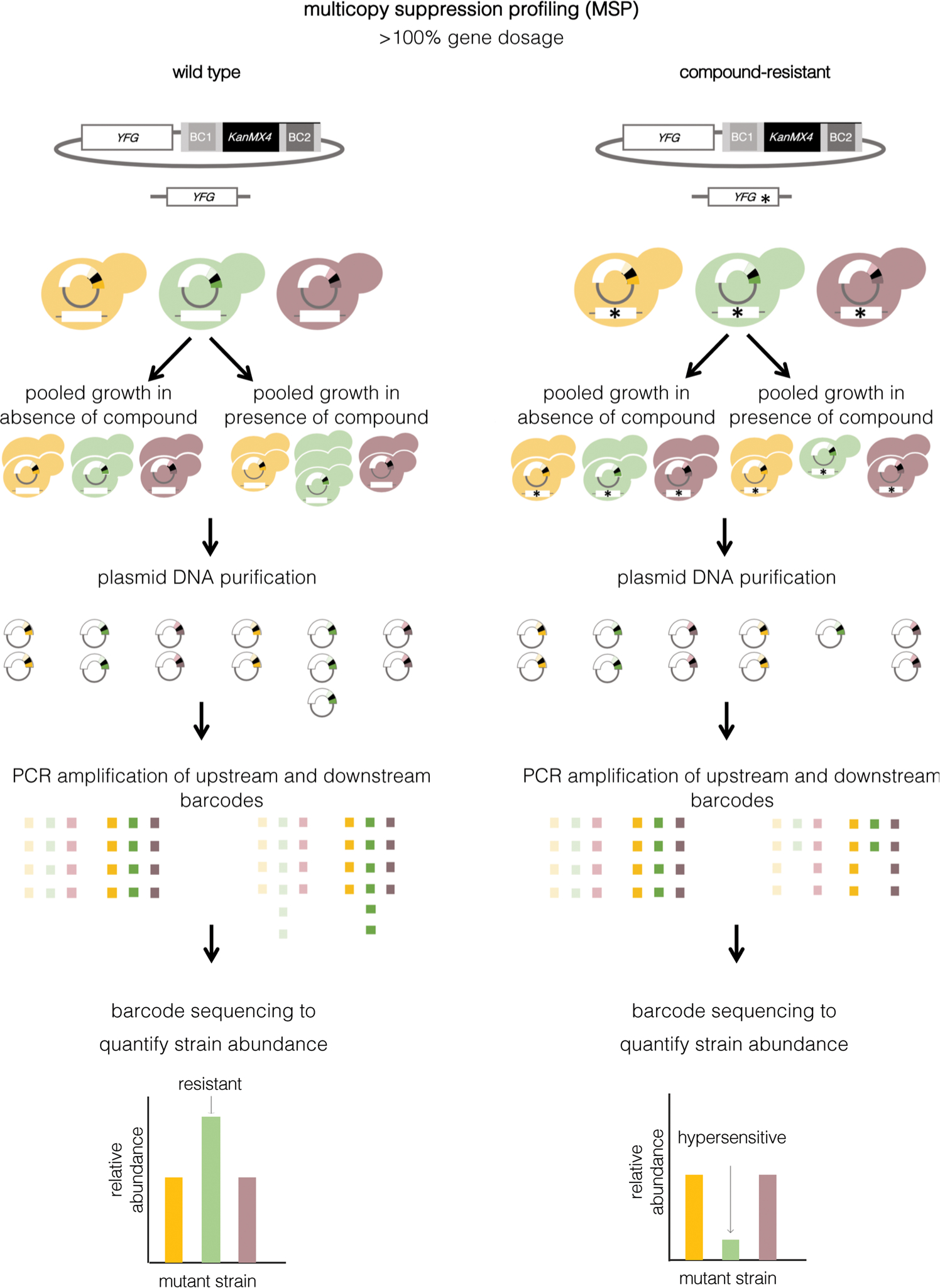
Multicopy suppression profiling. The MoBY ORF library is constructed such that each gene is tagged with two unique DNA barcodes, similar to the yeast deletion collection. This collection can be transformed into a wild-type strain (left panel) or a haploid drug-resistant strain that harbors a recessive mutation (right panel). The resulting pools can be grown with a compound of choice. Plasmid DNA is isolated. Strain-specific barcodes are amplified using plasmid primers that flank each insert. High-throughput barcode sequencing and normalization to the untreated pool is used to determine strain representation. In a wild-type background, increased expression of a compound’s target gene typically confers enhanced fitness in the presence of the compound relative to other genes. In a drug-resistant background that contains a recessive resistance mutation, a plasmid with significantly reduced barcode counts identifies the gene that harbors the mutation responsible for resistance to the compound of interest.
Haploinsufficiency, homozygous, and haploid deletion profiling
Haploinsufficiency profiling (HIP) operates under the principle that deletion of one copy of a target gene in a diploid organism confers hypersensitivity to chemical inhibition.60 Drug-induced loss of activity of the remaining gene product emulates a complete gene deletion and is exhibited as a quantifiable growth defect.55 This is particularly true when the compound target is an essential (or highly critical) gene. In this case, the heterozygous strain likely shows no growth defect in untreated conditions but becomes inviable and drops out of a population in the presence of the compound that has effectively inhibited the function of the remaining target protein. HIP enables all deletion strains in the S. cerevisiase genome to be pooled together in order to determine competitive growth differences in a single genome-wide experiment by quantification of the barcodes traditionally through microarray and currently via high-throughput sequencing (Fig. 3).26,55,60 This experimental approach provides a relative rank order of hypersensitive mutants, as well as information as to the genes whose perturbation is associated with significant phenotypic effects.55 Initial applications of HIP proved the accuracy of the experimental approach, as mutants of genes encoding well-characterized targets of antimicrobial agents were readily identified as the most sensitive from the pool of ~6000 heterozygous deletion strains.55,60,61 In addition, heterozygous deletion mutants in gene encoding products that belonged to related signaling or metabolic pathways, or factors that reduced compound availability, were often shown to be hypersensitive.26,55 For example, S. cerevisiae pooled screening with methotrexate identified hypersensitive mutants, including for the reported drug target dihydrofolate reductase gene, DFR1, as well as FOL1 and FOL2, which encode upstream components in the folic acid biosynthetic pathway.55 The utility of HIP in uncovering novel mechanisms of action for countless small molecules has been repeatedly demonstrated, including two independent studies that implicated ribosomal RNA processing as the mechanism of 5-fluorouracil,55,61 an antitumoural agent widely considered to act via inhibition of the DNA synthesis enzyme thymidylate synthetase.62 Furthermore, HIP identified new chemical probes targeting septin, actin, and tubulin,56 as well as numerous alternative eukaryotic targets for diverse psychoactive drugs.63
While these examples highlight the potential of HIP in uncovering the mechanism of small molecules, not all genes display haploinsufficiency and increased sensitivity upon a 50% reduction of dosage. Furthermore, gene expression in select heterozygous deletion mutants may in fact be greater than the expected 50% due to transcriptional upregulation, which can compensate for reduced copy number.64 The decreased abundance by mRNA perturbation (DAmP) strategy is an alternative to HIP that uses antibiotic resistance cassettes to systematically disrupt the 3′-untranslated region of essential genes in haploid yeast, destabilizing the corresponding transcripts to an estimated 10% of wild-type expression.65,66 This methodology was used to generate a barcoded collection encompassing ~1400 strains, including approximately 90% of all essential genes.59,65,66 This analogous approach is preferable to simply elevating drug concentrations in heterozygous deletion mutants, which may cause loss of target specificity and thus generalized cellular toxicity.26,55
Homozygous deletion profiling (HOP) or haploid deletion profiling (HAP) is conceptually and experimentally similar to HIP but employs full deletion of nonessential genes in either diploid (HOP) or haploid (HAP) S. cerevisiae strains (Fig. 3). These approaches provide complementary and powerful assays in order to probe the mechanism of action of compounds that lack an essential protein target, and instead target cellular factors, such as DNA or lipids, or for those compounds that have redundant protein targets.26,67 For example, HIP of the DNA-intercalating agent cisplatin proved unsuccessful at identifying a putative target as there was no single protein in the cell for which genetic reduction would confer sensitivity to a compound that binds DNA. However, parallel HOP analysis uncovered numerous DNA repair factors, supporting DNA itself as the target.55 Although these approaches can be performed in either haploid or diploid gene deletion libraries, diploid strains present the advantage of being less impacted by secondary-site mutations, which have the potential to confound data interpretation.53 HOP/HAP also allows for the identification of genes in target-related pathways or detoxification processes, including metabolism and efflux, which buffer the cellular response to chemical stress.26,64,68 Whereas HIP analysis often produces fewer than 10 genes of interest, HOP/HAP assays commonly identify in the tens-to-hundreds of significantly hypersensitive mutants.
