

Abstract
Vertical transmission of maternal microbes is a major route for establishing the gut microbiome in newborns. The impact of perinatal antibiotics on vertical transmission of microbes and antimicrobial resistance is not well understood. Using a metagenomic approach, we analyzed the fecal samples from mothers and vaginally delivered infants from a control group (10 pairs) and a treatment group (10 pairs) receiving perinatal antibiotics. Antibiotic-usage had a significant impact on the main source of inoculum in the gut microbiome of newborns. The control group had significantly more species transmitted from mothers to infants (P = .03) than the antibiotic-treated group. Approximately 72% of the gut microbial population of infants at 3–7 days after birth in the control group was transmitted from their mothers, versus only 25% in the antibiotic-treated group. In conclusion, perinatal antibiotics markedly disturbed vertical transmission and changed the source of gut colonization towards horizontal transfer from the environment to the infants.
Keywords: microbiome, gut microbiome, microbiome transmission, antimicrobial resistance genes, AMR, antibiotics, infant microbiome
Widely used perinatal antibiotics resulted in a significantly decreased vertical transmission of microbes from mothers to infants’ gastrointestinal tract after birth. Infants treated with antibiotics had more microbes deriving from other environmental sources.
Antibiotics have revolutionized the treatment of infectious diseases and enabled safe and modern clinical medicine. However, the emergence of antimicrobial resistance (AMR) has become a global threat to this success [1]. Antimicrobials are widely used after birth. During vaginal delivery, 20%–30% of mothers in most developed countries receive antimicrobials to prevent early-onset group B streptococcal sepsis in newborn infants [2, 3], and 2%–5% of newborns are treated with antibiotics owing to suspected sepsis [4, 5].
Over the last 15 years, the scientific community has gained extensive knowledge on human microbiome communities at different body sites [6–9]. Based on metagenomic sequencing and strain-level analysis of human microbiota in mother-to-child transmission, the maternal gut has been found to be the largest source of colonizing bacteria in the gastrointestinal tract of healthy infants [10]. During the neonatal period, mother-to-child microbiome transmission occurs at a much higher frequency in vaginally delivered newborn infants than in those born by cesarean delivery [11]. Vertical transmission of specific strains from mothers to infants is less well understood. Dominant strains in the maternal gut microbiome, comprising >70% of a particular species, are often inherited from mothers to infants [12]. However, infants also acquire strains that are less abundant in the maternal microbiome [12]. Besides strain-level metagenomic profiling, a metatranscriptomic approach was also applied to confirm the activity of transmitted strains [13].
Perinatal antibiotics given during delivery or after birth rapidly changes the colonization process of the gut in both term and preterm infants [14–17]. There are limited data, however, on the impact of perinatal antibiotics on the vertical transmission of microbes and AMR genes from mothers to infants.
Earlier, Tapiainen et al [15] reported the impact of perinatal antibiotics on the colonization of vaginally delivered infants in a prospective cohort. In the present study, we further investigate the impact of perinatal antibiotic on vertical microbe transmission from mother to infant using metagenomic analysis.
MATERIALS AND METHODS
Study Design and Study Population
The details are described elsewhere [15]. Briefly, this prospective controlled cohort included 149 term, vaginally delivered infants followed up from birth to age 12 months. Participants included 4 groups: the control group (neither mother nor infant received antibiotics), the intrapartum antibiotic prophylaxis (IAP) group (mother received IAP, infant did not receive antibiotics), the “postnatal” group (mother did not receive antibiotics, infants received postnatal antibiotics), and the IAP + postnatal group (both mother and infant received antibiotics). The Regional Ethics Committee of the Northern Ostrobothnia Hospital District, Oulu University Hospital, Oulu, Finland, reviewed and approved the study plan. All experiments were performed according to relevant regulations and guidelines. We enrolled only newborn infants whose parents gave written informed consent.
Sample Collection
The details of sample collection are described elsewhere [15]. Daily fecal samples were collected from diapers for infants until discharge. Mothers provided a fecal sample for analysis after birth or soon after discharge. Infants exposed to postnatal antibiotics received Lactobacillus reuteri probiotic until discharge, according to local clinical policy.
The present study investigated a subset of 56 samples from 10 mother-infant pairs in the IAP + postnatal group (the antibiotic-treated group) and 10 mother-infant pairs in the control group. The samples included 20 mothers’ samples collected after birth, 20 infants’ samples collected before discharge from the hospital (3–7 days after birth), and 16 children’s samples collected at 12 months (2 children dropped out per group). This study focused on the combined effects of antibiotics exposure from both mothers and infants. The IAP group and the postnatal group from the original cohort were not investigated.
Metagenomic Sequencing
For metagenomic sequencing, DNA was extracted from fecal samples using the MO BIO Powersoil DNA Extraction kit (MO BIO Laboratories). Paired-end bar-coded libraries were prepared from extracted DNA using the NexteraXT kit (Illumina) and sequenced on a NextSeq instrument (Illumina), generating approximately 1.78 GB of shotgun sequence data (150–base pair [bp] paired end reads) per sample (Supplementary Table 1).
Metagenomic Sequence Analysis
Raw reads were processed using Trimmomatic software [18] to remove low-quality bases and adapter sequences. High-quality reads longer than 100 bp were mapped to the human reference genome (hg38) with BWA-MEM version 0.7.12 [19] and removed if they mapped concordantly with an alignment score of ≥60. The remaining reads were mapped to a reference genome database, compiled and curated from the National Center for Biotechnology Information RefSeq genome database, consisting of genomes from viruses, bacteria, archaea, fungi, and other small microbial organisms. Mapping was performed with BWA-MEM. Top-scored (≥75), concordantly mapped alignments were used for taxonomic profiling, which calculated the relative species abundance at different taxonomic ranks using a voting scheme. High-quality nonhuman reads were assembled into scaffolds using metaSPAdes software version 3.11.0 [20]. Scaffolds shorter than 250 bp were removed. Protein-coding genes were predicted using Prodigal software version 2.6.3 [21]. Genes were annotated by comparison with an internally curated Kyoto Encyclopedia of Genes and Genomes (KEGG) [22] protein reference database using BLASTP+ version 2.7.1. Abundance of KEGG Ortholog clusters and pathways were calculated based on BLAST (Basic Local Alignment Search Tool) search. Protein-coding genes were processed using Resistance Gene Identifier software version 4.2.2 in “strict” mode to predict the antibiotic resistome of each metagenomic sample with the Comprehensive Antibiotic Resistance Database (CARD) database [23].
