

Abstract
Introduction
Our goal was to determine whether cognitive and cerebrospinal fluid (CSF) markers of tau and amyloid beta 1‐42 (Aβ42) differ between Vietnam‐era veterans with and without history of traumatic brain injury (TBI) and whether TBI moderates the association between CSF markers and neurocognitive functioning.
Methods
A total of 102 male participants (52 TBI, 50 military controls [MCs]; mean age = 68) were included. Levels of CSF Aβ42, tau phosphorylated at the threonine 181 position (p‐tau), and total tau (t‐tau) were quantified. Group differences in CSF markers and cognition as well as the moderating effect of TBI on CSF and cognition associations were explored.
Results
Relative to MCs, the TBI group showed significantly higher p‐tau (P = .01) and t‐tau (P = .02), but no differences in amyloid (P = .09). TBI history moderated the association between CSF tau and performance on a measure of processing speed (t‐tau: P = .04; p‐tau: P = .02).
Discussion
Tau accumulation may represent a mechanism of dementia risk in older veterans with remote TBI.
Keywords: cerebrospinal fluid, tau, traumatic brain injury
1. INTRODUCTION
Traumatic brain injury (TBI) has been identified as a risk factor for the development of neurodegenerative disorders and dementia (e.g., Alzheimer's disease [AD], chronic traumatic encephalopathy [CTE]) in late life. 1 , 2 , 3 Epidemiological studies have shown that individuals with a history of TBI demonstrate an earlier age of dementia onset relative to controls 1 , 4 and that this risk appears to be magnified within the context of repetitive head trauma and increasing injury severity. 1 , 2 , 5 The precise neuropathological mechanisms by which remote TBI contributes to late‐life neurodegeneration is poorly understood, but amyloid beta42 (Aβ42) and/or tau aggregation may play a pivotal role.
As a consequence of TBI, structural damage (e.g., axonal injury) initiates Aβ42 and tau pathogenesis within damaged tissue, and secondary neuroinflammatory cascades contribute to the failed clearance and accumulation of these protein aggregates. 5 , 6 , 7 , 8 , 9 Studies have shown protracted microglia for up to two decades in the aftermath of TBI, 10 and there is evidence for abnormal tau propagation and accumulation in tandem with these changes. 11 Abnormal Aβ42 and tau accumulation are not unique to neurotrauma, as these neuropathologic changes are also well‐established features of pathological aging and a primary driver of AD‐related degenerative processes. 12 However, studies examining these protein markers within the intersection of TBI and aging are limited, and the precise manner in which long‐term neurocognitive outcomes differ as a function of TBI and are ultimately influenced by each protein marker remains unclear.
The current study aimed to (1) understand the nature of cerebrospinal fluid (CSF) markers of Aβ42 and tau in older Vietnam‐era veterans with and without a TBI history, (2) explore potential group differences in cognitive outcomes, and (3) determine the extent to which TBI history may moderate associations between CSF markers and cognitive functioning in older adults.
RESEARCH IN CONTEXT
Systematic Review: The precise neuropathological mechanisms by which remote traumatic brain injury (TBI) contributes to dementia risk is currently poorly understood. Although amyloid beta42 (Aβ42) and/or tau aggregation has been posited to play a pivotal role, studies exploring these cerebrospinal fluid (CSF) protein markers in samples of older adults with history of remote TBI are limited.
Interpretation: Findings from the Department of Defense Alzheimer's Disease Neuroimaging Initiative (DOD‐ADNI) dataset highlight that Vietnam‐era veterans with remote TBI demonstrate greater levels of CSF tau, although no differences in CSF Aβ42 levels were observed. Additionally, elevated levels of tau were associated with poorer processing speed within the TBI group.
Future Directions: Tau pathology may contribute to the increased risk for dementia outlined by epidemiological studies characterizing neurodegenerative disorders after TBI. Additional work using tau positron emission tomography is needed to clarify TBI‐related drivers of tauopathy (e.g., axonal injury, neuroinflammation, repetitive trauma), as well as the spatio‐temporal patterns of tau pathology in the brain.
2. METHODS
2.1. Data, protocol approvals, and patient consent
Data used for the present study were obtained from the publicly available Brain Aging in Vietnam War Veterans/Department of Defense Alzheimer's Disease Neuroimaging Initiative (DOD‐ADNI) database (adni.loni.usc.edu). The study is directed by principal investigator Dr. Michael Weiner of the San Francisco VA Medical Center and University of California, San Francisco. The overarching goals of the DOD‐ADNI study are to characterize the long‐term neural and behavioral consequences of TBI and/or posttraumatic stress disorder (PTSD). The main aims and methods are described in detail elsewhere, 13 and up‐to‐date information can be found at www.adni‐info.org. This research was approved by the institutional review boards of all participating sites within ADNI and written informed consent was obtained for all study participants.
