

Summary
Therapeutic application of induced pluripotent stem cell (iPSC) derivatives requires comprehensive assessment of the integrity of their nuclear and mitochondrial DNA (mtDNA) to exclude oncogenic potential and functional deficits. It is unknown, to which extent mtDNA variants originate from their parental cells or from de novo mutagenesis, and whether dynamics in heteroplasmy levels are caused by inter- and intracellular selection or genetic drift. Sequencing of mtDNA of 26 iPSC clones did not reveal evidence for de novo mutagenesis, or for any selection processes during reprogramming or differentiation. Culture expansion, however, selected against putatively actionable mtDNA mutations. Altogether, our findings point toward a scenario in which intracellular selection of mtDNA variants during culture expansion shapes the mutational landscape of the mitochondrial genome. Our results suggest that intercellular selection and genetic drift exert minor impact and that the bottleneck effect in context of the mtDNA genetic pool might have been overestimated.
Keywords: induced pluripotent stem cells, mitochondrial genome, genomic integrity, small-scale mutations, reprogramming, prolonged expansion culture, selection
Graphical abstract
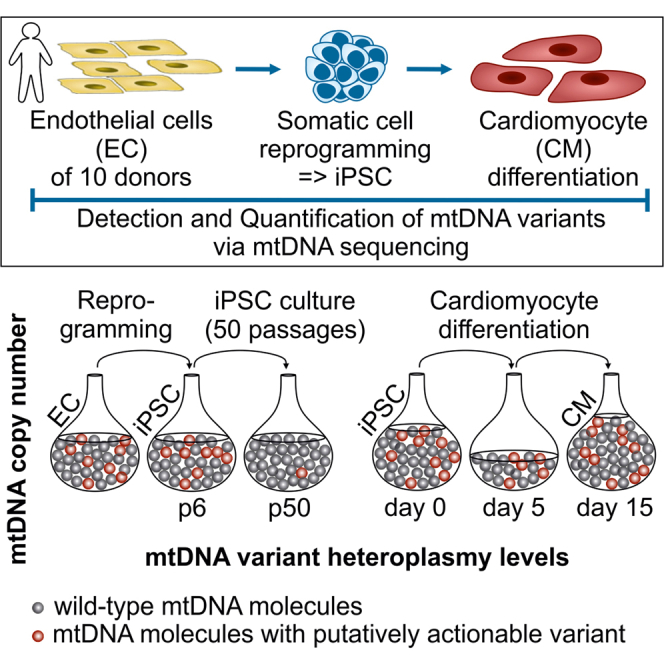
Highlights
-
•
Expansion culture selects against putatively actionable mtDNA mutations in iPSCs
-
•
Intracellular selection on mtDNA molecules shapes the mutational landscape
-
•
Random genetic drift and intercellular selection exert minor impact
-
•
Selection acts during culture expansion but not during reprogramming or differentiation
The results of Kosanke et al. point toward a scenario in which intracellular selection of mitochondrial DNA (mtDNA) variants during iPSC culture expansion, but not during reprogramming or targeted cardiomyocyte differentiation, shapes the mutational landscape of the mitochondrial genome in iPSCs. Other mechanisms, such as intercellular selection, genetic bottleneck, and random genetic drift appear to exert minor impact.
Introduction
Genomic instability of human induced pluripotent stem cells (iPSCs) that may cause loss of function and tumorigenic potential of their derivatives is considered as one major hurdle on the path toward clinical application (Andrews et al., 2017; Weissbein et al., 2014; Yoshihara et al., 2017). The majority of research addressing this issue is focusing on the investigation of genomic stability of the nuclear genome. However, a part of a cell’s genetic information, including 13 proteins of the electron transport chain (ETC) essential for oxidative phosphorylation (OXPHOS) and 24 RNAs, is encoded in the mitochondrial genome (Clayton, 1991). Mutations of those highly conserved genes are the cause of a variety of human diseases, especially affecting tissues with high energy demand (Park and Larsson, 2011).
The mitochondrial genome exhibits a polyploid set with a few hundred up to thousands of 16.6 kb large circular mitochondrial DNA (mtDNA) molecules that are continuously replicated independently of the cell cycle (Clayton, 1991). The existence of multiple mtDNA copies per cells allows a phenomenon called heteroplasmy, which describes the simultaneous existence of wild-type and mutated mtDNA molecules in a cell. Conversely, the state when only one mtDNA genotype is present in a cell is defined as homoplasmy. However, the mtDNA composition and mitochondria network are not fixed but subjected to permanent flux and also, during cell division, the segregation of mutated and wild-type mtDNA molecules to cell progenies can also be unequal. Hence, it is not surprising that, during reprogramming, clonal iPSC lines derived from the same parental cell population were observed to harbor different variants and heteroplasmy levels (Cherry et al., 2013; Folmes et al., 2013; Kang et al., 2016; Perales-Clemente et al., 2016; Prigione et al., 2011; Yokota et al., 2015; Zambelli et al., 2018). This unequal segregation of heteroplasmies during reprogramming might arise from three sources acting on an inter- and intracellular level with different forces depending on the external conditions, namely (1) de novo mutations (Deuse et al., 2019; Kang et al., 2016; Prigione et al., 2011), (2) genetic mosaicism in parental cell population leading to genetically distinct iPSC clones, and (3) random allele drift during genetic bottleneck (Floros et al., 2018; Perales-Clemente et al., 2016; Zambelli et al., 2018). Several recent studies report nuclear reprogramming as the cause of de novo mtDNA mutations in iPSCs (Deuse et al., 2019; Kang et al., 2016; Prigione et al., 2011). However, Payne et al. (2013) introduced the concept of “Universal Heteroplasmy” demonstrating that mosaicism of heteroplasmic mtDNA variants in somatic cells, albeit at low levels (<1%), appears to be a universal finding among healthy individuals. Accordingly, many reports show evidence that the majority if not all mtDNA variants in iPSCs are pre-existing in individual somatic parental cells and arise from this mosaicism in the corresponding parental cell population (Perales-Clemente et al., 2016; Zambelli et al., 2018).
During reprogramming, somatic mitochondria are largely replaced by immature mitochondria resembling organelle morphology and distribution in embryonic stem cells (ESCs), and metabolism is switching toward glycolysis (Ma et al., 2015; Prigione et al., 2010). During this process, mtDNA copy number per cell is reduced, which is assumed to lead to a genetic bottleneck (Cao et al., 2009; Hamalainen et al., 2013). Such a reduction in mtDNA pool increases the effect of random genetic drift on mtDNA segregation within a cell and between cell offspring during cell division (Aryaman et al., 2019; Roze et al., 2005). However, at the same time this bottleneck might expose mutations to selective forces, and subtle selective pressures can exert maximal impact (Floros et al., 2018; Hamalainen et al., 2013; Roze et al., 2005; Wei et al., 2019). As of yet, it is not understood to what extent the segregation is driven by random allele drift or selection (Zambelli et al., 2018).
In contrast to the nuclear genome, selection on mutated mtDNA molecules can act on an intercellular or intracellular level. On the intercellular level, genetically encoded inequalities in cell fitness of parental cells can lead to eliteness of cells to attain iPSC state and their dominance in the reprogramming niche (Kosanke et al., 2021; Shakiba et al., 2019). However, high mutational burden or pathogenic mutations in specific mtDNA regions can also hinder reprogramming (Floros et al., 2018; Hung et al., 2016; Kang et al., 2016; Latorre-Pellicer et al., 2016; Wahlestedt et al., 2014; Yokota et al., 2015).
On the intracellular level, certain mtDNA variants can be selected (Latorre-Pellicer et al., 2016). As mtDNA molecules are uniparentally inherited and the mutation rate is 6- to 20-fold higher than that of genomic DNA (Naue et al., 2015), theoretically, without a repair mechanism, mtDNA would continuously accumulate mutations ultimately ending in a “mutational meltdown” called Muller’s ratchet (Chinnery and Prudent, 2019; Floros et al., 2018). To avert this, during female germline development and early embryogenesis, fragmentation of mitochondria and mitochondrial selective autophagy (mitophagy) purges mutated mtDNA and mitochondria (Chinnery and Prudent, 2019; Floros et al., 2018; Kandul et al., 2016; Latorre-Pellicer et al., 2016; Lieber et al., 2019). As iPSCs experience rejuvenation to an ESC-like state (Prigione et al., 2010), a similar mechanism might act during reprogramming that leads to mitophagy of damaged organelles or reduced segregation of such (Floros et al., 2018).
