

ABSTRACT
Transcriptional reporters are reliable and time-tested tools to study gene regulation. In Staphylococcus aureus, β-galactosidase (lacZ)-based genetic screens are not widely used because of the necessity of selectable markers for strain construction and the production of staphyloxanthin pigment, which obfuscates results. We describe a series of vectors that allow for markerless insertion of codon-optimized lacZ-based transcriptional reporters. The vectors code for different ribosomal binding sites, allowing for tailored lacZ expression. A ΔcrtM::kanR deletion insertion mutant was constructed that prevents the synthesis of staphyloxanthin, thereby permitting blue-white screening without the interference of carotenoid production. We demonstrate the utility of these vectors to monitor aerobic and anaerobic transcriptional activities. For the latter, we describe the use of a ferrocyanide-ferricyanide redox system [Fe(CN)63−/4−] permitting blue-white screening in the absence of oxygen. We also describe additional reporter systems and methods for monitoring transcriptional activity during anaerobic culture, including an FAD-binding fluorescent protein (EcFbFP), alpha-hemolysin (hla), or lipase (geh). The systems and methods described are compatible with vectors utilized to create and screen high-density transposon mutant libraries.
IMPORTANCE Staphylococcus aureus is a human pathogen and a leading cause of infectious disease-related illness and death worldwide. For S. aureus to successfully colonize and invade host tissues, it must tightly control the expression of genes encoding virulence factors. Oxygen tension varies greatly at infection sites, and many abscesses are devoid of oxygen. In this study, we have developed novel tools and methods to study how and when S. aureus alters transcription of genes. A key advantage of these methods and tools is that they can be utilized in the presence and absence of oxygen. A better understanding of anaerobic gene expression in S. aureus will provide important insights into the regulation of genes in low-oxygen environments.
KEYWORDS: Staphylococcus aureus, β-galactosidase, anaerobic, aerobic, lacZ, staphyloxanthin, fluorescence, reporter, hemolysin, lipase, facultative anaerobes, transcriptional regulation, transcriptional reporter
INTRODUCTION
Staphylococcus aureus is a Gram-positive pathogen causing morbidity and mortality worldwide. S. aureus produces numerous virulence factors that contribute to bacterial pathogenesis. Understanding how and when S. aureus alters transcription of genes encoding virulence factors is key to understanding pathogenesis.
β-Galactosidase (lacZ) assays have been widely used to study the function of bacterial gene regulatory elements by allowing for quantification of promoter activity on gene expression. Escherichia coli LacZ (120 kDa, 1,024 amino acids) has β-galactosidase activity, which catalyzes the hydrolysis of β-galactosides into monosaccharides. β-Galactosidase has high stability, is resistant to proteolytic degradation, and does not significantly degrade or bleach (as occurs with fluorescent reporters), which promotes successful usage of β-galactosidase as a transcriptional reporter (1, 2). Miller described a standardized protocol for measuring β-galactosidase activity using the synthetic substrate o-nitrophenyl-β-d-galactoside (ONPG) (3). The hydrolysis of ONPG leads to production of the colored compound o-nitrophenol (ONP), which can be measured spectrophotometrically; moreover, monitoring the hydrolysis of ONPG is fast, inexpensive, consistent, and sensitive. The compound X-Gal (5-bromo-4-chloro-3-indolyl β-d-galactopyranoside) is hydrolyzed by LacZ to produce 5-bromo-4-chloro-3-hydroxyindole, which can be oxidized to form 5,5-dibromo-4,4′dichloro-indigo, an insoluble indigo precipitate (4). Dimerization of 5-bromo-4-chloro-3-hydroxyindole is widely used to detect β-galactosidase activity on solid media during aerobic culture where dioxygen serves as the oxidant.
lacZ-based technologies have been used in S. aureus previously. O’Neill et al. developed a β-galactosidase leakage assay to assess the ability of molecules to cause membrane damage (5). To this end, they created an S. aureus strain carrying the E. coli lacZ gene under the transcriptional control of a strong staphylococcal promoter (cap1A). The strains were exposed to various membrane-damaging substances, and leakage was detected by monitoring the activity of β-galactosidase in the cell-free supernatant using a fluorescence assay with 4-methylumbelliferyl-β-d-galactoside as a substrate. Similarly, Ranjit et al. used β-galactosidase leakage assays to examine the role of disulfide bond formation in cell lysis and oligomerization of a membrane-associated holin protein CidA (6). Baum et al. constructed S. aureus strains in which chromosomal insertions contained lacZ under the transcriptional control of the msrA1 or msrB promoters (7). These strains were used to monitor lacZ expression after generating additional chromosomal mutations and after the addition of the cell wall-active antibiotic oxacillin.
In addition to quantitative β-galactosidase activity assays, lacZ-based colorimetric screening experiments have also been previously employed in S. aureus. Nielsen et al. developed an assay to screen compounds that influence virulence factor production in S. aureus using transcriptional lacZ reporters fused to the promoter sequences of virulence factor genes hla, rnaIII, and spa (8). These strains were placed as top agar overlays in medium containing X-Gal substrate, and cell-free fungal lysates containing potential compound(s) of interest were spotted upon the overlay. Blue-white color development in the agar overlay was monitored to indicate changes in promoter activity. More recently, Bojer et al. used a similar approach to investigate the effects of antimicrobial peptides on virulence gene expression (9). Another work by Ding et al. demonstrated the influence of a citrate-responsive catabolite control protein E (CcpE) on the promoter activity of aconitase gene (the second enzyme of the tricarboxylic acid cycle) citB (10). They created transcriptionally fused citB-lacZ and found that not only was the promoter activity of citB considerably reduced in the ΔccpE mutant than in the wild-type strain but also the mutation of box-I sequence in citB promoter completely abolished the promoter activity. LacZ-based technologies have not been widely applied to nonbiased genetic screens in S. aureus.
To our knowledge, anaerobic monitoring of lacZ expression using X-Gal has not been utilized, because the dimerization of the reaction intermediate monomers (5-bromo-4-chloro-3-hydroxyindol) requires oxidation to form blue precipitate (5,5′-dibromo-4,4’-dichloro-indigo). Other reporter systems, including luciferase- and GFP-based fluorescent proteins, also require oxygen for signal output. Recently, a new class of oxygen-independent flavin mononucleotide-based fluorescent proteins (FbFPs) has been characterized (11, 12). Drepper et al. engineered a set of FbFPs that are derivatives of bacterial blue-light receptors from Bacillus subtilis and Pseudomonas putida (12). These proteins were used to generate fluorescent reporter systems that are functional under both aerobic and anaerobic conditions in E. coli (EcFbFP).
Alpha-hemolysin (alpha-toxin), encoded by hla, is a prototypic β-barrel toxin and one of the key virulence factors of S. aureus. Upon secretion, it forms a pore in the membranes of target host red blood cells, resulting in cell lysis (13). The S. aureus genome encodes several secreted lipase enzymes, which serve to break down host-derived lipids into free fatty acids for nutrient acquisition (14). Of these, the glycerol ester hydrolase lipase is encoded by the geh locus and is specific for long-chain fatty acids (15, 16).
Here, we describe vectors that allow for markerless transcriptional reporters utilizing lacZ, EcFbFP, hla, or geh expression to monitor promoter activity. The vectors allow for expression to be driven by different ribosome binding sequences (RBS) of varied strength. Importantly, when the vectors are resolved after making mutants, they do not leave behind genetic determinants that provide antibiotic resistance and therefore can be used for additional genetic manipulations, including the generation of transposon mutant libraries. We also describe a crtM::kan deletion insertion mutation that prevents staphyloxanthin production and aids in mutant identification during blue-white screening. We outline methods to use the vectors for both aerobic and anaerobic screening.
RESULTS
Creation of S. aureus lacZ transcriptional reporters.
We envisioned a series of plasmids containing transcriptional reporters that could be used to generate markerless S. aureus chromosomal insertions. We chose to use the pJB38 vector as a backbone, which encodes ampicillin and chloramphenicol resistance in E. coli and S. aureus, respectively (17). It also encodes a temperature-sensitive origin of replication in S. aureus and for an anhydrotetracycline-inducible antisense RNA to S. aureus secY, allowing for inducible counterselection.