2.2. Participants
Currently, a total of 299 Vietnam War veterans between the ages of 50 and 90 have been recruited into DOD‐ADNI. This study leveraged 102 veterans (TBI: n = 52; military controls [MCs]: n = 50) with available data for the following variables (downloaded on January 1, 2019): CSF Aβ42, tau phosphorylated at the threonine 181 position (p‐tau), and total tau (t‐tau) values; apolipoprotein E (APOE) genotyping; cognitive scores; ADNI clinical group assignments, and key demographic information. Participants were excluded if they endorsed a head injury, but could not provide details surrounding the presence or duration of a loss of consciousness (LOC), alteration of consciousness (AOC), or posttraumatic amnesia (PTA). The larger DOD‐ADNI study also excludes subjects with a diagnosis of mild cognitive impairment or dementia as defined by Mini‐Mental Status Examination scores <24 and Clinical Dementia Rating score of 0.5 or higher. 13
2.3. TBI diagnosis
Self‐reported details about whether a head injury resulted in a hospitalization and the presence and duration of any LOC, AOC, or PTA were recorded for each subject. This information was downloaded from the RECTBIINJ.csv file and the Veterans Affiars (VA)/DoD criteria 2011 14 for TBI was used to determine whether each reported injury met clinical criteria for TBI, as well as to classify the severity of the reported injury. An injury was classified as mild if the participant sustained an LOC <30 minutes, or AOC or PTA <24 hours; moderate if LOC >30 minutes but <24 hours, AOC >24 hours, or PTA >1 day but <7 days; or severe if the participant sustained a LOC ≥24 hours, AOC >24 hours, or PTA ≥7 days.
2.4. Biofluid and genetic markers
CSF samples were collected through lumbar punctures and baseline levels of Aβ42, t‐tau, and p‐tau were measured using Elecsys electrochemi‐luminescence immunoassays on a fully automated Cobas e601 platform. APOE ε4 positivity was determined by the possession of at least one APOE ε4 allele.
2.5. Psychiatric and cognitive variables
The Clinician‐Administered PTSD Scale (CAPS) 15 was used to assess current and lifetime symptoms of posttraumatic stress in accordance with the Diagnostic and Statistical Manual of Mental Disorders—Fourth Edition, Text Revision (DSM‐IV‐TR). 16 Cognitive measures that have previously been associated with TBI‐associated deficits in older adult samples 17 , 18 were selected. All raw scores on measures of processing speed (Trail Making Test Part A), executive functioning (Trail Making Test Part B), verbal learning (Logical Memory I; Rey Auditory Verbal Learning Test [RAVLT] Trials 1 to 5 Total), and verbal memory (Logical Memory II, Total Delayed Recall, and Recognition Total) were converted to z‐scores for analyses. Given there were multiple measures for the verbal learning and memory domains, z‐scores were averaged to create composites scores. Poorer cognitive performance was indicated by higher z‐scores on the processing speed and executive functioning measures, or lower z‐scores on verbal learning and memory composites.
2.6. Statistical analyses
Analyses of variance (ANOVAs) were used to determine whether the groups (MCs vs. TBI) differed on continuous demographic and psychiatric variables. Chi‐squared analyses were used to examine group differences on categorical demographic variables.
Analyses of covariance (ANCOVAs) adjusting for (1) age and APOE ε4 positivity examined group differences in CSF levels of Aβ42, t‐tau, and p‐tau, and (2) age, education, and APOE ε4 positivity were used to examine group differences in cognitive performance. Preliminary analyses revealed that the residuals for Aβ42 were normally distributed, but the tau variables were non‐normally distributed and strongly positively skewed (t‐tau and p‐tau Shapiro‐Wilk's P’s < .001, skewness for t‐tau = 1.16 and p‐tau = 1.29). Therefore, prior to data analysis, Box‐Cox transformations were conducted to normalize tau variables ([x^λ–1]/λ where λ = –0.4 for t‐tau and –0.6 for p‐tau). Multiple linear regressions controlling for age, education, and APOE ε4 positivity were used to determine whether TBI group status moderated CSF and cognitive associations. Follow‐up ANCOVAs and regressions were performed with the current CAPS total included as a covariate. Degrees of freedom slightly differ across analyses: p‐tau data were degraded for two subjects; there were two outliers (z‐scores >3) on Trails A and one outlier on Trails B removed from cognitive analyses; and CAPS data were missing for five subjects.
Reported statistics (i.e., parameter estimates) reflect the difference between estimated marginal means of Box‐Cox transformed values, but untransformed and unadjusted group means are presented within the text and figures to facilitate interpretation. All analyses were performed with the SPSS version 26 19 and R version 3.5.0 (https://cran.r‐project.org/). 20 The Bejamini‐Hochberg method was used control the false discovery rate and adjusted P‐values are reported alongside significant results within the text.
3. RESULTS
3.1. Sample characteristics
Participant demographics and TBI injury characteristics are presented in (Table 1). The TBI group was ≈ 1.4 years older on average than the MCs group (P = .04. ηp 2 = 0.04), and the majority of the TBI group endorsed an injury that was determined to be mild in severity. Clinical data for the groups are presented in (Table 2). There were no group differences in combat exposure, psychiatric symptoms, or health variables (Ps > .05).
TABLE 1.