After metagenome assembly and gene prediction, the nucleotide sequences of all the genes from all samples were pooled and clustered using cd-hit-est version 4.8.1 [24, 25] at 100% sequence identity (command line parameters: -c 1.00 -n 10 -g 1 -G 0 -aS 0.8 -aL 0.8). The gene clusters that are only found in 2 different subjects (not >2) from the same species were considered exclusively shared genes (ESGs). The statistical analysis comparing the number of transmitted species and ESGs between different groups was performed with Wilcoxon rank sum tests, using the R package (R release 3.6.1). (All metagenomes were deposited and are available at the National Center for Biotechnology Information Sequence Read Archive under BioProject no. PRJNA543520.)
RESULTS
Metagenomic sequencing and analysis were performed on a subset of subjects from a previously reported study cohort [15], including 10 mother-infant pairs in the antibiotic-treated group (both mothers and infants received antibiotics) and 10 pairs in the control group (neither mothers nor infants received antibiotics). In the antibiotic-treated group, mothers received antimicrobial prophylaxis, mainly penicillin, during delivery, and infants received postnatal antibiotics, mainly a combination of penicillin and aminoglycoside started within 24 hours after birth because of suspected sepsis. Fifty-six stool samples were studied, including 20 samples from mothers that were collected immediately after birth, 20 newborn samples collected before discharge from the hospital (3–7 days after birth), and 16 infant samples collected at 12 months. Clinical background data for the samples are presented in Supplementary Table 1.
Source of Microbiome Transmission
After bioinformatic processing of each sample, the genes identified from all 56 samples were analyzed to identify the bacterial species and genes shared between samples (see Methods). ESGs, defined as genes that were found in only 2 individuals of all subjects, were compared. In this study, the nucleotide sequences of ESGs were required to be 100% identical. As such, ESGs are strong indicators of the transmission of species and genes from one subject to another. If a single species is found in 2 individuals (1 mother, 1 infant) who have ≥1 ESG that belongs to this species, the species is considered a transmitted species. Similarly, if 1 species and ≥1 ESG belonging to this species are found in a child’s samples at birth and at 12 months, the species is considered persistent.
We found that by 3–7 days after birth, the number of transmitted species in mother-infant pairs was significantly higher in the control group, unexposed to antibiotics (mean, 26 species), than in the antibiotic-treated group (5 species; P = .03) (Table 1) or in unrelated woman-infant pairs in the control group (7 species; P = .005) or the antibiotic-treated group (1 species; P < .001) (Figure 1A). The number of ESGs in the mother-infant pairs in the control group (mean, 1091 ESGs) was also significantly higher than in the antibiotic-treated group (38 ESGs; P = .01) (Table 1) or in unrelated woman-infant pairs in the control group (49 ESGs; P = .002) or the antibiotic-treated group (9 ESGs; P = .001) (Figure 1B). This was also true for the transmitted species with AMR genes or the ESGs that are AMR genes (Table 1 and Figure 1C and 1D). None of the AMR genes in the infants in the antibiotic-treated group originated from their mother’s microbiome.
Table 1.
Numbers of Transmitted Species and Exclusively Shared Genes in Mother-Infant Pairs From Control and Antibiotic-Treated Groups
| Study ID (Control Group; Antibiotic Group) | Transmitted Species or ESGs (AMR Genesa), No. | |||||||
|---|---|---|---|---|---|---|---|---|
| Age 3–7 d | Age 12 mo | |||||||
| Control Group | Antibiotic Group | Control Group | Antibiotic Group | |||||
| Transmitted Species | ESGs | Transmitted Species | ESGs | Transmitted Species | ESGs | Transmitted Species | ESGs | |
| 13; 6 | 20 (1) | 466 (2) | 1 (0) | 2 (0) | 32 (1) | 1656 (1) | 59 (0) | 298 (0) |
| 26; 31 | 8 (1) | 270 (2) | 6 (0) | 26 (0) | 38 (1) | 1297 (2) | 42 (0) | 221 (0) |
| 35; 32 | 0 (0) | 0 (0) | 0 (0) | 0 (0) | NA | NA | ||
| 37; 70 | 37 (0) | 495 (0) | 10 (0) | 74 (0) | 36 (0) | 125 (0) | 68 (0) | 873 (0) |
| 43; 74 | 43 (1) | 2114 (23) | 0 (0) | 0 (0) | 69 (0) | 1344 (0) | 22 (0) | 98 (0) |
| 45; 88 | 18 (1) | 1223 (2) | 0 (0) | 0 (0) | NA | 11 (0) | 32 (0) | |
| 89; 95 | 24 (1) | 1786 (11) | 1 (0) | 1 (0) | 68 (0) | 3144 (0) | 40 (0) | 257 (0) |
| 93; 98 | 0 (0) | 0 (0) | 1 (0) | 1 (0) | 34 (0) | 327 (0) | 18 (0) | 159 (0) |
| 105; 136 | 58 (4) | 2080 (14) | 27 (0) | 274 (0) | 62 (2) | 1278 (3) | NA | |
| 123; 148 | 47 (2) | 2471 (2) | 0 (0) | 0 (0) | 40 (0) | 552 (0) | 62 (1) | 585 (1) |
| Mean; mean | 26 (1) | 1091 (5) | 5 (0) | 38 (0) | 47 (1) | 1215 (1) | 40 (0) | 315 (0) |
Abbreviations: AMR, antimicrobial-resistant; ESGs, exclusively shared genes; ID, identification number; NA, dropped out.
aParenthetical numbers represent species with AMR genes or ESGs that are AMR genes.