The complementary application of these chemical-genetic approaches has proved invaluable to deciphering the mode of action of bioactive agents in the context of a living eukaryotic cell. One of the first large-scale chemogenomic screening efforts performed 1144 full-genome HIP experiments and 418 HOP experiments.54 This study revealed a phenotype for 97% of genes in S. cerevisiae, suggesting that virtually the entire yeast genome is conditionally essential and therefore accessible to inhibition with small molecules;54 this is a critical observation considering the difficulty in identifying novel single-agent antifungals. A more recent large-scale analysis profiled 3250 small molecules and identified 317 compounds that specifically targeted the function of 121 genes. This work suggested that the cellular response to small molecules is limited by a network of 45 major chemogenomic signatures, and coupled such cellular processes to specific chemical moieties.56 Finally, recent advances in chemical-genetic screening platforms coupled with comprehensive sequence databases have further expanded our ability to profile compounds in a rapid and systematic manner, such that 13,524 molecules were investigated in a single study.69 This was achieved by using a diagnostic set of viable yeast gene deletion mutants spanning all major biological processes in a drug-sensitized genetic background, coupled with a highly multiplexed (768-plex) barcode sequencing protocol. For data analysis, the authors also generated a computational platform to functionally annotate compounds to specific biological processes and pathways.69–71
With recent technological advances that have enabled large-scale data generation, sophisticated computational platforms have proven imperative for the analysis of complex chemical–genetic interactions. Correlations between distinct chemical-genetic fitness profiles can be used for “guilt-by-association” to infer the mode of action of novel compounds, as compounds exhibiting similar chemical–genetic interactions tend to target similar processes (Fig. 2C).56,67,69,72 Likewise, the phenotypes of uncharacterized targets with known small molecules can glean insight into their biological roles. To make these comparisons, chemical-genetic profiles can be mapped onto the global genetic interaction profile similarity network.47 Many of these genetic, chemical, and chemical–genetic relationships can be explored using online resources, such as the publicly accessible HIP-HOP chemogenomics database56 and the MOSAIC database.69,71
Multicopy suppression profiling
Multicopy suppression profiling (MSP) is an orthogonal gene dosage–based approach built upon the concept that target gene overexpression enables increased tolerance to drug exposure (Fig. 4).53 Traditional suppressor screens based on such ideology have been employed for decades. Such studies typically use a high-copy plasmid library carrying randomly generated yeast genomic inserts to identify genes that, when overexpressed, confer resistance. Notably, these screens involve cumbersome plating techniques and clone characterization where plasmids are isolated from resistant colonies and sequenced in order to identify the gene(s) responsible for conferring the resistance phenotype. This approach was employed to identify the target of tunicamycin as Alg7.73 With the creation of advanced functional genomic resources, this approach was adapted in order to culture pools of strains in liquid medium in a manner analogous to the HIP assay.53 One of the first examples of MSP being employed in a pooled manner involved a high-copy plasmid collection containing yeast genomic DNA fragments with genes expressed from native promoters (Table 1). This library was screened in whole cells at high concentrations of compounds that inhibited a wild-type control by approximately 90%. Under such conditions, only one or a few resistant strains were selected from the population.68 Plasmids were then isolated from resistant cells, inserts were amplified by PCR and hybridized to a microarray carrying probes complementary to each yeast open reading frame in order to determine gene abundance in compound-treated relative to an untreated control pool.53 This approach correctly identified Dfr1, Erg11, and Tor1 as the targets of methotrexate, fluconazole, and rapamycin, respectively.53 Similarly, the Yeast Genome Tiling collection contains overlapping fragments of the yeast genome, which are each approximately 10 kb in size, cloned into high-copy vectors (Table 1).74 The ends of each insert of this library have been sequenced, and the plasmids organized in a tiling fashion across the yeast genome, ensuring near-saturation (97.2%) coverage of the yeast genome. In both examples, each insert contains multiple genes, and thus once a fragment that confers resistance is identified, the specific gene target must be cloned and its effect on the resistance confirmed.
Table 1.