Sample demographics and clinical characteristics, mean (SD)
| Military controls (n = 50) | TBI (n = 52) | F or χ2 | P | Effect size | |
|---|---|---|---|---|---|
| Age, years | 67.5 (3.5) | 68.9 (3.5) | 4.3 | .04c | ηp 2 = 0.04 |
| Education, years | 14.9 (2.1) | 15.4 (2.5) | 1.0 | .31 | ηp 2 = 0.01 |
| MMSE total score | 28.4 (1.4) | 28.3 (1.8) | 0.1 | .73 | ηp 2 = 0.001 |
| Sex a , male | 100% | 98% | 1.4 | .24 | φ = 0.09 |
| APOE ε4 positivity b , yes | 22% | 23% | 0.2 | .89 | φ = 0.01 |
| Race a | 2.6 | .76 | φ = 0.15 | ||
| American Indian/Alaskan Native | 2% | 2% | |||
| Asian | 2% | 0% | |||
| Black | 6% | 4% | |||
| White | 82% | 90% | |||
| Multi‐racial | 4% | 2% | |||
| Unknown | 4% | 2% | |||
| Highest rank during military service a | – | – | 4.2 | .24 | φ = 0.17 |
| Enlisted | 78% | 75% | |||
| Warrant officer | 0% | 4% | |||
| Officer | 22% | 19% | |||
| Unknown | 0% | 2% | |||
| CSF markers | |||||
| Aβ42, pg/mL | 1197.2(415.7) | 1334.4(540.4) | 2.9 | .09 | ηp 2 = 0.03 |
| Total tau, pg/mL | 192.0 (57.1) | 231.0 (84.5) | 5.9 | .02 c | ηp 2 = 0.06 |
| P‐tau, pg/mL | 16.3 (5.4) | 20.3 (8.2) | 6.7 | .01 c | ηp 2 = 0.07 |
| TBI injury characteristics | |||||
| Total TBI count | 1.4 (0.8), range 1 to 5 | ||||
| Time since last TBI, years | 31.0 (18.8), range 0 to 65 | ||||
| % of individuals with mild versus moderate/severe for worst injury | 63%, 37% | ||||
| % of mild TBI group with a single versus multiple injuries | 64% 36% | ||||
| % of individuals that endorsed an injury that required hospitalization, Yes: No | 54%, 46% | ||||
| % of individuals that endorsed an injury that resulted in a LOC, Yes: No | 69%, 31% | ||||
| % of individuals that endorsed an injury that resulted in an AOC, Yes: No | 85%, 15% | ||||
| % of individuals that endorsed an injury that resulted in PTA, Yes: No | 33%, 67% |
Abbreviations: Aβ, amyloid beta; AOC, alteration of consciousness; APOE, apolipoprotein E; CSF, cerebrospinal fluid; LOC, loss of consciousness; MC, military controls; MMSE, Mini‐Mental Status Examination; PTA, posttraumatic amnesia; SD, standard deviation; TBI, traumatic brain injury.
Denotes likelihood ratio.
Denotes chi‐squared test.
Denotes P < .05.
TABLE 2.
Sample demographics and clinical characteristics
| Military controls (n = 50) | TBI (n = 52) | F or χ2 | P | Effect size | |
|---|---|---|---|---|---|
| Combat exposure and psychiatric symptom severity | |||||
| Served in combat a , yes | 86% | 85% | 0.04 | .84 | φ = 0.02 |
| CAPS current total score | 31.0 (30.8) | 33.7 (26.2) | 0.2 | .65 | ηp 2 = 0.002 |
| CAPS lifetime total score | 43.3 (40.7) | 48.2 (30.1) | 0.4 | .51 | ηp 2 = 0.005 |
| Geriatric Depression Scale total score | 2.8 (3.5) | 2.6 (2.6) | 0.09 | .76 | ηp 2 = 0.001 |
| Medical history and substance use | |||||
| History of diabetes, yes | 44% | 42% | 0.03 | .86 | φ = 0.02 |
| History of high blood pressure, yes | 66% | 56% | 1.1 | .29 | φ = 0.11 |
| History of alcohol use disorder, yes | 37% | 45% | 0.72 | .40 | φ = 0.09 |
| History of substance use disorder, yes | 12% | 10% | 0.15 | .69 | φ = 0.04 |
Abbreviations: CAPS, Clinician Administered PTSD Scale; MCs, military controls; MMSE, Mini‐Mental Status Examination; TBI, traumatic brain injury.
Denotes chi‐squared test
‡Denotes P < .05.
Please note that five subjects (three from the MCs and two from the TBI group) were missing CAPS data; two TBI subjects (one from the MCs and one from the TBI group) were missing alcohol or substance use information.
3.2. Group comparisons on CSF markers
ANCOVAs adjusting for age and APOE ε4 positivity revealed that the TBI group displayed significantly higher levels of p‐tau (F [1, 96] = 6.86, P = .01, adjusted P = .03, ηp 2 = 0.07) and t‐tau (F [1, 98] = 5.91, P = .02, adjusted P = .04, ηp 2 = 0.06). In contrast, there was no main effect of group for Aβ42 (F [1, 98] = 2.91, P = .09, ηp 2 = 0.03), although CSF levels of Aβ42 were higher on average within the TBI group versus controls (see Table 1 and Figure 1).
FIGURE 1.
