

Abstract
Introduction
Physical activity (PA) is associated with better cognitive and brain health. However, it remains unclear whether PA relates to accumulation of disease pathology (“resistance”) or indirectly moderates adverse effects of pathology on cognition (“cognitive resilience”).
Methods
Five hundred thirteen Rush Memory and Aging Project (MAP) decedents completed longitudinal actigraphy monitoring, cognitive testing, and neuropathological examination. Cross‐sectional models tested the relationship between average PA and pathology, and the moderating effect of baseline PA on the association between pathology and cognition. Longitudinal models examined whether changes in PA moderated associations between pathology and cognition.
Results
PA was negatively associated with Lewy body disease (LBD), but positively associated with Alzheimer's disease (AD) burdens. Baseline PA attenuated the association between cerebrovascular pathology and cognition, whereas longitudinal change in PA attenuated associations between AD, cerebral amyloid angiopathy, TAR DNA‐binding protein 43, and atherosclerosis on cognitive decline.
Discussion
Whereas PA relates to “cognitive resilience” against cerebrovascular disease, AD, and other neuropathologies, “resistance” effects were limited.
Keywords: Alzheimer's disease, cognition, physical activity, resilience, resistance
1. INTRODUCTION
Physical activity (PA) is a modifiable factor associated with positive effects on cognition, brain structure, and psychological function. In older adults, greater PA is associated with slower rates of cognitive decline 1 and lower rates of dementia. 2 , 3 , 4 , 5 , 6 , 7 , 8 However, it remains unclear whether the protective effects of PA are conferred through a reduction in the accumulation of disease‐related brain pathology (“resistance”) or by moderating adverse effects of pathology on cognition (“resilience”). 9 , 10 Further, these relationships may differ depending on the specific neurodegenerative or cerebrovascular pathology.
In support of a resistance model, PA has been linked with lower age‐related amyloid burden, particularly in those at greater genetic risk for Alzheimer's disease (AD). 11 , 12 , 13 , 14 , 15 However, evidence is mixed 16 , 17 and most prior studies of the association between PA and amyloid burden relied on cross‐sectional approaches 12 , 13 , 14 (but see also Stillman et al. 11 and Müller et al. 15 ), and use self‐report measures of PA, 11 , 12 , 13 , 14 , 15 leaving questions about the accuracy and duration of PA effects. PA was also associated with reduced cerebrovascular disease, including reduced severity of white matter lesions, 18 and lower likelihood of macroinfarcts in a prior study from the Rush Memory and Aging Project (MAP). 19 In this latter study, objectively quantified movement, including PA, proximate to death were not strongly associated with other cerebrovascular measures (arteriosclerosis, atherosclerosis, microinfarcts), global AD pathology, TAR DNA‐binding protein 43 (TDP‐43), hippocampal sclerosis, cerebral amyloid angiopathy (CAA), or Lewy body disease (LBD). 19 Yet, animal models demonstrate exercise‐related reductions in soluble amyloid beta (Aß) and extracellular Aß plaques, 20 , 21 hippocampal tau pathology, 22 and genes involved in cholesterol trafficking. 22 Therefore, existing evidence is mixed, but may support a role for direct effects of PA on certain aspects of AD pathology and cerebrovascular structure.
In addition to directly reducing the accumulation of brain pathology, PA may modify the toxicity of disease pathology on cognition. Indeed, higher self‐reported PA attenuated the negative association between white matter hyperintensities and cognitive decline in functionally normal older adults 23 and patients with stroke/transient ischemic attack, 24 and reduced the impact of frontotemporal atrophy on cognitive decline in autosomal dominant frontotemporal dementia. 23 In prior work from Rush MAP, higher levels of objectively measured PA were associated with better global cognition, controlling for AD and other pathologies, 25 suggesting pathology‐wide resilience against cognitive decline. Taken together, PA also appears to be related to a reduced negative impact of a multitude of neurodegenerative pathologies on cognition at various stages of cognitive impairment.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (e.g., PubMed) sources and cited existing studies on direct associations between physical activity (PA) and disease pathology, as well as evidence for a moderating effect of PA on the association between disease burden and cognition.
Interpretation: Greater late‐life PA attenuated the negative association between multiple pathological markers and cognition over time, supporting “resilience” models. A novel association between late‐life PA and Lewy body disease at death was also identified, suggesting multiple pathways for PA effects.
Future directions: Given the use of post mortem pathology markers, future studies are needed to identify associations between PA and disease pathology during life. Studies that use in vivo measurement of multiple disease pathologies would clarify how changes in PA impact disease pathology, and support the identification of the ideal dose and frequency of PA based on individual factors and a precision‐medicine approach.
Building on prior cross‐sectional analyses modeling metrics proximate to death from the Rush MAP, 19 , 25 the current study used all available actigraphy data across multiple longitudinal time points to measure late‐life PA and determine whether late‐life PA related to protective effects on brain health through an association with disease accumulation or through moderation of the negative effects of pathology on global cognition. We tested cross‐sectional “resistance” modeling of the relationship between pathology (single time point variable) and late‐life PA. For longitudinal “cognitive resilience” models, we evaluated the interaction (moderating effect) between within‐person changes in PA on the association between individual pathological markers and global cognition over time. These models allowed for the identification of direct and moderating pathology‐specific effects of PA that may inform future intervention and therapeutic approaches. Given prior work demonstrating PA effects on resistance and cognitive resilience against AD pathology and cerebrovascular factors, we hypothesized a negative association between late‐life PA and pathology, and a moderating effect of PA on the association between these pathologies and cognitive decline. However, based on additional evidence from animal studies of more widespread effects of PA on brain health through increased clearance of toxins through improved glymphatic, microglial, and synaptic function, 26 we additionally anticipated a more global impact of PA across pathology markers.
2. METHODS
2.1. Participants
Five hundred thirteen decedents enrolled in the Rush MAP who completed at least one actigraphy visit, cognitive assessment, and post mortem neuropathological examination were included (Table 1). 27 Participants completed an average of four actigraphy and cognitive visits (range = 1–12). The study was approved by the Rush University Medical Center Institutional Review Board. Written informed consent and signed Uniform Anatomical Gift Act were obtained.
TABLE 1.