During prolonged culture of iPSCs, genetic drift and selection continue to form the mutational landscape of the mitochondrial genome. Although most studies report that mtDNA variants did not experience any substantial change of heteroplasmy level during long-term culture, there is also evidence for both positive (Deuse et al., 2019; Zambelli et al., 2018) and negative selection of mtDNA variants (Cherry et al., 2013; Folmes et al., 2013; Perales-Clemente et al., 2016), without clear comprehension of the causes or mechanisms. On one hand, mitochondrial heteroplasmic variants were observed to arise in or dominate human iPSC and ESC lines during prolonged culture (Deuse et al., 2019; Maitra et al., 2005; Zambelli et al., 2018). Notably, single-cell analysis has revealed heterogeneous heteroplasmies among cell populations and there was no indication of selection against potentially pathogenic variants during culture (Zambelli et al., 2018). On the other hand, various studies reported that extended passaging of iPSC clones with high level of heteroplasmy affecting specific genes led to reduction of mutant mtDNA (Cherry et al., 2013; Folmes et al., 2013; Hamalainen et al., 2013; Latorre-Pellicer et al., 2016; Perales-Clemente et al., 2016).
It is, however, currently unknown on which scale the diverse mechanisms shape the mutational landscape of mtDNA. We have, therefore, evaluated to what extent and at which stage random genetic drift and selection might act. To this end, we have analyzed 26 clonal iPSC lines and their corresponding parental cell populations by mtDNA sequencing and determination of their copy number to gain insight into the dynamics and mechanism behind mtDNA segregation during reprogramming, prolonged culture, and differentiation.
Results
No evidence for clonal eliteness or mutation clearance mechanisms during reprogramming
To analyze the effect of reprogramming on mtDNA segregation and mutational landscape, we generated 26 clonal iPSC lines of early passages of endothelial cells (ECs) of 6 neonatal (human umbilical vein EC [hUVEC]) and 4 aged donors (human saphenous vein ECs [hSVEC]; age 64–88 years). To assess inter-clonal differences, 3 iPSC clones were derived per donor, with the exception of D#6 hUVEC and D#40 hSVEC, for which only one clone each was analyzed. In contrast to prior assumptions, mtDNA copy number in early passage iPSCs (p4-9) was, with, on average, 1,252 mtDNA copies per cell, similar to ECs of the corresponding parental cell populations (∼1,114 copies per cell at ∼ p4) and ESCs (∼934 mtDNA copies per cell) (Figure 1A). Although there was no distinct reduction and, hence, bottleneck in mtDNA copy number in iPSCs compared with their parental cells, the mitochondria network was remodeled to a more immature state with mitochondria being punctual and located in the perinuclear region (Figure S1), as described previously (Prigione et al., 2010).
Figure 1.

Characterization of mtDNA copy numbers and mtDNA variants in iPSCs and their parental cells
(A) Average mtDNA copy number per cell in parental endothelial cells (ECs), EC-derived iPSC clones, and ESC lines. EC N = 10 donors; iPSC N = 26 early passage clones; ESC N = 3 lines; each with 1–5 biological replicates. One-way ANOVA with Tukey's multiple comparison test.
(B) Corresponding allele frequencies (AFs) of all variants in iPSC clones and parental cell populations detected by mtDNA sequencing of 26 early passage iPSC clones and their corresponding 10 parental cell populations. Group A non-transmitted variants were detected only in parental cell populations (AF > 0.02). Group B homoplasmic variants in parental cell population and iPSCs derived thereof. Group C heteroplasmic variants detected in an iPSC clone with AF > 0.02 but less frequent in parental cell population. Category 1 comprises haplotype variants, category 2 polymorphisms defined by a population frequency ≥ 0.02, and category 3 variants that are specific to a donor. N = 10 donors.
(C) Proportion of variant groups and categories of all variants. N = 10 donors.
(D) Average number of variants per group and category in iPSC clones and/or corresponding parental cell population of neonatal and aged donors. Neonatal donors N = 6 donors with 16 iPSC clones; aged donors N = 4 donors with 10 iPSC clones. Displayed is mean with SD. ∗Group C (heteroplasmic) variants with heteroplasmy level above 10% (AF > 0.1) in iPSCs.
(E) Effect of variant on gene functionality was predicted based on a consensus of in silico prediction algorithms (snpEff impact, CADD, Condel, and HmtVar). Graph displays ratio of variants with neutral and putatively actionable prediction within group A (non-transmitted), group B (homoplasmic), and group C (heteroplasmic). N = 10 donors.
mtDNA of early passages of iPSCs (mean p6.5) and the corresponding parental cell populations, at a similar passage as subjected to reprogramming (mean p4.5), was sequenced. Analysis of mtDNA variants in iPSCs and ECs revealed three groups. Group A, non-transmitted variants, includes variants detected only within the parental cell populations that were not transmitted to any iPSC clone of the representative donor (detection limit allele frequency [AF] 0.02). In contrast, groups B and C contain transmitted variants with group B variants being homoplasmic in iPSC clones, and their corresponding parental cell populations and group C variants being heteroplasmic and detected in one iPSC clone per donor only (Figure 1B; Tables 1 and S1). The heteroplasmies are defined for every variant separately as the percentage of variant allele count per total allele count at the variant locus (= AF × 100). Although, in most cases the AF of those variants within the parental cell population was below the general detection limit of 0.02, careful assessment of variants within their genomic context taking local error rates in account allowed us to confirm pre-existence of more than half of the variants with statistical confidence (p = 0.05) (Table S2) (see experimental procedures). We could not prove pre-existence of the other half of variants with statistical confidence (Table S2, indicated by value in brackets), but previous reports also support our assumption that most of these variants arose from the parental cell population (Perales-Clemente et al., 2016; Zambelli et al., 2018).
Table 1.
Number of mtDNA variants in iPSC clones and corresponding parental cell populations
| Donor | Haplogroup | Non-transmitted (group A) |
Transmitted (groups B + C) |
|||||
|---|---|---|---|---|---|---|---|---|
| Homoplasmic (group B) |
Heteroplasmic (group C) |
|||||||
| Polymorphisms (cat. 2) | Donor specifi (cat. 3) | Haplogroup variants (cat. 1) | Polymorphisms (cat. 2) | Donor specific (cat. 3) | Polymorphisms (cat. 2) | Donor specific (cat. 3) | ||
| D#1 hUVEC | H7a1 | 0 | 0 | 9 | 3 | 0 | 0 | 1 |
| D#2 hUVEC | H1c3 | 0 | 0 | 10 | 5 | 1 | 0 | 0 |
| D#3 hUVEC | H2a1c | 0 | 1 | 7 | 3 | 0 | 2 | 2 |
| D#4 hUVEC | J1c8a | 0 | 0 | 28 | 3 | 1 | 1 | 1 |
| D#5 hUVEC | J1b1a3 | 0 | 0 | 34 | 8 | 1 | 0 | 0 |
| D#6 hUVEC | H87 | 0 | 0 | 7 | 4 | 0 | 0 | 1 |
| Neonatal donors | mean | 0.0 | 0.2 | 15.8 | 4.3 | 0.5 | 0.5 | 0.8 |
| D#31 hSVEC | H1c2 | 4 | 0 | 9 | 4 | 0 | 0 | 2 |
| D#37 hSVEC | H10b | 0 | 0 | 8 | 3 | 0 | 4 | 6 |
| D#38 hSVEC | I3 | 3 | 0 | 32 | 4 | 0 | 1 | 0 |
| D#40 hSVEC | J1c5a | 1 | 1 | 29 | 4 | 0 | 0 | 0 |
| Aged donors | mean | 2.0 | 0.3 | 19.5 | 3.8 | 0.0 | 1.3 | 2.0 |
| Total | mean | 0.8 | 0.2 | 17.3 | 4.1 | 0.3 | 0.8 | 1.3 |
| sum | 8 | 2 | 73 | 23 | 3 | 8 | 13 | |
Variants of each group can be further categorized: category 1, haplotype variants, are variants that define the haplogroup of every donor. Category 2 variants comprise polymorphisms defined by a population frequency of the variant (NCBI GenBank frequency [GB]) > 0.02. Category 3 variants are unique to a donor and rare in human populations and, are, therefore, called hereafter donor-specific variants (Tables 1 and S1).
Group B (homoplasmic) variants, mainly haplotype variants or polymorphisms (category 1 and 2), represent the vast majority of all variants (>87%) detected in mtDNA. Almost 9% of variants in iPSCs were heteroplasmic (group C) variants that were typically rare among the parental cell population and mainly categorized as donor specific (category 3) (Figure 1C; Table 1).