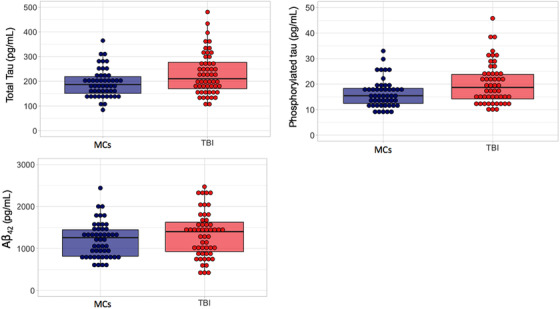
Group comparisons on cerebrospinal fluid (CSF) protein markers of amyloid beta (Aβ)42 and total tau (t‐tau) or tau phosphorylated at the threonine 181 positionp‐tau. Top left panel depicts significant group differences in CSF levels of t‐tau (left) versus right top right shows p‐tau (right). Bottom row depicts Aβ levels between the two groups
3.3. Group comparisons on cognitive performance
ANCOVAs adjusting for age, education, and APOE ε4 positivity revealed that the TBI group performed significantly worse than the MC group on the delayed memory composite (F [1, 96] = 7.90, P = .006, adjusted P = .02, ηp 2 = 0.08). There were no significant group differences in performance on the verbal learning composite (F [1, 96] = 1.24, P = .27, ηp 2 = 0.01), Trails A (F [1, 94] = .11, P = .74, ηp 2 = 0.001), or Trails B (F [1, 92] = 0.78, P = .37, ηp 2 = 0.009). See Table 3.
TABLE 3.
MCs versus TBI group comparisons on cognitive variables of interest
| Military controls (n = 50) | TBI (n = 52) | F | P | Effect size | |
|---|---|---|---|---|---|
| Verbal learning composite z‐score | 0.05 (0.85) | −0.05 (0.82) | 1.24 | .27 | ηp 2 = 0.01 |
| Verbal memory composite z‐score | 0.22 (0.69) | −0.21 (0.81) | 7.90 | .006 | ηp 2 = 0.08 |
| Trails A total time z‐score | −0.15 (0.66) | −0.05 (0.63) | 0.11 | .74 | ηp 2 = 0.001 |
| Trails B total time z‐score | −0.16 (0.77) | 0.04 (0.86) | 0.79 | .37 | ηp 2 = 0.009 |
Abbreviations: AVLT, Auditory Verbal Learning Test; MCs, military controls; TBI, traumatic brain injury.
Note: One MC subject was missing AVLT data for the verbal learning and memory composites; one MC subject was missing data for Trails A and two TBI subjects were determined to be outliers for Trails A data; three MCs and one TBI were missing Trails B data and one TBI subject was determined to be an outlier.
3.4. Group by CSF interactions on cognitive performance
With respect to Trails A performance, there were significant group × p‐tau (unstandardized β = 5.08, t = 2.39, P = .02, adjusted P = .06, rpart 2 = 0.24) and group × t‐tau (unstandardized β = 6.93, t = 2.06, P = .04, adjusted P = .08, rpart 2 = 0.20) interactions. Examination of main effects revealed that poorer processing speed was associated with higher levels of p‐tau (unstandardized β = 3.74, t = 2.74, P = .009, rpart 2 = 0.37) and t‐tau (unstandardized β = 4.20, t = 1.91, P = .06, rpart 2 = 0.27); however, there were no such significant associations observed within the MC group (all P's > .41). See Figure 2. No significant group × Aβ42 interactions were observed (unstandardized β = 0.0001, t = –.51, P = .62, rpart 2 = –0.05) for Trails A performance.
FIGURE 2.
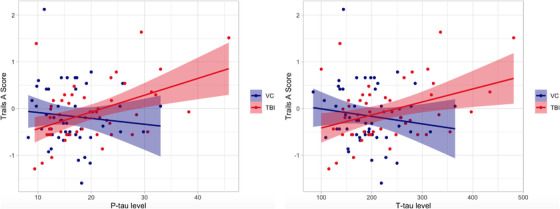
Group (military controls [MCs] vs. traumatic brain injury [TBI]) x tau interactions on processing speed. Left panel depicts the significant associations between p‐tau and Trails Making Test A performance within TBI and MCs groups. Right panel depicts between t‐tau and Trails A performance within TBI and MCs groups
With regard to Trails B performance, no significant interactions for the group × p‐tau (unstandardized β = 5.89, t = 2.00, P = .05, rpart 2 = 0.21) and group x t‐tau (unstandardized β = 8.21, t = 1.80, P = .08, rpart 2 = 0.18). No significant group × Aβ42 interaction was observed (unstandardized B = 0.0001, t = 0.36, P = .72,rpart 2 = 0.04) for Trails B. Additionally, there were no significant group × CSF t‐tau, p‐tau, or Aβ42 interactions for verbal learning (Ps > .51) or memory composites (Ps > .18).
3.5. Sensitivity analyses adjusting for PTSD
A series of follow‐up analyses were conducted with current CAPS total score included as a covariate in the significant models described above. Results from the original ANCOVAs were retained for the p‐tau (F [1, 90] = 4.56, P = .04, ηp 2 = 0.05) analyses, but were attenuated for t‐tau (F [1, 92] = 3.91, P = .05, ηp 2 = 0.04). Notably, current CAPS total score was not associated with t‐tau (F = 0.43, P = 0.51, ηp 2 = 0.005) or p‐tau (F = 0.11, P = .739, ηp 2 = 0.001) in the models.