We created a series of four vectors (see Table S1 in the supplemental material) that integrate into the geh locus (SAUSA300_0320), which is commonly used as an episome integration site (18). The vectors have one of four ribosomal binding sites (RBS) that drive expression of Escherichia coli lacZ that was codon optimized for S. aureus expression: hld, sarA, TIR (transcription initiation region), and sodM (Fig. 1A) (19). Altering the ribosomal binding site can alter gene expression in S. aureus (20). The transcription initiation region (TIR) RBS was recently described to drive a constitutively high level of expression (21, 22). The vectors contain the yeast 2μ origin of replication and URE3, allowing for the selection for uracil prototrophs using Saccharomyces cerevisiae strain FY2, thereby permitting yeast recombinational cloning. The vectors contain a polylinker region upstream of the promoter as well as KpnI and MluI restriction sites downstream of the promoter yet upstream of the individual RBS (Table S5). These restriction sites allow for the interchanging of promoter sequences while simultaneously preserving the location and presence of the RBS. Importantly, once the vector backbone is excised from the S. aureus genome, the integrated elements do not leave a gene encoding antibiotic resistance. This allows for strains containing the chromosome-resolved transcriptional reporter to be used for further genetic manipulation.
FIG 1.

Ribosomal binding site alters lacZ expression. (A) The DNA sequences of the four ribosomal binding sites (RBS) utilized to generate the transcriptional reporters. (B) β-Galactosidase activity in the wild-type (JMB 1100; no lacZ), geh::suf _TIR RBS_lacZ (JMB 9741), suf _sodM RBS_lacZ (JMB 9739), suf _sarA RBS_lacZ (JMB 9740), and suf _hld RBS_lacZ (JMB 9738) strains during logarithmic or stationary growth phases. (C) Overnight cultures of the wild type (JMB 1100), cap5a_TIR RBS_lacZ (JMB 9765), cap5a_sodM RBS_lacZ (JMB 9764), cap5a _sarA RBS_lacZ (JMB 9754), and cap5a _hld RBS_lacZ (JMB 9777) strains were serially diluted by 10-fold dilutions, and 5 μl of each strain was spotted (10−2 through 10−5) on TSA containing 100 μg ml−1 X-Gal. The data displayed in panel B are averages from biological triplicates with the standard deviations shown. A representative image is shown in panel C.
We first constructed a series of strains containing the promoter for the sufCDSUB operon driving expression of lacZ (23). Each of the four strains contained a different RBS sequence driving lacZ expression. β-Galactosidase activity was quantified after liquid growth and visualized on solid media. The different sufC::lacZ strains had varied β-galactosidase activity after liquid growth, resulting in the following expression pattern: TIR>hld>sarA>sodM (Fig. 1B). The alterations in RBS-driven lacZ expression were independent of growth phase. As predicted, the wild-type strain (JMB 1100) did not display significant β-galactosidase activity. We could not visually distinguish a difference between these reporter strains when they were spot plated on solid medium containing X-Gal (data not shown).
We also examined lacZ expression driven by the cap5a promoter on solid medium containing X-Gal. In this case, we could visualize variation in intensity of blue color of the cap5a::lacZ strains, which displayed a pattern of indole formation that was consistent with the liquid assay results: TIR>hld>sarA-sodM (Fig. 1C).
Examining the effect of chromosomal manipulations on transcriptional reporter activity.
SaeRS is a two-component regulatory system in S. aureus (24). SaeR and SaeS are the response regulator and the histidine kinase, respectively. SaeS also interacts with SaeP, which stimulates the phosphatase activity of SaeS (25). The S. aureus sae locus is comprised of four open reading frames (saePQRS), which have two promoters. The first promoter, denoted P1, lies upstream of saeP and is responsive to the phosphorylation status of SaeR (25).
We created a strain containing the P1 promoter driving lacZ expression (geh::P1 sarA RBS_lacZ; P1sae::lacZ). Because the sarA RBS showed intermediate levels of β-galactosidase activity (Fig. 1), we selected the sarA RBS for construction of subsequent reporter strains used for blue-white screening to facilitate visualization of alterations in reporter activity and variations in levels of indole precipitate formation. We hypothesized that intermediate lacZ expression would allow us to identify genes that positively and negatively impact P1sae transcriptional activity. We transduced this strain with saeR::Tn and saeP::Tn insertional inactivation mutations and examined lacZ expression on solid medium by spot plating the P1sae::lacZ strains with various concentrations of X-Gal. The strains displayed a concomitant increase in indole formation as a function of X-Gal concentration (Fig. 2A). The strain containing the saeR::Tn mutant displayed a visual decrease in indole formation (Fig. 2A) compared to the parent strain. The strain containing the saeP::Tn mutation behaved similarly to the parent, suggesting that SaeP was inactive under the growth conditions utilized. These data suggest that the concentration of X-Gal could be varied to be effectively used to examine both activators and repressors of a locus.
FIG 2.

Directed chromosomal manipulations alter P1sae::lacZ expression. (A) Overnight cultures of the wild-type (JMB 1100), P1sae::lacZ (JMB 9709), P1sae::lacZ saeR::Tn (JMB 9727), and P1sae::lacZ saeP::Tn (JMB 9728) strains were serially diluted by 10-fold dilutions and spot plated (10−1 through 10−6) on TSA with various concentrations of X-Gal. (B) Quantitative β-galactosidase assays using the strains examined in panel A. All strains utilized the sarA RBS to drive lacZ expression. Representative images are displayed in panel A. The data depicted in panel B represent the averages from biological triplicates with the standard deviations shown.
We analyzed β-galactosidase expression after aerobic liquid growth. As expected, the saeR::Tn mutant was required to activate transcription of P1sae::lacZ. The saeP::Tn mutant had increased β-galactosidase activity compared to the parent strain, suggesting the SaeP was stimulating phosphatase activity under this growth condition.
Generation of ΔcrtM::kan allele to improve the resolution of blue-white screening.
We built a mariner-based transposon mutant library in a P1sae::lacZ (sarA_RBS) reporter strain and screened the library for strains with altered P1 promoter activity. Several strains were isolated that visually appeared to have altered indole formation after aerobic growth. When the strains were plated on TSA medium that did not contain X-Gal, they had no pigmentation (Fig. 3A). The pigment staphyloxanthin provides most S. aureus strains with a characteristic golden color (26). We determined the chromosomal location of one of the insertions that generated a nonpigmented S. aureus strain and found that it was in the crtP gene locus (SAUSA300_2501) (Fig. 3A). CrtP is one of the enzymes required for staphyloxanthin biosynthesis (27). We determined β-galactosidase activity in the parent and the crtP mutant after liquid growth and found that there was not a significant difference in either logarithmic or stationary phases of growth (Fig. 3B).
FIG 3.
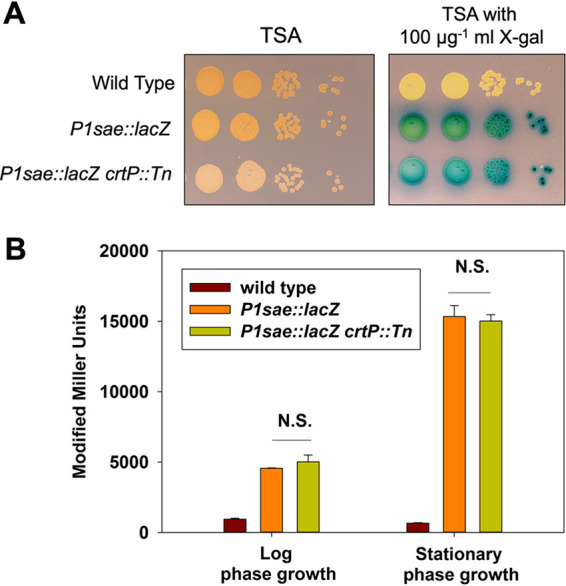
Staphyloxanthin production obfuscates visualizing indole formation. (A) Overnight cultures of the wild-type (JMB 1100, no lacZ), P1sae::lacZ (JMB 9740), and P1sae::lacZ crtP::Tn (JMB 9832) strains were serially diluted by 10-fold dilutions and spot plated (10−3 through 10−6) on TSA with and without 100 μg ml−1 X-Gal. (B) β-Galactosidase activity in the wild-type (JMB 1100), P1sae::lacZ (JMB 9709), and P1sae::lacZ crtP::Tn (JMB 9832) strains during logarithmic or stationary growth phases. N.S., not significant. All strains utilized the sarA RBS to drive lacZ expression. A representative photo is displayed in panel A. The data displayed in panel B represent the averages from biological triplicates with the standard deviations shown.
We hypothesized that staphyloxathin accumulation was interfering with the intensity of blue color and complicating our screening process. To facilitate better resolution for solid medium lacZ-based blue-white screening with S. aureus, we constructed a strain that lacked the ability to produce staphyloxanthin. CrtM catalyzes the first committed step in staphyloxanthin production (27). We constructed a ΔcrtM::kan mutant because the vectors needed for building transposon mutant libraries do not utilize kanamycin resistance determinants and kanamycin resistance is not routinely used to manipulate the S. aureus genome.