In contrast to Kang et al. (2016), but in accordance with Payne et al. (2013), we did not observe a dependency of overall mitochondrial mutational load of iPSCs with donor age (Figure 1D; Table 1), which might be partly attributed to different source tissue. Moreover, on average, aged donors tended toward a slightly higher number of group A (non-transmitted) and C (heteroplasmic) variants, indicating higher mosaicism as reported by previous studies (Naue et al., 2015).
Next, we analyzed the functional consequences of variants to evaluate to which extent clonal eliteness or negative selection might impact their transmission from parental cells to iPSC clones. Variant effect prediction based on a consensus of in silico prediction algorithms revealed that all group B (homoplasmic) variants and 90% of group A (non-transmitted) variants are neutral (Figure 1E). Strikingly, almost 48% of the group C (heteroplasmic) variants in iPSCs are putatively actionable, suggesting that mtDNA segregation during reprogramming does not act selectively against potentially damaging mutations. However, while we cannot exclude positive selection, we also found no direct evidence for enrichment of individual mutations or any mutational hotspot in specific genes during reprogramming.
Notably, the heteroplasmy level of the group C (heteroplasmic) variants was generally low (below 10%; AF < 0.1) in iPSCs with the exception of five variants (Figure 1B, marked with ∗). While two of these five variants were neutral polymorphisms with very high heteroplasmy levels >90% (AF > 0.9) in individual iPSC clones, the other three variants were putatively actionable donor specific with intermediate heteroplasmy levels of 16%–38% (AF 0.16–0.38) in single iPSC clones, affecting NADH dehydrogenase (ND) ND4 and ND5 genes of the ETC complex I. Interestingly, complex I, in particular ND5, is frequently mutated in diverse diseases and cancer (Copeland et al., 2002). However, our data do not allow any conclusion whether the affected iPSC clones simply represent stochastic events, where iPSC clones were derived from rare parental cell clones with increased heteroplasmy levels, or whether positive selection processes occurred. Anyway, we could not observe altered mitochondrial features or metabolic functionality of the ND5 mutations in affected iPSC clones (D#3 hUVEC C2 with 38% m.13099G > A and D#37 hSVEC C10 with 23% m.12686T > C) (Figure S2, marked with ∗) compared with their sister clones (D#3 hUVEC C1 and D#37 hSVEC C4) derived from the same parental cell population. All iPSC clones displayed a similar proliferation rate measured as population doubling, mtDNA copy number per cell, specific yield coefficient of lactate per glucose molecule, mitochondrial content, mitochondrial membrane potential, and reactive oxygen species (ROS) content, as well as similar expression of genes involved in metabolism (mTOR, ND5, PDK1, PGC1a, PGC1b, PRKAA1) (Figure S2). Also, genes of mtDNA replication and transcription (POLG2, TFAM, NRF1), mitochondria dynamics (MFN1, OPA1, DNML1), and mitophagy (PINK1, PRKN) were not differentially expressed between non-mutated and mutated iPSC clones, although they were overall significantly higher expressed in iPSCs compared with their parental cells (Figure S2F). As iPSC metabolism is not predominantly based on OXPHOS and the heteroplasmy rates were only 23% and 38%, this result is not unexpected and, in accordance to observations of previous work (Hamalainen et al., 2013; Perales-Clemente et al., 2016; Wahlestedt et al., 2014; Yokota et al., 2015), also decreases the likelihood of a clonal eliteness on the intercellular level during reprogramming.
Prolonged culture expansion leads to clearance of putatively actionable mtDNA mutations
There are contradictory reports regarding the propagation of mtDNA mutations during prolonged culture. While some studies report increase and dominance (Deuse et al., 2019; Maitra et al., 2005; Zambelli et al., 2018), other studies observed a reduced proportion of mutated mtDNA during culture (Cherry et al., 2013; Folmes et al., 2013; Perales-Clemente et al., 2016). Seven of the iPSC clones that had been analyzed in an early passage were also sequenced in later passages p30 and p50 to trace the dynamic of mtDNA heteroplasmies during culture expansion.
It is noteworthy that 86% of the group C (heteroplasmic) variants detected in later passages were also detectable in early iPSC passages and/or the corresponding parental cell population with statistical confidence (p = 0.05). The remaining variants were apparently present in the parental cells and early passages, too, but could not be confirmed with statistical confidence (p = 0.05). Hence, although we cannot exclude it, we also did not observe substantial evidence for de novo mutagenesis during prolonged culture expansion (Table 2).
Table 2.
Group C mtDNA variant heteroplasmy levels change during iPSC culture expansion
 
|
(); existence of variant in parental cell population or iPSC clone not confirmed with statistical confidence (p > 0.05).
Investigation of the regions affected by mutations showed that around 60% of group C (heteroplasmic) variants were located in coding regions (Figure 2A, black) and, therefore, affected coding regions more frequently than group A (non-transmitted) and group B (homoplasmic) variants. Furthermore, compared with group A and B variants which constitute almost entirely nucleotide transitions (transition/transversion ratio [Ts/Tv] > 30), group C (heteroplasmic) variants are characterized by lower T > C transitions compared with C > T and by higher proportion of transversion events (Ts/Tv 3.4) (Figure 2B). Both C > T and T > C substitutions are described as the predominant type of substitution in homoplasmic variants originating from germline transmission (Naue et al., 2015; Wei et al., 2019). The mutational signatures of heteroplasmic group C variants seems to be mainly shaped by the same mechanism; however, abundance of other substitution events suggest an additional origin, such as EC culture expansion or tissue mosaicism of those variants. Consequently, group C mutations would, therefore, never have been subjected to the control and repair mechanisms acting during mtDNA transmission in germ cells.
Figure 2.

Transmitted heteroplasmic group C variants affect coding regions more frequently and are characterized by a higher transversion rate
Twenty-six iPSC clones at an early passage, with 7 of these additionally at later passages, as well as the corresponding parental cell population of the total 10 donors, were subjected to mtDNA sequencing. All variants were classified into three groups. Variants of group A, non-transmitted variants, were detected only in parental cell populations. Group B variants were homoplasmic in parental cell population and iPSCs derived thereof. Group C, heteroplasmic variants, were detected in iPSC clones with AF > 0.02 at any passage but were less frequent in the corresponding parental cell population. N = 10 donors.
(A) Total number of non-transmitted, homoplasmic, and heteroplasmic variants that affected regulatory, RNA coding, or coding regions.
(B) Mutational spectra. Ts, transition; Tv, transversion. Transitions are marked in bold.
Depending on the dynamic of mtDNA mutation progression during culture expansion, all group C variants in iPSCs were divided into four groups, namely enriched, stable, depleted, and fluctuating variants (Table 2, illustrated by the changes in blue shades). While enriched variants are defined as such that increased in AF over iPSC expansion culture more than 10-fold (average 76-fold; range 11- to 250-fold), stable variants did not change their AF during iPSC expansion culture (average fold change between passages 1.03; range 0.67–1.49; SD 0.25). Depleted variants were reduced in AF from the early or intermediate passage to the late one by more than 10-fold (average 64-fold; range 15- to 225-fold) (Table 2). The group of fluctuating variants incorporates basically variants that might be also added to one of the above-mentioned groups, but as their fold change over culture only ranged from 2.7- to 8.8-fold (average 4.7; SD 2.2), we decided not to include them. A last group of variants (named “nd in p30-50”) shown in Table 2 comprises those iPSC-enriched variants that were found in iPSC clones that were only analyzed at early passage.
Among the entirety of group C (heteroplasmic) variants (AF > 0.02), all enriched variants were predicted to be neutral as well as 83% of the stable variants. The group of fluctuating variants or those not analyzed in high passages (nd in p30-50) contained 43%–50% putatively actionable variants (Figure 3A). Most interestingly, 88% of putatively actionable variants was observed among the depleted variants. Focusing only on group C (heteroplasmic) variants with intermediate heteroplasmy levels (AF > 0.1, which equals an averaged heteroplasmy level of >10%), yielded similar results, although the sample size of such was quite small (Figure 3B). In general, donor age did not affect the genetic stability or clearance mechanisms as, in all groups (enriched, stable, depleted, and fluctuating), heteroplasmic group C variants are equally apportioned in neonatal (hUVEC) and aged (hSVEC) donors (Table 2). Furthermore, karyotype analysis performed for D#3 hUVEC C2, D#37 hSVEC C10, and control clone D#3 hUVEC C1 demonstrated that clones maintained normal karyotypes over time in culture (Table S3). However, we did not analyze for smaller chromosomal aberration.
Figure 3.