With regard to cognition, ANCOVAs revealed that the TBI group performed significantly worse than the MC group on the verbal memory composite (F [1, 90] = 6.15, P = .02, ηp 2 = 0.06) and current CAPS total score was not associated cognition (F = 0.88, P = .35, ηp 2 = 0.01) in the model.
Finally, findings from the group x CSF interactions regressions demonstrated that the group x p‐tau interactions for Trails A (unstandardized β = 4.50, t = 2.13, P = .04, rpart 2 = 0.22) remained significant, but the group x t‐tau interaction for Trails A (unstandardized β = 6.10, t = 1.81, P = .07, rpart 2 = 0.19) was slightly attenuated.
4. DISCUSSION
Our study sought to clarify whether group differences in CSF markers of AD pathologic processes (Aβ42, p‐tau, and t‐tau) and cognitive functioning could be observed between older Vietnam War–era veterans with and without history of TBI. We also tested whether TBI history moderated the association between CSF protein markers and cognitive functioning. Results showed that, relative to MCs, older Veterans with history of TBI displayed elevated levels of both CSF p‐tau and t‐tau, although there were no group differences in Aβ42. Additionally, the TBI group performed significantly worse than the MC group on verbal memory measures, although higher levels of CSF tau were associated with slower processing speed and worse executive functioning within the TBI group only. Findings suggest that prior TBI may lead to elevated levels of CSF tau but not Aβ42 many years after initial injury. Results provide preliminary evidence that tau‐related disease activity negatively influences cognition in late‐life and highlight potential pathologic changes that may explain higher rates of dementia after head trauma. 1 , 2 , 3
Our findings of elevated levels of t‐tau and p‐tau in older veterans with TBI provide some support for the biophysical model of tau pathogenesis. 21 , 22 Tau is a microtubule‐associated protein involved in regulating axonal structure 23 and high tensile strain during neurotrauma causes stretching and shearing of the axon. This damage causes tau to detach, phosphorylate, and aggregate into neurofibrillary tangles that cannot be cleared. Importantly, tau pathogenesis also appears to be partially mediated by neuroinflammatory processes 24 and continued Wallerian degeneration may act as a conduit for abnormal tau propagation and accumulation at sites distal from initial injury. 25 , 26 Although the temporal course of these pathophysiological processes remains unclear, increased t‐tau is thought to reflect ongoing axonal injury and degeneration, whereas higher p‐tau is more reflective of tangle pathology. 27 , 28 Thus, our results provide evidence of both an evolving and consequential pathological disease state in older adults with remote TBI histories.
Tau protein aggregation is not unique to neurotrauma and occurs across the continuum of healthy to pathologic aging. 29 , 30 , 31 , 32 Although elevated levels CSF, 28 plasma, 33 , 34 and PET tau 35 , 36 in older adults across, our study suggests that primary age‐related processes alone do not fully account for elevated CSF tau in older adults with remote TBI histories. Our work also extends findings demonstrating elevated levels of tau in middle‐aged TBI samples 37 , 38 , 39 , 40 and aligns with a recent study illustrating that central nervous system‐enriched blood‐based exosomal markers of tau differentiated older adults with and without TBI histories. 41 Nevertheless, additional longitudinal tau PET imaging, and histopathological studies, are needed to disentangle the spatio‐temporal patterns of tau pathology within the brain and the what neurodegenerative process (e.g., AD, CTE) tau elevations represent. Moreover, as illustrated by Peltz et al., 41 there is no obvious single pathology underlying TBI history in older adults and use of multiple biomarkers in tandem may further aid in identifying older adults with cognitive impairment after TBI.
We failed to find group differences in CSF Aβ42 pathology, which somewhat contrasts with existing literature demonstrating lower CSF Aβ42 (indicative of greater cerebral protein accumulation and plaque formation) in other mixed severity TBI samples 42 , 43 , 44 as well as diffuse Aβ plaque accumulation on histopathological examinations of acute and long‐term TBI survivors. 5 , 45 , 46 Differences in sample characteristics (e.g., time since injury, severity of injury) partially explain discrepant findings across studies. Most existing CSF studies have examined moderate‐to‐severe TBI samples within weeks of injury and temporal variations in CSF Aβ42 have been noted in the acute phase of injury. 44 , 47 Nevertheless, a recent ADNI‐DoD study showed increased Aβ deposition detected by [18F]‐AV45‐PET in a subset of individuals with TBI and comorbid PTSD compared to controls, 48 suggesting that CSF protein markers of amyloid may be a less sensitive biomarker of TBI relative to amyloid neuroimaging techniques.