Participant demographics, clinical characteristics, and pathological disease burden
| Measure | Mean (SD) | Range |
|---|---|---|
| Age at baseline actigraphy visit | 84.85 (5.59) | (62, 100) |
| Age at death | 91.24 (6.02) | (66, 108) |
| Sex (M/F) | 143/368 | – |
| Education | 14.84 (2.86) | (5, 25) |
| Mini‐Mental State Examination at baseline visit | 26.87 (3.45) | (7, 30) |
| Mini‐Mental State Examination at last actigraphy visit | 25.60 (4.40) | (7, 30) |
| Average late‐life daily PA | 1.97 (1.07) | (0.15, 9.13) |
| Motor composite | 0.90 (0.20) | (0.31, 1.50) |
| Global cognition, z‐score | −0.13 (0.67) | (−3.13, 1.32) |
| Interval from baseline actigraphy visit until death | 6.39 (3.51) | (0.13, 14.16) |
| Interval from last actigraphy visit until death | 2.70 (2.62) | (0.02, 13.13) |
| Pathological markers | ||
| Global AD pathology | 0.71 (0.58) | |
| AD Reagan, percent meets criteria for AD | 64% | |
| Hippocampal sclerosis, present | 9% | |
| TDP‐43, moderate–severe | 38% | |
| Lewy bodies, moderate–severe | 22% | |
| CAA, moderate–severe | 34% | |
| Atherosclerosis, moderate–severe | 24% | |
| Arteriosclerosis, moderate–severe | 29% | |
| Macroscopic infarcts present | 38% | |
| Microscopic infarcts present | 32% |
Notes: Unless otherwise specified, time‐varying variables are reported at baseline actigraphy visit. Global AD is a summative variable averaging across multiple standardized metrics of AD pathologies (neuritic plaques, diffuse plaques, neurofibrillary tangles) with no established cut‐off for meeting criteria for AD. Therefore, the quantitative variable is reported here.
Abbreviations: AD, Alzheimer's disease; CAA, cerebral amyloid angiopathy; PA, physical activity; TDP‐43, TAR DNA‐binding protein 43.
2.2. Physical activity
Total daily PA was measured continuously for 24 hours/day for up to 10 days with an omnidirectional accelerometer worn on the non‐dominant wrist (Actical; Mini Mitter). Devices captured all movement in 360 degrees. Records were visually examined for periods of suspected device removal. In addition, any period of 4 hours or greater with no activity counts at all was considered suspicious for device removal. Actical estimates daily raw activity counts every 15 seconds from the device. As previously described, 25 total daily PA was the average of daily activity counts for each 15‐second epoch for full days of data. 3 We did not use intensity measures for the purposes of this article. For cross‐sectional models examining the direct relationship with pathology, rather than relying on a single time point of PA data, which may be impacted by physical or emotional state or disease severity, we used all data by averaging across actigraphy visits as a proxy for “late‐life PA.”
2.3. Cognition
Annual cognitive testing was administered by trained technicians. A global cognition composite was derived from z‐scores from 19 cognitive tests, 28 including seven measures of episodic memory, three language measures, three measures of auditory attention/working memory, two measures of visuoperceptual skills, and four measures of processing speed.
2.4. Post mortem histopathology indices
A standard protocol was used for brain removal, tissue sectioning, and preservation, and a uniform gross and microscopic examination with quantification of post mortem indices. 29 Staff was blinded to clinical data. Post mortem indices included global burden of AD pathology, hippocampal sclerosis (HS), LBD, TDP‐43, CAA, and several measures of cerebrovascular disease (macroinfarcts, microinfarcts, arteriosclerosis, atherosclerosis). For additional information, see the supporting information and prior Rush MAP works. 3 , 19 , 25 , 29
2.5. Motor function
Multiple measures of gait (time and number of steps to cover a distance of 8 feet and turn 360 degrees) were averaged to create a composite of motor function. An average of motor function across visits was included in cross‐sectional models, and visit‐specific motor function was included in longitudinal models to account for objective motor capacity (i.e., possible disease‐related motor restrictions on PA).
2.6. Statistical analyses
“Resistance” Models. The direct relationship between PA and pathology was examined in a single cross‐sectional model that tested the association between average late‐life PA (actigraphy averaged across all visits) and each pathological marker at death entered simultaneously, along with sex, age at death, time from last actigraphy visit to death, and education as covariates (all covariates mean‐centered); to adjust for disease‐related motor changes, we further covaried for the motor function composite. In follow‐up analyses, to test for reverse causality (e.g., those with dementia engaging in less PA due to their impairment and driving significant effects), we first covaried for clinical diagnosis at death (i.e., normal, mild cognitive impairment [MCI], dementia). Next, we entered the interaction between clinical diagnosis at death and each of the pathological markers on PA in separate models controlling for all other pathological markers. If reverse causality was significantly impacting our findings, we expected to find significant interactions between clinical diagnosis at death and pathological markers on PA with stronger associations observed between pathology and PA in those with more advanced clinical/functional decline (dementia > MCI > normal).
2.6.1. Post hoc analyses of AD
To further understand the results of the cross‐sectional “resistance” model, we investigated the association between PA and the three major components of the global AD measure—diffuse plaques, neuritic plaques, and neurofibrillary tangles, controlling for all other pathological variables and covariates.
2.6.2. Sensitivity analyses
After plotting the models, a small subset of participants demonstrated disproportionately high PA levels. To confirm that our results were not driven by this small subset of participants, we conducted sensitivity analyses excluding participants with > 90th percentile of daily movement (average daily Actical count > 3.3; n = 45). Further, to ensure that our findings were not driven by those with only one visit or participants who remained in the study significantly longer than the average (> 6 visits), we conducted sensitivity analyses restricting the sample to those with at least two but fewer than seven visits.