Figure 1.

Transmitted species and exclusively shared genes of different adult/mother-infant groups. P values (Wilcoxon rank sum test) for unrelated woman-infant antibiotic-treated group, unrelated woman-infant control group, and paired mother-infant antibiotic-treated group all represent comparisons with the paired mother-infant/child control group. Abbreviations: ESGs, exclusively shared genes.
For the infants at 12 months of age, there was no statistically significant difference between the control and the antibiotic-treated groups in terms of transmitted species in mother-infant pairs. However, our analysis showed that the number of ESGs in mother-infant pairs for infants at 12 months of age, was still significantly higher in the control group (mean, 1215 ESGs) than in the antibiotic-treated group (315 ESGs; P = .02) (Table 1) or in unrelated woman-infant pairs in the control group (20 ESGs; P < .001) or the antibiotic-treated group (26 ESGs; P < .001) (Figure 1E and 1F).
Although a transmitted species is defined when a single ESG exists in a mother-infant pair, the actual detected transmitted species, as listed in Table 1, have multiple ESGs per species. On average, 1 transmitted species has 43 ESGs between the mother-infant pairs in the control group and 8 ESGs in the treatment group. Therefore, the identified transmitted species are supported by multiple ESGs.
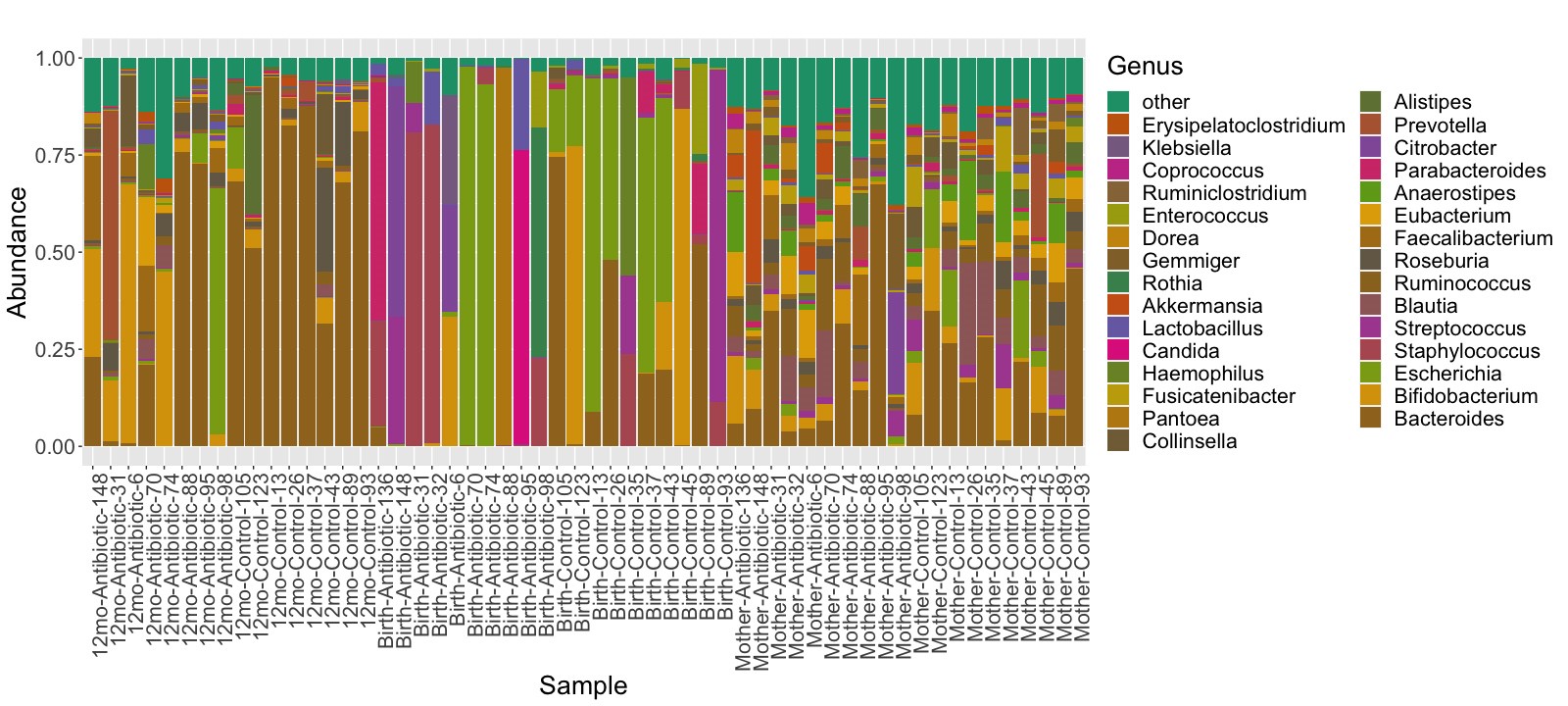
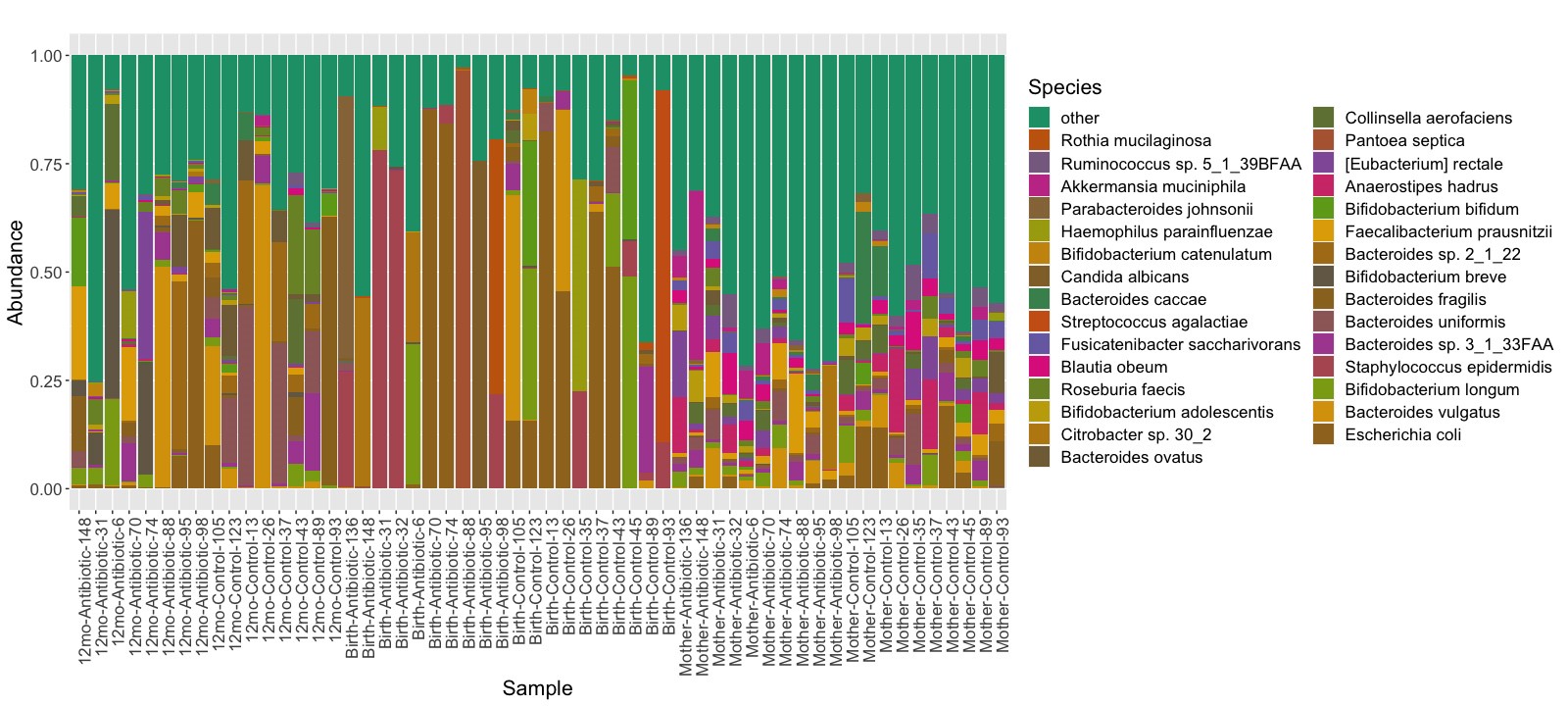
Microbial species composition profiles were calculated at different taxonomic ranks (Figure 2 and Supplementary Figures 1 and 2). The relative abundance data is also available in Supplementary Tables 2–4. For each individual sample from the infants, we identified which species were transmitted from their mother, and which from unknown sources, such as horizontally transferred from the environment or other people (Figure 3). By 3–7 days after birth, on average 72% (σ = 39%) of the species population in the infants’ samples in the control group were shared with their mothers’ samples, compared to only 25% (σ = 38%) in the infants in the antibiotic-treated group (P = .04). At 12 months, the relative abundance of total bacteria species in the fecal samples of infants transmitted from their mothers was 82% (σ = 7%) in the control group and 72% (σ = 21%) in the antibiotic-treated group (P = .38).
Figure 2.

Relative abundance at phylum level.