SrrAB is a two-component regulatory system utilizing the SrrA DNA-binding response regulator and the membrane-associated histidine kinase SrrB (28, 29). The srrA promoter responds to the phosphorylation status of SrrA. We created the pJB38_srrAp_sarA RBS_lacZ plasmid and used it to create a srrA::lacZ reporter strain. The levels of expression of β-galactosidase from the P1sae::lacZ and srrA::lacZ reporters in wild type and isogenic ΔcrtM::kan strains were not significantly different (Fig. 4A). When we serially diluted and spot plated these strains alongside isogenic strains containing saeR::Tn or srrA::Tn, we noted an increase in visual differentiation in indole formation in the strains lacking staphyloxanthin production (Fig. 4B). Interestingly, we also observed that strains lacking SrrA displayed sensitivity to X-Gal.
FIG 4.

crtM::kan allele prevents staphyloxanthin expression and aids in blue-white screening. (A) β-Galactosidase activity in the P1sae::lacZ (JMB 9709), P1sae::lacZ crtM::kanR (JMB 10021), srrA::lacZ (JMB 9742), and srrAp::lacZ crtM::kanR (JMB 10025) strains during logarithmic or stationary growth phases. (B) The following strains were cultured overnight before serial diluting by 10-fold dilutions and spot plating (10−2 through 10−5) on TSA and TSA with 50 μg ml−1 X-Gal: wild type (JMB 1100), srrAp::lacZ (JMB 9742), srrAp::lacZ srrA::Tn (JMB 10064), srrA::lacZ ΔcrtM::kan (JMB 10025), srrA::lacZ ΔcrtM::kan srrA::Tn (JMB 10067), P1sae::lacZ (JMB 9709), P1sae::lacZ saeR::Tn (JMB 9727), P1sae::lacZ ΔcrtM::kan (JMB 10021), and P1sae::lacZ ΔcrtM::kan saeR::Tn (JMB 10236). All strains utilized the sarA RBS to drive lacZ expression. The data displayed in panel A are averages from biological triplicates with the standard deviations shown. Representative images are displayed in panel B.
The finding that the growth of the srrA::lacZ srrA::Tn strain was negatively affected by the presence of X-Gal led us to examine whether the geh locus is important for cellular fitness. We conducted a series of competition experiments using the WT, geh::Tn mutants, or the strains containing the srrA::lacZ construct within geh. We found that disrupting geh with either a transposon or the srrA::lacZ transcriptional reporter had no effect on fitness under the culture conditions utilized (Table S6). Likewise, the growth of the geh::Tn and srrA::lacZ strains were identical in TSB medium (Fig. S1). Taken together, these data indicate the srrA::lacZ strain containing the srrA::Tn mutation are adversely effected by the presence of X-Gal and that the geh locus does not play a critical role in fitness of S. aureus USA300_LAC under the growth conditions utilized.
Using lacZ transcriptional reporter strains for anaerobic screening.
Traditionally, lacZ-dependent X-Gal hydrolysis screens have only been conducted aerobically because an oxidant (i.e., dioxygen) is necessary for dimerization of the hydrolysis by-product 5-bromo-4-chloro-3-hydroxyindole, which is visualized as indigo color. S. aureus is a facultative anaerobe (30), and blue-white screens have previously been performed after anaerobic growth using X-Gal spray overlays (31). We sought to apply this method to visualize β-galactosidase activity in S. aureus grown anaerobically on solid medium. We monitored lacZ expression in the srrA::lacZ strain as well as the isogenic srrB::Tn and ΔsrrAB::tet mutants that had been spot plated and cultured anaerobically. The agar plates were colorless after incubation. One plate was sprayed with a 25-mg ml−1 X-Gal solution and removed from the anaerobic chamber. Another plate also sprayed with a solution of 25 mg ml−1 X-Gal and 3.3 mg ml−1 tetracycline to prevent new β-galactosidase synthesis. Upon exposure to oxygen and indole color development, we observed that the srrB::Tn mutant exhibited decreased srrAp::lacZ reporter activity compared to the parent strain (Fig. 5A). The addition of tetracycline had no impact on chromatic development suggesting that the presence of oxygen was not significantly altering lacZ expression under the time frame utilized.
FIG 5.

Monitoring lacZ expression during anaerobic growth. (A) Overnight cultures of the wild type (JMB 1100), srrAp::lacZ (JMB 9742), srrAp::lacZ srrB::Tn (JMB 10066), and srrAp::lacZ ΔsrrAB::tetM (JMB 10065) were serially diluted, spotted on TSA, and cultured anaerobically. One plate was sprayed with a 25-mg ml−1 solution of X-Gal, and another was sprayed with a solution of 25 mg ml−1 X-Gal and 3.3 mg ml−1 tetracycline before all three plates were removed from the anaerobic chamber and allowed to develop. (B) Overnight cultures of wild type (JMB 1100), P1sae::lacZ (JMB 9709), and P1sae::lacZ saeR::Tn (JMB 9727) were serially diluted and spotted on TSA with and without 100 μg ml−1 X-Gal and with and without 5 mM Fe(CN)63− and Fe(CN)64− before incubating in aerobic or anaerobic conditions. (C) Overnight cultures of the same strains used in panel B were serially diluted and spotted on plates with or without S-gal. The plates were incubated anaerobically. Representative images are shown and depict 10-fold serial dilutions (10−3 through 10−6).
We next developed a method for monitoring X-Gal hydrolysis on solid medium in the absence of dioxygen. The addition of a ferrocyanide-ferricyanide redox system [Fe(CN)63−/4–] has been shown to act as an electron acceptor to increase the rate of 5,5′-dibromo-4,4′-dichloro-indigo development when monitoring lacZ expression for histochemical analyses (32, 33). We examined whether the addition of these chemicals to solid media would allow us to visualize X-Gal hydrolysis anaerobically. The inclusion of Fe(CN)63–/4− resulted in the formation of an indigo precipitate anaerobically (Fig. 5B). Addition of 5 mM Fe(CN)63− and Fe(CN)64− resulted in optimal visualization and little to no growth inhibition. The wild-type and the P1sae::lacZ saeR::Tn strains did not display noticeable chromophore development, consistent with the blue color arising from lacZ expression and X-Gal cleavage.
3,4-Cyclohexeneoesculetin-β-d-galactopyranoside sodium salt (S-gal) is a chromogenic substrate for β-galactosidase that can be utilized to examine lacZ expression in the absence of oxygen. When hydrolyzed, the by-product can chelate iron resulting in a black precipitate (34). Importantly, S-gal staining does not require oxygen and therefore can be utilized to visualize β-galactosidase activity under anaerobic conditions. To visualize anaerobic promoter activity, we spotted P1sae::lacZ reporter strains on plates containing S-gal and Fe3+ (Fig. 5C). As expected, the P1sae::lacZ strain displayed a black precipitant after anaerobic incubation. The wild-type and P1sae::lacZ saeR::Tn strains displayed little to no black precipitant, suggesting that lacZ expression was leading to this phenotype.
Using a FbFP reporter system to assay anaerobic transcriptional activity.
Flavin mononucleotide-based fluorescent proteins (FbFPs) have been used to develop fluorescent reporter systems that are functional under both aerobic and anaerobic conditions in E. coli (12). We generated a multicopy plasmid FbFP-based transcriptional reporter for expression in S. aureus. The E. coli-derived (EcFbFP) genetic sequence was codon optimized for S. aureus expression and placed under the transcriptional control of the P1sae promoter. S. aureus cells harboring the P1sae::EcFbFP reporter displayed significantly greater fluorescence than wild-type cells after aerobic culture (Fig. 6A). Fluorescence was greatly diminished in a ΔsaePQRS::spec mutant containing P1sae::EcFbFP reporter, suggesting that the fluorescence signal was specific to P1sae promoter activity. Importantly, the P1sae::EcFbFP reporter system was functional in the absence of oxygen (Fig. 6B).
FIG 6.

Using EcFbFP expression to monitor transcriptional activity. Fluorescence was monitored in the wild type (JMB 1100) and ΔsaePQRS::spec (JMB 1335) strains containing either pCM28 or pCM28_ P1sae::EcPbFP after aerobic (A) or anaerobic (B) culture. (C) Fluorescence was monitored in the wild-type (JMB 1100), P1sae::EcFbFP (JMB 10207), and P1sae::EcFbFP saeR::Tn (JMB 10217) strains after aerobic culture. The data represent the averages from biological triplicates, and standard deviations are shown.