“Cognitive Resilience” Models. The moderating effect of PA on the association between pathology and cognition was modeled through baseline cross‐sectional and longitudinal analyses. A cross‐sectional model examined the interaction between baseline PA and pathology on baseline cognition. Longitudinal models examined the moderating effect of change in PA on the relationship between pathology and cognition over time using separate linear mixed effects models (LME) for each pathology marker. Actigraphy was decomposed into between‐person effects (baseline PA levels) and within‐person effects (visit‐specific change in PA compared to an individual's baseline PA) to avoid estimation bias from incorrectly assuming common within‐ and between‐subject effects. 30 , 31 Specifically, we entered the interaction term between within‐person changes in PA and pathology on global cognition over time, adjusting for covariates (age at death, sex, education, time from last actigraphy visit until death, motor function) and baseline actigraphy levels. All predictors of interest were mean centered in LME to facilitate interpretation. All LME analyses were modeled with random slopes and intercepts. Unstandardized regression coefficients (b) are reported. Significant interactions were probed by plotting predicted slopes at 10th, 50th, and 90th percentiles of actigraphy. Paralleling the “resistance” models, we tested for reverse causality by first covarying for clinical diagnosis at death (i.e., normal, MCI, dementia), and next, entering the three‐way interaction among cognitive diagnosis at death, within‐person change in PA, and pathology on cognition in each of the above models.
3. RESULTS
Participant demographics and clinical characteristics are reported in Table 1.
Greater average late‐life PA was associated with younger age (r = –.15, P < .001), lower education (r = –.10, P = .021), higher Mini‐Mental State Examination (MMSE; r = .14, P = .001), and better motor function (r = .45, P < .001).
3.1. Direct relationship between physical activity and pathology
In regression analyses controlling for demographics, time from last actigraphy visit to death, and motor function, average late‐life PA was independently associated with lower LBD and macroinfarct (marginally), but greater global AD burdens (Table 2). The relative associations between PA and LBD or AD pathologies were equal in magnitude though opposite in directionality (see Figure 1). The effect of PA on macroinfarcts, though marginal, was ≈75% the size of the effect on LBD. Average late‐life PA did not significantly relate to hippocampal sclerosis, TDP, CAA, microinfarcts, arteriosclerosis, and atherosclerosis.
TABLE 2.
Linear regression model examining the association between pathology markers and average PA, controlling for age at death, sex, education, and motor function
| Standardized betas | Unstandardized coefficient | St. error | P | 95% CI | |
|---|---|---|---|---|---|
| Constant | .774 | .078 | <.001 | [.62, .93] | |
| Age at death | −.064 | −.004 | .003 | .124 | [−.009, .001] |
| Time from last actigraphy visit to death | .172 | .029 | .007 | <.001 | [.02, .04] |
| Education | −.104 | −.014 | .005 | .010 | [−.02, −.003] |
| Sex | −.077 | −.064 | .034 | .064 | [−.13, .004] |
| Motor function | .395 | .672 | .072 | <.001 | [.53, .81] |
| Alzheimer's disease | .109 | .121 | .048 | .013 | [.03, .22] |
| Hippocampal sclerosis | −.050 | −.066 | .057 | .249 | [−.18, .05] |
| TDP‐43 | .004 | .001 | .013 | .923 | [−.02, .03] |
| LBD | −.107 | −.035 | .013 | .008 | [−.06, −.01] |
| CAA | .027 | .011 | .017 | .520 | [−.02, .04] |
| Macroinfarcts | −.075 | −.057 | .032 | .075 | [−.12, .01] |
| Microinfarcts | −.015 | −.012 | .033 | .704 | [−.08, .05] |
| Atherosclerosis | −.020 | −.009 | .020 | .642 | [−.05, .03] |
| Arteriosclerosis | .030 | .013 | .018 | .485 | [−.02, .05] |
Note: Significant predictors are bolded.
Abbreviations: CAA, cerebral amyloid angiopathy; CI, confidence interval; LBD, Lewy body disease; PA, physical activity; TDP‐43, TAR DNA‐binding protein 43.
FIGURE 1.
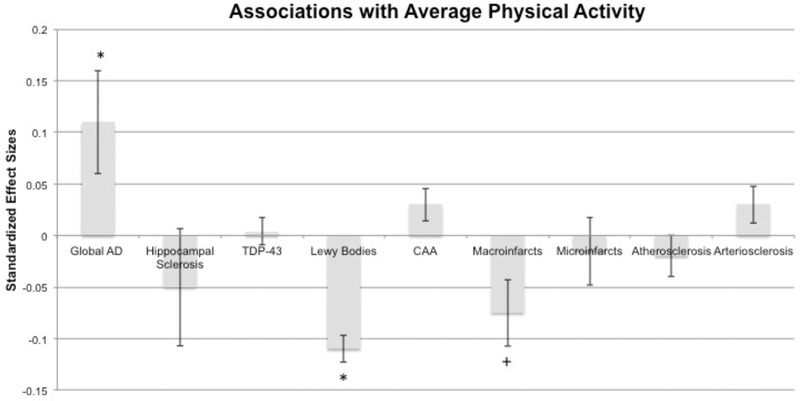
Associations between pathological markers and late‐life physical activity (PA) from the linear regression model (standardized effect sizes). Bars represent 95% confidence interval. *adjusted for age at death, education, sex, average motor function, and time from last actigraphy visit until death. AD, Alzheimer's disease; CAA, cerebral amyloid angiopathy; TDP‐43, TAR DNA‐binding protein 43
To address issues of reverse causality, we entered clinical diagnosis at death (i.e., normal, MCI, dementia) to the above model, which did not change the results. Next, we entered the interaction between cognitive diagnosis at death and pathology on PA, which did not reach statistical significance for any of the pathological markers (Bs < .16, P‐values > .071), except macroinfarcts. The negative association between late‐life PA and macroinfarcts was more robust in the dementia compared to normal group (B = −.14, b = −.16, P = .034, 95% confidence interval [CI; −.30, −.01]); the association did not differ between the MCI and normal groups (B = −.09, b = −.12, P = .141, 95% CI [−.28, .04]). Overall, except for macroinfarcts, these findings do not support reverse causality (i.e., reduced PA in those with more advanced disease pathology/dementia driving the effects), as the associations were weakest in the dementia group.
3.1.1. Post hoc analyses of global AD burden
There was a marginal, positive association between late‐life PA and post mortem diffuse plaques (B = .08, b = .04, standard error [SE] = .02, P = .062, 95% CI [−.002, .09]). Late‐life PA demonstrated minimal associations with neuritic plaques (B = .002, b = .001, SE = .02, P = .968, 95% CI [−0.05, 0.05]) or neurofibrillary tangles (B = .06, b = .03, SE = .02, P = .207, 95% CI [−0.02, 0.08]).