Figure 3.

Relative species abundance of children’s fecal samples. Infant samples obtained 3–7 days after birth are shown on the bottom, and samples obtained at 12 months on the top. Species on the left side of the x-axis are species transmitted from the maternal gut microbiome. Only species that with ≥5% relative abundance in any sample are shown. (Individual species and their abundances are available in Supplementary Table 5.)
Transmitted Species and AMR Genes
The species that were vertically transmitted were not always the species with a high abundance in the maternal microbiome. In about 75% of cases, the relative abundance of these transmitted species is <1% in the maternal samples. The mean relative abundance of the transmitted species in mothers is 1.3% (σ = 3.0%) (Supplementary Table 6). The most frequently transmitted species from mother to infants in the control group, including the species found in either infants’ stool samples at 3–7 days after birth or at 12 months of age, were Bacteroides sp. 3_1_33FAA (9 occurrences among 10 children), Bacteroides vulgatus (8 occurrences), and Bacteroides fragilis, Bacteroides ovatus, Bacteroides salanitronis, Bifidobacterium longum, and Parabacteroides distasonis (7 occurrences each) (Supplementary Table 7). In the antibiotic-treated group, the most frequently transmitted species were quite different and included Faecalibacterium prausnitzii (8 occurrences among 10 children), B. vulgatus (7 occurrences), and Eubacterium rectale, Anaerostipes hadrus, B. ovatus, Bacteroides sp. 2_1_22, Flavonifractor plautii, Roseburia hominis, Roseburia intestinalis, and Ruminococcus sp. 5_1_39BFAA (6 occurrences each) (Supplementary Table 7).
In the control group, the most abundant species transmitted from mothers to infants at either 3–7 days after birth or at 12 months of age were Escherichia coli (mean relative abundance, 0.28), B. vulgatus (0.13), B. longum (0.10), Bifidobacterium bifidum (0.09), Bacteroides uniformis (0.08), B. fragilis (0.06), and Bacteroides. sp. 2_1_22 (0.05) (Supplementary Table 8). In the antibiotic-treated group, these species were E. coli (mean relative abundance, 0.15), Parabacteroides johnsonii (0.12), Bifidobacterium breve (0.10), Collinsella aerofaciens (0.08), B. longum (0.07), F. prausnitzii (0.07), Citrobacter sp. 30_2 (0.07), B. vulgatus (0.06), and Bacteroides sp. 2_1_22 (0.05) (Supplementary Table 8).