In addition to the plasmid-based EcFbFP reporter system, we also generated a chromosomally integrated P1sae::EcFbFP reporter strain, which allows for markerless insertion of the reporter sequence into geh of the S. aureus genome. The integrated P1sae::EcFbFP strain displayed weaker fluorescence signal than a strain carrying the plasmid-based reporter (Fig. 6C). This decrease in signal output likely reflects transcriptional reporter copy number.
Creation of vectors to utilize hemolysin or lipase expression to monitor transcriptional activity.
We generated two vectors that can be used to screen aerobic or anaerobic cultures for altered promoter transcriptional activity using the endogenous S. aureus enzymes alpha-hemolysin (hla) and lipase (geh). Both reporters utilize the TIR ribosomal binding site because of its constitutively high level of expression. The vectors provide markerless integration into the S. aureus genome, allowing for further genetic manipulation. We used the vectors to replace the hla or geh promoters with the P1sae promoter. Notably, the gene products of both hla and geh are secreted proteins, which allows for screening on solid media.
When examined on rabbit blood agar, S. aureus cells containing the P1sae::hla reporter produced a zone of hemolysis (Fig. 7A). Introduction of an saeP::Tn or saeR::Tn mutation increased and decreased the zone of hemolysis, respectively, suggesting that the changes in hla expression were specific to the P1sae promoter (Fig. 7B). Importantly, alpha-hemolysis activity was observed under both aerobic and anaerobic conditions.
FIG 7.
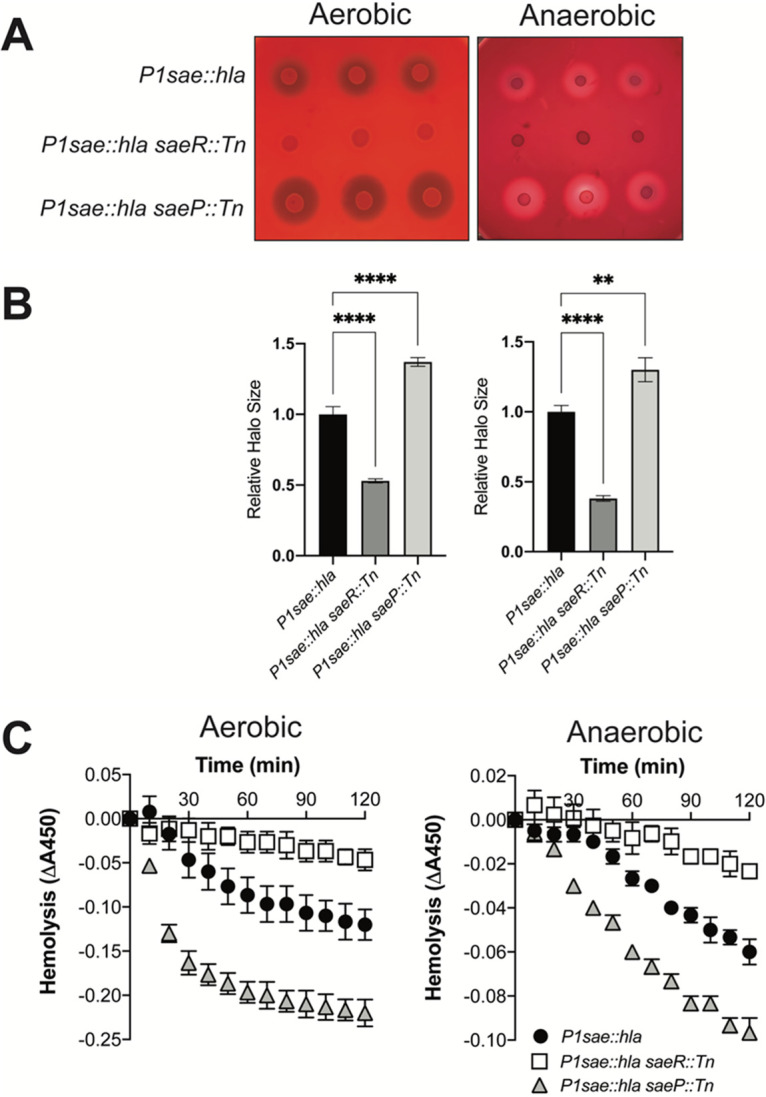
Using hla expression to monitor transcriptional activity. (A) Overnight cultures of the P1sae::hla (JMB 10341), P1sae::hla saeR::Tn (JMB 10350), and P1sae::hla saeP::Tn (JMB 10351) strains were spotted in triplicate on TSA plates containing 5% defibrinated rabbit blood. (B) Quantification of hla expression by measuring the clearance zones from the plate image shown in panel A. Relative halo size was normalized to the average clearance zone of P1sae::hla (JMB 10341). (C) Quantification of hla expression by monitoring hemolysis. Hemolysis of rabbit red blood cells was assessed by monitoring absorbance at 450 nm and incubating with spent medium from overnight cultures. The strains utilized are the same as the strains utilized in panel A. The data represent averages from biological triplicates, and the error bars represent standard deviations.
To complement the plate-based screening, we quantified alpha-hemolysin activity (Fig. 7C). We assessed hemolysis of rabbit red blood cells incubated with cell-free spent media from overnight cultures. We observed significantly more hemolysis in the spent media from the saeP::Tn mutant than the parent strain. This result suggests that SaeP plays a role in regulating SaeRS activity in both the presence and absence of oxygen. As expected, we noted significantly decreased hemolysis in the saeR::Tn mutant.
We next examined geh expression by supplementing the solid medium with Tween 80 substrate and calcium salt, which forms an insoluble precipitate when bound to free fatty acids generated by lipase. S. aureus cells harboring the P1sae::geh reporter produced a zone of fatty acid precipitate (Fig. 8). The precipitate zone was significantly smaller and larger upon the introduction of saeR::Tn or saeP::Tn mutations, respectively. These data suggest that the witnessed geh expression was specifically controlled by the transcriptional activity of the P1sae promoter. As observed with the alpha-hemolysin reporter system, the P1sae::geh reporter was active under both aerobic and anaerobic conditions.
FIG 8.
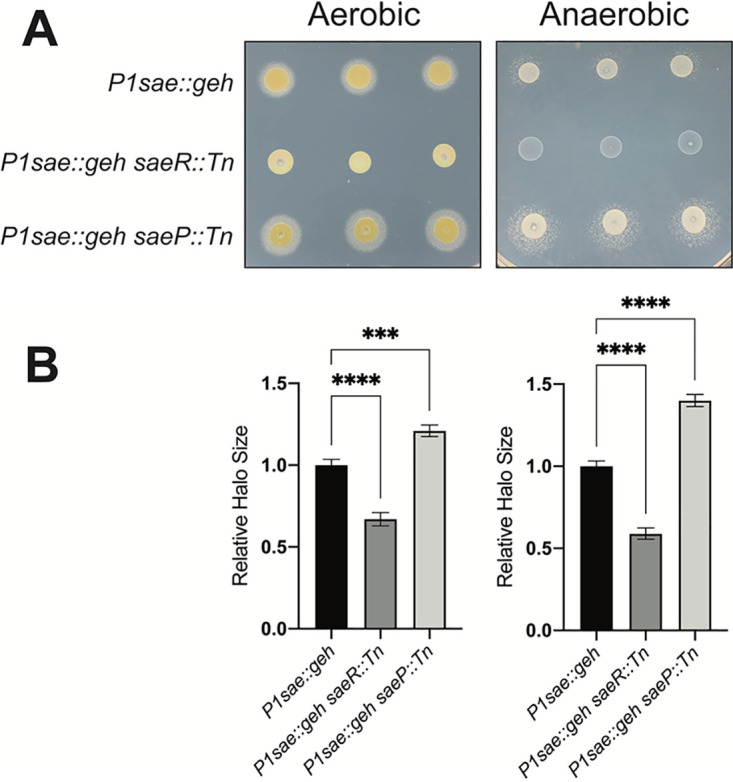
Using geh expression to monitor transcriptional activity. (A) Overnight cultures of the P1sae::geh (JMB 10337), P1sae::geh saeR::Tn (JMB 10346), and P1sae::geh saeP::Tn (JMB 10347) strains were spotted in triplicate on lipase activity plates containing 1% Tween 80 and calcium salt. (B) Quantification of geh expression by measuring the precipitate zones from the plate image is shown in panel A. Relative halo size was normalized to the average precipitate zone of P1sae::geh (JMB 10337).