3.1.2. Sensitivity analyses
Excluding participants with > 90th percentile of PA (average daily Actical count > 3.3; n = 45), the associations between average late‐life PA and AD (B = .11, b = .10, P = .019) and LBD remained significant (B = −.10, b = −.03, P = .018). The marginal association between average late‐life PA and macroinfarcts weakened (B = −.02, b = −.02, P = .597). Restricting the sample to participants with two visits (N = 385) did not alter the significant associations between late‐life PA and global AD (B = .11, b = .12, P = .021) or LBD (B = −.12, b = −.04, P = .007), though the association with macroinfarcts was not significant (B = −.07, b = −.05, P = .114). After further restricting the sample to those with at least one follow‐up and fewer than seven visits (N = 299), the directionality of the associations remained unchanged with differences in statistical significance likely related to reduced sample size and power (LBD: B = −.13, b = −.04, P = .01; AD: B = .09, b = .10, P = .097; macroinfarcts: B = −.09, b = −.07, P = .103).
3.2. Cross‐sectional model of physical activity as a moderator of the relationship between pathology and baseline cognition
Examining individual models, baseline PA interacted with atherosclerosis, arteriolosclerosis, and macroinfarcts (marginally) on baseline cognition (see Table 3). For these three cerebrovascular pathologies, greater baseline PA attenuated the negative association between pathology and baseline cognition. Interactions were not significant for other pathological markers (Bs < .13, P‐values > .20). Controlling for all additional pathological markers did not attenuate significant interactions. Instead, the interaction effects were strengthened (atherosclerosis x PA: B = .24; arteriosclerosis x PA: B = .24; macroinfarcts x PA: B = .20). When the three‐way interaction among clinical diagnosis, baseline PA, and pathology was included in the above models, it did not reach statistical significance for any of the pathological markers (Bs < .23, P‐values > .129).
TABLE 3.
Baseline regression models examining the interaction between baseline PA and pathology on baseline cognition
| PA × Pathology | ||||
|---|---|---|---|---|
| Standardized beta | Estimate (SE) | P | 95% CI | |
| Atherosclerosis x PA | .18 | .05 (.03) | .039 | [.003, .10] |
| Arteriolosclerosis x PA | .22 | .05 (.02) | .019 | [.008, .09] |
| Macroinfarcts x PA | .16 | .08 (.04) | .072 | [−.007, .16] |
| Microinfarcts x PA | .07 | .03 (.04) | .462 | [−.05, .11] |
| AD x PA | −.12 | −.06 (.06) | .300 | [−.18, .05] |
| TDP‐43 x PA | −.01 | −.002 (.02) | .877 | [−.03, .03] |
| CAA x PA | −.07 | −.01 (.02) | .528 | [−.05, .03] |
| LBD x PA | .06 | .01 (.02) | .468 | [−.02, .05] |
| Hippocampal sclerosis x PA | .13 | .11 (.09) | .200 | [−.06, .28] |
Notes: Pathology interaction term of interest illustrated. All models adjusted for age at death, education, sex, motor function, and time from last actigraphy visit until death. Significant interactions are shown in bold.
Abbreviations: AD, Alzheimer's disease; CAA, cerebral amyloid angiopathy; CI, confidence interval; LBD, Lewy body disease; PA, physical activity; TDP‐43, TAR DNA‐binding protein 43.
3.3. Longitudinal physical activity as a moderator of the relationship between pathology and cognition
Examining individual models, within‐person changes in PA across visits interacted with global AD, CAA, TDP‐43, and atherosclerosis on global cognitive changes (see Tables 4). Increases in PA attenuated the adverse relationships between each pathology and cognitive decline over time. In participants who increased their PA between study visits (at the 90% within‐percentile of person change), the effect of pathology on cognition was near zero, compared to −0.67 in those with stable PA and average levels of AD, and approximately −0.10 in those with stable PA and average levels of CAA, TDP‐43, or atherosclerosis. Further, changes in PA had a larger impact on cognitive trajectories at higher levels of pathology burden. For example, the effect on cognitive decline between those who increased their PA (90th percentile) versus those who decreased their PA (10th percentile) was much greater (≈0.40 standard deviation [SD] better cognition on average) at higher levels of AD, CAA, TDP, and atherosclerosis (90th percentile), compared to the difference in groups based on PA at lower levels of pathology (≈0.2 SD better cognition at average (50th percentile) of pathology. Said differently, the beneficial effect of maintaining or increasing PA was double in participants with high pathology at death compared to those with average levels of CAA, TDP‐43, atherosclerosis, or AD pathology at death. Within‐person changes in PA did not significantly interact on the relationship between LBD, macroinfarcts, microinfarcts, arteriosclerosis, or HS and global cognition over time (see Figure 2). Greater baseline PA was associated with better cognition across all models (unstandardized effects ≥ .06, P‐values < .002). After including all additional pathological markers, the significant interactions between within‐person change in PA and AD, CAA and TDP‐43 on cognitive trajectories remained significant (unstandardized effects ≥ .03, P‐values < .005); the interaction between within‐person change in PA and atherosclerosis did not (b = .02, P = .191).
TABLE 4.
Linear mixed effects models examining within‐ and between‐person changes in PA, and the interaction between PA and pathology on global cognition. Full models reported in supporting information
| Global AD | CAA | TDP‐43 | Atherosclerosis | |||||
|---|---|---|---|---|---|---|---|---|
| Estimate (SE) | P | Estimate (SE) | P | Estimate (SE) | P | Estimate (SE) | P | |
| Baseline PA | 0.09 (0.02) | <.001 | 0.08 (0.02) | <.001 | 0.07 (.02) | <.001 | 0.06 (0.02) | .002 |
| Pathology | −0.67 (0.08) | <.001 | −0.10 (.03) | <.001 | −0.09 (0.02) | <.001 | −0.10 (0.03) | .003 |
| Within visit change in PA | 0.07 (0.01) | <.001 | 0.08 (0.01) | <.001 | 0.08 (0.01) | <.001 | 0.09 (0.01) | .001 |
| Within PA × Pathology | 0.20 (0.03) | <.001 | 0.04 (0.01) | <.001 | 0.03 (0.01) | <.001 | 0.04 (0.02) | .021 |
Abbreviations: AD, Alzheimer's disease; CAA, cerebral amyloid angiopathy; PA, physical activity; SE, standard error; TDP‐43, TAR DNA‐binding protein 43.
FIGURE 2.