Among the species that were not transmitted from the mother’s gut (Supplementary Table 9), the most commonly seen was Staphylococcus epidermidis. S. epidermidis, an ubiquitous skin colonizer [26], is among the antibiotic-resistant pathogens that cause healthcare-associated infections [27]. Our analysis, which lacks skin samples from mothers, could not confirm the source of S. epidermidis. However, it has been known that human skin and hospital room surfaces share common species [28] and contaminated surfaces in hospitals contribute to the transmission of nosocomial pathogens [29]. We found S. epidermidis in 5 and 4 infants in the antibiotic and the control groups, respectively, at 3–7 days after birth. The relative abundance of S. epidermidis in the antibiotic-treated group (41%) was higher than in the control group (10%; P = .1) (Supplementary Table 9). L. reuteri, which was administered to the infants in the antibiotic-treated group, was found in 4 infants from this group. Several species from the genus Klebsiella were found to be horizontally transmitted from the environment only in the antibiotic-treated group (Supplementary Table 9).
No statistically significant difference was found in the abundance of total AMR genes in infants’ samples between the 2 groups (Figure 4). However, when we separated the AMR genes according to whether they were vertically transmitted from mothers’ gut microbiome, we saw clear differences between the 2 groups. In the antibiotic-treated group, except for an outlier (Sample ID: Birth-antibiotic-70), the main source of AMR genes was not the maternal gut microbiome, especially at 3–7 days after birth, while in the control group, most AMR genes were inherited from the species transmitted from mothers.
Figure 4.

Antimicrobial-resistant (AMR) gene abundance and source of AMR genes in infants’ gut microbiome. The unit of abundance unit is copies per million (the number of reads mapped to the AMR gene per million reads mapped to all genes in the sample). AMR genes in infants transmitted from mothers are not necessarily exclusively shared genes (as listed in Table 1); exclusively shared genes must exist in both mother and infant and must be 100% identical in full gene length. In this figure, an AMR gene in an infant’s sample is considered transmitted if the gene is from a species that can be traced back to the mother’s sample, even if the AMR gene was not found in the mother’s sequencing data.
Persistence of Transmitted Species From Birth Until 12 Months
At 12 months, the control group shared more species with their corresponding 3–7-day samples than the antibiotic-treated group. The control group had significantly higher numbers of persistent bacterial species (mean, 21 species) and ESGs (mean, 1128 species) between 3–7 days and 12 months than the antibiotic-treated group (2 persistent species [P = .02]) and 27 persistent ESGs [P = .02]) (Table 2). This suggests that, in the first week of life, healthy infants vertically inherit a maternal microbiome and AMR signatures, which largely persist at 12 months after birth.
Table 2.
Numbers of Persistent Species and Exclusively Shared Genes Between Samples Collected at 3–7 Days and 12 Months After Birth in the Same Child
| Study ID | Transmitted Species or ESGs (AMR Genesa), No. | |
|---|---|---|
| Persistent Species | ESGs | |
| Control group | ||
| 13 | 20 (0) | 473 (0) |
| 26 | 7 (0) | 443 (0) |
| 37 | 11 (0) | 1608 (0) |
| 43 | 46 (0) | 1187 (0) |
| 89 | 27 (1) | 2769 (0) |
| 93 | 0 (0) | 0 (0) |
| 105 | 44 (0) | 2219 (0) |
| 123 | 16 (0) | 324 (0) |
| Mean | 21 (0) | 1128 (0) |
| Antibiotic group | ||
| 6 | 4 (0) | 170 (0) |
| 31 | 2 (0) | 2 (0) |
| 70 | 10 (0) | 40 (0) |
| 74 | 0 (0) | 0 (0) |
| 88 | 0 (0) | 0 (0) |
| 95 | 0 (0) | 0 (0) |
| 98 | 0 (0) | 0 (0) |
| 148 | 0 (0) | 0 (0) |
| Mean | 2 (0) | 27 (0) |
Abbreviations: AMR, antimicrobial-resistant; ESGs, exclusively shared genes; ID, identification number.
aNumbers in parenthesis are species with AMR genes or ESGs that are AMR genes.
Shift of Microbiome Composition From Birth to 12 Months
Microbial species composition profiles were calculated at different taxonomic ranks, and the relative abundances are provided in Supplementary Tables 2–4. We used a nonmetric multidimensional scaling approach to visualize the similarities between all the samples, using Bray-Curtis distance on species relative abundance (Figure 5A). Infants in the antibiotic-treated group have gut taxonomic composition that is more dissimilar from their mothers (Figure 5A and 5B) at 3–7 days after birth then the control group. However, during the first 12 months, the gut microbiome of the infants in the antibiotic-treated group underwent a bigger shift and mostly returned to normal by age 12 months. Most stool samples from infants at 12 months, in both groups, had an almost adultlike taxonomic composition (Figure 5A).
Figure 5.

Nonmetric multidimensional scaling (MDS) plot of samples and taxonomic distance between groups. A, X-axis and y-axis are the two MDS dimensions. Each point is a sample. A dashed line connects a mother’s sample to an infant’s sample at 3–7 days after birth, then to that child’s sample at 12 months. The 2 overlapping ellipses cover the mothers’ samples in the control and the antibiotic-treated groups. B, Taxonomic distance (range, 0.0–1.0) between different groups. Both plots were based on Bray-Curtis distance on species relative abundance.
DISCUSSION
We show that widely used perinatal antibiotics resulted in a significantly decreased vertical transmission of microbes from mothers to infants at 3–7 days after birth. At the same time, infants in the antibiotic-treated group had an increased proportion of microbes deriving from other environmental sources. Furthermore, infants in the antibiotic-treated group obtained their AMR species from their environment instead from their mothers. This emphasizes the potential role of the hospital microbial environment in the spread of AMR in infants exposed to antibiotics at birth.