Features of plasmid libraries used for multicopy suppression profiling
| MoBY-ORF library | Yeast tiling collection | Galactose inducible libraries | Random genomic fragment libraries | |
|---|---|---|---|---|
| PCR free | ✓ | ✓ | ||
| Promoter type | Native | Native | Inducible | Native |
| Plasmid copy number | Low | High | High | High |
| Barcoded | ✓ | |||
| Percent genome coverage | 90% | 97.2% | 80% | ~85% |
| Average fragment size | 2 kb | 10 kb | 1.5 kb | 5 kb |
| Number of genes per fragment | 1 | 4–6 | 1 | 2–3 |
| References | 69 | 66 | 67, 68 | 48 |
Several alternative libraries offer advantages over the traditional randomly generated yeast genomic inserts collections described above. Two such resources consist of thousands of yeast strains, each carrying a vector harboring a single yeast open reading frame under the expression of the GAL1 promoter (Table 1).75,76 A proof-of-principle experiment with one of these inducible collections successfully identified vectors carrying TOR1 as the target of rapamycin.75 Finally, the generation of the molecular barcoded yeast (MoBY) ORF collection, which comprises over 90% of the yeast genome, was completed in order to further enhance the utility and efficiency of this experimental approach (Table 1).77 The MoBY ORF library is constructed with a centromere-based vector where each gene is tagged with two unique DNA barcodes, similar to the yeast deletion collection.77 Once pools of strains are cultured in the presence of compound, resistant strains are identified by amplifying the strain-specific barcodes and employing microarrays or high-throughput sequencing for strain identification (Fig. 4). This approach has been successful at predicting the mode-of-action of multiple molecules.77 For example, chitosan oligosaccharide was known to exert fungal cell membrane stress, similar to the azoles and polyenes.78 However, the MoBY-ORF collection determined that overexpression of the Ras superfamily GTPase ARL1 confers resistance to chitosan oligosaccharide, highlighting a previously unappreciated cellular target.79
An additional application of the MoBY-ORF library is the identification of recessive genes responsible for drug resistance (Fig. 4).77 After a recessive drug-resistant mutant is identified, it can be transformed with the MoBY-ORF collection and complementation by one or more wild-type alleles from the collection that restores drug sensitivity, enables the identification of the gene harboring the recessive resistance mutation.77 This assay enables the identification of drug targets in cases where the compound must interact with another protein to become toxic. Complementation of recessive drug-resistant alleles can also be used to systematically uncover general and specific resistance mechanisms. For example, this approach was employed to further define the mechanism of action of the natural product theopalauamide.77 Theopalauamide-resistant mutants were transformed with the MoBY-ORF collection, and the clone harboring MVD1, a gene encoding an essential enzyme in an early step of the ergosterol biosynthesis pathway, was observed to restore drug sensitivity.77 The theopalauamide-resistant strain was confirmed to contain a single amino acid substitution within the active site of Mvd1 and subsequent analyses indicated that theopalauamide binds to ergosterol, defining a novel class of sterol-binding compounds.77
As is the case with all experimental approaches, MSP has certain limitations. For example, overexpression of a target gene may not impart an observable drug resistance phenotype if the encoded protein product resides within a larger complex with distinct stoichiometric requirements.26,68 Increased dosage of certain genes may also itself interfere with cellular fitness, as an estimated 15% of yeast protein-coding genes are deleterious upon overexpression.76 The use of centromere-based vectors for the MoBY-ORF collection minimizes overexpression-induced toxicity; however, it is certainly a limitation for other overexpression collections. In addition, an exceptionally sensitive drug target expressed at wild-type levels could mask the suppressor effects of another less sensitive overexpressed target, which would therefore escape detection.60 Finally, drug pumps or other indirect targets may dominate in the set of strains resistant to compound. Creating similar overexpression libraries in diverse drug pump-deficient mutants can alleviate this challenge.68
Variomics
Several years ago, an alternative to gene dosage–based approaches was reported for the systematic discovery of drug target genes, as well as resistance genes, in S. cerevisiae.80 This experimental approach involved a genome-wide compendium of mutant libraries constructed by high complexity random mutagenesis. The allelic variants are carried on low-copy centromeric plasmids, with 5847 available sets representing 90% of the S. cerevisiae genome.80 Each variomic library contains >2.0 × 105 independent alleles, including both single and multiple mutated variants, to ensure high genetic diversity to maximize the likelihood of mutations spanning all encoded amino acid residues.80 Similar to deletion and overexpression profiling, pooled yeast growth and variant allele quantification through high-throughput barcode sequencing are performed with follow-up validation of individual resistant mutants. This variomics tool was first tested against rapamycin, validating its known target genes FPR1, TOR1, and TOR2, as well as the previously described resistance gene NPR1.80 Notably, the TOR1 and TOR2 resistance alleles all incorporated mutations within their known drug-binding domain.80 Both heterozygous and haploid variomic libraries have been constructed to determine if different types of mutations are able to confer resistance when the genetic background contains a wild-type copy of the desired allele. It was observed that haploid variomic pools tend to contain less genetic diversity at later time points during selection, given that any resistant mutations must also retain viability.81 By contrast, variomic libraries expressed in heterozygous deletion mutants can in principle achieve separation-of-function. This library enabled the identification of dfr1 hypomorphic alleles in the diploid state, which modulate methotrexate resistance, as well as previously characterized dominant dfr1 mutations in the haploid background.81
A strength of the variomics approach is that it defines key amino acid residues modulating compound interactions and selectivity, providing further resolution into drug–target engagement. As well, mutant allele preconstruction obviates the reliance on spontaneous mutation rates, which can otherwise be inconsistent. However, spontaneous resistant mutants may still emerge during selection, leading to false positives and demanding functional validation.80 Finally, variomics-based analysis is unable to delineate resistance mechanisms that involve multiple genes.80
Ultimately, the complementary approaches highlighted above are best applied in parallel to present a more powerful strategy to elucidate compound mode of action. Congruent results lend higher confidence to target identification. For instance, mutants of the targets of fluconazole, rapamycin, and methotrexate, although consistently hypersensitive in HIP assays with the cognate inhibitor, are also not always the most hypersensitive strains, but the corresponding target genes also confer resistance when overexpressed in MSP analysis.53
Chemical genomics in fungal pathogens
While the direct extension of findings from S. cerevisiae chemical-genetic analyses to pathogenic species, like C. albicans, has been fairly successful in determining the mode of action of antifungal molecules, significant limitations regarding genetic comparisons have also arisen due to phylogenetic divergence.82 In particular, fungal pathogens harbor mechanisms and pathways required for virulence that either do not exist in the model yeast or have been substantially repurposed over evolutionary time.83 Moreover, the list of probable drug targets in fungal pathogens is predicted to be even greater than the druggable S. cerevisiae genome.84,85 Thus, the ability to dissect virulence attributes and drug responses in these organisms in a comprehensive manner requires the development of genetic resources directly in the fungal pathogens. Fortunately, recent advances in the generation of functional genomic tools in pathogenic fungi have enabled experimentation in the pathogens of interest.82,83,86,87 Despite the challenges of genetic intractability and cryptic life cycles, the availability of complete or draft genome sequences in pathogenic fungal species,88–90 coupled with advances in genetic manipulation,91–93 has enabled progress with the production of comprehensive mutant collections and permitted the systematic discovery of compound–target connections.