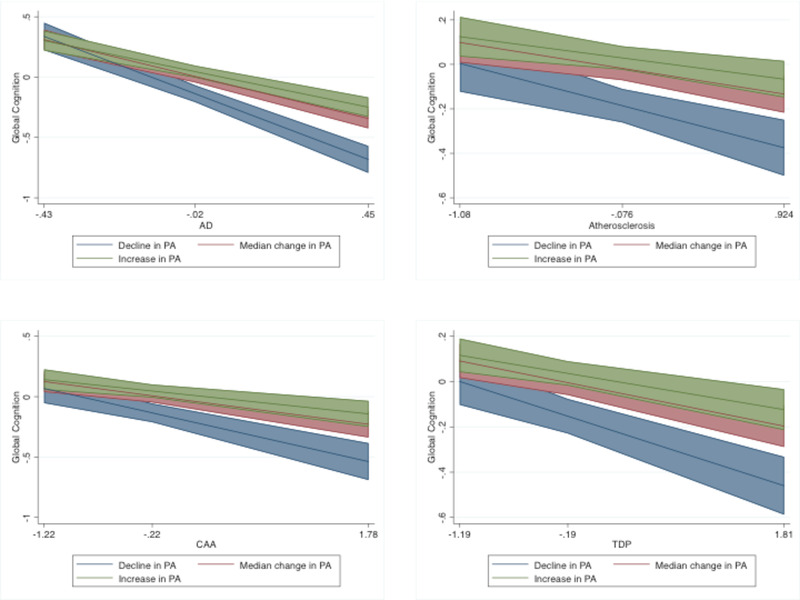
Interactions between within‐person variation in PA and pathology (AD, TDP‐43, CAA, and atherosclerosis) on global cognition. For purposes of illustration, a decline in PA is depicted at the 10th percentile of within‐person changes, and an increase in PA is depicted at the 90th percentile of within‐person changes. Median change reflects the 50th percentile. AD, Alzheimer's disease; CAA, cerebral amyloid angiopathy; PA, physical activity; TDP‐43, TAR DNA‐binding protein 43
Similarly, controlling for cognitive diagnosis at death did not impact the significant interactions between within‐person change in PA and AD, CAA, or TDP‐43 on cognition (unstandardized effects > 0.03, P‐values < .002), suggesting that effects were not driven by impairment levels (i.e., possible reverse directionality). However, the interaction between PA and atherosclerosis was not statistically significant after controlling for cognitive diagnosis at death (b = .02, P = .140). When clinical diagnosis was entered in three‐way interactions with within‐person change in PA, and pathology on cognition, none of the interactions reached statistical significance (unstandardized effects < .12, P‐values < .080). Therefore, the moderating effect of change in PA on the association between pathology and cognition did not vary based on clinical diagnosis, suggesting that PA may show protective relationships across the clinical spectrum.
4. DISCUSSION
In a sample of more than 500 community‐dwelling older adults, we examined whether the protective relationships of PA were conferred directly through an association with brain pathology burden (“resistance”) or by moderating adverse relationships of pathology on cognition (“cognitive resilience”). The protective relationships with PA differed based on pathology. Greater late‐life PA was associated with fewer Lewy bodies, mildly lower macroinfarcts, but greater diffuse plaques. At baseline, PA interacted with multiple cerebrovascular disease markers (atherosclerosis, arteriosclerosis, and macroinfarcts), such that greater baseline PA attenuated the negative association between pathology and baseline cognition. In longitudinal analyses, within‐person increases in PA over time attenuated the negative association between AD, CAA, and TDP‐43 with cognitive trajectories. Of note, these are observational data and effects related to reverse directionality must be considered (e.g., adults with unhealthy brains may inherently exercise less); however, models (with the exception of atherosclerosis) remained when adjusting for objective motor (gait) capacity, other neuropathology burdens, and clinical diagnosis at death. Taken together, PA relationships appear to be pathology‐specific, with different pathways underlying protective effects against different pathologies. These data have important implications for primary (“resistance”) and secondary (“cognitive resilience”) exercise intervention approaches for dementia prevention.
Regarding resistance models, we identified a novel association between PA and LBD burden, and replicated prior work demonstrating lower cerebrovascular burden in those with greater PA. In contrast to hypothesized global effects of PA on pathology, particularly on the accumulation of toxic proteinopathies (amyloid, TDP‐43), our findings demonstrated specificity in the direct associations between PA and LBD pathology. Little is known regarding the factors that directly contribute to Lewy body aggregation, and to our knowledge, this is the first study to identify an association between PA and LBD itself, though there have been several works indicating a beneficial relationship between PA and neurobehavioral outcomes in clinical Parkinson's disease. 32 , 33 , 34 These data may indicate that greater PA maintains health of motor networks most vulnerable to Lewy body aggregation and helps prevent or delay pathological accumulation. Alternatively, or in conjunction, motor deficits associated with LBD may contribute to reductions in PA. The beneficial effects of PA on macroinfarcts were marginal, and appeared driven by participants with very high PA (> 90%). Aging is associated with elevated central arterial stiffness, 35 , 36 which impacts cerebrovascular function through endothelial dysfunction,vasoconstriction, reduced cerebral blood flow, and ultimately, increased susceptibility to macroinfarcts. PA may counteract these age‐related cerebrovascular changes by decreasing central arterial stiffness and restoring endothelial function, 35 increasing regional cerebral blood flow 37 , 38 (but see also Guiney et al. 39 and van der Kleij et al. 40 ), and promoting glial homeostasis. 26
Given prior work in human 11 , 12 , 13 , 14 , 15 and animal models 20 , 21 , 22 demonstrating reduced AD pathology with greater PA, our finding of a positive association between PA and AD burden was surprising. Follow‐up analyses indicated that this effect was not seemingly related to cognitive/clinical diagnosis at death, and driven by diffuse amyloid plaques, an amyloid isoform that is not associated with neuronal death or inflammation. 41 However, given that the association between PA and AD appears to be driven by the less harmful form of amyloid (diffuse plaques), AD is the most common pathology with advanced age, and we found that PA moderates the association between AD and cognitive trajectories, it is possible that the positive association between PA and AD burden represents a confounding survival (vs. iatrogenic) effect. That is, adults who exercise may both live longer and therefore have greater opportunity to accumulate AD pathology. Although we did not find that age at death/longevity impacted this unexpected relationship (data not shown), our cohort has somewhat restricted age variance given the clinicopathologic (vs. true epidemiologic) design, which may limit this type of analysis. While the positive association of our direct effect was unexpected, it adds to a body of literature based on self‐report measures of PA that have found no direct effect of PA on amyloid pathology, 16 , 17 , 42 and contributes to ongoing uncertainty regarding the effects of PA on AD biomarkers. Importantly, again, we found that regardless of the direct effect, greater PA resulted in better clinical outcomes accounting for AD burden (i.e., resilience models). PA intervention studies including AD biomarkers are importantly needed to more precisely tease apart these relationships.