The present study demonstrates the vertical transmission of AMR genes from mothers to infants. In an earlier systematic review of 4839 articles [30], only 5 studies about vertical transmission of AMR pathogens or genes were identified, including studies about the transmission of penicillin-resistant Streptococcus pneumoniae [31], antibiotic-resistant group B streptococcus [32], extended β-lactamase producing E. coli [33], ampicillin-resistant E. coli [34], and tetracycline resistance genes [35]. Our study extends the earlier findings and presents the magnitude of the vertical transmission process using metagenome sequencing in a controlled study.
Our results of maternal gut microbiome as the source of infant gut microbiome are analogous to those of the previous studies. Ferretti et al [10] have shown that the maternal gut provides a larger contribution of colonizing bacteria in the gastrointestinal tract in young infants than the skin, oral cavity, or vagina. Although we did not collect skin, oral or vaginal samples from mothers, our study demonstrated that 82% and 72% of bacterial species in children’s samples at 12 months of age originated from the mother’s gut, indicating that the maternal gut microbiome is the major source for gut colonization in newborns. Several transmitted organisms identified in our study, such as species in the genera Bacteroides and Bifidobacterium and in E. coli, are in alignment with transmitted species observed in other studies [10, 11]. Similar to the observations by Yassour et al [12], showing that both primary strains and secondary strains are inherited from mothers to infants, our present study shows that infants acquire both abundant and less abundant species from their mother.
Perinatal antibiotics led to an increased environmental source in the newborn colonization process, which has clinical implications. In classic studies in the 1980s [36], Tullus et al [36] showed that newborn infants treated in certain neonatal wards subsequently had a long-term increased risk of E. coli infections, most likely owing to the early acquisition of nosocomially spread E. coli clones in the gut microbiome. Exposure to perinatal antibiotics may thus result in an increased risk of infections due to strains acquired from the hospital environment instead of the maternal microbiome. Furthermore, prenatal consumption of probiotics has shown some promise in preventing long-term diseases in offspring [37, 38]. Prenatal interventions based on the vertical transmission of microbes from mothers to infants may fail, however, if perinatal antibiotics, interfering with the source of colonization, are used. Finally, nosocomial spread of AMR is a known threat in neonatal wards [39–41]. The nosocomial acquisition of AMR from a hospital environment seems to be more likely in newborn infants exposed to antibiotics owing to the changed colonization process.
Our study is one of the first controlled studies that used metagenomic sequencing to investigate the impact of perinatal antibiotics on newborn microbiome transmission. One of its major strength is the controlled prospective cohort that was followed up for a year. Although our study cohort (n = 10 per group for each group) is not very large, it still enabled us to find significant differences in vertical microbiome transmission between the antibiotic-exposed group and the control group. Cohorts in other related studies, owing to different designs and configurations, are not as well suited to answer the question of how antibiotics affect vertical microbiome transmission, as we have been able to accomplish in our study. For example, although the large cohort by Shao et al [11] included 596 subjects, they only had fecal samples for 5 mother-infant pairs in which both mothers and infants received antibiotic treatment and multiple samples were collected from infants [11], [supplementary table 1].
The antibiotic and microbiome study by Korpela et al [42] included 236 children, but only 14 stool samples were sequenced using a metagenomic approach and no samples were collected from mother. In the study by Yassour et al [12], samples from mothers and infants in 44 mother-infant pairs were sequenced using metagenomic approaches, and multiple samples were collected for the infants until they were 3 month of age. Clear patterns in vertical microbiome transmission were observed in that study, but it did not track antibiotic usage and the cohort did not have an antibiotic usage group. In addition, the infants were followed up until only 3 months of age. The study by Ferretti et al [10] focused on vertical microbiome transmission of different body sites in healthy populations. Only 4 infants received antepartum or intrapartum antibiotic treatments in that cohort [10], [supplementary table 1].
Breastfeeding has been shown to result in marked and persistent differences in the infant gut microbiome [43]. Bacteria in maternal breast milk are likely to be an important source of the infant gut microbiome [44]. In the current study, perinatal antibiotics seemed to disturb the mother-to-infant colonization even in the presence of breastfeeding, because all vaginally delivered infants, including those exposed to perinatal antibiotics, received maternal breast milk. The differences between groups might thus have been even larger in settings with low breastfeeding rates in sick infants receiving antibiotics.
Our cohort study does have some limitations, however. The results may not be generalizable to all infant populations, because the cohort was from a homogenic Finnish population with a rather low background prevalence of clinically significant AMR genes as compared to that in many other countries. We only recruited vaginally delivered infants, so the possible effects in infants born by cesarean delivery were not studied in this population. The relatively small cohort size did not allow comparison of different antibiotics or other factors on the outcome. Therefore, more studies with larger cohorts that include different populations and cesarean-delivered infants are needed to further investigate how antibiotic treatments affect vertical microbiome transmission.
The identification of transmitted species and genes can be influenced by sequencing depth. It is expected that a higher sequencing depth will provide better genome coverage, enabling more transmitted genes to be found. The actual transmitted species and genes may be missed if the sequencing depth is too low for these species to be detected in samples from either mothers or infants. Mothers have more diverse microbiomes than infants. Therefore, the average per-species sequencing depth is much lower in mothers than in infants, so variations in the sequencing depths of samples from mothers have a bigger impact in the analysis of transmitted species or genes. Furthermore, the abundance of transmitted species in mothers’ samples can have a wide range and can be low (Supplementary Table 6). Therefore, in our data set, the number of ESGs is not correlated with the sequencing depth of samples from either mothers or infants. As in typical microbiome studies, the sequencing depths of the samples in this study have a wide range (Supplementary Table 1B). However, there is no significant difference in sequencing depth between control and antibiotic groups in mothers or infants (both P = .81; t test). Therefore, variation in sequencing depth will not affect the significant findings in this study.