DISCUSSION
We have designed a suite of vectors and methods to monitor transcriptional activity in S. aureus under both aerobic and anaerobic growth conditions. These vectors will enable researchers to identify physiological conditions and genetic loci that alter promoter activity. Importantly, these vectors have several key advantages over previously described S. aureus vectors. The presence of a yeast cloning cassette and restriction sites that flank the promoter region allow for simple replacement of the promoter of interest using restriction enzyme-based cloning or recombinational cloning. The shuttle vectors can be moved easily between S. cerevisiae, E. coli, and S. aureus. The vectors have one of four RBS, which allows researchers to tailor reporter gene transcription. There are restriction sites upstream and downstream of the reporter gene, allowing for replacement with alternate reporter genes (gfp, yfp, mCherry, etc.). Many of the vectors integrate into the nonessential geh locus and, to our knowledge, do not hamper the fitness of S. aureus under standard laboratory culture conditions (see Table S6 and Fig. S1 in the supplemental material). Lastly, the vectors enable researchers to construct markerless reporter strains, which allows for further genetic manipulation, such as the building of transposon mutant libraries using plasmids requiring extensive selection for antibiotic resistance.
S. aureus does not show an inherent β-galactosidase activity like other coagulase-positive staphylococci, which allows for the monitoring of X-Gal hydrolysis (18). A wide array of β-galactosidase substrates is available for detection of β-galactosidase activity; however, nearly all the substrates described require dioxygen for development. Here, we demonstrate that anaerobic S. aureus gene expression studies can be successfully conducted on solid media with lacZ-based reporters using S-gal, an X-Gal spray, or the inclusion of an Fe(CN)63−/4− redox cycling system.
After building transposon mutant libraries in our reporter strains, we found that nearly all the mutants isolated contained mutations that altered staphyloxanthin production, which obfuscated the results from blue-white screening. To circumvent this problem, we created a ΔcrtM::kanR mutation which prevented staphyloxanthin production and afforded better clarity in plate screening. Kanamycin is rarely used in S. aureus molecular biology; therefore, the ΔcrtM::kanR mutation allows for further genetic manipulation and for locus movement between strains via transduction. Staphyloxanthin production has been shown to protect cells from reactive oxygen species (ROS) and to have a role in pathogenesis. The presence of staphyloxanthin did not alter the activities of the promoters studied here. That said, researchers should keep the role of crtM in mind when examining promoter activity under conditions of high ROS production and avoid using the crtM::kanR allele for in vivo pathogenesis studies.
Three non-lacZ-based reporter systems allowed us to monitor gene transcriptional activity in the absence of oxygen. Monitoring P1sae transcriptional activity by quantifying EcFbFP expression worked well when the reporter system was provided as a multicopy plasmid. The sensitivity of this reporter was diminished when provided in single copy via chromosomal integration even when using the TIR RBS to drive expression. The hla and geh reporters provided robust expression patterns on solid media. Importantly, the activities of both alpha-hemolysin and lipase do not require oxygen, making these reporter vectors ideal for monitoring anaerobic gene transcription.
As a facultative anaerobe, S. aureus can use respiration or fermentation to generate energy and maintain redox homeostasis. Most infection sites are low-oxygen or anaerobic environments. Not surprisingly, S. aureus alters the transcription of many virulence genes as a variable of oxygen tension. A better understanding of anaerobic gene expression will provide insight into S. aureus pathogenesis. The vectors, strains, and methods described here provide an advanced toolkit to dissect aerobic and anaerobic gene regulation in S. aureus.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Tryptic soy broth (TSB) was purchased from VWR. X-Gal was purchased from VWR, and 3,4-cyclohexeneoesculetin-β-d-galactopyranoside sodium salt (S-gal) was purchased from Sigma-Aldrich. For solid medium (TSA), TSB was supplemented with 1.5% agar. For aerobic spotting assays, individual strains were grown in 5 ml of TSB in 30-ml culture tubes and shaken at 220 rpm at 37°C to an optical density at 600 nm (OD600) of 1. Strains were serially diluted, and 5-μl samples were spotted as 10-fold dilutions on TSA plates containing various concentrations of X-Gal or S-gal. For plates containing S-gal, the agar was also supplemented with ferric ammonium citrate (Sigma-Aldrich) at a concentration of 62.5 μg ml−1. For anaerobic spotting experiments, plates were incubated at 37°C within a COY anaerobic chamber for 36 h. Overlay plates were sprayed carefully in the chamber with X-Gal (25 mg ml−1 prepared in dimethyl sulfoxide [DMSO]) supplemented with tetracycline (3.3 mg ml−1) until the agar surface was completely covered. Sprayed plates were removed from the chamber and exposed to oxygen. Plates were dried in a fume hood and were developed for an hour.
When selecting for plasmids, episomes, or chromosomal insertions, antibiotics were added at the following final concentrations: 150 μg ml−1 ampicillin, 30 μg ml−1 chloramphenicol (Cm), 10 μg ml−1 erythromycin (Erm), 50 μg ml−1 kanamycin (Kan), or 3 μg ml−1 tetracycline (Tet).
Growth curves and fitness assays.
Liquid growth curve analysis was conducted in a 96-well microtiter plate using a BioTek 808E visible absorption spectrophotometer. Plates were continually shaken at approximately 200 rpm at 37°C for 12 h, and culture densities were read at 600 nm every 30 min. Cells used for inoculation were cultured for 18 h in TSB medium before washing with phosphate-buffered saline (PBS). Prior to inoculation, the optical densities of the cell suspensions were adjusted to 1 (OD600), and 5 μl was added to 195 μl of TSB medium per well. Biological triplicates of each strain were assayed.
For growth competition assays, triplicate cocultures of the desired pairs of strains were grown at a 1:1 ratio in TSB medium for approximately 18 h at 37°C. Cells used for inoculation were cultured for 18 h in TSB medium before washing with PBS. Prior to coculture inoculation, the optical densities of the cell suspensions were adjusted to ∼1, and 10 μl of each strain was added to 2 ml of TSB medium per coculture tube. After 18 h of growth at 37°C, cocultures were serially diluted in PBS by a factor of 1 × 10−6, and 100 μl of each dilution was spread on TSB agar plates supplemented with or without 10 μg ml−1 Erm. After overnight incubation at 37°C, the relative proportion of both strains in each coculture was determined by counting CFU on each plate. The fraction of the coculture containing each strain was expressed relative to total CFU.
Plasmid and strain construction.
All transductions were conducted using bacteriophage 80α (35). All bacterial strains were PCR verified before use. Plasmids were sequenced at Genewiz (South Plainfield, NJ) (Tables 1 and 2). Synthetic DNA was synthesized by Twist Biosciences (San Francisco, CA) or Integrated DNA Technologies (Coralville, IA). DNA primers were purchased from Integrated DNA Technologies (Coralville, IA). Phusion DNA polymerase was purchased from New England Biolabs. lacZ and EcFbFP were codon optimized for expression in S. aureus using the online Integrated DNA Technologies codon optimization tool.
TABLE 1.
Plasmids utilized in this study
| Plasmid | Construct | Source or reference |
|---|---|---|
| pJB38 | 22 | |
| pJB38_ΔcopBL | YCC amplification | 41 |
| pJB185 | lacZ amplification | 42 |
| pMG020 | Encoding transposase | 38 |
| pBursa | Bursa transposon | 39 |
| pCM28 | 43 | |
| pJB38_ΔcrtM::kanR | crtM mutant | This study |
| pJB38_P1sae_TIR RBS_lacZ | P1 transcriptional reporter | This study |
| pJB38_P1sae_sodM RBS_lacZ | P1 transcriptional reporter | This study |
| pJB38_P1sae_sarA RBS_lacZ | P1 transcriptional reporter | This study |
| pJB38_P1sae_hld RBS_lacZ | P1 transcriptional reporter | This study |
| pJB38_P1sae_sarA RBS_ EcFbFP | P1 transcriptional reporter | This study |
| pCM28_P1sae_sarA RBS_ EcFbFP | P1 transcriptional reporter | This study |
| pJB38_P1sae_TIR RBS_geh | P1 transcriptional reporter | This study |
| pJB38_P1sae_TIR RBS_hla | P1 transcriptional reporter | This study |
| pJB38_suf_TIR RBS_lacZ | suf transcriptional reporter | This study |
| pJB38_suf_sodM RBS_lacZ | suf transcriptional reporter | This study |
| pJB38_suf_sarA RBS_lacZ | suf transcriptional reporter | This study |
| pJB38_suf_hld RBS_lacZ | suf transcriptional reporter | This study |
| pJB38_cap5a_TIR RBS_lacZ | cap5a transcriptional reporter | This study |
| pJB38_cap5a_sodM RBS_lacZ | cap5a transcriptional reporter | This study |
| pJB38_cap5a_sarA RBS_lacZ | cap5a transcriptional reporter | This study |
| pJB38_cap5a_hld RBS_lacZ | cap5a transcriptional reporter | This study |
| pJB38_srrAp_sarA RBS_lacZ | srrA transcriptional reporter | This study |
TABLE 2.