Our study demonstrated that veterans with a history of TBI perform more poorly than MCs on verbal memory measures, although there was no association between CSF protein markers and memory performance within this sample. These findings suggest that there may be another biological mechanism responsible for poor memory performance within our TBI group and certainly warrants further exploration. Importantly, compared to veterans with no TBI, similar deficits in processing speed and executive functioning have been noted in an independent sample of slightly older veterans (mean age = 79) with remote TBI. 17 Although we failed to find group differences in performance within these domains, we observed that higher levels of CSF tau appear to be particularly deleterious for the cognitive domains of processing speed and executive functioning performance for older adults with TBI. Additional tau PET imaging techniques are needed to target regional specificity of these findings, as results suggest a potential regional vulnerability of frontal‐subcortical regions which has been shown across many studies to be characteristic of neurotrauma that may become more pronounced with advancing age. 49
Although our sample consisted mostly of individuals with mild TBI, we conducted a series of exploratory analyses to better understand the potential role of TBI injury severity in the pattern of observed results (see supporting information). Results revealed that elevated levels of tau were largely driven by those with mild TBI, although the smaller sample size of the moderate/severe TBI group likely contributed to low power, which potentially hindered the detection of group differences. Additionally, cognitive analyses revealed no significant differences in performance between the moderate/severe and mild TBI groups. Findings suggest elevated levels of tau and the observed associations with cognition are not merely the byproduct of increasing injury severity. Finally, psychiatric symptoms have been independently linked to elevated levels of tau accumulation in younger military TBI samples. 50 However, our results were only somewhat attenuated by the inclusion of CAPS total scores within the model; PTSD symptoms were not a significant predictor of cognitive performance within our models. We suspect that TBI rather than PTSD is the primary moderator of tau and cognitive associations among older veterans and results highlight the need to take into account remote TBI history in older adult samples and provide evidence of potentially long‐lasting brain changes that are not simply the byproduct of comorbid psychiatric distress.
There are many strengths of the study including the application of VA/DoD TBI criteria for enhanced reliability of TBI diagnosis, robust Elecsys CSF analysis methods, and consideration of PTSD and TBI injury severity. However, limitations that warrant careful consideration include: retrospective self‐report and potential recall bias of TBI injury details; the cross‐sectional nature and associated study observations could be the result of pre‐injury differences; and while ADNI‐DOD is a robust dataset for exploration of the long‐term consequences of head‐trauma, peripheral markers of neurodegeneration or inflammation (e.g., neurofilament light) that could provide insight into relevant mechanisms of injury are not captured. Future longitudinal studies are needed to better elucidate the link between CSF biomarkers of neurodegeneration and cognitive decline in older adults with histories of neurotrauma. Moreover, given the limited racial/ethnic and sex representation of these samples, there is a critical need to better understand whether and how these pathologic processes may differ in more diverse samples.
5. CONCLUSIONS
This study demonstrated that CSF tau is elevated in Vietnam War–era veterans with remote TBI histories and higher tau is associated with poorer neurocognitive performance. Results dovetail with previous work suggesting that TBI may be associated with pathological brain changes that persist well into late‐life and provide a potential mechanism for increased dementia risk. Future research studies should replicate these findings in larger samples and explore the spatio‐temporal course of tau accumulation after TBI across the lifespan.
AUTHOR CONTRIBUTIONS
Alexandra L. Clark: design and conceptualization of study design, data analysis, data interpretation, drafting of manuscript for intellectual content, statistical analysis. Alexandra J. Weigand: data analysis, data interpretation, drafting of manuscript for intellectual content, statistical analysis. Katherine J. Bangen: data interpretation, drafting of manuscript for intellectual content. Kelsey R. Thomas: data interpretation, drafting of manuscript for intellectual content. Graham M.L. Eglit: data interpretation, drafting of manuscript for intellectual content. Mark W. Bondi: data interpretation, revision of manuscript for intellectual content. Lisa Delano‐Wood: data interpretation, revision of manuscript for intellectual content.
Supporting information
SUPPLEMENTAL MATERIAL
ACKNOWLEDGMENTS
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI; National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. The authors thank all participants of the Alzheimer's Disease Neuroimaging Initiative for providing data for this manuscript, as well as the individuals who work to make these data available for public use. This work was further supported by Veterans Affairs and Department of Defense grants awarded to Dr. Delano‐Wood and a National Institutes of Health (National Institute of Neurological Disorders and Stroke Ruth‐Kirschstein Fellowship) and a Veterans Affairs Advanced Polytrauma and TBI Rehabilitation Research Fellowship awarded to Dr. Clark. Dr. Eglit reports no disclosures. Drs. Clark, Bangen, Thomas, and Ms. Weigand are members of the International Society to Advance Alzheimer's Research and Treatment. Drs. Clark, Bangen, and Thomas have received funding from the Shiley‐Marcos Alzheimer's Disease Research Center. Dr. Bangen serves as an Associate Editor of the Journal of Alzheimer's Disease and has previously received grant funding from the Alzheimer's Association. Dr. Thomas has previously received grant funding from the Alzheimer's Association. Dr. Bondi receives royalties from Oxford University Press and serves as a consultant for Eisai, Novartis, and Roche Pharmaceutical companies. Dr. Delano‐Wood has received honoria from Takeda and is a board member of the San Diego Alzheimer's Association chapter.