The method we developed based on clustering analysis of genes for the detection of transmitted species between microbiome is very effective and sensitive. Several computational tools that offer strain-level analysis on microbiome samples can be used to detect transmission of species. For example, StrainPhlAn [45] is a commonly used program for strain analysis. StrainPhlAn is now part of the MetaPhlAn2 package [46], which analyzes a set of conserved and unique species marker genes to obtain taxonomic profiles for metagenomic samples. StrainPhlAn uses the results from MetaPhlAn2 to detect single-nucleotide polymorphisms (SNPs) so that the SNPs can be compared to find strain-level differences between samples. Because only a limited set of marker genes are analyzed, StrainPhlAn may miss transmission SNPs on other genes.
StrainEST is another program for bacteria strain analysis in microbiome samples [47]. It can perform full-length genome SNP calling, using 1 representative genome per species to build multiple genome alignment for multiple strains in a species before SNP calling. For species with very diverse strains (ie, those with a large pangenome), it is difficult for the single representative genome to cover the whole pangenome. Therefore, this method may fail to detect transmitted species or genes.
MIDAS software is another method for strain level analysis [48]. Like MetaPhlAn2, MIDAS relies on a set of marker genes for taxonomy profiling. But like StrainEST, it performs genome-wide SNP calling. MIDAS was also used for tracking the transmission of species from mother to infant in its original publication [48]. In the publication of MIDAS [48], the transmission of species from mother to infant (healthy population) was studied and it was found that, on average, approximately 40 species were transmitted at birth and approximately 60 species were transmitted at age 12 months. This is very comparable to our findings in the control group at these time points (Table 1). Compared with the above methods, our method for detection of transmitted species and genes has several advantages.
The above-mentioned methods usually require relatively high sequencing depth for SNP calling. Our method does not have this requirement and can work with low-abundance species. Because our method is based on the clustering analysis of all the genes from the pangenomes, it can detect transmission events based on information beyond a subset of marker genes or the representative genome used in the above-mentioned methods. In addition, our analysis is gene focused and can therefore track the transmission of genes and AMR genes, which is an important aspect of our study.
In summary, this study demonstrated that antibiotic use markedly affected the vertical transmission of maternal microbes to infants during early colonization. Antibiotics changed the main source of colonization and led to more horizontally transmitted species from the environment, some of which are potentially pathogenic species common to hospital environments. Our study suggests that vertical transmission is a continuous process. Despite the initial interruption of vertical transmission at birth caused by antibiotics, the children in the antibiotic-treated group still reached adultlike microbiome composition at 12 months of age. However, at 12 months of age, there were still detectable differences between the antibiotic-treated group and the control group, showing the possible long-term effect due to antibiotic use at birth.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
Notes
Acknowledgments. We would like to thank the study nurse, Leena Okkonen, Oulu University Hospital, and the study coordinator, Helena Moilanen, NordLab, Oulu, Finland.
Author contributions. T. T., M. T., and J. S. planned the clinical study design and organized stool sample collection and clinical data collection. K. M. performed metagenomic sequencing. W. L. performed bioinformatic analysis. W. L., L. B., and H. A. L. analyzed the data. W. L., T. T., and K. E. N. wrote the manuscript. All authors approved the final version of the manuscript.
Financial support. The work performed in Finland was supported by the Academy of Finland and Pediatric Research Foundation, Finland. The work performed at the J. Craig Venter Institute, in the United States, was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, US Department of Health and Human Services (grant U19AI110819 to H. A. L. and K. E. N.).
Potential conflicts of interest. All authors: No reported conflicts. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Marston HD, Dixon DM, Knisely JM, Palmore TN, Fauci AS. Antimicrobial resistance. JAMA 2016; 316:1193–204. [DOI] [PubMed] [Google Scholar]
- 2. Schrag SJ, Zywicki S, Farley MM, et al. . Group B streptococcal disease in the era of intrapartum antibiotic prophylaxis. N Engl J Med 2000; 342:15–20. [DOI] [PubMed] [Google Scholar]
- 3. Moore MR, Schrag SJ, Schuchat A. Effects of intrapartum antimicrobial prophylaxis for prevention of group-B-streptococcal disease on the incidence and ecology of early-onset neonatal sepsis. Lancet Infect Dis 2003; 3:201–13. [DOI] [PubMed] [Google Scholar]
- 4. Escobar GJ, Puopolo KM, Wi S, et al. . Stratification of risk of early-onset sepsis in newborns ≥ 34 weeks’ gestation. Pediatrics 2014; 133:30–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fjalstad JW, Stensvold HJ, Bergseng H, et al. . Early-onset sepsis and antibiotic exposure in term infants: a nationwide population-based study in Norway. Pediatr Infect Dis J 2016; 35:1–6. [DOI] [PubMed] [Google Scholar]
- 6. Gill SR, Pop M, Deboy RT, et al. . Metagenomic analysis of the human distal gut microbiome. Science 2006; 312:1355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qin J, Li R, Raes J, et al. ; MetaHIT Consortium . A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012; 486:207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lloyd-Price J, Mahurkar A, Rahnavard G, et al. . Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 2017; 550:61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ferretti P, Pasolli E, Tett A, et al. . Mother-to-infant microbial transmission from different body sites shapes the developing infant gut microbiome. Cell Host Microbe 2018; 24:133–45.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shao Y, Forster SC, Tsaliki E, et al. . Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature 2019; 574:117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yassour M, Jason E, Hogstrom LJ, et al. . Strain-level analysis of mother-to-child bacterial transmission during the first few months of life. Cell Host Microbe 2018; 24:146–54.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Asnicar F, Manara S, Zolfo M, et al. . Studying vertical microbiome transmission from mothers to infants by strain-level metagenomic profiling. mSystems 2017; 2:e00164-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dardas M, Gill SR, Grier A, et al. . The impact of postnatal antibiotics on the preterm intestinal microbiome. Pediatr Res 2014; 76:150–8. [DOI] [PubMed] [Google Scholar]