S. aureus strains utilized in this study
| Name | Genotype | Source or reference |
|---|---|---|
| RN4220 | Restriction minus | 44 |
| JMB 1100 | Wild type | 43 |
| JMB 2122 | ΔbshA::kanR | 45 |
| JMB 1335 | ΔsaePQRS::spec | 30 |
| JMB 9741 | geh::suf _TIR RBS_lacZ | This study |
| JMB 9739 | geh::suf _sodM RBS_lacZ | This study |
| JMB 9740 | geh::suf _sarA RBS_lacZ | This study |
| JMB 9738 | geh::suf _hld RBS_lacZ | This study |
| JMB 9765 | geh::cap5a_TIR RBS_lacZ | This study |
| JMB 9764 | geh::cap5a_sodM RBS_lacZ | This study |
| JMB 9754 | geh::cap5a _sarA RBS_lacZ | This study |
| JMB 9777 | geh::cap5a _hld RBS_lacZ | This study |
| JMB 9709 | geh::P1sae_sarA RBS_lacZ | This study |
| JMB 9727 | geh::P1sae_sarA RBS_lacZ saeR::Tn (ermB) | This study, 46 |
| JMB 9774 | geh::P1sae_sarA RBS_lacZ Tn library | This study |
| JMB 9728 | geh::P1sae_sarA RBS_lacZ saeP::Tn (ermB) | This study, 46 |
| JMB 9832 | geh::P1sae_sarA RBS _lacZ SAUSA300_2501::Tn (crtP) (ermB) | This study |
| JMB 9964 | crtM::kanR | This study |
| JMB 9742 | geh::srrAp _sarA_RBS_lacZ | This study |
| JMB 10025 | geh::srrAp_sarA_RBS_lacZ ΔcrtM::kanR | This study |
| JMB 10021 | geh:: P1sae_sarA RBS_lacZ ΔcrtM::kanR | This study |
| JMB 10067 | geh::srrAp_sarA_RBS_lacZ ΔcrtM::kanR srrA::Tn (ermB) | This study, 46 |
| JMB 10236 | geh::P1sae_sarA RBS_lacZ ΔcrtM::kanR saeR::Tn (ermB) | This study, 46 |
| JMB 10064 | geh::srrAp_sarA RBS_lacZ srrA::Tn (ermB) | This study, 46 |
| JMB 10065 | geh::srrAp_sarA RBS_lacZ ΔsrrAB::tetM | This study, 28 |
| JMB 10207 | geh:: P1sae_sarA RBS_EcFbFP | This study |
| JMB 10217 | geh:: P1sae_sarA RBS_EcFbFP saeR::Tn (ermB) | This study, 46 |
| JMB 10341 | P1sae_TIR RBS::hla | This study |
| JMB 10350 | P1sae_TIR RBS::hla saeR::Tn (ermB) | This study, 46 |
| JMB 10351 | P1sae_TIR RBS::hla saeP::Tn (ermB) | This study, 46 |
| JMB 10337 | P1sae_TIR RBS::geh | This study |
| JMB 10346 | P1sae_TIR RBS::geh saeR::Tn (ermB) | This study, 46 |
| JMB 10347 | P1sae_TIR RBS::geh saeP::Tn (ermB) | This study, 46 |
| JMB 10621 | lacB::Tn (ermB) | This study, 46 |
| JMB 10623 | geh::srrAp _sarA_RBS_lacZ lacB::Tn (ermB) | This study, 46 |
| JMB 1886 | geh::pLL39 | This study |
| JMB 10624 | geh::pLL39 lacB::Tn (ermB) | This study, 30, 46 |
| JMB 10622 | geh::Tn (ermB) | This study, 46 |
Yeast homologous recombination was used to construct plasmids as previously described (17, 36). To begin, portions of DNA were synthesized containing (i) a 3′ portion homologous to an upstream portion of geh, (ii) a polylinker, (iii) the P1sae or suf promoter, (iv) KpnI, NheI, MluI, and SalI restriction sites, (v) a sodM, sarA, hld, or TIR ribosomal binding site, and (vi) the 5′ portion of the codon-optimized lacZ. The sequences of the synthesized DNA constructs used for construction of plasmids are listed in Table S1 in the supplemental material. The sequences of the DNA primers utilized to generate PCR amplicons are listed in Table S2. The yeast cloning cassette and lacZ sequences were amplified using pJB38_ΔcopBL and pJB185 as templates, respectively (19). The plasmids that were created using yeast recombinational cloning along with the DNA primers and DNA templates used to generate the amplicons are listed in Table S3. The amplicons were combined with EcoRI-digested pJB38 and transformed into Saccharomyces cerevisiae strain FY2. S. cerevisiae colonies containing the plasmid of interest were identified by colony PCR and further propagated. Plasmids were recovered from yeast and electroporated into E. coli PX5 (Protein Express) cells selecting for Amp resistance. The plasmids were then transformed into S. aureus RN4220 and selected for Cm resistance. The vectors were transduced into JMB1100, and the integrates were constructed as previously described (37).
In order to create the ΔcrtM::kanR deletion strain, approximately 500 bp upstream and downstream of the crtM gene (SAUSA300_2499) was PCR amplified using JMB1100 or JMB2122 (kanR) chromosomal DNA as a template and the following primer pairs: YCC_crtM_for and kanR_up_crtM_rev; up kanR_crtM_for and down_kanR_crtM_rev; and kanR_down_crtM_for and pJB38_crtM_rev. pJB38_rseE::tet was digested with MluI and NheI and gel purified. The vector and amplicons were combined and transduced into S. cerevisiae strain FY2, resulting in pJB38_ΔcrtM::kanR. The plasmid was recovered using E. coli PX5 and transformed into S. aureus strain RN4220. The mutant was created in JMB 1100, resulting in strain JMB9964, and then the crtM::kanR allele was transduced into strains of interest.
To generate the pJB38_ P1sae_sarA RBS_EcFbFP vector, the pJB38_ P1sae_TIR RBS_lacZ vector was digested with MluI and PstI. MluI cuts upstream of lacZ. PstI cuts downstream of the geh downstream fragment. The vector backbone was gel purified and combined with the amplicons generated using the following primer pairs: gehpJB38 pstI and EcFbFPgehfor and P1TIR EcFbFP for and EcFbFPgehrev. Chromosomal DNA and the synthesized EcFbFP fragment were used as templates for PCR. To generate the pCM28_ P1sae_sarA RBS_EcFbFP vector, pCM28 was digested with BamHI and SalI. The gel-purified vector backbone was combined with amplicons generated using pJB38_ P1sae_sarA RBS_EcFbFP as a PCR template and the following primer pairs: pCM28_YCC_for and YCC_P1_rev and YCC_P1_for and EcFbFP_pCM28_3. Note that the upstream and downstream primers did not contain the BamHI and SalI restriction sites found in pCM28, yielding a plasmid lacking these sites.
To create the pJB38_ P1sae_TIR RBS_hla pJB38_ P1sae_TIR RBS_geh vectors, pJB38 was linearized with EcoRI. The native hla or geh promoters and RBS were replaced with the P1sae promoter and either the sodM or TIR RBS. This was flanked by approximately 500 bp of chromosomal DNA from upstream of the promoter and 500 bp of downstream chromosomal DNA that initiates the translational start site. The vectors integrate at the native hla or geh loci and replace the native promoter with the P1sae promoter and the selected RBS.
Several additional plasmids were created using restriction enzyme-based cloning. These plasmids, as well as the primers, vector backbones, and restriction enzymes that were utilized to create them, are listed in Table S4. Representative vector sequences and maps are shown in Table S5.
Quantitative β-galactosidase assay.