Chemical genomics in Candida
Thus far, the most comprehensive chemical-genetic resources among pathogenic fungi have been generated for C. albicans. Two primary functional genomics tools have been instrumental in advancing our understanding of gene function in this species. First, the C. albicans double barcoded (DBC) heterozygous deletion library covers ~90% of the C. albicans genome.82 Similar to the S. cerevisiae heterozygous deletion collection, the DBC library also includes strain-specific molecular sequences that are flanked by universal priming sequences. These strain-specific barcodes surround a HIS3 auxotrophic marker and serve as unique molecular identifiers for each strain in the C. albicans genome.82 Second, building upon the DBC collection, a library of C. albicans mutants was generated to facilitate large-scale functional analysis of its genome. To do so, a gene replacement and conditional expression (GRACE) strategy was employed where a heterozygous deletion mutant was transformed such that the expression of the remaining allele was conditionally controlled by replacing the native promoter with a tetracycline-repressible promoter.86 This GRACE collection currently consists of ~2400 mutants representing approximately ~40% of the C. albicans genome. Furthermore, this methodology has enabled a direct comparison of phenotypes under nonrepressing and repressing conditions in order to define those genes essential for C. albicans growth under laboratory conditions, as well as in vivo.86,94 Defining the compendium of essential genes in C. albicans is imperative to the elucidation of novel antifungal targets.
Additional genomic resources have expanded the repertoire of experimental approaches to interrogate gene function in C. albicans. The transposon insertion-based TagModule collection is another open-access, barcoded set of heterozygous deletion mutants, which comprises 59% of predicted C. albicans open reading frames.95 This collection was used to identify novel regulators of filamentous growth and new genes involved in essential processes, as well as verify the target of brefeldin A as Sec7.95 This TagModule toolkit is universal, meaning that it can be applied to diverse microorganisms to support antimicrobial research. As well, a barcoded homozygous deletion library spanning 11% of annotated C. albicans coding genes was generated in order to profile genes important for growth, morphogenesis, virulence, and commensalism.96,97 Several resources have also been developed for focused gene overexpression studies in C. albicans,98,99 albeit with limited application outside of functional annotation of specific genetic networks.
In comparison, there is a dearth of existing genetic tools in non-albicans Candida species. The haploid genomes of C. glabrata and C. auris are refractory to HIP, though a barcoded deletion mutant library covering 12% of the C. glabrata genome was recently employed to identify genes that influence azole and echinocandin susceptibility.100 Likewise, recent completion of annotated C. auris genome assemblies101 and advances in CRISPR-Cas9 technology, particularly the use of RNA-Cas9 complexes for expression-free gene editing systems in C. glabrata and C. auris,91 can be envisioned as guiding large-scale loss-of-function mutant collections and drug target determination in the near future.
Chemical genomics in Cryptococcus
Genetic tool development for C. neoformans can be considered arduous, in large part due to inefficient transformation that is achieved by either electroporation or the bombardment of DNA-coated particles (biolistic transformation), low homologous recombination rates, as well as complex genomic structural features.90,102,103 However, the development of cloning-free fusion PCR methods for the addition of long homology arms for gene targeting, optimization of transformation parameters, the development of multiple dominant drug-selectable marker systems, and the optimization of rapid screening for genotyping transformants made it feasible to construct large numbers of gene deletions in C. neoformans.104 Specifically, an open-access library of 1201 signature-tagged deletion mutants, biased for genes lacking S. cerevisiae orthologs, was constructed.105 Each deletion mutant harbored one of 48 DNA oligonucletoide barcodes that enabled quantitative detection of each mutant in a pooled sample.104,105 Furthermore, this collection along with another barcoded gene deletion library105 that together encompassed 1448 gene knockouts, was used to generate a chemogenomic atlas where each homozygous deletion mutant was cultured in the presence of one of 439 small molecules.87 Eighty-three percent of the mutants were found to be associated with chemical phenotypes (either hypersensitive or resistant), and each compound induced one or more genetic responses. Importantly, a comparison of these results with large-scale studies in S. cerevisiae54,67 highlighted that chemical-genetic profiles from C. neoformans were largely distinct from those in S. cerevisiae, emphasizing the relevance of pathogen-focused studies.87 In particular, chemical-genetic screens with this deletion mutant collection were pivotal in determining that the azole synergizing agent dicyclomine increases cell permeability and decreases nutrient uptake in C. neoformans.106 Additional gene deletion collections have also been generated for functional genomic profiling, including a 322 signature-tagged gene deletion collection encompassing 155 putative transcription factor genes, as well as 264 signature-tagged gene deletion mutants for 129 putative kinases.107,108 These collections were profiled to unveil key regulators of virulence and drug resistance in order to map potential anticryptococcal targets to be exploited for therapeutic development. Finally, the genome editing strategy transient CRISPR-Cas9 coupled with electroporation system (TRACE) was recently developed that dramatically improves the efficiency of targeted mutagenesis in the Cryptococcus genome,109,110 thus providing great promise for the generation of future mutant collections in this species.
Chemical genomics in Aspergillus
The major obstacles to targeted genetic manipulation in A. fumigatus are poor homologous recombination and prolific genetic exchange via vegetative cell fusion.111 To date, systematic mutant collections for chemical-genetic fitness profiling are lacking; however, the outlook is bright with improvements in gene deletion and overexpression methods. Transposon insertion mutagenesis, as well as parasexual genetics, has been deployed to identify essential A. fumigatus genes.112,113 Moreover, a conditional promoter replacement strategy was implemented to distinguish and prioritize essential and putative antifungal drug target genes among 54 genes with essential C. albicans or S. cerevisiae orthologs.114 This included the generation of a conditional ALG7 mutant, which was confirmed to elicit specific hypersensitivity to its known inhibitor tunicamycin, upon genetic depletion in a whole cell.114 More recently, a study demonstrating minimal induced cell fusion under controlled culture conditions served as proof-of-principle for barcode-free, high-throughput, competitive fitness profiling in A. fumigatus.115 A mini-library of 46 heterozygous deletion strains of known drug target genes and/or essential genes was created by a rapid allelic replacement technique, with cyp51A and arf2 mutants rediscovered as the only significantly hypersensitive outliers for their cognate inhibitors itraconazole and brefeldin A, respectively.115 In addition, multiple groups have recently reported advances with CRISPR-Cas9 to promote homologous recombination for targeted gene disruption and loss-of-function studies in Aspergillus species.92,116,117
Characterization of novel antifungals using chemical genomics
With the vast array of functional genomics resources available in S. cerevisiae, C. albicans, and C. neoformans, it is not surprising that the application of chemical-genomic approaches for antifungal mode of action analysis is expanding. For example, the natural product parnafungin was shown to display potent and broad spectrum activity against diverse fungal pathogens by inhibiting fungal poly(A) polymerase,118 and the antifungal plant defensin, RsAFP2, was demonstrated through HIP to target the C. albicans glucosylceramides, key components of the fungal cell wall.119 Similar research identified structurally related synthetic molecules that induced hypersensitivity in an OLE1 heterozygous mutant, implicating the biosynthesis of unsaturated fatty acids as a potential antifungal target.120 In addition, synthetic compounds with antifungal activity against C. neoformans were shown to target sphingolipid biosynthesis using S. cerevisiae HIP-HOP to elucidate mode of action.121 Finally, recent studies unveiled great potential of targeting either glycosylphosphatidylinositol precursor biosynthesis,122 or the fungal casein kinase Yck2,123 through their use of C. albicans HIP to identify the mechanism of action of novel bioactive compounds.