Prolonged culture expansion selects against putatively actionable mtDNA mutations in iPSCs
Twenty-six iPSC clones at early passage (on average p6.5) and seven of them additionally at intermediate (p30) and late passage (p50) were analyzed by mtDNA sequencing. Heteroplasmic variants (group C) were divided into the groups of enriched variants (defined as >10-fold increase in heteroplasmy level during culture), stable variants (with fold change ∼1), depleted variants (decreased by >10-fold), and fluctuating variants (change of heteroplasmy level did not allow confident classification into one of the above-mentioned groups). The group of “nd in p30-50” comprises variants that were detected in iPSC clones that were only analyzed in the early passage. The ratio of neutral and putatively actionable iPSC-enriched variants within each group was determined. Effect prediction is based on a consensus of in silico prediction algorithms (snpEff impact, CADD, Condel, and HmtVar).
(A) Analysis including all heteroplasmic group C variants with minimum AF 0.02 at any passage of iPSC expansion culture. N = 10 donors.
(B) Analysis including all group C variants with minimum AF 0.1 at any passage. N = 10 donors.
(C) Average mtDNA copy number per iPSC clone in different passages of culture expansion. N = 9 clones. One-way ANOVA with Tukey's multiple comparison test.
(D) Modeling of heteroplasmy variance over passages, exemplarily for three variants with high, intermediate, and low heteroplasmy levels for each group. The model for genetic drift was adapted from Aryaman et al. (2019) and allows prediction of variance of the heteroplasmy level and, therefore, possible change of the heteroplasmy levels over time. As a neutral model, it predicts heteroplasmy variance only based on random gene drift, while selection forces can be incorporated by an additional coefficient. For the different heteroplasmy levels in iPSCs with N = 1,000 mtDNA molecules, the heteroplasmy variances in a neutral situation (green line) and with selection acting with various strength (μ = 2, 5, 10, and 50) are shown.
It is assumed that iPSCs are subjected to a genetic bottleneck when the mtDNA copy number per cell is reduced in the pluripotent state compared with somatic cells. It is still controversially discussed, however, whether random genetic drift during this bottleneck or selection processes are responsible for the observed changes in heteroplasmy levels during prolonged iPSC culture expansion (Aryaman et al., 2019; Hamalainen et al., 2013). Notably, we did not observe any strong reduction in the mtDNA copy number in iPSCs compared with ECs of the parental cell populations (Figure 1A). Furthermore, the mtDNA copy number per iPSCs stayed constant during prolonged iPSC expansion culture (Figure 3C).
Here, we applied a neutral model of mtDNA genetic dynamics of Aryaman et al. (2019) to understand how distribution of heteroplasmies evolves over time without selection. Therefore, this model was utilized as a null hypothesis to assess whether random genetic drift by itself is sufficient to explain the observed changes in heteroplasmy levels, or whether additional selection factors play a role. The model of Aryaman et al. was developed for prediction of a possible change in heteroplasmy level of one mutated allele in a post-mitotic cell with mtDNA undergoing turnover. This means that degradation and replication of mtDNA occurs to the same extent over time, allowing the ratio of mutated allele to fluctuate. Although our culture consists of mitotic iPSCs and requires frequent cell culture splitting, we applied this model here under the assumptions that (1) iPSCs experience symmetric division and equal segregation and expansion of all mtDNA molecules and (2) the effect of random gene drift during iPSC culture passaging is negligible. A more complex model predicting dynamics and selection in a unicellular life cycle with bottleneck and expansion in a multi-cellular organism (Roze et al., 2005) confirms that these assumptions prove valid for our population size (cells in culture before and after splitting). The model of Aryaman et al. includes, in addition, a fragmentation factor, which indicates the ratio of fused mitochondria. As unfused, punctuated mitochondria are a typical feature of iPSCs (Figure S1), we excluded this factor in our modeling. The rate of turnover of mtDNA molecules and also selective mitophagy is set in this model by the factor μ (mitophagy rate). This factor is equivalent to a selection coefficient in other models (Roze et al., 2005).
Determination of mtDNA content has shown that the mtDNA copy number stayed constant over time during iPSC culture expansion (on average ∼1,000 copies/cell) (Figure 3C). Thus, we simulated the possible heteroplasmy variance over time for three different heteroplasmic group C variants exemplarily for each group of enriched, stable, and depleted variants, assuming that cells contain 1,000 mtDNA molecules and no selection is acting (Figure 3D, green line). The variance in heteroplasmy level in cultured iPSCs increases with time according to random genetic drift. However, the modeling also indicates that expectable changes in heteroplasmy due to random genetic drift without computing an additional selection coefficient are very low even over the course of 50 iPSC passages (Figure 3D, green line). Consequently, this result is consistent with the observation made in the group of stable group C variants, which proved to be unchanged over the course of time (fold change on average 1.03, Table 2). However, this simulation without additional selection factor cannot explain the observed increase or decrease in heteroplasmy levels of the enriched or depleted variants. In contrast, incorporating a selection coefficient (included as the μ factor) in the model can explain our observation. For depleted variants, a selection coefficient of μ = 10–50 (Figure 3D, dark and light red lines) was, in general, sufficient to simulate the reduction of heteroplasmies. In contrast, to reach the change in heteroplasmy level observed by enriched variants, μ > 50 would even be needed in most of the cases. Taken together, the analysis of the mtDNA copy number and modeling revealed two aspects: (1) the mtDNA copy number in iPSCs is high enough (no strong bottleneck in the pluripotent state) to prevent noteworthy effects by random genetic drift and (2) enrichment of neutral and clearance of putatively actionable group C (heteroplasmic) variants is likely caused by active selection during iPSC expansion culture.
Dynamics of mtDNA copy number during differentiation does not change variant heteroplasmy levels
To investigate the development of heteroplasmy levels during differentiation and the consequences of mtDNA mutations for differentiated iPSC derivatives, the two iPSC clones harboring intermediate level of putatively actionable ND5 mutations (D#3 hUVEC C2 with 38% m.13099G > A and D#37 hSVEC C10 with 23% m.12686T > C) and their sister iPSC clones (D#3 hUVEC C1 and D#37 hSVEC C4) derived from the same parental cell population were differentiated into cardiomyocytes (CMs). Monitoring of the mtDNA copy number during the differentiation process, surprisingly, revealed a more complex dynamic than expected. Instead of a simple increase of mtDNA copies from iPSCs to CMs with higher metabolic demands, the general dynamic followed a process through three phases (Figure 4A). During the first phase (differentiation day [dd] 0 to dd1), starting with WNT pathway activation by CHIR99021 treatment for 24 h, mtDNA copy number per cell increased. After subsequent WNT pathway inhibition by Wnt-C59 for 48 h (dd1–dd3), mtDNA copy number per cell was reduced reaching its minimum around dd5 or dd6. After dd6 mtDNA copy number increased again to ∼dd10. The mtDNA copy number from dd10 onward stayed unchanged with, on average, 1,532 mtDNA copies per cell (Figure 4A). During reprogramming, such a bottleneck effect is suspected to exert influence on mtDNA segregation. However, analysis of heteroplasmy levels during the differentiation process by mtDNA sequencing at dd0, dd5, and dd15 of differentiation demonstrated that the heteroplasmy level of all variants stayed constant during differentiation, with an average fold change of 1.00 (range 0.83–1.15; SD 0.08) (Figure 4B), a result supported by the observations of previous work (Hamalainen et al., 2013; Zambelli et al., 2018).
Figure 4.

mtDNA content experiences reduction during cardiomyocyte differentiation without change in variant heteroplasmy levels
Two iPSC clones (D#3 hUVEC C2 and D#37 hSVEC C10) each harboring a different putatively actionable mutation in ND5 (m.13099G > A and m.12686T > C, respectively) at intermediate heteroplasmy level and their sister iPSC clones generated from the same parental cell population were differentiated into cardiomyocytes (CMs). Mutated iPSC clones are marked with ∗.
(A) Average mtDNA copy number per cell during CM differentiation. N = 1–2 differentiations.
(B) At different time points during CM differentiation (differentiation day [dd] 0, dd5, and dd15), mtDNA was sequenced. Plot displays the dynamic of heteroplasmy levels during CM differentiation of heteroplasmic variants with AF > 0.02. N = 4 clones.