The bacterial strains were grown to an OD600 of approximately 1. Cell culture (1 ml) was pelleted by centrifugation. Cell pellets were resuspended in 1.2 ml of Z-buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, pH 7.0) in 2-ml screw-cap tubes containing 0.1-mm silica glass beads (MP Biomedicals). Cells were lysed by bead beating (3 cycles, 40 s each, 6.0 m/s) using a FastPrep homogenizer (MP Biomedicals). Material was centrifuged at 13,000 × g for 2 min to remove unlysed cells and insoluble debris. Supernatant lysate (20 μl) was added to 680 μl of Z-buffer, and 140 μl of ONPG (4 mg ml−1 [wt/vol]) was added to the samples. The reactions were quenched by adding 200 μl stop solution (1 M Na2CO3) as soon as the samples turned light yellow, and the reaction time was recorded. The A420 of the samples were measured using a UV spectrophotometer (Beckman Coulter DU 530 Life Science UV/Vis spectrophotometer). The corresponding protein concentrations of the samples were measured using a Bradford assay as previously described (15). The modified Miller units (specific activity) were calculated as the following:
For anaerobic β-galactosidase assays, the reporter strains were grown aerobically to an OD600 of ∼1 and then transferred to a 37°C incubator in an anaerobic chamber and grown statically overnight. The cells were pelleted inside the anaerobic chamber by centrifugation, resuspended in 1.2 ml of Z-buffer, and transferred to glass bead-containing screw-cap tubes prior to removal from the anaerobic chamber. The remaining procedure after cell lysis was carried out as described above.
Transposon library construction.
Transposon mutant libraries were constructed in the P1sae_sarA RBS_lacZ (JMB 9709) reporter strain as previously described (38, 39). Briefly, the plasmid pMG020 (encoding transposase) was freshly transformed into RN4220 and incubated on TSA Tet (10 μg ml−1) at 30°C. Single colonies were selected and grown in TSB Tet (10 μg ml−1) at 30°C, and lysates were generated. Reporter strains carrying pBursa were transduced with pMG020 and selected on TSA Cm-Tet plates at 30°C. Cells grown from individual colonies struck on TSA Cm-Tet plates were diluted and suspended in 200 μl PBS buffer, and 15-μl aliquots were spread onto TSA plates containing 10 μg ml−1 Erm and then incubated at 42°C for 24 h to allow for transposition. In total, colonies from 22 large petri plates (containing approximately 3,000 colonies each) were pooled using TSB 10 μg ml−1 Erm supplemented with 25% glycerol. Aliquots were thoroughly mixed by vortexing and combined into a single pool of transposon mutants. Aliquots (1 ml each) were then frozen and stored at −80°C. Aliquots of the transposon libraries were plated on TSA Erm plates containing 50 μg ml−1 X-Gal to select single-colony mutants with altered lacZ expression. Mutants with altered lacZ expression were reconstructed by transforming the Tn lesion back into the parent, followed by qualitative and quantitative β-galactosidase assays.
Mapping the locations of chromosomal insertions.
The genomic locations of transposon insertions were mapped as previously described, with slight modifications (38, 39). Briefly, genomic DNA was isolated from the colonies that displayed a variation in X-Gal hydrolysis using a Lucigen Gram-positive DNA purification kit. Chromosomal DNA (1 μg) was digested with 10 U (1 μl) of restriction enzyme AciI (New England Biolabs [NEB]) for 1 h at 37°C, heat inactivated at 65°C for 30 min, and ligated using 1 μl Quick ligase (NEB) at room temperature for 15 min. PCRs were then performed in a final volume of 50 μl containing the ligated DNA, Phusion polymerase, and Tn-Buster and Martn-ermR DNA primers. DNA was amplified using a three-step PCR cycle, denaturation (98°C for 30s), annealing (50°C for 30s), and elongation (72°C for 2 min), repeated 25 times. The PCR products were separated on a 1% agarose gel, and DNA bands were gel extracted (Qiagen) and submitted for Sanger sequencing using either the Tn-Buster or Martn-ermR primers.
Examining expression of EcFbFP reporter constructs.
Fluorescence measurements of whole-cell liquid cultures were carried out photometrically on a Variskan Lux plate reader (Thermo Scientific). Aliquots of cell cultures (1 ml) grown under aerobic or anaerobic conditions were pelleted by centrifugation and resuspended in 1 ml of PBS. Samples of 200 μl were used for quantification of the fluorescence intensity in a black 96-well plate at room temperature. Measurements were taken with an excitation wavelength of 450 nm, an emission wavelength of 495 nm, and a 12-nm path length. Triplicate samples from each strain were averaged and normalized against cell density.
Hemolysin and lipase activity assays.
For plate-based hemolysis and lipase assays, overnight cultures grown in TSB were diluted and grown to an optical density of 1 (A600). For hemolysis assays, 2-μl aliquots of the cell suspension were spotted onto TSA plates containing 5% defibrinated rabbit blood (HemoStat Laboratories). For lipase assays, 2-μl aliquots of cell suspension were spotted onto lipase activity plates (1% peptone, 85 mM NaCl, 8.8 mM CaCl2, 1.5% agar) containing 1% Tween 80 (VWR). Plates were incubated at 37°C aerobically or in a COY anaerobic chamber until halos surrounding the spotted cells appeared.
Quantitative hemolysis assays were performed as previously described, with slight modifications (40). Briefly, cultures were incubated at 37°C with shaking at 220 rpm for 18 h. For anaerobic samples, strains were grown aerobically to an OD600 of ∼1 and then transferred to a 37°C incubator in an anaerobic chamber and grown statically overnight. The cultures were diluted with TSB to equalize the OD600 to ∼0.05, pelleted by centrifugation, and sterilized through a 0.2-μm filter. Samples (100 μl) were incubated at 37°C with a 3% solution of PBS-washed rabbit blood cells in a BioTek Epoch 2 microplate reader, and hemolysis was assessed with absorbance measurements at 450 nm taken every 4 min for 2 h. Biological triplicates were assayed in duplicate and averaged.
Footnotes
Supplemental material is available online only.
Contributor Information
Jeffrey M. Boyd, Email: jeffboyd@sebs.rutgers.edu.
Jeremy D. Semrau, University of Michigan-Ann Arbor
REFERENCES
- 1.Rohlfing SR, Crawford IP. 1966. Purification and characterization of the beta-galactosidase of Aeromonas formicans. J Bacteriol 91:1085–1097. 10.1128/jb.91.3.1085-1097.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mandelstam J. 1958. Turnover of protein in growing and non-growing populations of Escherichia coli. Biochem J 69:110–119. 10.1042/bj0690110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 4.Horwitz JP, Chua J, Curby RJ, Tomson AJ, Darooge MA, Fisher BE, Mauricio J, Klundt I. 1964. Substrates for cytochemical demonstration of enzyme activity. I. Some substituted 3-indolyl-beta-D-glycopyranosides. J Med Chem 7:574–575. 10.1021/jm00334a044. [DOI] [PubMed] [Google Scholar]
- 5.O'Neill AJ, Miller K, Oliva B, Chopra I. 2004. Comparison of assays for detection of agents causing membrane damage in Staphylococcus aureus. J Antimicrob Chemother 54:1127–1129. 10.1093/jac/dkh476. [DOI] [PubMed] [Google Scholar]
- 6.Ranjit DK, Endres JL, Bayles KW. 2011. Staphylococcus aureus CidA and LrgA proteins exhibit holin-like properties. J Bacteriol 193:2468–2476. 10.1128/JB.01545-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baum KR, Ahmad Z, Singh VK. 2015. Regulation of expression of oxacillin-inducible methionine sulfoxide reductases in Staphylococcus aureus. Int J Microbiol 2015:617925. 10.1155/2015/617925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nielsen A, Nielsen KF, Frees D, Larsen TO, Ingmer H. 2010. Method for screening compounds that influence virulence gene expression in Staphylococcus aureus. Antimicrob Agents Chemother 54:509–512. 10.1128/AAC.00940-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bojer MS, Baldry M, Ingmer H. 2017. Protocols for screening antimicrobial peptides that influence virulence gene expression in Staphylococcus aureus. Methods Mol Biol 1548:387–394. 10.1007/978-1-4939-6737-7_28. [DOI] [PubMed] [Google Scholar]