In addition to the examples highlighted above, several studies have also leveraged high-throughput approaches to determine the cellular targets of diverse compounds that display antifungal activity. Two such studies performed high-throughput screens to identify antifungal potentiators and then characterized the mode of action of top candidates using S. cerevisiase chemical genomic resources, implicating effects on membrane permeability or sphingolipid metabolism.124,125 Furthermore, efforts from industry to screen ~1800 natural product extracts identified and characterized a number of natural products with antifungal activity, including: yefafungin, which targets the fungal-specific translation initiation factor YEF3; campafungin, predicted to inhibit adenylate cyclase activity and/or cAMP regulation; and fellutamides C and D, which were predicted to inhibit the fungal proteasome.126
One final alternative application of fungal chemical genomic resources to combat fungal infection is to identify genes and genetic networks important for fungal virulence and identify virulence factors targeted by novel small molecules. Targeting fungal virulence provides a complementary approach to the development of antifungal agents, as the goal is to occlude the ability of a microbe to cause harm to its host. Targeting virulence factors offers many benefits, including expanding the repertoire of antifungal targets, minimizing effects on the host myco-biome, and reducing selection pressure for the evolution of drug resistance.127,128 One such virulence trait in C. albicans is the ability to transition between yeast and filamentous morphologies. This developmental transition is not only imperative for the establishment of systemic infection,96,129 but is also required for the formation of drug-resistant biofilms on surfaces, such as medical devices.130 Chemical-genetic resources in fungal pathogens can aid in elucidating novel virulence pathways that can be targeted by small molecules, as well as identify the precise virulence factor(s) that are targeted by such compounds. For example, one such study employed the C. albicans GRACE collection to identify 102 negative morphogenetic regulators and 872 positive regulators, implicating ergosterol biosynthesis and N-linked glycosylation in morphogenesis.94 Another study using the GRACE library implicated the Arp2/3 complex in C. albicans adherence and biofilm formation, due to its role in modulating cell surface hydrophobicity and remodeling of chitin and β-glucans in the fungal cell wall.131 Finally, an approach combining genome-wide C. albicans transcriptional analysis and mining S. cerevisiae chemical genomic data successfully pinpointed inhibition of the mitochondrial retrograde response via MGE1 as the probable mechanism of niclosamide, a repurposed antihelmintic agent that disrupts C. albicans filamentation and biofilm formation.132
Concluding thoughts: steady steps forward on the long road ahead
This review has highlighted the tremendous progress that has been made over the past few decades in the development of functional genomic resources that have advanced our understanding of how small molecules impact the fungal cell. While several hurdles remain to translate what we as a community have learned into development of the next antifungal drug that can be deployed in the clinic, the diverse array of cellular targets identified provides hope that there remains a plethora of cellular targets to be exploited to combat mycotic disease with much needed single agent and combination therapy treatments. The continued expansion of chemical genetic resources in diverse fungal pathogens will greatly empower inference of mode of action and resistance mechanisms for novel candidate antifungals. Likewise, elucidation of pathogen-specific drug–target pairs will further empower structure–activity relationship analysis and optimization of more species-selective and effective analogs. Beyond human health applications, chemical genetic approaches are also being implemented directly in fungal plant pathogens for agricultural fungicide development, which is similarly facing unprecedented resistance pressures.133 Overall, although the generation of functional genomic resources in diverse fungal species requires substantial investment of time and resources, it is exquisitely clear that these advances will catalyze the development of much-needed antifungal drugs.
Acknowledgments
We thank all members of the Cowen lab for helpful discussions. L.E.C. is supported by the Canadian Institutes of Health Research Foundation Grant (FDN-154288), two National Institutes of Health NIAID R01 Grants (1R01AI127375-01 and 1R01AI120958 - 01A1), and a National Institutes of Health NIAID R21 Grant (1R21AI141080-01). L.E.C. is a Canada Research Chair (Tier 1) in Microbial Genomics & Infectious Disease and Co-Director of the CIFAR Fungal Kingdom: Threats & Opportunities program.
Footnotes
Competing interests
L.E.C. is a cofounder and shareholder in Bright Angel Therapeutics, a platform company for development of novel antifungal therapeutics; and a consultant for Boragen, a small molecule development company focused on leveraging the unique chemical properties of boron chemistry for crop protection and animal health.
References
- 1.Brown GD, Denning DW, Gow NA, et al. 2012. Hidden killers: human fungal infections. Sci. Transl. Med 4: 165rv113. [DOI] [PubMed] [Google Scholar]
- 2.Pfaller MA & Diekema DJ. 2010. Epidemiology of invasive mycoses in North America. Crit. Rev. Microbiol 36: 1–53. [DOI] [PubMed] [Google Scholar]
- 3.Benedict K, Jackson BR, Chiller T, et al. 2019. Estimation of direct healthcare costs of fungal diseases in the United States. Clin. Infect. Dis 68: 1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robbins N, Caplan T & Cowen LE. 2017. Molecular evolution of antifungal drug resistance. Annu. Rev. Microbiol 71: 753–775. [DOI] [PubMed] [Google Scholar]
- 5.Berman J & Krysan DJ. 2020. Drug resistance and tolerance in fungi. Nat. Rev. Microbiol 18: 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lockhart SR 2019. Candida auris and multidrug resistance: defining the new normal. Fungal Genet. Biol 131: 103243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Revie NM, Iyer KR, Robbins N, et al. 2018. Antifungal drug resistance: evolution, mechanisms and impact. Curr. Opin. Microbiol 45: 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wisplinghoff H, Bischoff T, Tallent SM, et al. 2004. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis 39: 309–317. [DOI] [PubMed] [Google Scholar]
- 9.Pfaller MA & Diekema DJ. 2007. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev 20: 133–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lockhart SR, Etienne KA, Vallabhaneni S, et al. 2017. Simultaneous emergence of multidrug-resistant Candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin. Infect. Dis 64: 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denning DW & Bromley MJ. 2015. Infectious disease. How to bolster the antifungal pipeline. Science 347: 1414–1416. [DOI] [PubMed] [Google Scholar]
- 12.Pfaller MA, Diekema DJ, Turnidge JD, et al. 2019. Twenty years of the SENTRY antifungal surveillance program: results for Candida species from 1997–2016. Open Forum Infect. Dis 6: S79–S94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park BJ, Wannemuehler KA, Marston BJ, et al. 2009. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS 23: 525–530. [DOI] [PubMed] [Google Scholar]