Moreover, the mtDNA copy number dynamic was not influenced by the presence of the ND5 mutations in iPSC clones D#3 hUVEC C2 or D#37 hSVEC 37 C10 (Figure 4A, marked with ∗). Taking into account the relatively low AF, it is, however, not unexpected that differentiation efficiency, final mtDNA copy number in CMs, and the phenotype of the mitochondria network were similar in the iPSC clones with and without the respective ND5 mutations (Figure S3). Similarly, metabolic features measured as mitochondrial content (MitoTracker), membrane potential (tetramethylrhodamine methyl ester [TMRM]), and ROS (Brite 670) in mutated relative to wild-type iPSC-derived CMs was not significantly affected (Figure S4A). Notably, gene expression quantification demonstrated that, surprisingly, ND5 expression was not upregulated in iPSC-derived CMs (Figure S4B). Hence, beside the relatively low heteroplasmy level of the mutations, additional low expression level of ND5 could explain the absence of any molecular phenotype, at least under our differentiation conditions. Finally, bioartificial cardiac tissues (BCTs) (Figure S4C) were generated out of iPSC-derived CMs. Assessment of BCT diameter did not reveal any difference between tissues generated of CMs from different clones (Figure S4D). However, the analysis of the functionality assessed by active forces of the BCTs demonstrated inter-clonal differences (Figures S4E and S4F). Overall, it seems that the m.13099G > A mutation in ND5 with a heteroplasmy level of 38% in D#3 hUVEC C2-derived CMs resulted in a higher active force of BCTs compared with BCTs generated from the sister iPSC clone D#3 hUVEC C1.
Discussion
Random genetic drift, positive selection, or clearance of mutated mtDNA, are suspected to shape the mutational landscape of mtDNA in iPSCs, but to which extent and at what stage those partly contradicting processes might act is widely unknown. In any case, occurrence of mtDNA mutations that alter the functionality of iPSC derivatives would exclude their clinical application.
Here, we have analyzed mtDNA variants in iPSCs and their corresponding parental cell populations during reprogramming, prolonged culture, and differentiation. We neither observed a distinct bottleneck nor any selection for or against mtDNA variants during reprogramming. Overall, the number of mtDNA molecules per iPSC was similar to the one in parental ECs. All mtDNA variants detected in iPSC clones have also been detected at different levels in the parental cells, although not in all cases with statistical confidence. Thus, we did not observe any evidence for substantial de novo mutagenesis, and inter-clonal differences in iPSCs apparently arose from the mitochondrial variant mosaicism within the parental cell population.
In contrast, during prolonged culture of iPSCs the heteroplasmy level of variants changed, while the mtDNA copy number per cell stayed uniform. Importantly, modeling the effect of random genetic drift on heteroplasmies revealed that a neutral model without selection cannot explain the observed changes. Interestingly, the selection acts pivotally against putatively actionable mutations, while neutral variants are accepted or even enriched. On a functional level, no difference between sister iPSC clones with and without ND5 mutation at intermediate heteroplasmy level derived from the same parental cell population was observed. Although heterogeneity among single iPSCs or karyotypic abnormalities (Zambelli et al., 2018) might have led to elimination of damaged cells or dominance of aberrant cell clones within the iPSC clone culture, this scenario is unlikely as simultaneously other variants (stable heteroplasmic group C variants) were maintained at a constant heteroplasmy level. It is more likely that even minor functional impact of mutations on mitochondria level led to selective mitophagy of damaged mitochondria and mtDNA via PINK1 and Parkin (PRKN) pathways, similarly as described for mtDNA integrity maintenance during germline transition (Kandul et al., 2016; Latorre-Pellicer et al., 2016; Lieber et al., 2019). In particular, PRKN was expressed at significantly higher levels in iPSCs compared with the somatic parental cells. Hence, the clearance of mtDNA mutations is presumably rather executed by an intrinsic process on the intracellular level than on an intercellular level.
The finding that the heteroplasmy levels of individual variants can increase by selection from the parental cell population or culture expansion highlights the need for careful consideration of variants with low heteroplasmy level. Although low heteroplasmy levels are generally regarded as not being clinically relevant, those levels could be augmented reaching the heteroplasmy threshold and impact functionality. Moreover, the heteroplasmy threshold at which a molecular phenotype manifests differs depending on variant and context (Sercel et al., 2021).
Importantly, our results demonstrate that, during targeted CM differentiation, the mtDNA copy number per cell is subjected to a distinct dynamic including an increase-reduction-increase cycle. Interestingly, this dynamic does not influence mtDNA average heteroplasmy levels. This observation leads to two conclusions: (1) the heteroplasmy level in iPSCs dictates the heteroplasmy level of the iPSC-derived CMs and (2) the observed dynamics of the mtDNA copy number during the course of the CM differentiation seems to be not yet strong enough to cause unequal mtDNA segregation.
While we did not observe any functional differences between sister iPSC clones with and without ND5 mutation, CMs generated from D#3 hUVEC C2 with the m.13099G > A mutation at a heteroplasmy level of 38% resulted in BCTs exhibiting higher active forces than BCTs generated from the non-mutated sister clone. Although we cannot exclude further inter-clonal differences between both clones (Mannhardt et al., 2020), reduced levels of mitochondrial proteins (including ND5) can lead to reduction of ATP/ADP ratio, mitochondrial membrane potential, and mitochondrial Ca2+ uptake, followed by increment in cytosolic Ca2+, activation of different signaling pathways, and altered cardiac functions, including hypertrophic cardiomyopathy-specific electrophysiological abnormalities, and elevated force (Bonora et al., 2019; Laude and Simpson, 2009; Li et al., 2018; Mannhardt et al., 2020). Therefore, depending on the (culture) conditions and maturation state, putatively actionable mtDNA variants, even at an intermediate heteroplasmy level, may eventually alter the functional characteristics of iPSC derivatives as reported previously (Hamalainen et al., 2013; Klein Gunnewiek et al., 2020; Wahlestedt et al., 2014; Yokota et al., 2015).
In conclusion, our results suggest that bottleneck effects in iPSC cultures might have been overestimated in the context of the mtDNA genetic pool. Our findings point toward a scenario in which intracellular positive and negative selection of mtDNA molecules mainly shapes the mutational landscape of the mitochondrial genome in iPSCs, while other mechanisms, such as random genetic drift and intercellular selection, exert minor impact.
Experimental procedures
Reprogramming and iPSC culture
Derivation and culture of ECs, virus production, retroviral reprogramming, iPSC characterization, and cultivation were performed as described previously (Haase et al., 2009) (supplemental experimental procedures). In total, 26 EC-derived iPSC clones were derived from 10 donors.
Total DNA extraction and determination of mitochondrial DNA copy number using quantitative real-time PCR
Total DNA was isolated using QIAamp DNA Blood mini kit (QIAGEN). Quantitative real-time PCR was performed on 25 ng total DNA per reaction in duplicates on Mastercycler ep realplex2 (Eppendorf) and with Absolute QPCR SYBR Green Mix (Thermo Scientific). The primer designing is described in the supplemental experimental procedures and primers are listed in Table S4. Sizes of amplicons and absence of nonspecific byproducts were confirmed via melting curve analysis using realplex software (Eppendorf). Quantification was obtained by both calculation of ddCt values and standard curve comparison. mtDNA copy number per cell was calculated as average mtDNA copy number relative to average gDNA copy number/2.
mtDNA extraction and mtDNA sequencing
The workflow for mtDNA sequencing and data processing is displayed in Figure S5A. mtDNA was extracted via QIAprep Spin Miniprep kit (QIAGEN) from early passage iPSC clones (mean p6.5), late passages (p30 and p50) of seven clones, and parental cell populations (mean p4.5). Entire mtDNA was amplified using two primer pairs (F2480A/R10858A [3:1] and F10653B/R2688B [1:1]) (Table S4) generating two overlapping PCR products (McElhoe et al., 2014). Two hundred nanograms of isolate were used as input, which equals ∼5 × 108 mtDNA molecules of, on average, 5 × 106 iPSCs or 2.5 × 106 parental cells. Overall, mtDNA isolation via Miniprep kit and PCR amplification yielded, on average, a 12- and 15,000-fold increase of mtDNA over normal cellular content (Figure S5B). See also supplemental experimental procedures. Amplicons were purified by applying AMPure XP beads (Beckman Coulter) in a 0.6× ratio, sheared by focused ultrasonication (Covaris), and quantified using a Qubit fluorimeter (Invitrogen). Sheared fragments were purified with AMPure XP beads (0.9× ratio), and 150 ng of each sample was fed to a library preparation using a NEBNext Ultra II DNA Library Preparation kit for Illumina (New England Biolabs) according to the manufacturer's instructions. Final concentration, fragment distribution (mean fragment length 600 bp), and quality were assessed on a Qubit fluorimeter and a Agilent Bioanalyzer. Libraries were sequenced as paired-end 250 bp reads using MiSeq Reagent Kit v.2 (Illumina).