- 15. Tapiainen T, Koivusaari P, Brinkac L, et al. . Impact of intrapartum and postnatal antibiotics on the gut microbiome and emergence of antimicrobial resistance in infants. Sci Rep 2019; 9:10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yassour M, Vatanen T, Siljander H, et al. . Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med 2016; 8:343ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bokulich NA, Chung J, Battaglia T, et al. . Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 2016; 8:343ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Ilmina sequence data. Bioinformatics 2014; 30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013; 3:13033997. [Google Scholar]
- 20. Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res 2017; 27:824–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 1999; 27:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jia B, Raphenya AR, Alcock B, et al. . CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res 2017; 45:D566–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 2012; 28:3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006; 22:1658–9. [DOI] [PubMed] [Google Scholar]
- 26. Otto M. Staphylococcus epidermidis—the ‘accidental’ pathogen. Nat Rev Microbiol 2009; 7:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Widerström M. Significance of Staphylococcus epidermidis in health care-associated infections, from contaminant to clinically relevant pathogen: this is a wake-up call! J Clin Microbiol 2016; 54:1679–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lax S, Sangwan N, Smith D, et al. . Bacterial colonization and succession in a newly opened hospital. Sci Transl Med 2017; 9:eaah6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Otter JA, Yezli S, French GL. The role played by contaminated surfaces in the transmission of nosocomial pathogens. Infect Control Hosp Epidemiol 2011; 32:687–99. [DOI] [PubMed] [Google Scholar]
- 30. Seale J, Millar M. Perinatal vertical transmission of antibiotic-resistant bacteria: a systematic review and proposed research strategy. BJOG 2014; 121:923–8. [DOI] [PubMed] [Google Scholar]
- 31. McDonald LC, Bryant K, Snyder J. Peripartum transmission of penicillin-resistant Streptococcus pneumoniae. J Clin Microbiol 2003; 41:2258–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Strus M, Pawlik D, Brzychczy-Wloch M, et al. . Group B streptococcus colonization of pregnant women and their children observed on obstetric and neonatal wards of the University Hospital in Krakow, Poland. J Med Microbiol 2009; 58:228–33. [DOI] [PubMed] [Google Scholar]
- 33. Dubois V, De Barbeyrac B, Rogues AM, et al. . CTX-M-producing Escherichia coli in a maternity ward: a likely community importation and evidence of mother-to-neonate transmission. J Antimicrob Chemother 2010; 65:1368–71. [DOI] [PubMed] [Google Scholar]
- 34. Tamelienė R, Barčaitė E, Stonienė D, et al. . Escherichia coli colonization in neonates: prevalence, perinatal transmission, antimicrobial susceptibility, and risk factors. Medicina 2012; 48:71–6. [PubMed] [Google Scholar]
- 35. Alicea-Serrano AM, Contreras M, Magris M, Hidalgo G, Dominguez-Bello MG. Tetracycline resistance genes acquired at birth. Arch Microbiol 2013; 195:447–51. [DOI] [PubMed] [Google Scholar]
- 36. Tullus K, Hörlin K, Svenson SB, Källenius G. Epidemic outbreaks of acute pyelonephritis caused by nosocomial spread of P fimbriated Escherichia coli in children. J Infect Dis 1984; 150:728–36. [DOI] [PubMed] [Google Scholar]
- 37. Osendarp SJ, van Raaij JM, Darmstadt GL, Baqui AH, Hautvast JG, Fuchs GJ. Zinc supplementation during pregnancy and effects on growth and morbidity in low birthweight infants: a randomised placebo controlled trial. Lancet 2001; 357:1080–5. [DOI] [PubMed] [Google Scholar]
- 38. Kukkonen K, Savilahti E, Haahtela T, et al. . Probiotics and prebiotic galacto-oligosaccharides in the prevention of allergic diseases: a randomized, double-blind, placebo-controlled trial. J Allergy Clin Immunol 2007; 119:192–8. [DOI] [PubMed] [Google Scholar]
- 39. Sarvikivi E, Lyytikäinen O, Salmenlinna S, et al. . Clustering of Serratia marcescens infections in a neonatal intensive care unit. Infect Control Hosp Epidemiol 2004; 25:723–9. [DOI] [PubMed] [Google Scholar]
- 40. Maragakis LL, Winkler A, Tucker MG, et al. . Outbreak of multidrug-resistant Serratia marcescens infection in a neonatal intensive care unit. Infect Control Hosp Epidemiol 2008; 29:418–23. [DOI] [PubMed] [Google Scholar]
- 41. Schmithausen RM, Sib E, Exner M, et al. . The washing machine as a reservoir for transmission of extended-spectrum-beta-lactamase (CTX-M-15)-producing Klebsiella oxytoca ST201 to newborns. Appl Environ Microbiol 2019; 85:e01435-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Korpela K, Salonen A, Virta LJ, et al. . Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat Commun 2016; 7:10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ho NT, Li F, Lee-Sarwar KA, et al. . Meta-analysis of effects of exclusive breastfeeding on infant gut microbiota across populations. Nat Commun 2018; 9:4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pannaraj PS, Li F, Cerini C, et al. . Association between breast milk bacterial communities and establishment and development of the infant gut microbiome. JAMA Pediatr 2017; 171:647–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res 2017; 27:626–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Truong DT, Franzosa EA, Tickle TL, et al. . MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 2015; 12:902–3. [DOI] [PubMed] [Google Scholar]
- 47. Albanese D, Donati C. Strain profiling and epidemiology of bacterial species from metagenomic sequencing. Nat Commun 2017; 8:2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nayfach S, Rodriguez-Mueller B, Garud N, Pollard KS. An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res 2016; 26:1612–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.