Consistent with prior work from Rush MAP, greater average PA was associated with better cognition after covarying for all the pathological markers, suggesting that PA confers global cognitive resilience against disease pathology. Baseline models demonstrated an attenuation of the negative effects of cerebrovascular disease (atherosclerosis, arteriolosclerosis, and macroinfarcts) on baseline cognition with greater PA. Further, increases in PA over time attenuated the negative association among AD, TDP‐43, CAA, and atherosclerosis and longitudinal cognitive trajectories, such that among those with greater pathology burden (i.e., greatest risk for cognitive declines), adults who increased or maintained their PA demonstrated disproportionately better cognition compared to adults with declining PA. From a precision medicine perspective, this finding is important in establishing the etiologies and optimal timing of PA intervention. These findings also suggest that particularly in the context of amyloid or TDP‐43, PA may function via other brain pathways (e.g., reduced microglial inflammation, synaptic homeostasis) to support cognition versus proteinopathy clearance, per se. Building on aerobic intervention trials that identified positive associations between improved cardiorespiratory fitness and clinical outcomes in AD 43 , 44 and amnestic MCI, 45 , 46 only a handful of prior studies examined the interacting (i.e., modifying) effect of PA on pathology and cognition. 24 , 25 , 47 , 48 Similar to our findings, the majority of these studies identified that PA is particularly beneficial at sustaining cognition in those with higher levels of AD 47 , 48 and vascular burden. 24 To our knowledge, this is the first study to demonstrate beneficial effects of PA in those with greater TDP‐43 and CAA pathology. Prior work from our group demonstrated an attenuation of the negative association between frontotemporal atrophy and cognition in highly active patients with autosomal dominant frontotemporal dementia. 23 Given that TDP‐43 is one of the primary proteinopathies in frontotemporal dementia, it is possible that these findings were partially attributable to the beneficial effect of PA on TDP‐43 aggregation. Taken together, an increase in PA as AD, TDP‐43, and CAA pathology are aggregating may help sustain clinical functioning, despite significant proteinopathy burden.
From a public health standpoint, these findings highlight the need for in vivo biomarkers to identify both etiology and burden of pathology in older adults to determine who is most likely to benefit from increased PA and at what time. Our resistance models suggest that older adults with a family history of parkinsonism or multiple risk factors for stroke may benefit from engagement in PA prior to disease accumulation, whereas our resilience models suggest that greater PA in late life may suffice to preserve cognition in older adults at risk for cerebrovascular disease (atherosclerosis, arteriosclerosis), amyloid accumulation, or TDP‐43–related diseases (frontotemporal lobar degeneration, limbic‐predominant age‐related TDP‐43 encephalopathy).
Although the present highly characterized sample provides insights into pathways of PA for disease progression and cognition, there are several notable limitations of the current findings. First and foremost, disease pathology was measured at autopsy, rather than at each actigraphy time point, limiting our ability to demonstrate time‐locked associations between within‐person change and disease outcomes. Ideally, positron emission tomography, cerebrospinal fluid, and other fluid biomarkers could circumvent these issues, and future intervention studies should strive to incorporate longitudinal metrics of pathology to better understand how these factors interact over time and with lifestyle manipulation. Although we attempted to account for reverse causality by controlling for motor functioning and cognitive diagnosis at death, and by examining interactions with cognitive diagnosis, the relationship between PA and pathology is likely bidirectional. Further, a prior large‐scale prospective study (N = 10,308) found a decline in PA up to 9 years before dementia diagnosis 49 highlighting the complexity of teasing apart the directionality of relationships among PA, cognitive/functional decline, and pathology accumulation. Habitual PA was measured in the present study without experimental manipulation, resulting in correlational findings and limited understanding of causality. Further, the last actigraphy measurement was an average of 2.7 years from death, and the degree to which PA changed in the interim and impacted pathology or cognitive decline is unknown. An additional challenge in actigraphy measurement is understanding the effects of external factors (e.g., weather, physical health, lifestyle changes) on PA engagement. Finally, there is some debate regarding the best placement of actigraphy devices with evidence that hip‐worn devices may more accurately correlate with energy expenditure than wrist‐worn devices, 50 , 51 though each approach has noted strengths and limitations.
Taken together, our findings demonstrate pathology‐specific protective effects of PA on age‐related decline in brain structure and function. PA contributes to robust resilience against arteriosclerosis, atherosclerosis, amyloid, and TDP‐43, and may relate to resistance against LBD and cerebrovascular dysfunction, suggesting multiple pathways are differentially involved in PA‐to‐brain benefits. The identified associations between PA and less studied disease pathologies (TDP‐43, CAA, LBD) provide novel insights into the diverse neurobiological benefits of PA. These findings support a precision medicine approach, and highlight the need to further specify the appropriate timing, dose, and frequency of PA necessary to maximize positive outcomes on brain health and cognition.
CONFLICTS OF INTEREST
The current authors have no conflicts of interest to report.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
This study was supported by NIH‐NIA grants R01AG17917 (PI: Bennett), K23AG058752 and R01AG072475 (PI: KBC). Our work was also supported by the Alzheimer's Association (AARG‐20‐683875, PI: KBC). MAP data can be requested at https://www.radc.rush.edu. MM received funding from the San Francisco VA for conference attendance. KC received funding from UCSF and NIH for conference attendance. DB received funding from NIH Neurovision, consulting fees from AbbVie DSMB Takeda and Origent SBIR, government funding for presentations and travel, and materials from Rush philanthropy. He served on the advisory board for AbbVie.