Read alignment, variant calling and refinement
Post-run fastq files were imported into Galaxy (v.17.05) (Afgan et al., 2018) instance of RCU Genomics, Hannover Medical School, Germany, for read trimming, quality assessment, alignment, and variant calling. Quality and adapter trimming, as well as read mapping, are presented in the supplemental experimental procedures and as Galaxy workflow in Data S1. Reads were aligned to the human genome GRCh38 chromosome MT (the Cambridge Reference sequence rCRS [NC_012,920]). The coverage was 19,000 on average.
Variants were called utilizing FreeBayes (v.1.0.2; Galaxy implementation v.1.0.2.29-3) with filters set to ploidy 10, minimum coverage 100, and minimum AF (alternative frequency) 0.02. AF ≥ 0.02 as filter criterion was established by manually reviewing called variants within their read context (supplemental experimental procedures). A receiver operating characteristic curve with AF as discrimination threshold (Figure S5C) showed that AF 0.02 retrieves very high specificity while maintaining most true positive variants. False variants with AF ≥ 0.02 comprised the remaining adapter sequences and presumable sequencing errors of C-stretch or C-rich genomic regions and were manually removed. Comparison of dd0 samples of the CM differentiations of three iPSC clones (D#3C1, D#3C2, and D#37C10) and early iPSC passages (p6) of the respective clones proved that variants with AF ≥ 0.02 can be detected in sequencing replicates with repetitious accuracy. The dd0 samples represent independent replicates (separate sample preparation and sequencing) of the sequencing of the corresponding p6 iPSC clones. All heteroplasmic variants (AF 0.02–0.97) showed very similar AFs in both replicates (difference in AF: mean 0.01, median 0.01, range 0.00–0.03) (Table S5). Furthermore, we did not observe large variations in the heteroplasmy levels between dd0, dd5, and dd15 samples of the CM differentiation process of different iPSC clones (fold change ∼1 independent of the variant heteroplasmy level), underlining the high accuracy of our approach (Table S5).
Detection of variants at very low AF
Furthermore, to investigate presence of a choice of variants at very low AF (≤0.02) in iPSC clones or assess their pre-existence in parental cell populations, a total of 128 variants at 38 different genetic regions were examined individually within their genetic context as described previously (Kosanke et al., 2021) (supplemental experimental procedures). The mean error rate and detection limit were calculated individually for every variant, and the presence of variant was confirmed with p = 0.05 against background of errors (Figure S5D; Table S2). Overall, the average coverage of the variant regions was 22,000 and detection limit was 1 read in ∼340 (SD 450), which equals, on average, a detection sensitivity down to AF 0.003.
Variant annotation and prediction of functional consequences
Variant annotation was performed using the web interface of the Ensembl Variant effect predictor (VEP) (v.95) (assembly GRCh38.p12) (McLaren et al., 2016). Haplogroups and corresponding haplotype variants were identified using the HaploGrep algorithm (Weissensteiner et al., 2016). MITOMAP (a human mitochondrial genome database, r103) was utilized to retrieve population frequencies (NCBI GenBank frequency [GB]) of variants. Variants with GB frequency ≥ 0.02 were considered to be polymorphic.
Prediction of functional consequences was obtained from Ensembl VEP and HmtVar (Preste et al., 2019). The functional consequence of a variant was classified by a consensus based on the in silico predictions of snpEff impact prediction, Condel (consensus of SIFT and Polyphen2), CADD, and HmtVar, which comprises predictions of MutPred, Panther, PhD SNP, Polyphen2, and SNPs&GO, and contains information about variants in protein and RNA coding regions. If a variant had a harmful designation by snpEff (high impact = frameshift) or at least by two of the other algorithms (Condel = deleterious, CADD phred > 12, HmtVar = pathogenic), it was considered as putatively actionable.
Modeling heteroplasmy variance over time
A model by Aryaman et al. (2019) was adapted for modeling heteroplasmy variance over time.
where fs is a factor for fragmentation, which indicates the ratio of fused mitochondria, μ is a factor for mitophagy rate, and h is the heteroplasmy level. As iPSCs are featured by unfused mitochondria, fs was excluded in our modeling. n represents the mtDNA copy number/cell. Determination of the mtDNA copy number in iPSCs (n) in p6, p30, and p50 showed that n stayed constant over time in culture and was, on average, 1,000. Heteroplasmy variance (V(h)) was calculated for different heteroplasmy levels with different μ values to assess effect of genetic drift (without selection, μ = 1) and selection (μ = 2, 5, 10, and 50).
Karyotype analysis
Preparation of metaphases, fluorescence R-banding, and analysis of at least 10 metaphases were performed according to standard procedures (supplemental experimental procedures).
Differentiation of CM, and BCT generation and analysis
For embryoid body formation (dd −3), 1 × 106 iPSCs/well were aggregated in 3 mL E8 medium containing 10 μM Rho-Kinase inhibitor Y-27632 (RI) on low attachment six-well plates (Greiner Bio-one) and an orbital shaker (70 rpm; Infors). CM differentiation, and BCT generation and analysis, were performed as described earlier (Halloin et al., 2019; Kensah et al., 2011, 2013) (supplemental experimental procedures).
Live-cell staining and flow cytometric analysis
Live-cell staining was performed of adherent cells with MitoTracker, ROS Brite 670, and TMRM (25 nM; Thermo Fisher Scientific) diluted in Hank’s solution (Gibco) with 20 mM HEPES (Sigma-Aldrich) (HHBS) according to the manufacturer’s recommendation for 45 min. A list of antibodies and dyes is provided in Table S6. Cells were dissociated by Versene (Gibco) or trypsin/EDTA treatment for iPSCs or ECs, respectively, resuspended in HHBS buffer, filtered, and analyzed by flow cytometry. Flow cytometry was executed on a MACSQuant analyzer (Miltenyi Biotec) and data were analyzed with FlowJo (v.10).
Immunofluorescence staining and microscopic analysis
ECs, iPSCs, and CMs were seeded on cover slides and, after a culture period of 1–2 days for ECs and iPSCs, and 3 days for CMs, cells were fixed, permeabilized, and stained with anti-ATPIF1 antibody (Table S6) (supplemental experimental procedures). Cells were analyzed using an Axioserver A1 fluorescent microscope (Zeiss) and Axiovision (v.4.71) software.
Measurement of glucose consumption and lactate production rate
iPSCs were cultured as monolayers in house-made E8 medium. On day 1 after cell seeding, the medium was exchanged and, on the subsequent 2 days, lactate and glucose concentrations from cell-free supernatant were measured employing Biosen C-Line glucose and lactate analyzer (EKF Diagnostics). On each day of analysis, the cell numbers were counted using a Neubauer chamber. Technical triplicates initiated from the same starting cell population were analyzed for each sample and time point. Yield coefficient of lactate from glucose was calculated as described previously (Kropp et al., 2016).
Gene expression analysis via quantitative real-time PCR
Total RNA was isolated via RNeasy Kit (Macherey-Nagel, Düren, Germany) and reverse transcribed utilizing Superscript II (Life Technologies) and random primers according to the manufacturer's instructions. Quantitative real-time PCR was performed on 5 ng cDNA per reaction in duplicates on a CFX Connecet Real-time system (Bio-Rad) and with Absolute QPCR SYBR Green Mix (Thermo Scientific). Primers are listed in Table S4. Absence of nonspecific byproducts was confirmed via melting curve analysis. Expression levels of target genes were normalized to GAPDH and RPL13A transcript levels. The analysis was performed using Bio-Rad CFX Maestro 1.1 (v.4.1.2433.1219) software.
Statistical analyses
If not stated otherwise, N refers to independent batches of cells. GraphPad Prism (v.6.07), RStudio (v.1.1.463; R v.3.5.1), or Bio-Rad CFX Maestro 1.1 software were employed for statistical analysis and visualization. Results are presented as mean and SD. The D’Agostino-Pearson omnibus normality test was executed to check if samples were normally distributed. In cases where the sample size was not sufficiently large, the Shapiro-Wilk normality test and Kolmogorov-Smirnov test were applied. Unpaired two-tailed t test and non-parametric two-tailed Mann Whitney test were employed as appropriate to test for significant differences between groups. One-way ANOVA with post hoc Tukey’s multiple comparisons test and Kruskal-Wallis test with post hoc Dunn multiple comparison test, respectively, were used to test for significant differences between multiple groups.
Comparison of metabolic features of mutated against wild-type iPSC clones or of CMs derived thereof, was underlined by non-parametric Wilcoxon signed-rank test for derivation from hypothetical value of 1.
Local error rates of the amplicon sequencing was calculated and (pre-)existence of variants assessed (p < 0.05) as described previously (Kosanke et al., 2021) (supplemental experimental procedures).
Code availability
Sequencing data analysis was mainly performed on the Galaxy (v.17.05) (Afgan et al., 2018) instance of the RCU Genomics, Hannover Medical School, Germany. The workflow is available in Data S1.
Data availability
Data are deposited in the European Genome-phenome Archive at the European Bioinformatics Institute. The accession number for the study reported in this paper is EGA: EGAS00001005560.
Author contributions
M.K. contributed to designing and coordinating the study, performed, experiments, collected and analyzed data, and wrote the manuscript. C.D. and L.W. performed sequencing and contributed to analyzing bioinformatics data. T.K. performed the experiments. M.D. provided technical assistance in fragment library construction and sequencing. J.G., K.M., and M. Sievert provided technical assistance in performing experiments. M. Szepes, I.G., and A.M. coordinated and performed cardiomyocyte differentiation and analysis. G.G. provided karyotype analysis. U.M. designed and coordinated the study.
Conflicts of interest
The authors declare no competing interests.
Acknowledgments
Many thanks to K. Osetek for isolation of hUVEC and hSVEC, and their reprogramming into iPSCs, to P. Stiefel for several HSVEC isolations, and to T. Scheper for providing bFGF. We are also thankful to S. Rojas, T. Goecke, and other surgical colleagues, who provided human tissue samples. We thank J. Thomson for providing the reprogramming plasmids via Addgene and D. Trono for providing the lentiviral packaging and transfer plasmids psPAX2 and pMD2.G. Many thanks as well to A. Haase and S. Merkert for a critical reading of the manuscript. This work was funded by the German Federal Ministry of Education and Research (CARPuD, 01GM1110A-C; iCARE 01EK1601A), the German Center for Lung Research (DZL, 82DZL00201, 82DZL00401) the German Research Foundation (Cluster of Excellence REBIRTH, EXC 62/3), the Ministry for Science and Culture Lower Saxony (Niedersächsisches Vorab, REBIRTH, ZN 3440), and by the Sächsische AufbauBank/Vita34.
Published: September 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.08.016.
Supplemental information
References
- Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Gruning B.A. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544. doi: 10.1093/nar/gky379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews P.W., Ben-David U., Benvenisty N., Coffey P., Eggan K., Knowles B.B., Nagy A., Pera M., Reubinoff B., Rugg-Gunn P.J. Assessing the safety of human pluripotent stem cells and their derivatives for clinical applications. Stem Cell Reports. 2017;9:1–4. doi: 10.1016/j.stemcr.2017.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aryaman J., Bowles C., Jones N.S., Johnston I.G. Mitochondrial network state scales mtDNA genetic dynamics. Genetics. 2019;212:1429–1443. doi: 10.1534/genetics.119.302423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora M., Wieckowski M.R., Sinclair D.A., Kroemer G., Pinton P., Galluzzi L. Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019;16:33–55. doi: 10.1038/s41569-018-0074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L., Shitara H., Sugimoto M., Hayashi J., Abe K., Yonekawa H. New evidence confirms that the mitochondrial bottleneck is generated without reduction of mitochondrial DNA content in early primordial germ cells of mice. PLoS Genet. 2009;5:e1000756. doi: 10.1371/journal.pgen.1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry A.B., Gagne K.E., McLoughlin E.M., Baccei A., Gorman B., Hartung O., Miller J.D., Zhang J., Zon R.L., Ince T.A. Induced pluripotent stem cells with a mitochondrial DNA deletion. Stem Cells. 2013;31:1287–1297. doi: 10.1002/stem.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery P.F., Prudent J. De-fusing mitochondria defuses the mtDNA time-bomb. Cell Res. 2019;29:781–782. doi: 10.1038/s41422-019-0206-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991;7:453–478. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- Copeland W.C., Wachsman J.T., Johnson F.M., Penta J.S. Mitochondrial DNA alterations in cancer. Cancer Invest. 2002;20:557–569. doi: 10.1081/cnv-120002155. [DOI] [PubMed] [Google Scholar]
- Deuse T., Hu X., Agbor-Enoh S., Koch M., Spitzer M.H., Gravina A., Alawi M., Marishta A., Peters B., Kosaloglu-Yalcin Z. De novo mutations in mitochondrial DNA of iPSCs produce immunogenic neoepitopes in mice and humans. Nat. Biotechnol. 2019;37:1137–1144. doi: 10.1038/s41587-019-0227-7. [DOI] [PubMed] [Google Scholar]
- Floros V.I., Pyle A., Dietmann S., Wei W., Tang W.C.W., Irie N., Payne B., Capalbo A., Noli L., Coxhead J. Segregation of mitochondrial DNA heteroplasmy through a developmental genetic bottleneck in human embryos. Nat. Cell Biol. 2018;20:144–151. doi: 10.1038/s41556-017-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmes C.D., Martinez-Fernandez A., Perales-Clemente E., Li X., McDonald A., Oglesbee D., Hrstka S.C., Perez-Terzic C., Terzic A., Nelson T.J. Disease-causing mitochondrial heteroplasmy segregated within induced pluripotent stem cell clones derived from a patient with MELAS. Stem Cells. 2013;31:1298–1308. doi: 10.1002/stem.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase A., Olmer R., Schwanke K., Wunderlich S., Merkert S., Hess C., Zweigerdt R., Gruh I., Meyer J., Wagner S. Generation of induced pluripotent stem cells from human cord blood. Cell Stem Cell. 2009;5:434–441. doi: 10.1016/j.stem.2009.08.021. [DOI] [PubMed] [Google Scholar]
- Halloin C., Schwanke K., Lobel W., Franke A., Szepes M., Biswanath S., Wunderlich S., Merkert S., Weber N., Osten F. Continuous WNT control enables advanced hPSC cardiac processing and prognostic surface marker identification in chemically defined suspension culture. Stem Cell Reports. 2019;13:366–379. doi: 10.1016/j.stemcr.2019.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamalainen R.H., Manninen T., Koivumaki H., Kislin M., Otonkoski T., Suomalainen A. Tissue- and cell-type-specific manifestations of heteroplasmic mtDNA 3243A>G mutation in human induced pluripotent stem cell-derived disease model. Proc. Natl. Acad. Sci. 2013;110:E3622–E3630. doi: 10.1073/pnas.1311660110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung S.S., Van Bergen N.J., Jackson S., Liang H., Mackey D.A., Hernandez D., Lim S.Y., Hewitt A.W., Trounce I., Pebay A. Study of mitochondrial respiratory defects on reprogramming to human induced pluripotent stem cells. Aging (Albany NY) 2016;8:945–957. doi: 10.18632/aging.100950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandul N.P., Zhang T., Hay B.A., Guo M. Selective removal of deletion-bearing mitochondrial DNA in heteroplasmic Drosophila. Nat. Commun. 2016;7:13100. doi: 10.1038/ncomms13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang E., Wang X., Tippner-Hedges R., Ma H., Folmes C.D., Gutierrez N.M., Lee Y., Van Dyken C., Ahmed R., Li Y. Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell. 2016;18:625–636. doi: 10.1016/j.stem.2016.02.005. [DOI] [PubMed] [Google Scholar]
- Kensah G., Gruh I., Viering J., Schumann H., Dahlmann J., Meyer H., Skvorc D., Bar A., Akhyari P., Heisterkamp A. A novel miniaturized multimodal bioreactor for continuous in situ assessment of bioartificial cardiac tissue during stimulation and maturation. Tissue Eng. Part C Methods. 2011;17:463–473. doi: 10.1089/ten.tec.2010.0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensah G., Roa Lara A., Dahlmann J., Zweigerdt R., Schwanke K., Hegermann J., Skvorc D., Gawol A., Azizian A., Wagner S. Murine and human pluripotent stem cell-derived cardiac bodies form contractile myocardial tissue in vitro. Eur. Heart J. 2013;34:1134–1146. doi: 10.1093/eurheartj/ehs349. [DOI] [PubMed] [Google Scholar]
- Klein Gunnewiek T.M., Van Hugte E.J.H., Frega M., Guardia G.S., Foreman K., Panneman D., Mossink B., Linda K., Keller J.M., Schubert D. m.3243A > G-induced mitochondrial dysfunction impairs human neuronal development and reduces neuronal network activity and synchronicity. Cell Rep. 2020;31:107538. doi: 10.1016/j.celrep.2020.107538. [DOI] [PubMed] [Google Scholar]
- Kosanke M., Osetek K., Haase A., Wiehlmann L., Davenport C., Schwarzer A., Adams F., Schambach A., Merkert S., Wunderlich S. Reprogramming enriches for somatic cell clones with small scale mutations in cancer-associated genes. Mol. Ther. 2021;S1525-0016:00194–00195. doi: 10.1016/j.ymthe.2021.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kropp C., Kempf H., Halloin C., Robles-Diaz D., Franke A., Scheper T., Kinast K., Knorpp T., Joos T.O., Haverich A. Impact of feeding strategies on the scalable expansion of human pluripotent stem cells in single-use stirred tank bioreactors. Stem Cells Transl. Med. 2016;5:1289–1301. doi: 10.5966/sctm.2015-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre-Pellicer A., Moreno-Loshuertos R., Lechuga-Vieco A.V., Sánchez-Cabo F., Torroja C., Acín-Pérez R., Calvo E., Aix E., González-Guerra A., Logan A. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature. 2016;535:561–565. doi: 10.1038/nature18618. [DOI] [PubMed] [Google Scholar]
- Laude A.J., Simpson A.W. Compartmentalized signalling: Ca2+ compartments, microdomains and the many facets of Ca2+ signalling. FEBS J. 2009;276:1800–1816. doi: 10.1111/j.1742-4658.2009.06927.x. [DOI] [PubMed] [Google Scholar]
- Li S., Pan H., Tan C., Sun Y., Song Y., Zhang X., Yang W., Wang X., Li D., Dai Y. Mitochondrial dysfunctions contribute to hypertrophic cardiomyopathy in patient iPSC-derived cardiomyocytes with MT-RNR2 mutation. Stem Cell Reports. 2018;10:808–821. doi: 10.1016/j.stemcr.2018.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber T., Jeedigunta S.P., Palozzi J.M., Lehmann R., Hurd T.R. Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nature. 2019;570:380–384. doi: 10.1038/s41586-019-1213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T., Li J., Xu Y., Yu C., Xu T., Wang H., Liu K., Cao N., Nie B.M., Zhu S.Y. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat. Cell Biol. 2015;17:1379–1387. doi: 10.1038/ncb3256. [DOI] [PubMed] [Google Scholar]
- Maitra A., Arking D.E., Shivapurkar N., Ikeda M., Stastny V., Kassauei K., Sui G., Cutler D.J., Liu Y., Brimble S.N. Genomic alterations in cultured human embryonic stem cells. Nat. Genet. 2005;37:1099–1103. doi: 10.1038/ng1631. [DOI] [PubMed] [Google Scholar]
- Mannhardt I., Saleem U., Mosqueira D., Loos M.F., Ulmer B.M., Lemoine M.D., Larsson C., Ameen C., de Korte T., Vlaming M.L.H. Comparison of 10 control hPSC lines for drug screening in an engineered heart tissue format. Stem Cell Reports. 2020;15:983–998. doi: 10.1016/j.stemcr.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhoe J.A., Holland M.M., Makova K.D., Su M.S., Paul I.M., Baker C.H., Faith S.A., Young B. Development and assessment of an optimized next-generation DNA sequencing approach for the mtgenome using the Illumina MiSeq. Forensic Sci. Int. Genet. 2014;13:20–29. doi: 10.1016/j.fsigen.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naue J., Horer S., Sanger T., Strobl C., Hatzer-Grubwieser P., Parson W., Lutz-Bonengel S. Evidence for frequent and tissue-specific sequence heteroplasmy in human mitochondrial DNA. Mitochondrion. 2015;20:82–94. doi: 10.1016/j.mito.2014.12.002. [DOI] [PubMed] [Google Scholar]
- Park C.B., Larsson N.G. Mitochondrial DNA mutations in disease and aging. J. Cell Biol. 2011;193:809–818. doi: 10.1083/jcb.201010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne B.A., Wilson I.J., Yu-Wai-Man P., Coxhead J., Deehan D., Horvath R., Taylor R.W., Samuels D.C., Santibanez-Koref M., Chinnery P.F. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 2013;22:384–390. doi: 10.1093/hmg/dds435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perales-Clemente E., Cook A.N., Evans J.M., Roellinger S., Secreto F., Emmanuele V., Oglesbee D., Mootha V.K., Hirano M., Schon E.A. Natural underlying mtDNA heteroplasmy as a potential source of intra-person hiPSC variability. EMBO J. 2016;35:1979–1990. doi: 10.15252/embj.201694892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preste R., Vitale O., Clima R., Gasparre G., Attimonelli M. HmtVar: a new resource for human mitochondrial variations and pathogenicity data. Nucleic Acids Res. 2019;47:D1202–D1210. doi: 10.1093/nar/gky1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigione A., Fauler B., Lurz R., Lehrach H., Adjaye J. The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells. 2010;28:721–733. doi: 10.1002/stem.404. [DOI] [PubMed] [Google Scholar]
- Prigione A., Hossini A.M., Lichtner B., Serin A., Fauler B., Megges M., Lurz R., Lehrach H., Makrantonaki E., Zouboulis C.C. Mitochondrial-associated cell death mechanisms are reset to an embryonic-like state in aged donor-derived iPS cells harboring chromosomal aberrations. PLoS One. 2011;6:e27352. doi: 10.1371/journal.pone.0027352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roze D., Rousset F., Michalakis Y. Germline bottlenecks, biparental inheritance and selection on mitochondrial variants: a two-level selection model. Genetics. 2005;170:1385–1399. doi: 10.1534/genetics.104.039495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sercel A.J., Carlson N.M., Patananan A.N., Teitell M.A. Mitochondrial DNA dynamics in reprogramming to pluripotency. Trends Cell Biol. 2021;31:311–323. doi: 10.1016/j.tcb.2020.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakiba N., Fahmy A., Jayakumaran G., McGibbon S., David L., Trcka D., Elbaz J., Puri M.C., Nagy A., van der Kooy D. Cell competition during reprogramming gives rise to dominant clones. Science. 2019;364:eaan0925. doi: 10.1126/science.aan0925. [DOI] [PubMed] [Google Scholar]
- Wahlestedt M., Ameur A., Moraghebi R., Norddahl G.L., Sten G., Woods N.B., Bryder D. Somatic cells with a heavy mitochondrial DNA mutational load render induced pluripotent stem cells with distinct differentiation defects. Stem Cells. 2014;32:1173–1182. doi: 10.1002/stem.1630. [DOI] [PubMed] [Google Scholar]
- Wei W., Tuna S., Keogh M.J., Smith K.R., Aitman T.J., Beales P.L., Bennett D.L., Gale D.P., Bitner-Glindzicz M.A.K., Black G.C. Germline selection shapes human mitochondrial DNA diversity. Science. 2019;364:eaau6520. doi: 10.1126/science.aau6520. [DOI] [PubMed] [Google Scholar]
- Weissbein U., Benvenisty N., Ben-David U. Quality control: genome maintenance in pluripotent stem cells. J. Cell Biol. 2014;204:153–163. doi: 10.1083/jcb.201310135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissensteiner H., Pacher D., Kloss-Brandstatter A., Forer L., Specht G., Bandelt H.J., Kronenberg F., Salas A., Schonherr S. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016;44:W58–W63. doi: 10.1093/nar/gkw233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota M., Hatakeyama H., Okabe S., Ono Y., Goto Y. Mitochondrial respiratory dysfunction caused by a heteroplasmic mitochondrial DNA mutation blocks cellular reprogramming. Hum. Mol. Genet. 2015;24:4698–4709. doi: 10.1093/hmg/ddv201. [DOI] [PubMed] [Google Scholar]
- Yoshihara M., Araki R., Kasama Y., Sunayama M., Abe M., Nishida K., Kawaji H., Hayashizaki Y., Murakawa Y. Hotspots of de novo point mutations in induced pluripotent stem cells. Cell Rep. 2017;21:308–315. doi: 10.1016/j.celrep.2017.09.060. [DOI] [PubMed] [Google Scholar]
- Zambelli F., Mertens J., Dziedzicka D., Sterckx J., Markouli C., Keller A., Tropel P., Jung L., Viville S., Van de Velde H. Random mutagenesis, clonal events, and embryonic or somatic origin determine the mtDNA variant type and load in human pluripotent stem cells. Stem Cell Reports. 2018;11:102–114. doi: 10.1016/j.stemcr.2018.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are deposited in the European Genome-phenome Archive at the European Bioinformatics Institute. The accession number for the study reported in this paper is EGA: EGAS00001005560.