- 10.Ding Y, Liu X, Chen F, Di H, Xu B, Zhou L, Deng X, Wu M, Yang CG, Lan L. 2014. Metabolic sensor governing bacterial virulence in Staphylococcus aureus. Proc Natl Acad Sci USA 111:E4981–E4990. 10.1073/pnas.1411077111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman S, Faulkner C, Kaiserli E, Garcia-Mata C, Savenkov EI, Roberts AG, Oparka KJ, Christie JM. 2008. The photoreversible fluorescent protein iLOV outperforms GFP as a reporter of plant virus infection. Proc Natl Acad Sci USA 105:20038–20043. 10.1073/pnas.0807551105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drepper T, Eggert T, Circolone F, Heck A, Krauss U, Guterl J-K, Wendorff M, Losi A, Gärtner W, Jaeger K-E. 2007. Reporter proteins for in vivo fluorescence without oxygen. Nat Biotechnol 25:443–445. 10.1038/nbt1293. [DOI] [PubMed] [Google Scholar]
- 13.Dinges MM, Orwin PM, Schlievert PM. 2000. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev 13:16–34. 10.1128/CMR.13.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenstein R, Götz F. 2000. Staphylococcal lipases: biochemical and molecular characterization. Biochimie 82:1005–1014. 10.1016/s0300-9084(00)01180-9. [DOI] [PubMed] [Google Scholar]
- 15.Rollof J, Hedstrom SA, Nilsson-Ehle P. 1987. Purification and characterization of a lipase from Staphylococcus aureus. Biochim Biophys Acta 921:364–369. 10.1016/0005-2760(87)90038-5. [DOI] [PubMed] [Google Scholar]
- 16.Cadieux B, Vijayakumaran V, Bernards MA, McGavin MJ, Heinrichs DE. 2014. Role of lipase from community-associated methicillin-resistant Staphylococcus aureus strain USA300 in hydrolyzing triglycerides into growth-inhibitory free fatty acids. J Bacteriol 196:4044–4056. 10.1128/JB.02044-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mashruwala AA, Boyd JM. 2016. De novo assembly of plasmids using yeast recombinational cloning. Methods Mol Biol 1373:33–41. 10.1007/7651_2015_275. [DOI] [PubMed] [Google Scholar]
- 18.Luong TT, Lee CY. 2007. Improved single-copy integration vectors for Staphylococcus aureus. J Microbiol Methods 70:186–190. 10.1016/j.mimet.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krute CN, Rice KC, Bose JL. 2017. VfrB is a key activator of the Staphylococcus aureus SaeRS two-component system. J Bacteriol 199:e00828-16. 10.1128/JB.00828-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malone CL, Boles BR, Lauderdale KJ, Thoendel M, Kavanaugh JS, Horswill AR. 2009. Fluorescent reporters for Staphylococcus aureus. J Microbiol Methods 77:251–260. 10.1016/j.mimet.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bose JL. 2014. Genetic manipulation of staphylococci. Methods Mol Biol 1106:101–111. 10.1007/978-1-62703-736-5_8. [DOI] [PubMed] [Google Scholar]
- 22.Bose JL, Fey PD, Bayles KW. 2013. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl Environ Microbiol 79:2218–2224. 10.1128/AEM.00136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roberts CA, Al-Tameemi HM, Mashruwala AA, Rosario-Cruz Z, Chauhan U, Sause WE, Torres VJ, Belden WJ, Boyd JM. 2017. The Suf iron-sulfur cluster biosynthetic system is essential in Staphylococcus aureus, and decreased Suf function results in global metabolic defects and reduced survival in human neutrophils. Infect Immun 85:e00100-17. 10.1128/IAI.00100-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mashruwala AA, Gries CM, Scherr TD, Kielian T, Boyd JM. 2017. SaeRS is responsive to cellular respiratory status and regulates fermentative biofilm formation in Staphylococcus aureus. Infect Immun 85:e00157-17. 10.1128/IAI.00157-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeong DW, Cho H, Jones MB, Shatzkes K, Sun F, Ji Q, Liu Q, Peterson SN, He C, Bae T. 2012. The auxiliary protein complex SaePQ activates the phosphatase activity of sensor kinase SaeS in the SaeRS two-component system of Staphylococcus aureus. Mol Microbiol 86:331–348. 10.1111/j.1365-2958.2012.08198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. 2008. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319:1391–1394. 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pelz A, Wieland KP, Putzbach K, Hentschel P, Albert K, Gotz F. 2005. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J Biol Chem 280:32493–32498. 10.1074/jbc.M505070200. [DOI] [PubMed] [Google Scholar]
- 28.Mashruwala AA, Boyd JM. 2017. The Staphylococcus aureus SrrAB regulatory system modulates hydrogen peroxide resistance factors, which imparts protection to aconitase during aerobic growth. PLoS One 12:e0170283. 10.1371/journal.pone.0170283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yarwood JM, McCormick JK, Schlievert PM. 2001. Identification of a novel two-component regulatory system that acts in global regulation of virulence factors of Staphylococcus aureus. J Bacteriol 183:1113–1123. 10.1128/JB.183.4.1113-1123.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mashruwala AA, Van De Guchte A, Boyd JM. 2017. Impaired respiration elicits SrrAB-dependent programmed cell lysis and biofilm formation in Staphylococcus aureus. Elife 6:e23845. 10.7554/eLife.23845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lie TJ, Leigh JA. 2007. Genetic screen for regulatory mutations in Methanococcus maripaludis and its use in identification of induction-deficient mutants of the euryarchaeal repressor NrpR. Appl Environ Microbiol 73:6595–6600. 10.1128/AEM.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lojda Z. 1970. Indigogenic methods for glycosidases. II. An improved method for beta-D-galactosidase and its application to localization studies of the enzymes in the intestine and in other tissues. Histochemie 23:266–288. 10.1007/BF00306428. [DOI] [PubMed] [Google Scholar]
- 33.Trifonov S, Yamashita Y, Kase M, Maruyama M, Sugimoto T. 2016. Overview and assessment of the histochemical methods and reagents for the detection of beta-galactosidase activity in transgenic animals. Anat Sci Int 91:56–67. 10.1007/s12565-015-0300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heuermann K, Cosgrove J. 2001. S-Gal: an autoclavable dye for color selection of cloned DNA inserts. Biotechniques 30:1142–1147. 10.2144/01305pf01. [DOI] [PubMed] [Google Scholar]
- 35.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- 36.Joska TM, Mashruwala A, Boyd JM, Belden WJ. 2014. A universal cloning method based on yeast homologous recombination that is simple, efficient, and versatile. J Microbiol Methods 100:46–51. 10.1016/j.mimet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pang YY, Schwartz J, Bloomberg S, Boyd JM, Horswill AR, Nauseef WM. 2014. Methionine sulfoxide reductases protect against oxidative stress in Staphylococcus aureus encountering exogenous oxidants and human neutrophils. J Innate Immun 6:353–364. 10.1159/000355915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grosser MR, Paluscio E, Thurlow LR, Dillon MM, Cooper VS, Kawula TH, Richardson AR. 2018. Genetic requirements for Staphylococcus aureus nitric oxide resistance and virulence. PLoS Pathog 14:e1006907. 10.1371/journal.ppat.1006907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bae T, Glass EM, Schneewind O, Missiakas D. 2008. Generating a collection of insertion mutations in the Staphylococcus aureus genome using bursa aurealis. Methods Mol Biol 416:103–116. 10.1007/978-1-59745-321-9_7. [DOI] [PubMed] [Google Scholar]
- 40.Bose JL, Daly SM, Hall PR, Bayles KW. 2014. Identification of the Staphylococcus aureus vfrAB operon, a novel virulence factor regulatory locus. Infect Immun 82:1813–1822. 10.1128/IAI.01655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Tameemi H, Beavers WN, Norambuena J, Skaar EP, Boyd JM. 2021. Staphylococcus aureus lacking a functional MntABC manganese import system has increased resistance to copper. Mol Microbiol 115:554–573. 10.1111/mmi.14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Austin CM, Garabaglu S, Krute CN, Ridder MJ, Seawell NA, Markiewicz MA, Boyd JM, Bose JL. 2019. Contribution of YjbIH to virulence factor expression and host colonization in Staphylococcus aureus. Infect Immun 87:e00155-19. 10.1128/IAI.00155-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mashruwala AA, Pang YY, Rosario-Cruz Z, Chahal HK, Benson MA, Mike LA, Skaar EP, Torres VJ, Nauseef WM, Boyd JM. 2015. Nfu facilitates the maturation of iron-sulfur proteins and participates in virulence in Staphylococcus aureus. Mol Microbiol 95:383–409. 10.1111/mmi.12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 45.Rosario-Cruz Z, Chahal HK, Mike LA, Skaar EP, Boyd JM. 2015. Bacillithiol has a role in Fe-S cluster biogenesis in Staphylococcus aureus. Mol Microbiol 98:218–242. 10.1111/mmi.13115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537-12. 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 to S6, Fig. S1. Download AEM.01108-21-s0001.pdf, PDF file, 0.5 MB (467.3KB, pdf)