APPENDIX 1. ADNI investigators
1.1.
| Name | Location | Role | Contribution |
|---|---|---|---|
| Michael W. Weiner, MD | University of California, San Francisco | Principal investigator | Administrative core |
| Andrew J. Saykin, PsyD | Indiana University | Principal investigator | Genetics core |
| John Q. Trojanowski, MD, PhD | University of Pennsylvania | Principal investigator | Biomarker core |
| Leslie Shaw, PhD | University of Pennsylvania | Principal investigator | Biomarker core |
| Arthur W. Toga, PhD | University of Southern California | Principal investigator | Informatics core |
| Laurel Beckett, PhD | University of California, Davis | Principal investigator | Biostatistics core |
| Clifford Jack, MD | Mayo Clinic | Principal investigator | MRI core |
| Paul Aisen | University of Southern California | Principal investigator | Clinical core |
| Ronald Petersen, MD, PhD | Mayo Clinic | Principal investigator | Clinical core |
| John C. Morris | Washington University | Principal investigator | Neuropathology core |
Clark AL, Weigand AJ, Bangen KJ, et al., Higher cerebrospinal fluid tau is associated with history of traumatic brain injury and reduced processing speed in Vietnam‐era veterans: A Department of Defense Alzheimer's Disease Neuroimaging Initiative (DOD‐ADNI) study. Alzheimer's Dement. 2021;13:e12239. 10.1002/dad2.12239
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wpcontent/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
DATA AVAILABILITY STATEMENT
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf
REFERENCES
- 1. Gardner RC, Burke JF, Nettiksimmons J, et al. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. 2014;71(12):1490‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barnes DE, Byers AL, Gardner RC, et al. Association of mild traumatic brain injury with and without loss of consciousness with dementia in US military veterans. JAMA Neurol. 2018;75(9):1055‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barnes DE, Kaup A, Kirby KA, et al. Traumatic brain injury and risk of dementia in older veterans. Neurology. 2014;83(4):312‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schaffert J, LoBue C, White CL, et al. Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy‐confirmed Alzheimer's disease. Neuropsychology. 2018;32(4):410‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnson VE, Stewart W, Smith DH. Widespread τ and amyloid‐β pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22(2):142‐149. https://pubmed.ncbi.nlm.nih.gov/21714827/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith DH, Johnson VE, Stewart W. Chronic neuropathologies of single and repetitive TBI: substrates of dementia?. Nat Rev Neurol. 2013;9(4):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shively S, Scher AI, Perl DP, Diaz‐Arrastia R. Dementia resulting from traumatic brain injury: what is the pathology?. Arch Neurol. 2012;69(10):1245‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeKosky ST, Blennow K, Ikonomovic MD, Gandy S. Acute and chronic traumatic encephalopathies: pathogenesis and biomarkers. Nat Rev Neurol. 2013;9(4):192‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Faden AI, Loane DJ. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation?. Neurotherapeutics. 2015;12(1):143‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramlackhansingh AF, Brooks DJ, Greenwood RJ, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70(3):374‐383. [DOI] [PubMed] [Google Scholar]
- 11. Cherry JD, Tripodis Y, Alvarez VE, et al. Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun. 2016;4(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci. 2015;16(2):109‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weiner MW, Veitch DP, Hayes J, et al. Effects of traumatic brain injury and posttraumatic stress disorder on Alzheimer's disease in veterans, using the Alzheimer's disease neuroimaging initiative. Alzheimers Dement. 2014;10:S226‐235. 3 Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. The Management of Concussion/mTBI Working Group (2016). VA/DoD clinical practice guideline for the management of concussion/mild traumatic brain injury (mTBI): Guideline summary. Washington, DC: Department of Veterans Affairs, Department of Defense. https://www.va.gov/covidtraining/docs/mTBICPGFullCPG50821816.pdf [Google Scholar]
- 15. Blake DD, Weathers FW, Nagy LM, et al. The development of a clinician‐administered PTSD scale. J Trauma Stress. 1995;8(1):75‐90. [DOI] [PubMed] [Google Scholar]
- 16. American Psyciatric Association . Diagnostic and Statistical Manual of Mental Disorders. Fourth edition. Washington, DC: American Psychiatic Association; 2000. Text revision. [Google Scholar]
- 17. Kaup AR, Peltz C, Kenney K, et al. Neuropsychological profile of lifetime traumatic brain injury in older veterans. J Int Neuropsychol Soc. 2017;23(1):56‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peltz CB, Gardner RC, Kenney K, et al. Neurobehavioral characteristics of older veterans with remote traumatic brain injury. J Head Trauma Rehabil. 2017;32(1):E8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. IBM SPSS Statistics for Windows, Version 25.0. In. Armonk, NY. IBM Corp; 2017. [Google Scholar]
- 20. R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3‐900051‐07‐0, http://www.R-project.org/. [Google Scholar]