- 14.Maziarz EK & Perfect JR. 2016. Cryptococcosis. Infect. Dis. Clin. North Am 30: 179–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kidd SE, Hagen F, Tscharke RL, et al. 2004. A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc. Natl. Acad. Sci. USA 101: 17258–17263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verweij PE, Snelders E, Kema GH, et al. 2009. Azole resistance in Aspergillus fumigatus: a side-effect of environmental fungicide use? Lancet Infect. Dis 9: 789–795. [DOI] [PubMed] [Google Scholar]
- 17.Ostrosky-Zeichner L, Casadevall A, Galgiani JN, et al. 2010. An insight into the antifungal pipeline: selected new molecules and beyond. Nat. Rev. Drug Discov 9: 719–727. [DOI] [PubMed] [Google Scholar]
- 18.Robbins N, Wright GD & Cowen LE. 2016. Antifungal drugs: the current armamentarium and development of new agents. Microbiol. Spectr 4. 10.1128/microbiolspec.FUNK-0002-2016. [DOI] [PubMed] [Google Scholar]
- 19.Perfect JR 2017. The antifungal pipeline: a reality check. Nat. Rev. Drug Discov 16: 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson TM, Clay MC, Cioffi AG, et al. 2014. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat. Chem. Biol 10: 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vincent BM, Lancaster AK, Scherz-Shouval R, et al. 2013. Fitness trade-offs restrict the evolution of resistance to amphotericin B. PLoS Biol. 11: e1001692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Day JN, Chau TTH, Wolbers M, et al. 2013. Combination antifungal therapy for cryptococcal meningitis. N. Engl. J. Med 368: 1291–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pappas PG, Kauffman CA, Andes DR, et al. 2016. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin. Infect. Dis 62: e1–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pouliot M & Jeanmart S. 2016. Pan assay interference compounds (PAINS) and other promiscuous compounds in antifungal research. J. Med. Chem 59: 497–503. [DOI] [PubMed] [Google Scholar]
- 25.Roemer T & Krysan DJ. 2014. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harbor Perspect. Med 4: a019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roemer T, Davies J, Giaever G, et al. 2012. Bugs, drugs and chemical genomics. Nat. Chem. Biol 8: 46–56. [DOI] [PubMed] [Google Scholar]
- 27.Smith AM, Durbic T, Oh J, et al. 2011. Competitive genomic screens of barcoded yeast libraries. J. Vis. Exp 2011: 2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schenone M, Dancik V, Wagner BK, et al. 2013. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol 9: 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goffeau A, Barrell BG, Bussey H, et al. 1996. Life with 6000 genes. Science 274: 546, 563–567. [DOI] [PubMed] [Google Scholar]
- 30.Winzeler EA, Shoemaker DD, Astromoff A, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. [DOI] [PubMed] [Google Scholar]
- 31.Shoemaker DD, Lashkari DA, Morris D, et al. 1996. Quantitative phenotypic analysis of yeast deletion mutants using a highly parallel molecular bar-coding strategy. Nat. Genet 14: 450–456. [DOI] [PubMed] [Google Scholar]
- 32.Giaever G, Chu AM, Ni L, et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391. [DOI] [PubMed] [Google Scholar]
- 33.Li Z, Vizeacoumar FJ, Bahr S, et al. 2011. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat. Biotech 29: 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ben-Aroya S, Coombes C, Kwok T, et al. 2008. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol. Cell 30: 248–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davierwala AP, Haynes J, Li Z, et al. 2005. The synthetic genetic interaction spectrum of essential genes. Nat. Genet 37: 1147–1152. [DOI] [PubMed] [Google Scholar]
- 36.Mnaimneh S, Davierwala AP, Haynes J, et al. 2004. Exploration of essential gene functions via titratable promoter alleles. Cell 118: 31–44. [DOI] [PubMed] [Google Scholar]
- 37.Krogan NJ, Cagney G, Yu H, et al. 2006. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643. [DOI] [PubMed] [Google Scholar]
- 38.Ghaemmaghami S, Huh WK, Bower K, et al. 2003. Global analysis of protein expression in yeast. Nature 425: 737–741. [DOI] [PubMed] [Google Scholar]
- 39.Ito T, Chiba T, Ozawa R, et al. 2001. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 98: 4569–4574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Costanzo M, VanderSluis B, Koch EN, et al. 2016. A global genetic interaction network maps a wiring diagram of cellular function. Science 353: aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Costanzo M, Baryshnikova A, Myers CL, et al. 2011. Charting the genetic interaction map of a cell. Curr. Opin. Biotechnol 22: 66–74. [DOI] [PubMed] [Google Scholar]
- 42.Costanzo M, Kuzmin E, van Leeuwen J, et al. 2019. Global genetic networks and the genotype-to-phenotype relationship. Cell 177: 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mani R, St Onge RP, Hartman JL, et al. 2008. Defining genetic interaction. Proc. Natl. Acad. Sci. USA 105: 3461–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stearns T & Botstein D. 1988. Unlinked noncomplementation: isolation of new conditional-lethal mutations in each of the tubulin genes of Saccharomyces cerevisiae. Genetics 119: 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tong AH, Lesage G, Bader GD, et al. 2004. Global mapping of the yeast genetic interaction network. Science 303: 808–813. [DOI] [PubMed] [Google Scholar]
- 46.Tong AH, Evangelista M, Parsons AB, et al. 2001. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294: 2364–2368. [DOI] [PubMed] [Google Scholar]
- 47.Costanzo M, Baryshnikova A, Bellay J, et al. 2010. The genetic landscape of a cell. Science 327: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spitzer M, Robbins N & Wright GD. 2017. Combinatorial strategies for combating invasive fungal infections. Virulence 8: 169–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sandovsky-Losica H, Shwartzman R, Lahat Y, et al. 2008. Antifungal activity against Candida albicans of nikkomycin Z in combination with caspofungin, voriconazole or amphotericin B. J. Antimicrob. Chemother 62: 635–637. [DOI] [PubMed] [Google Scholar]
- 50.Verwer PE, van Duijn ML, Tavakol M, et al. 2012. Reshuffling of Aspergillus fumigatus cell wall components chitin and β-glucan under the influence of caspofungin or nikkomycin Z alone or in combination. Antimicrob. Agents Chemother 56: 1595–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wildenhain J, Spitzer M, Dolma S, et al. 2015. Prediction of synergism from chemical–genetic interactions by machine learning. Cell Syst. 1: 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nijman SM 2015. Functional genomics to uncover drug mechanism of action. Nat. Chem. Biol 11: 942–948. [DOI] [PubMed] [Google Scholar]
- 53.Hoon S, Smith AM, Wallace IM, et al. 2008. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat. Chem. Biol 4: 498–506. [DOI] [PubMed] [Google Scholar]
- 54.Hillenmeyer ME, Fung E, Wildenhain J, et al. 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320: 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giaever G, Flaherty P, Kumm J, et al. 2004. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc. Natl. Acad. Sci. USA 101: 793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee AY, St Onge RP, Proctor MJ, et al. 2014. Mapping the cellular response to small molecules using chemogenomic fitness signatures. Science 344: 208–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ho CH, Piotrowski J, Dixon SJ, et al. 2011. Combining functional genomics and chemical biology to identify targets of bioactive compounds. Curr. Opin. Chem. Biol 15: 66–78. [DOI] [PubMed] [Google Scholar]
- 58.Roemer T & Boone C. 2013. Systems-level antimicrobial drug and drug synergy discovery. Nat. Chem. Biol 9: 222–231. [DOI] [PubMed] [Google Scholar]
- 59.Yan Z, Costanzo M, Heisler LE, et al. 2008. Yeast Bar-coders: a chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nat. Methods 5: 719–725. [DOI] [PubMed] [Google Scholar]
- 60.Giaever G, Shoemaker DD, Jones TW, et al. 1999. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat. Genet 21: 278–283. [DOI] [PubMed] [Google Scholar]
- 61.Lum PY, Armour CD, Stepaniants SB, et al. 2004. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell 116: 121–137. [DOI] [PubMed] [Google Scholar]
- 62.Parker WB & Cheng YC. 1990. Metabolism and mechanism of action of 5-fluorouracil. Pharmacol. Ther 48: 381–395. [DOI] [PubMed] [Google Scholar]
- 63.Ericson E, Gebbia M, Heisler LE, et al. 2008. Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genet. 4: e1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoon S, St Onge RP, Giaever G, et al. 2008. Yeast chemical genomics and drug discovery: an update. Trends Pharmacol. Sci 29: 499–504. [DOI] [PubMed] [Google Scholar]
- 65.Breslow DK, Cameron DM, Collins SR, et al. 2008. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat. Methods 5: 711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schuldiner M, Collins SR, Thompson NJ, et al. 2005. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123: 507–519. [DOI] [PubMed] [Google Scholar]
- 67.Parsons AB, Lopez A, Givoni IE, et al. 2006. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126: 611–625. [DOI] [PubMed] [Google Scholar]
- 68.Smith AM, Ammar R, Nislow C, et al. 2010. A survey of yeast genomic assays for drug and target discovery. Pharmacol. Ther 127: 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Piotrowski JS, Li SC, Deshpande R, et al. 2017. Functional annotation of chemical libraries across diverse biological processes. Nat. Chem. Biol 13: 982–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Simpkins SW, Deshpande R, Nelson J, et al. 2019. Using BEAN-counter to quantify genetic interactions from multiplexed barcode sequencing experiments. Nat. Protoc 14: 415–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nelson J, Simpkins SW, Safizadeh H, et al. 2018. MOSAIC: a chemical–genetic interaction data repository and web resource for exploring chemical modes of action. Bioinformatics 34: 1251–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Parsons AB, Brost RL, Ding H, et al. 2004. Integration of chemical–genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat. Biotech 22: 62–69. [DOI] [PubMed] [Google Scholar]
- 73.Rine J, Hansen W, Hardeman E, et al. 1983. Targeted selection of recombinant clones through gene dosage effects. Proc. Natl. Acad. Sci. USA 80: 6750–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jones GM, Stalker J, Humphray S, et al. 2008. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat. Methods 5: 239–241. [DOI] [PubMed] [Google Scholar]
- 75.Butcher RA, Bhullar BS, Perlstein EO, et al. 2006. Microarray-based method for monitoring yeast overexpression strains reveals small-molecule targets in TOR pathway. Nat. Chem. Biol 2: 103–109. [DOI] [PubMed] [Google Scholar]
- 76.Sopko R, Huang D, Preston N, et al. 2006. Mapping pathways and phenotypes by systematic gene overexpression. Mol. Cell 21: 319–330. [DOI] [PubMed] [Google Scholar]
- 77.Ho CH, Magtanong L, Barker SL, et al. 2009. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat. Biotech 27: 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zakrzewska A, Boorsma A, Brul S, et al. 2005. Transcriptional response of Saccharomyces cerevisiae to the plasma membrane-perturbing compound chitosan. Eukaryot. Cell 4: 703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jaime MD, Lopez-Llorca LV, Conesa A, et al. 2012. Identification of yeast genes that confer resistance to chitosan oligosaccharide (COS) using chemogenomics. BMC Genomics 13: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang Z, Chen K, Zhang J, et al. 2013. A functional variomics tool for discovering drug-resistance genes and drug targets. Cell Rep. 3: 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wong LH, Sinha S, Bergeron JR, et al. 2016. Reverse chemical genetics: comprehensive fitness profiling reveals the spectrum of drug target interactions. PLoS Genet. 12: e1006275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xu D, Jiang B, Ketela T, et al. 2007. Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans. PLoS Pathog. 3: e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goranov AI & Madhani HD. 2014. Functional profiling of human fungal pathogen genomes. Cold Spring Harbor Perspect. Med 5: a019596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hopkins AL & Groom CR. 2002. The druggable genome. Nat. Rev. Drug Discov 1: 727–730. [DOI] [PubMed] [Google Scholar]
- 85.Calderone R, Sun N, Gay-Andrieu F, et al. 2014. Antifungal drug discovery: the process and outcomes. Future Microbiol. 9: 791–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roemer T, Jiang B, Davison J, et al. 2003. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol. Microbiol 50: 167–181. [DOI] [PubMed] [Google Scholar]
- 87.Brown JC, Nelson J, VanderSluis B, et al. 2014. Unraveling the biology of a fungal meningitis pathogen using chemical genetics. Cell 159: 1168–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nierman WC, Pain A, Anderson MJ, et al. 2005. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature 438: 1151–1156. [DOI] [PubMed] [Google Scholar]
- 89.Jones T, Federspiel NA, Chibana H, et al. 2004. The diploid genome sequence of Candida albicans. Proc. Natl. Acad. Sci. USA 101: 7329–7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Loftus BJ, Fung E, Roncaglia P, et al. 2005. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science 307: 1321–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Grahl N, Demers EG, Crocker AW, et al. 2017. Use of RNA–protein complexes for genome editing in non-albicans Candida species. mSphere 2: e00218–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morio F, Lombardi L & Butler G. 2020. The CRISPR tool-box in medical mycology: state of the art and perspectives. PLoS Pathog. 16: e1008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vyas VK, Barrasa MI & Fink GR. 2015. A CRISPR system permits genetic engineering of essential genes and gene families. Sci. Adv 1: e1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O’Meara TR, Veri A, Ketela T, et al. 2015. Global analysis of fungal morphology exposes mechanisms of host cell escape. Nat. Commun 6: 6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oh J, Fung E, Schlecht U, et al. 2010. Gene annotation and drug target discovery in Candida albicans with a tagged transposon mutant collection. PLoS Pathog. 6: e1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Noble SM, French S, Kohn LA, et al. 2010. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet 42: 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Witchley JN, Penumetcha P, Abon NV, et al. 2019. Candida albicans morphogenesis programs control the balance between gut commensalism and invasive infection. Cell Host Microbe 25: 432–443.e436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chauvel M, Nesseir A, Cabral V, et al. 2012. A versatile overexpression strategy in the pathogenic yeast Candida albicans: identification of regulators of morphogenesis and fitness. PLoS One 7: e45912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ramirez-Zavala B, Weyler M, Gildor T, et al. 2013. Activation of the Cph1-dependent MAP kinase signaling pathway induces white-opaque switching in Candida albicans. PLoS Pathog. 9: e1003696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schwarzmuller T, Ma B, Hiller E, et al. 2014. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog. 10: e1004211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chatterjee S, Alampalli SV, Nageshan RK, et al. 2015. Draft genome of a commonly misdiagnosed multidrug resistant pathogen Candida auris. BMC Genomics 16: 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Toffaletti DL, Rude TH, Johnston SA, et al. 1993. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J. Bacteriol 175: 1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nelson RT, Pryor BA & Lodge JK. 2003. Sequence length required for homologous recombination in Cryptococcus neoformans. Fungal Genet. Biol 38: 1–9. [DOI] [PubMed] [Google Scholar]
- 104.Chun CD & Madhani HD. 2010. Applying genetics and molecular biology to the study of the human pathogen Cryptococcus neoformans. Methods Enzymol. 470: 797–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu OW, Chun CD, Chow ED, et al. 2008. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell 135: 174–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wambaugh MA, Denham ST, Ayala M, et al. 2020. Synergistic and antagonistic drug interactions in the treatment of systemic fungal infections. elife 9: e54160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee KT, So YS, Yang DH, et al. 2016. Systematic functional analysis of kinases in the fungal pathogen Cryptococcus neoformans. Nat. Commun 7: 12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jung KW, Yang DH, Maeng S, et al. 2015. Systematic functional profiling of transcription factor networks in Cryptococcus neoformans. Nat. Commun 6: 6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lin J, Fan Y & Lin X. 2020. Transformation of Cryptococcus neoformans by electroporation using a transient CRISPR-Cas9 expression (TRACE) system. Fungal Genet. Biol 138: 103364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fan Y & Lin X. 2018. Multiple applications of a transient CRISPR-Cas9 coupled with electroporation (TRACE) system in the Cryptococcus neoformans species complex. Genetics 208: 1357–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Brakhage AA & Langfelder K. 2002. Menacing mold: the molecular biology of Aspergillus fumigatus. Annu. Rev. Microbiol 56: 433–455. [DOI] [PubMed] [Google Scholar]
- 112.Carr PD, Tuckwell D, Hey PM, et al. 2010. The transposon impala is activated by low temperatures: use of a controlled transposition system to identify genes critical for viability of Aspergillus fumigatus. Eukaryot. Cell 9: 438–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Firon A, Villalba F, Beffa R, et al. 2003. Identification of essential genes in the human fungal pathogen Aspergillus fumigatus by transposon mutagenesis. Eukaryot Cell 2: 247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hu W, Sillaots S, Lemieux S, et al. 2007. Essential gene identification and drug target prioritization in Aspergillus fumigatus. PLoS Pathog. 3: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Macdonald D, Thomson DD, Johns A, et al. 2019. Inducible cell fusion permits use of competitive fitness profiling in the human pathogenic fungus Aspergillus fumigatus. Antimicrob. Agents Chemother 63: e01615–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Al Abdallah Q, Ge W & Fortwendel JR. 2017. A simple and universal system for gene manipulation in Aspergillus fumigatus: in vitro-assembled Cas9-guide RNA ribonucleoproteins coupled with microhomology repair templates. mSphere 2: e00446–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leynaud-Kieffer LMC, Curran SC, Kim I, et al. 2019. A new approach to Cas9-based genome editing in Aspergillus niger that is precise, efficient and selectable. PLoS One 14: e0210243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jiang B, Xu D, Allocco J, et al. 2008. PAP inhibitor with in vivo efficacy identified by Candida albicans genetic profiling of natural products. Chem. Biol 15: 363–374. [DOI] [PubMed] [Google Scholar]
- 119.Thevissen K, de Mello Tavares P, Xu D, et al. 2012. The plant defensin RsAFP2 induces cell wall stress, septin mis-localization and accumulation of ceramides in Candida albicans. Mol. Microbiol 84: 166–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Xu D, Sillaots S, Davison J, et al. 2009. Chemical genetic profiling and characterization of small-molecule compounds that affect the biosynthesis of unsaturated fatty acids in Candida albicans. J. Biol. Chem 284: 19754–19764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mor V, Rella A, Farnoud AM, et al. 2015. Identification of a new class of antifungals targeting the synthesis of fungal sphingolipids. mBio 6: e00647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mann PA, McLellan CA, Koseoglu S, et al. 2015. Chemical genomics-based antifungal drug discovery: targeting glycosylphosphatidylinositol (GPI) precursor biosynthesis. ACS Infect. Dis 1: 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Caplan T, Lorente-Macias A, Stogios PJ, et al. 2020. Overcoming fungal echinocandin resistance through inhibition of the non-essential stress kinase Yck2. Cell Chem. Biol 27: 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Robbins N, Spitzer M, Yu T, et al. 2015. An antifungal combination matrix identifies a rich pool of adjuvant molecules that enhance drug activity against diverse fungal pathogens. Cell Rep. 13: 1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Spitzer M, Griffiths E, Blakely KM, et al. 2011. Cross-species discovery of syncretic drug combinations that potentiate the antifungal fluconazole. Mol. Syst. Biol 7: 499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Roemer T, Xu D, Singh SB, et al. 2011. Confronting the challenges of natural product-based antifungal discovery. Chem. Biol 18: 148–164. [DOI] [PubMed] [Google Scholar]
- 127.Clatworthy AE, Pierson E & Hung DT. 2007. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol 3: 541–548. [DOI] [PubMed] [Google Scholar]
- 128.Gauwerky K, Borelli C & Korting HC. 2009. Targeting virulence: a new paradigm for antifungals. Drug Discov. Today 14: 214–222. [DOI] [PubMed] [Google Scholar]
- 129.Noble SM, Gianetti BA & Witchley JN. 2017. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat. Rev. Microbiol 15: 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Blankenship JR & Mitchell AP. 2006. How to build a biofilm: a fungal perspective. Curr. Opin. Microbiol 9: 588–594. [DOI] [PubMed] [Google Scholar]
- 131.Lee JA, Robbins N, Xie JL, et al. 2016. Functional genomic analysis of Candida albicans adherence reveals a key role for the Arp2/3 complex in cell wall remodelling and biofilm formation. PLoS Genet. 12: e1006452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Garcia C, Burgain A, Chaillot J, et al. 2018. A phenotypic small-molecule screen identifies halogenated salicylanilides as inhibitors of fungal morphogenesis, biofilm formation and host cell invasion. Sci. Rep 8: 11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cools HJ & Hammond-Kosack KE. 2013. Exploitation of genomics in fungicide research: current status and future perspectives. Mol. Plant Pathol 14: 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cowen LE 2008. The evolution of fungal drug resistance: modulating the trajectory from genotype to phenotype. Nat. Rev. Microbiol 6: 187–198. [DOI] [PubMed] [Google Scholar]