Memel M, Buchman AS, Bennett DA, Casaletto K. Relationship between objectively measured physical activity on neuropathology and cognitive outcomes in older adults: Resistance versus resilience? Alzheimer's Dement. 2021;13:e12245. 10.1002/dad2.12245
REFERENCES
- 1. Blondell SJ, Hammersley‐Mather R, Veerman JL. Does physical activity prevent cognitive decline and dementia?: a systematic review and meta‐analysis of longitudinal studies. BMC Public Health. 2014;14. 10.1186/1471-2458-14-510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hörder H, Johansson L, Guo X, et al. Midlife cardiovascular fitness and dementia. Neurology. 2018;90:e1298‐e1305. 10.1212/WNL.0000000000005290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Buchman AS, Boyle PA, Yu L, Shah RC, Wilson RS, Bennett DA. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology. 2012;78:1323‐1329. 10.1212/WNL.0b013e3182535d35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scarmeas N, Luchsinger JA, Schupf N, et al. Physical activity, diet, and risk of Alzheimer disease. JAMA ‐ J Am Med Assoc. 2009;302:627. 10.1001/jama.2009.1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer's disease prevalence. Lancet Neurol. 2011;10:819‐828. 10.1016/S1474-4422(11)70072-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer's disease: an analysis of population‐based data. Lancet Neurol. 2014;13:788‐794. 10.1016/S1474-4422(14)70136-X [DOI] [PubMed] [Google Scholar]
- 7. Santos‐Lozano A, Pareja‐Galeano H, Sanchis‐Gomar F, et al. Physical activity and Alzheimer disease: a protective association. Mayo Clin Proc. 2016;91:999‐1020. 10.1016/j.mayocp.2016.04.024 [DOI] [PubMed] [Google Scholar]
- 8. Tan ZS, Spartano NL, Beiser AS, et al. Physical activity, brain volume, and dementia risk: the framingham study. J Gerontol A Biol Sci Med Sci. 2017:glw130. 10.1093/gerona/glw130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arenaza‐Urquijo EM, Vemuri P. Resistance vs resilience to Alzheimer disease. Neurology. 2018;90:695‐703. 10.1212/WNL.0000000000005303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montine TJ, Cholerton BA, Corrada MM, et al. Concepts for brain aging: resistance, resilience, reserve, and compensation. Alzheimer's Res Ther. 2019;11. 10.1186/s13195-019-0479-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stillman CM, Lopez OL, Becker JT, et al. Physical activity predicts reduced plasma β amyloid in the Cardiovascular Health Study. Ann Clin Transl Neurol. 2017;4:284‐291. 10.1002/acn3.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okonkwo OC, Schultz SA, Oh JM, et al. Physical activity attenuates age‐related biomarker alterations in preclinical AD. Neurology. 2014;83:1753‐1760. 10.1212/WNL.0000000000000964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brown BM, Peiffer JJ, Taddei K, et al. Physical activity and amyloid‐β plasma and brain levels: results from the Australian imaging, biomarkers and lifestyle study of ageing. Mol Psychiatry. 2013;18:875‐881. 10.1038/mp.2012.107 [DOI] [PubMed] [Google Scholar]
- 14. Head D, Bugg JM, Goate AM, et al. Exercise engagement as a moderator of the effects of APOE genotype on amyloid deposition. Arch Neurol. 2012;69:636. 10.1001/archneurol.2011.845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Müller S, Preische O, Sohrabi HR, et al. Relationship between physical activity, cognition, and Alzheimer pathology in autosomal dominant Alzheimer's disease. Alzheimer's Dement. 2018;14:1427‐1437. 10.1016/j.jalz.2018.06.3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brown BM, Peiffer J, Rainey‐Smith SR. Exploring the relationship between physical activity, beta‐amyloid and tau: a narrative review. Ageing Res Rev. 2019;50:9‐18. 10.1016/j.arr.2019.01.003 [DOI] [PubMed] [Google Scholar]
- 17. Vemuri P, Lesnick TG, Przybelski SA, et al. Effect of lifestyle activities on alzheimer disease biomarkers and cognition. Ann Neurol. 2012;72(5):730‐738. 10.1002/ana.23665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sexton CE, Betts JF, Demnitz N, Dawes H, Ebmeier KP, Johansen‐Berg H. A systematic review of MRI studies examining the relationship between physical fitness and activity and the white matter of the ageing brain. Neuroimage. 2016;131:81‐90. 10.1016/j.neuroimage.2015.09.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buchman AS, Dawe RJ, Yu L, et al. Brain pathology is related to total daily physical activity in older adults. Neurology. 2018;90:e1911‐e1919. 10.1212/WNL.0000000000005552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Moore KM, Girens RE, Larson SK, et al. A spectrum of exercise training reduces soluble Aβ in a dose‐dependent manner in a mouse model of Alzheimer's disease. Neurobiol Dis. 2016;85:218‐224. 10.1016/j.nbd.2015.11.004 [DOI] [PubMed] [Google Scholar]
- 21. Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:4217‐4221. 10.1523/JNEUROSCI.0496-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Belarbi K, Burnouf S, Fernandez‐Gomez FJ, et al. Beneficial effects of exercise in a transgenic mouse model of Alzheimer's disease‐like Tau pathology. Neurobiol Dis. 2011;43:486‐494. 10.1016/j.nbd.2011.04.022 [DOI] [PubMed] [Google Scholar]
- 23. Casaletto KB, Staffaroni AM, Wolf A, et al. Active lifestyles moderate clinical outcomes in autosomal dominant frontotemporal degeneration. Alzheimer's Dement. 2020;16:91‐105. 10.1002/alz.12001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wong A, Yiu S, Lam BYK, et al. Physical activities attenuate the negative cognitive impact from white matter hyperintensities in stroke and TIA patients with low education. Int J Geriatr Psychiatry. 2019;34:1792‐1798. 10.1002/gps.5194 [DOI] [PubMed] [Google Scholar]
- 25. Buchman AS, Yu L, Wilson RS, et al. Physical activity, common brain pathologies, and cognition in community‐dwelling older adults. Neurology. 2019;92:e811‐e822. 10.1212/WNL.0000000000006954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He XF, Liu DX, Zhang Q, et al. Voluntary exercise promotes glymphatic clearance of amyloid beta and reduces the activation of astrocytes and microglia in aged mice. Front Mol Neurosci. 2017;10. 10.3389/fnmol.2017.00144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bennett DA, Buchman AS, Boyle PA, Barnes LL, Wilson RS, Schneider JA. Religious orders study and rush memory and aging project. J Alzheimer's Dis. 2018;64:S161‐S189. 10.3233/JAD-179939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wilson RS, Boyle PA, Yu L, et al. Temporal course and pathologic basis of unawareness of memory loss in dementia. Neurology. 2015;85:984‐991. 10.1212/WNL.0000000000001935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buchman AS, Leurgans SE, Nag S, Bennett DA, Schneider JA. Cerebrovascular disease pathology and parkinsonian signs in old age. Stroke. 2011;42:3183‐3189. 10.1161/STROKEAHA.111.623462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neuhaus JM, McCulloch CE. Separating between‐ and within‐cluster covariate effects by using conditional and partitioning methods. J R Stat Soc Ser B Stat Methodol. 2006;68:859‐872. 10.1111/j.1467-9868.2006.00570.x [DOI] [Google Scholar]
- 31. Neuhaus JM, Kalbfleisch JD. Between‐ and within‐cluster covariate effects in the analysis of clustered data. Biometrics. 1998;54:638. 10.2307/3109770 [DOI] [PubMed] [Google Scholar]
- 32. Fiorelli CM, Ciolac EG, Simieli L, et al. Differential acute effect of high‐intensity interval or continuous moderate exercise on cognition in individuals with Parkinson's disease. J Phys Act Heal. 2019;16:157‐164. 10.1123/jpah.2018-0189 [DOI] [PubMed] [Google Scholar]
- 33. Murray DK, Sacheli MA, Eng JJ, Stoessl AJ. The effects of exercise on cognition in Parkinson's disease: a systematic review. Transl Neurodegener. 2014;3. 10.1186/2047-9158-3-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. David FJ, Robichaud JA, Leurgans SE, et al. Exercise improves cognition in Parkinson's disease: the PRET‐PD randomized, clinical trial. Mov Disord. 2015;30:1657‐1663. 10.1002/mds.26291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seals DR, DeSouza CA, Donato AJ, Tanaka H. Habitual exercise and arterial aging. J Appl Physiol. 2008;105:1323‐1332. 10.1152/japplphysiol.90553.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Benetos A, Waeber B, Izzo J, et al. Influence of age, risk factors, and cardiovascular and renal disease on arterial stiffness: clinical applications. Am J Hypertens. 2002;15:1101‐1108. 10.1016/S0895-7061(02)03029-7 [DOI] [PubMed] [Google Scholar]
- 37. Chapman SB, Aslan S, Spence JS, et al. Shorter term aerobic exercise improves brain, cognition, and cardiovascular fitness in aging. Front Aging Neurosci. 2013;5. 10.3389/fnagi.2013.00075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tarumi T, Zhang R. Cerebral blood flow in normal aging adults: cardiovascular determinants, clinical implications, and aerobic fitness. J Neurochem. 2018;144:595‐608. 10.1111/jnc.14234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guiney H, Lucas SJE, Cotter JD, Machado L. Investigating links between habitual physical activity, cerebrovascular function, and cognitive control in healthy older adults. Neuropsychologia. 2019;125:62‐69. 10.1016/j.neuropsychologia.2019.01.011 [DOI] [PubMed] [Google Scholar]
- 40. van der Kleij LA, Petersen ET, Siebner HR, et al. The effect of physical exercise on cerebral blood flow in Alzheimer's disease. NeuroImage Clin. 2018;20:650‐654. 10.1016/j.nicl.2018.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. D'Andrea MR, Nagele RG. Morphologically distinct types of amyloid plaques point the way to a better understanding of Alzheimer's disease pathogenesis. Biotech Histochem. 2010;85:133‐147. 10.3109/10520290903389445 [DOI] [PubMed] [Google Scholar]
- 42. de Souto Barreto P, Andrieu S, Rolland Y. Physical activity and β‐amyloid brain levels in humans: a systematic review. J Prev Alzheimer's Dis. 2015;2(1):56‐63. 10.14283/jpad.2015.34 [DOI] [PubMed] [Google Scholar]
- 43. Morris JK, Vidoni ED, Johnson DK, et al. Aerobic exercise for Alzheimer's disease: a randomized controlled pilot trial. PLoS One. 2017;12:e0170547. 10.1371/journal.pone.0170547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vidoni ED, Perales J, Alshehri M, Giles AM, Siengsukon CF, Burns JM. Aerobic exercise sustains performance of instrumental activities of daily living in early‐stage Alzheimer disease. J Geriatr Phys Ther. 2019;42:E129‐E134. 10.1519/JPT.0000000000000172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cammisuli DM, Innocenti A, Franzoni F, Pruneti C. Aerobic exercise effects upon cognition in mild cognitive impairment: a systematic review of randomized controlled trials. Arch Ital Biol. 2017;155:54‐62. 10.12871/000398292017126 [DOI] [PubMed] [Google Scholar]
- 46. Baker LD, Frank LL, Foster‐Schubert K, et al. Effects of aerobic exercise on mild cognitive impairment: a controlled trial. Arch Neurol. 2010;67:71‐79. 10.1001/archneurol.2009.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rabin JS, Klein H, Kirn DR, et al. Associations of physical activity and β‐amyloid with longitudinal cognition and neurodegeneration in clinically normal older adults. JAMA Neurol. 2019;76:1203. 10.1001/jamaneurol.2019.1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schultz SA, Boots EA, Almeida RP, et al. Cardiorespiratory fitness attenuates the influence of amyloid on cognition. J Int Neuropsychol Soc. 2015;21:841‐850. 10.1017/S1355617715000843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sabia S, Dugravot A, Dartigues J‐F, et al. Physical activity, cognitive decline, and risk of dementia: 28 year follow‐up of Whitehall II cohort study. BMJ. 2017;357:j2709‐j2709. 10.1136/bmj.j2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sasaki JE, Hickey AM, Staudenmayer JW, John D, Kent JA, Freedson PS. Performance of activity classification algorithms in free‐living older adults. Med Sci Sports Exerc. 2016;48(5):941‐949. 10.1249/MSS.0000000000000844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schrack JA, Cooper R, Koster A, et al. Assessing daily physical activity in older adults: unraveling the complexity of monitors, measures, and methods. Journals Gerontol Ser A. 2016;71(8):1039‐1048. 10.1093/gerona/glw026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information