- 21. Ahmadzadeh H, Smith DH, Shenoy VB. Viscoelasticity of tau proteins leads to strain rate‐dependent breaking of microtubules during axonal stretch injury: predictions from a mathematical model. Biophys J. 2014;106(5):1123‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ahmadzadeh H, Smith DH, Shenoy VB. Mechanical effects of dynamic binding between tau proteins on microtubules during axonal injury. Biophys J. 2015;109(11):2328‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72(5):1858‐1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gabbita SP, Scheff SW, Menard RM, et al. Cleaved‐tau: a biomarker of neuronal damage after traumatic brain injury. J Neurotrauma. 2005;22(1):83‐94. [DOI] [PubMed] [Google Scholar]
- 25. Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid‐β pathology: a link to Alzheimer's disease?. Nature Reviews Neuroscience. 2010;11(5):361–370. 10.1038/nrn2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zetterberg H. Review: tau in biofluids—relation to pathology, imaging and clinical features. Neuropathol Appl Neurobiol. 2017;43(3):194‐199. [DOI] [PubMed] [Google Scholar]
- 28. Andreasen N, Sjögren M, Blennow K. CSF markers for Alzheimer's disease: Total tau, phospho‐tau and Aβ42. The World Journal of Biological Psychiatry. 2003;4 (4):147–155. 10.1080/15622970310029912 [DOI] [PubMed] [Google Scholar]
- 29. Orr ME, Sullivan AC, Frost B. A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol Sci. 2017;38(7):637‐648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maphis N, Xu G, Kokiko‐Cochran ON, et al. Loss of tau rescues inflammation‐mediated neurodegeneration. Front Neurosci. 2015;9:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70(11):960‐969. [DOI] [PubMed] [Google Scholar]
- 32. Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol (Berl). 2011;121(2):171‐181. [DOI] [PubMed] [Google Scholar]
- 33. Mattsson N, Zetterberg H, Janelidze S, et al. Plasma tau in Alzheimer disease. Neurology. 2016;87(17):1827‐1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zetterberg H, Wilson D, Andreasson U, et al. Plasma tau levels in Alzheimer's disease. Alzheimer's Res Ther. 2013;5(2):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gordon BA, Blazey TM, Christensen J, et al. Tau PET in autosomal dominant Alzheimer's disease: relationship with cognition, dementia and other biomarkers. Brain. 2019;142(4):1063‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brier MR, Gordon B, Friedrichsen K, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer's disease. Sci Transl Med. 2016;8(338):338ra366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bogoslovsky T, Wilson D, Chen Y, et al. Increases of plasma levels of glial fibrillary acidic protein, tau, and amyloid β up to 90 days after traumatic brain injury. J Neurotrauma. 2017;34(1):66‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Alosco ML, Tripodis Y, Fritts NG, et al. Cerebrospinal fluid tau, Aβ, and sTREM2 in Former National Football League Players: modeling the relationship between repetitive head impacts, microglial activation, and neurodegeneration. Alzheimer's Dement. 2018;14(9):1159‐1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rubenstein R, Chang B, Yue JK, et al. Comparing plasma phospho tau, total tau, and phospho tau–total tau ratio as acute and chronic traumatic brain injury biomarkers. JAMA Neurol. 2017;74(9):1063‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Olivera A, Lejbman N, Jeromin A, et al. Peripheral total tau in military personnel who sustain traumatic brain injuries during deployment. JAMA Neurol. 2015;72(10):1109‐1116. [DOI] [PubMed] [Google Scholar]
- 41. Peltz CB, Kenney K, Gill J, et al. Blood biomarkers of traumatic brain injury and cognitive impairment in older veterans. Neurology. 2020;95(9):e1126‐e1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shahim P, Tegner Y, Gustafsson B, et al. Neurochemical aftermath of repetitive mild traumatic brain injury. JAMA Neurol. 2016;73(11):1308‐1315. [DOI] [PubMed] [Google Scholar]
- 43. Mondello S, Buki A, Barzo P, et al. CSF and plasma amyloid‐β temporal profiles and relationships with neurological status and mortality after severe traumatic brain injury. Sci Rep. 2014;4:6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kay AD, Petzold A, Kerr M, Keir G, Thompson E, Nicoll JAR. Alterations in cerebrospinal fluid apolipoprotein E and amyloidβ‐Protein after Traumatic Brain Injury. Journal of Neurotrauma. 2003;20(10):943–952. 10.1089/089771503770195795 [DOI] [PubMed] [Google Scholar]
- 45. DeKosky ST, Abrahamson EE, Ciallella JR, et al. Association of increased cortical soluble Aβ42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol. 2007;64(4):541‐544. [DOI] [PubMed] [Google Scholar]
- 46. Chen X‐H, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid β plaques despite persistent accumulation of amyloid β in axons of long‐term survivors of traumatic brain injury. Brain Pathology. 2009;19(2):214–223. 10.1111/j.1750-3639.2008.00176.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ahmed F, Gyorgy A, Kamnaksh A, et al. Time‐dependent changes of protein biomarker levels in the cerebrospinal fluid after blast traumatic brain injury. Electrophoresis. 2012;33(24):3705‐3711. [DOI] [PubMed] [Google Scholar]
- 48. Mohamed AZ, Cumming P, Srour H, et al. Amyloid pathology fingerprint differentiates post‐traumatic stress disorder and traumatic brain injury. Neuroimage Clin. 2018;19:716‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. McAllister TW. Neurobiological consequences of traumatic brain injury. Dialogues Clin Neurosci. 2011;13(3):287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pattinson CL, Gill JM, Lippa SM, et al. Concurrent mild traumatic brain injury and posttraumatic stress disorder is associated with elevated tau concentrations in peripheral blood plasma. J Trauma Stress. 2019;32(4):546‐554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL MATERIAL
Data Availability Statement
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf