Abstract
We developed an Xrcc4M61R separation of function mouse line to overcome the embryonic lethality of Xrcc4-deficient mice. XRCC4M61R protein does not interact with Xlf, thus obliterating XRCC4-Xlf filament formation while preserving the ability to stabilize DNA ligase IV. X4M61R mice, which are DNA repair deficient, phenocopy the Nhej1-/- (known as Xlf -/-) setting with a minor impact on the development of the adaptive immune system. The core non-homologous end-joining (NHEJ) DNA repair factor XRCC4 is therefore not mandatory for V(D)J recombination aside from its role in stabilizing DNA ligase IV. In contrast, Xrcc4M61R mice crossed on Paxx-/-, Nhej1-/-, or Atm-/- backgrounds are severely immunocompromised, owing to aborted V(D)J recombination as in Xlf-Paxx and Xlf-Atm double Knock Out (DKO) settings. Furthermore, massive apoptosis of post-mitotic neurons causes embryonic lethality of Xrcc4M61R -Nhej1-/- double mutants. These in vivo results reveal new functional interplays between XRCC4 and PAXX, ATM and Xlf in mouse development and provide new insights into the understanding of the clinical manifestations of human XRCC4-deficient condition, in particular its absence of immune deficiency.
Research organism: Mouse
Introduction
Living organisms face DNA double-strand breaks (DSBs), the most toxic DNA lesions, from random or programmed (prDSBs) origins (Betermier et al., 2020), such as during the development of the adaptive immune system through V(D)J recombination. V(D)J recombination results in the somatic rearrangement of variable (V), diversity (D), and joining (J) elements of antigen receptor loci in T- and B-cell precursors (Jung et al., 2006). It is initiated by the domesticated transposase Recombination-Activating Genes 1 and 2 factors (RAG1/2), which introduce prDSBs at the border of V, D, and J elements within recombination signal sequences (RSS). The non-homologous end-joining (NHEJ) machinery is the sole DNA repair pathway to cope with these lymphoid-specific prDSBs. Briefly, the NHEJ pathway is signaled by the Ataxia-Telangiectasia mutated (ATM) protein (Arya and Bassing, 2017; Bassing et al., 2002) and prompted by the Ku70-Ku80 complex that recognizes broken DNA ends and recruits the DNA-dependent protein kinase-catalytic subunit (DNA-PKcs), which further activates the pathway (Sibanda et al., 2017). Broken DNA ends are processed by Artemis endo/exonuclease, which specifically opens the RAG1/2-generated hairpins at DNA ends during V(D)J recombination (Chang et al., 2017). DNA ligase IV (Lig4), X-ray repair cross-complementation 4 (XRCC4, or X4), XRCC4-like factor (Xlf, also known as Cernunnos), and Paralog of XRCC4 and Xlf (PAXX) (Craxton et al., 2015; Ochi et al., 2015; Xing et al., 2015) ensure ligation of broken ends (Lieber, 2010). Lig4 and XRCC4 are critical factors during development apart from their essential function during V(D)J recombination, and both Lig4-/- and Xrcc4-/- mice suffer late embryonic lethality caused by apoptosis of post-mitotic neurons (Barnes et al., 1998; Frank et al., 1998; Shull et al., 2009). Likewise, human patients with LIG4 syndrome present with dwarfism, microcephaly, and various degrees of immunodeficiency caused by hypomorphic mutations in the Lig4 gene (O’Driscoll et al., 2001; Staines Boone et al., 2018).
In most of the cases, defects in core NHEJ factors result in severe combined immunodeficiency (SCID), owing to aborted V(D)J recombination both in humans and animal models (de Villartay et al., 2003). NHEJ deficiency also results in genetic instability with the development of Pro-B cell lymphomas harboring chromosomal translocations when associated with Trp53-/- (Alt et al., 2013). Xlf/Cernunnos-deficient patients also present with developmental features including dwarfism, microcephaly, and combined immunodeficiency (CID) (Buck et al., 2006). However, V(D)J recombination is not severely affected nor in human (Buck et al., 2006; Recio et al., 2018; van der Burg and Gennery, 2011) or murine (Li et al., 2008; Roch et al., 2019; Vera et al., 2013) settings of Xlf deficiency. This paradoxical situation is a consequence of the functional redundancy between Xlf and several DNA repair factors, including PAXX, ATM, H2A.X, MDC1, MRI, and 53BP1 as revealed by the complete V(D)J recombination defect in combined deficient settings (Abramowski et al., 2018; Beck et al., 2019; Hung et al., 2018; Musilli et al., 2020; Oksenych et al., 2012; Zha et al., 2011) as well as RAG2 itself (Lescale et al., 2016a). Consistent with their overall efficient V(D)J recombination, Nhej1-Trp53 DKO mice indeed do not develop Pro-B cell lymphomas (Vera et al., 2013).
To account for this functional redundancy, we proposed a model in which prDSBs may benefit from evolutionary conserved DNA repair mechanisms as to avoid their intrinsic oncogenic potential (Betermier et al., 2020). In the particular context of V(D)J recombination, this mechanism would operate through a redundant ‘double DNA repair synapse,’ which strictly operates during V(D)J recombination to prevent genomic instability, but not in case of genotoxic-induced DNA damage (Abramowski et al., 2018; Lescale et al., 2016a). The ‘two-synapses’ model accounts for the absence of V(D)J recombination defect in the absence of Xlf. One essential actor of the ‘two-synapses’ model is the RAG2 factor itself, which together with RAG1 is known to remain on DNA broken ends during V(D)J recombination, forming the so-called post-cleavage complex (PCC) (Schatz and Swanson, 2011). We previously established that the C-terminus of RAG2 is determinant in complementing the lack of Xlf (Lescale et al., 2016a). Indeed, the combined absence of the C-terminus of RAG2 and Xlf results in SCID mice owing to a complete block of V(D)J recombination. In the ‘two-synapses’ model, the first synapse would be mediated by RAG2, PAXX, and ATM signaling as suggested by efficient V(D)J recombination in multiply deficient v-Abl transformed Pro-B cells (Lescale et al., 2016a; Lescale et al., 2016b);. The second synapse is constituted by the Xlf-X4 filament or the bona fide NHEJ core complexes, the structure of which was recently resolved through cryoelectron microscopy (cryo-EM) (Chaplin et al., 2021a; Chaplin et al., 2021b; Zhao et al., 2020). The V(D)J recombination-specific ‘two-synapses’ apparatus appears as a double-edged sword backup system to avoid genomic instability. Indeed, RAG2/Xlf double mutant mice develop typical NHEJ-deficient pro-B cell lymphomas when crossed onto a Trp53-/- background (Lescale et al., 2016a).
One question remains as to the contribution of X4 in this model given that classical KO strategies do not allow to directly assess the function of X4 since its absence results de facto in a complete Lig4 deficiency. Reminiscent to the Xlf-deficient condition, human patients identified with X4 mutations present DNA repair defect hallmarks, including dwarfism, microcephaly, increased cellular sensitivity to radiomimetic agents, but, strikingly, no immunodeficiency (de Villartay, 2015; Saito et al., 2016). The absence of noticeable immunophenotype argues for a dispensable role of X4 during V(D)J recombination, outside its general Lig4 stabilization function. It also interrogates the positioning of X4 within the ‘two-synapses’ model knowing its structural and functional relationship with Xlf (Callebaut et al., 2006).
XRCC4 is a keystone factor, playing two independent roles. First, X4 stabilizes Lig4 through its C-terminal coiled-coil domain (Grawunder et al., 1997). Indeed, Lig4 expression is abrogated in Xrcc4 Knock Out (KO) models both in vivo and in vitro (Gao et al., 1998). In addition, X4 and Xlf homodimers interact through their N-terminal globular head to form long polymeric ‘filaments’ (Reid et al., 2015; Ropars et al., 2011). X4-Xlf filaments generate a ‘DNA repair synapse’ to tether broken DNA ends (Brouwer et al., 2016; Reid et al., 2015). DNA-end synapsis is a central issue during NHEJ, and the recent development of in vitro single-molecule technologies has highlighted the dynamic formation of DNA end-to-end synapses (flexible/long range for DNA end tethering and close/short range for DNA ligation) in addition to the Xlf-X4 filament, in which the various core NHEJ DNA repair factors (Ku70/80, DNA-PKcs, Xlf, X4/L4, and PAXX) participate to various degrees, in particular the association of Xlf with both X4 and Ku (for a recent review, see Zhao et al., 2020). Several studies recently reported on the details of the structural assembly of these complexes using cryo-EM, thus improving our understanding on the composition of these complexes as well as the dynamics of the transition between various states during NHEJ-mediated DNA repair (Chaplin et al., 2021a; Chaplin et al., 2021b; Zhao et al., 2020). These studies support in particular the interaction of the L4X4 complex with that of Ku70/80 previously proposed by Costantini et al., 2007. The V(D)J recombination phenotype of Nhej1 KO mice would argue that the RAG2-mediated DNA tethering is also redundant with these DNA end-to-end synapses. Nevertheless, the intimate nature of DNA end joining during V(D)J recombination may not always strictly coincide with what we know for the repair of genotoxic DNA breaks, precisely because of the existence of the ‘two-synapses’ mechanism.
Since X4 is compulsory for Lig4 stabilization, Xrcc4-/- mice phenocopy Lig4-/- condition, with an embryonic lethality and SCID phenotype (Gao et al., 1998). The embryonic lethality is rescued on Trp53-/- background, but the SCID resulting from a complete block of lymphocyte development remains (Gao et al., 2000). Furthermore, Xrcc4-/-Trp53-/- DKO mice develop Pro-B cell lymphomas (Chen et al., 2016; Gao et al., 2000). To avoid disrupting the critical Lig4 stabilization function of X4, we engineered a Xrcc4 knock-in (KI) mouse model harboring the M61R missense mutation that abrogates X4-Xlf interaction (Ropars et al., 2011), while keeping the Lig4 interaction domain unperturbed. Xrcc4M61R mice are viable attesting for the stabilization of functional Lig4, thus allowing the study of lymphocyte development. Introduced on Atm-/-, Paxx-/-, and Nhej1-/- backgrounds, the Xrcc4M61R mutation allows to address the ‘two-synapses’ DNA repair model in V(D)J recombination, and to expand the picture of NHEJ apparatus in brain and lymphocyte development.
Results
Generation of XM61R mice
X4 and Xlf interact through a hydrophobic interface, which is disrupted by the X4M61R substitution, thus resulting in the loss of X4-Xlf filament (Ropars et al., 2011). We developed an Xrcc4M61R KI mouse model through CRISPR/Cas9 (Figure 1A). Homozygous Xrcc4M61R/M61R mice (Xrcc4M61R mice) were viable arguing against any harmful impact of the M61R substitution during embryonic development, as opposed to the embryonic lethality of Xrcc4 KOs.
Figure 1. X4 and Lig4 expression and impaired non-homologous end-joining (NHEJ) in Xrcc4M61R mice.
(A) Schematic representation of CRISPR/Cas9-driven homologous recombination strategy to generated Xrcc4M61R knock-in mouse model. (B) 5′UTR to 3′UTR Xrcc4 RT-PCR in mouse thymocyte extracts from four littermates. The lower transcript represents the splicing out of exon 3 as described (Gao et al., 1998). (C) WB analysis from mouse whole tissue extracts. (D) Western blot (WB) analysis from mouse embryonic fibroblast (MEF) extracts. (E) WB analysis from mouse whole tissue extracts. WB were performed at least three times with two animals per genotypes. (F) MEFs’ sensitivity to DNA double-strand break (DSB)-inducing agent phleomycin. Statistical tests for Xrcc4M61R vs. WT (gray *) and Xrcc4M61R vs. Xrcc4-/- (black *). (G) Relative survival of CD3/CD28-activated mature T cells following irradiation and 4 hr recovery. Statistical analysis for Xrcc4M61R vs. WT (gray *) and Xrcc4M61R vs. Nhej1-/- (green *).

Figure 1—figure supplement 1. Nucleotide sequence of two Xrcc4 transcripts in Xrcc4M61R mice.

X4M61R stabilizes Lig4
RT-PCR analysis of X4 mRNA expression unveiled two isoforms in M61R mutation-bearing cells (Figure 1B). The expression of the full-length transcript carrying the five mutations was reduced (Figure 1B, upper band) and was accompanied by a shorter isoform (Figure 1B, lower band). Sequencing this alternative transcript revealed the out-of-frame splicing-out of exon 3 (Figure 1—figure supplement 1). The same X4 transcript missing exon 3 was described in a previous Xrcc4 KO mouse model (Gao et al., 2000; Gao et al., 1998). A subsequent attempt to generate Xrcc4M61R KI mouse model using an ssODN template lacking the four silent mutations resulted in the same pattern of expression of the M61R allele (data not shown). At the protein level, X4 expression was barely detectable in spleen, brain, thymus, and mouse embryonic fibroblasts (MEFs) from Xrcc4M61R mice as opposed to their homozygous WT counterparts (Figure 1C and D). In summary, Xrcc4M61R mice express two X4 transcripts, one lacking exon 3 and resulting in a complete loss of function, and a second, full length, harboring the M61R missense variant encoding a weakly expressed X4 protein lacking its ability to interact with Xlf. Of note, Lig4 protein was readily detectable by WB in protein lysates from Xrcc4M61R mice spleen and thymus, attesting for its stabilization by the X4M61R protein (Figure 1E).
We conclude that the Xrcc4M61R mouse model represents a hypomorphic condition in which the X4M61R protein, although weakly expressed, retains the capacity to stabilize Lig4 and thus ensures proper embryonic development with viable animals at birth.
DSB repair defect in X4M61R cells
MEFs from Xrcc4M61R mice presented a statistically significant decrease in cell viability compared to WT cells upon phleomycin, a DSB inducer, treatment, attesting for a profound DNA repair defect (Figure 1F). Likewise, splenic T cells from Xrcc4M61R mice showed an increased IR sensitivity with an almost complete loss of viability at 2 Gy (Figure 1G). These experiments demonstrate that the X4M61R mutant protein, which preserves the stabilization of Lig4, is nevertheless impaired in its capacity to achieve full repair of DSB introduced by random genotoxic agents.
Lymphocyte development in Xrcc4M61R mice phenocopies that of Nhej1-/- mice
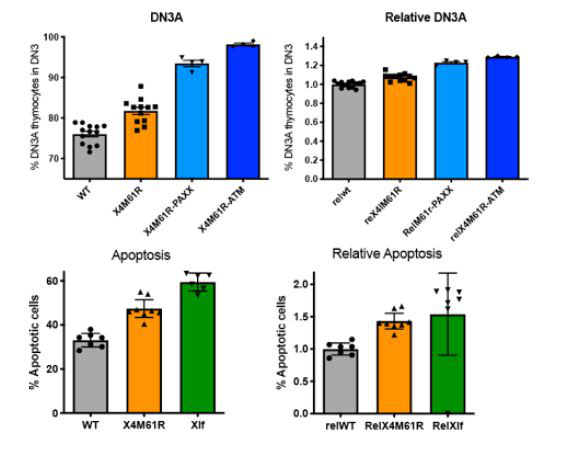
We analyzed the impact of X4M61R substitution on T cell development in the thymus. Thymocyte counts were significantly reduced in Xrcc4M61R thymus (Figure 2A), but remained in the range of Nhej1-/- thymus cellularity (respectively [*106] 26.8 ± 2.6 SEM vs. 115 ± 15 SEM in WT, p<0.0001, vs. 12.5 ± 2.6 SEM in Nhej1-/-, ns). The thymocyte maturation program, identified through the various CD4/CD8 stages, was similar in Xrcc4M61R and WT littermates (Figure 2B), yet with a mild accumulation of CD44-CD25+ CD28- DN3A Xrcc4M61R thymocytes (respectively 81.8% ± 0.9 SEM vs. 76.1% ± 0.7 SEM, p<0.0001) (Figure 2C), a trait shared by thymocytes from Nhej1-/- mice (81.1% ± 1.4 SEM) as described (Roch et al., 2019). Purified Xrcc4M61R thymocytes presented an increased apoptosis (7AAD+/AnnV+), similar to that of Nhej1-/- thymocytes (respectively 47.4% ± 1.7 SEM vs. 33.2% ± 1.3 SEM for WT, p=0.0003, vs. 59.5% ± 1.6 SEM, for Nhej1-/-, ns) (Figure 2D and E). This was accompanied by the expression of TP53 target genes Cdkn1a, Bbc3, and Bax, in both Xrcc4M61R and Nhej1-/- thymocytes (Figure 2F). Lastly, TCRα repertoire was skewed in Xrcc4M61R thymocytes (Figure 2G), with the decreased usage of distal VαJα rearrangements, a hallmark of reduced thymocyte viability or suboptimal V(D)J recombination.
Figure 2. Xrcc4M61R mice exhibit modest T cell development defect in the thymus phenocopying Nhej1-/- mice.

(A) Thymus cellularity of WT, Xrcc4M61R, and Nhej1-/- (Xlf) 6–9-week-old mice. (B) Immunostaining of thymus cellular populations. Various relevant populations are highlighted in black gates. (C) Quantification of DN3A thymocyte subpopulation (DN3 CD28-CD25+) gated from total DN3 thymocytes (CD4-CD8-CD44-CD25+). (D) Thymocyte apoptosis analysis after 20 hr of culture. AnnexinV+/7AAD+ apoptotic cells are highlighted in black gates. (E) Quantification of thymocyte apoptosis (AnnexinV+/7AAD+ cells) after 20 hr of culture. (F) Quantitative RT-PCR analysis of TP53 target genes Cdkn1a (encoding P21), Bbc3 (encoding PUMA), and Bax in thymocyte extracts. There is no statistical difference between Xrcc4M61R and Nhej1-/- in the expression of these TP53 target genes. (G) Illustrative representation of principal component analysis/unsupervised hierarchical clustering (PCA/HC) analysis of mTRAJ-mTRAV combinations in thymus according to PROMIDISα. (H) Immunostaining of bone marrow cellular populations. V(D)J recombination-dependent developmental stages are highlighted within red rectangles. Various relevant populations are highlighted in black gates. Quantification of B-cell population (B220+) (I) and immature B-cell population (CD19+ B220+ IgM+) (J) from total bone marrow.
The proportion of mature B lymphocytes in the bone marrow (BM) was not significantly altered in Xrcc4M61R mice (Figure 2H, I), while significantly decreased in Nhej1-/- mice (respectively 21.7% ± 1.8 SEM vs. 26.0% ± 1.1 SEM, ns, vs. 13.3% ± 0.6 SEM, p=.008). However, the proportions of immature B cells were decreased in Xrcc4M61R mice (Figure 2J), close to the already described decline of Nhej1-/- immature B cells (respectively 11.3% ± 3.3 SEM vs. 19.3% ± 2.6 SEM, p=0.0002, vs. 8.7% ± 2.5 SEM, ns).
These results indicate that B- and T-lymphocyte development is not severely affected in Xrcc4M61R mice, despite the reduced expression of X4M61R. The minor accumulation of DN3A thymocytes and immature B cells suggests a modest decreased V(D)J recombination efficiency, supported by the bias in TCRα usage. This is in sharp contrast with Xrcc4-/- and Lig4-/- mice, which present a complete arrest of lymphocyte maturation and a SCID phenotype. We conclude that when Lig4 is stabilized, the contribution of X4 through Xlf/X4 interaction, disrupted in the context of X4M61R, is not critical for V(D)J recombination in vivo.
PAXX and ATM are compensatory factors for X4 in lymphoid development
We previously proposed a model of ‘double DNA repair synapse’ during V(D)J recombination according to which the X4-Xlf ‘filament’ and the RAG1/2 PCC, together with ATM and PAXX, provide two complementary means of DNA end synapsis (Abramowski et al., 2018; Betermier et al., 2020; Lescale et al., 2016a). To address the role of X4 in this model, we crossed Xrcc4M61R onto Paxx-/- and Atm-/- mice (hereafter denoted as Xrcc4M61R-Paxx and Xrcc4M61R-Atm). Both double mutant mice were viable but exhibited severe growth retardation and facial dysmorphia as a consequence of either the X4M61R mutation and/or the overall reduction in X4/L4 expression (Figure 3—figure supplement 1).
The thymus and spleen cellularity was severely reduced in both X4M61R-Paxx and Xrcc4M61R-Atm mice (Figure 3A and B) as compared to their Xrcc4M61R single-mutant littermates (respectively [*106] 1.1 ± 0.2 SEM and 0.56 ± 0.15 SEM, p<0.0001, vs. 26.8 ± 2.6 SEM in thymus and 6.8 ± 1.2 SEM and 4.1 ± 1.0 SEM, p<0.0001, vs. 40.0 ± 3.8 SEM in spleen), and previously described Atm and Paxx single KO conditions (Abramowski et al., 2018). B cells expressing IgM were undetectable in BM from Xrcc4M61R-Atm mice and to a lesser extent in Xrcc4M61R-Paxx mice (Figure 3C). B220+ B cells were strongly reduced compared to Xrcc4M61R or WT littermates in both settings (respectively 10.9% ± 1.4 SEM and 7.2% ± 0.96 SEM, p=0.001, vs. 21.7 ± 1.79 SEM in Xrcc4M61R and 26.3 ± 1.08 SEM in WT) (Figure 3D). Likewise, Xrcc4M61R-Paxx and X4M61R-Atm mice presented with a substantial accumulation of DN3A thymocytes (93.4% ± 0.8 SEM and 98.2% ± 0.3 SEM, p=0.0001, respectively, vs. 76.1 ± 0.7 SEM in WT), which was significantly greater than the one observed for Xrcc4M61R (81.77 ± 1.4 SEM) or Nhej1-/- (81.1 ± 1.4 SEM) animals (Figure 3E and Abramowski et al., 2018). The immune phenotype of Xrcc4M61R-Paxx and X4M61R-Atm mice is highly reminiscent of the SCID condition experienced by Nhej1-Paxx, Nhej1-Atm, and Nhej1-Rag2c/c DKO mice (Abramowski et al., 2018; Lescale et al., 2016a; Musilli et al., 2020; Zha et al., 2011) and suggested a possible impaired V(D)J recombination in these double mutants. Indeed, while Dβ2-Jβ2 (upper panel) and Vβ10-Dβ2Jβ2 (lower panel) rearrangements of the Tcrb locus were present in thymocytes from Xrcc4M61R, Atm-/-, and WT mice (Figure 3F and G), as also previously observed in Paxx-/- mice (Abramowski et al., 2018), they were barely detectable in DNA from Xrcc4M61R-Paxx and Xrcc4M61R-Atm thymus.
Figure 3. PAXX and ATM are compensatory factors of X4M61R in immune development.
(A, B) Thymus and spleen cellularity of WT, Xrcc4M61R, Xrcc4M61R–Paxx, and Xrcc4M61R–Atm 6–9-week-old mice. (C) Immunostaining of BM and thymus cellular populations. Various relevant populations are highlighted in black gates. (D) Quantification of B220+ subpopulation of total BM. (E) Quantification of DN3A thymocyte subpopulation (DN3 CD28-CD25+) of total DN3 thymocytes (CD4-CD8-CD44-CD25+). (F) Schematic representation of PCR strategy to analyze Tcrb rearrangement according to Abramowski et al., 2018. (G) Autoradiogram of ladder of productive Dβ2-Jβ2 and Vβ10-Dβ2Jβ2 semi-quantitative PCR products revealed by the TCR-Jβ probe. Genomic DNA dilutions are represented with triangles. Germline allele configuration (GL) is revealed by the upper band of Dβ2-Jβ2 PCR. Southern were performed twice with two animals per genotype.

Figure 3—figure supplement 1. Growth retardations of Xrcc4M61R-Paxx and Xrcc4M61R-Atm mice.

We conclude that Xrcc4M61R crossed on Paxx-/- or Atm-/- phenocopy the SCID phenotype of Nhej1-Paxx and Nhej1-Atm DKO conditions owing to impaired V(D)J recombination. Therefore, aside from its essential role in Lig4 stabilization, X4 does participate in DNA coding-end tethering through its interaction with Xlf during V(D)J recombination, and PAXX/ATM are compensatory factors for this later function in vivo, as they are for Xlf. Alternatively, one may consider that the Xlf function being impaired in the context of X4M61R, PAXX/ATM complements this defect as already established.
Impaired brain development and immune deficiency in Xrcc4M61R Nhej1-/- mice
To further assess the function of X4M61R in vivo, we introduced Xrcc4M61R on Nhej1-/- background. No Xrcc4M61R-Nhej1-/- double homozygous mice (hereafter named as Xrcc4M61R-Nhej1) could be recovered out of 166 newborns obtained from various genotype combinations, when 23 were expected (Figure 4A), testifying the lethality of Xrcc4M61R-Nhej1 embryos. Nevertheless, lived Xrcc4M61R-Nhej1 fetuses could be recovered at E15.5 and E18.5. They were smaller than their Xrcc4M61R-Nhej1+/- littermates, confirming abnormal development (Figure 4B and data not shown). This is reminiscent of the late embryonic lethality of Lig4-/-, Xrcc4-/-, and Nhej1-Paxx DKO mice (Abramowski et al., 2018; Balmus et al., 2016; Frank et al., 1998; Gao et al., 1998; Liu et al., 2017). Massive neuronal apoptosis was detected in E15.5 brain slices through cleaved-caspase 3 (CC3) staining in upper layers of dorsal telencephalon corresponding to the intermediate zone (IZ) and the cortical plate (CP) in Xrcc4M61R-Nhej1 mice (Figure 4C–E). The IZ contains post-mitotic neurons migrating to populate the CP, while neural progenitors proliferate in lower layers: the ventricular zone (VZ) and sub-ventricular zone (SVZ). Thus, Xrcc4M61R-Nhej1 embryonic lethality recapitulates, although to a lesser extent, the massive post-mitotic neuron apoptosis observed in Nhej1-Paxx DKO and Xrcc4-/- brains (Figure 4C and E), which causes the previously described embryonic lethality of these conditions. In contrast, Nhej1-/- and Xrcc4M61R single mutants showed modest increases of CC3 staining (Figure 4D and E), while we showed previously that Paxx-/- are indistinguishable from WT (Abramowski et al., 2018).
Figure 4. Embryonic lethality and neuronal apoptosis of Xrcc4M61R-Nhej1 embryos.

(A) Impaired development of Xrcc4M61R-Nhej1 mice. Actual and expected numbers of Xrcc4M61R-Nhej1 alive pups, E18.5 and E15.5 embryos. (B) Pictures of Xrcc4M61R and Xrcc4M61R-Nhej1 E15.5 embryos. (C) Cleaved-caspase 3 (CC3, red) immunostaining of E15.5 brain slices. Scarce apoptotic cells are detected in the whole dorsal telencephalon from Nhej1-/- and Xrcc4M61R embryos. By contrast, massive neuronal apoptosis is observed in the upper layers of the developing cortex of Xrcc4M61R-Nhej1, Xrcc4-/-, and Nhej1-Paxx DKO embryos. The white boxes indicate the location of the standard window (100 µm wide) spanning from the ventricular to pial surface used to quantify pyknotic nuclei in the dorsal telencephalons (see below). Scale bar: 100 µm. (D) Number of CC3-positive cells in the VZ/SVZ (black)—containing the cycling neural progenitors—and intermediate zone (IZ)/cortical plate (CP) (gray)—containing post-mitotic neurons—of the whole dorsal telencephalons from WT, Nhej1-/-, and Xrcc4M61R embryos. (E) Percentage of apoptotic (pyknotic) nuclei in the ventricular zone (VZ)/sub-ventricular zone (SVZ) (left panel) and the IZ/CP (right panel) found in the standard windows shown in (C). Data were obtained from both hemispheres of three embryos per condition.
We next analyzed the adaptive immune system development of Xrcc4M61R-Nhej1 fetuses. B-lymphocyte development begins at E17.5 in the fetal liver (FL), with the rearrangement of the Igh locus and the intracellular expression of Ig-μH chains in CD19+ B220+ CD43+ Pro B cells, which is used as a proxy to evaluate Igh rearrangement completion (Kajikhina et al., 2016). Xrcc4M61R-Nhej1 Pro-B cells failed to express intracellular μH chain when compared to WT (0.90% ± 0.09 SEM, p=0.008), which contrasted with the slight reduction observed in Xrcc4M61R and Nhej1-/- mice (respectively 16.6% ± 1.2 SEM and 19.1% ± 2.2 SEM vs. 32.8% ± 1.3 SEM, p=0.0016) (Figure 5A and B), arguing for a severe block in B cell maturation at Pro-B cell stage in Xrcc4M61R-Nhej1 mice. Fetal thymocytes development of Xrcc4M61R and Nhej1-/- mice recapitulated young adult thymus phenotype with a modest block at DN3A stage compared to WT (respectively 83.3% ± 2.7 SEM and 86.2 ± 2.2 SEM vs. 70.3% ± 1.1 SEM, p=0.0004). In contrast, Xrcc4M61R-Nhej1 thymocyte development was severely compromised from the DN3A stage on when compared to Xrcc4M61R and Nhej1-/- (99.4% ± 0.13 SEM, p=0.0007), and DP thymocytes or mature T cells were virtually absent from these thymii (Figure 5A and C). Dβ2-Jβ2 and Vβ10-Dβ2Jβ2 Tcrb genes rearrangements, which begin at around E13 (Ramond et al., 2014), were undetectable in Xrcc4M61R-Nhej1 thymus, while readily observed in WT, Xrcc4M61R, and Nhej1-/- mice (Figure 5D and E), further attesting for an acute V(D)J recombination defect in Xrcc4M61R-Nhej1 cells.
Figure 5. Complete V(D)J recombination defect in E18.5 Xrcc4M61R-Xlf embryos.

(A) Immunostaining of E18.5 fetal CD19+ B220+ CD43+ hepatocytes and fetal thymocytes. Various relevant populations are highlighted in black gates. (B) Quantification of intracellular IgM expression in subpopulation of fetal Pro-B cells (CD19+ B220+ CD43+ surfaceIgM- hepatocytes). (C) Quantification of DN3A thymocyte subpopulation (DN3 CD28-CD25+) of total DN3 fetal thymocytes (CD4-CD8-CD44-CD25+). (D) Schematic representation of PCR strategy to analyze Tcrb rearrangement according to Abramowski et al., 2018. (E) Autoradiogram of ladder of productive Dβ2-Jβ2 and Vβ10-Dβ2Jβ2 direct PCR products revealed by the TCR-Jβ probe. Germline allele configuration (GL) is revealed by the upper band of Dβ2-Jβ2 PCR. Southern was performed once with two animal per genotype.
Altogether, Xrcc4M61R-Nhej1 embryonic development phenocopy Nhej1-Paxx DKO, with a profound defect in V(D)J recombination and the apoptosis of post-mitotic neurons leading to embryonic lethality.
Discussion
We created an Xrcc4 separation of function allele in mice, which harbor the M61R substitution previously described to abolish X4-Xlf interaction (Ropars et al., 2011). The X4M61R protein, although weakly expressed, preserves the stabilization of Lig4 but severely compromises the NHEJ DNA repair function in affected mice. Nevertheless, Xrcc4M61R mice are viable, arguing that the embryonic lethality of Xrcc4 KO mice is most likely the result of the de facto absence of Lig4 in this setting, thus phenocopying Lig4 KO condition. Interestingly, this also indicates that the formation of the X4-Xlf filament, abrogated by the M61R mutation, is dispensable for post-mitotic neuron viability, the loss of which causes lethality in Xrcc4 and Lig4 KO settings.
Immune development of Xrcc4M61R mice phenocopied that of Nhej1-/- animals, with a slight decreased thymic cellularity, an increased thymocyte apoptosis, and skewed TCRα repertoire, which we previously linked to a suboptimal Tcra rearrangement efficiency (Berland et al., 2019; Roch et al., 2019; Vera et al., 2013). In addition to their TCRα repertoire bias, Xrcc4M61R thymocytes also faced a developmental delay at DN3A both in fetal and adult thymus like Nhej1 KO condition, further attesting for a suboptimal V(D)J recombination activity in these mice. Therefore, although X4 is required for V(D)J recombination through Lig4 stabilization, it is not mandatory for coding-ends tethering during V(D)J recombination. These results confirm that X4 exerts a Lig4-independent function through its interaction with Xlf that is decisive for the repair of genotoxic-induced DSBs but compensated for by other DNA repair factors, and possibly RAG2 itself as shown for Xlf (Lescale et al., 2016a), during V(D)J recombination. Xrcc4M61R–Paxx and Xrcc4M61R–Atm double homozygous mice were severely immunocompromised, owing to aborted V(D)J recombination, as noticed in Nhej1-Paxx and Nhej1-Atm DKO. We conclude that PAXX and ATM are compensatory factors for X4M61R during V(D)J recombination. This result further supports our previously proposed model of ‘double DNA repair synapse’ in V(D)J recombination, mediated by the X4-Xlf and RAG2-PAXX-ATM axes, respectively (Abramowski et al., 2018; Betermier et al., 2020; Lescale et al., 2016a). In addition to their defect in V(D)J recombination, Xrcc4M61R-Nhej1 double homozygous mice were embryonic lethal owing to massive post-mitotic neuron apoptosis, recapitulating Xrcc4-/- setting. This observation may be the result of the lower expression of the X4M61R protein, which could have additive/synthetic effects with the loss of Xlf interaction during NHEJ. Indeed, Nhej1 deficiency causes synthetic lethality with several other NHEJ-deficient conditions, such as Prkdc-/-, Mri-/-, H2ax-/-, and Paxx-/- (Abramowski et al., 2018; Hung et al., 2018; Xing et al., 2017; Zha et al., 2011). Therefore, Xlf can rescue very different NHEJ defects, through an unknown mechanism, perhaps in relation with recovery of replication fork stalling as shown in the Nhej1-H2ax DKO mice (Chen et al., 2019).
Altogether, the Xrcc4M61R separation of function allele highlights for the first time at least two independent roles of X4 in NHEJ and establishes that X4 is not mandatory for coding ends tethering during V(D)J recombination. This study also unravels novel interplays between X4, PAXX, ATM, and Xlf during development of the brain and the immune system.
Several deleterious mutations in the XRCC4 gene have been reported in humans (see de Villartay, 2015 for review), most of which are associated with microcephalic primordial dwarfism (MPD), gonadal failure, early-onset metabolic syndrome, and cardiomyopathies. The DNA repair deficiency in these patients is manifest when tested in vitro retrospectively. Most surprisingly however, they do not present noticeable signs of immune dysfunction. Altogether, the clinical presentation of these patients was not evocative of an impaired NHEJ given the known impact of its deficiency on the development of the adaptive immune system. Indeed, X4 mutations were not identified in these patients through hypothesis-driven candidate gene sequencing but rather through unsupervised whole-exome sequencing. Our present study now provides some hints as to explain the absence of immunological features in X4-deficient patients; when Lig4 expression is spared to some extent by hypomorphic mutations, allowing birth, XRCC4 appears not critically required for the development of the adaptive immune system.
Materials and methods
Mice
Nhej1-/- (Vera et al., 2013), Paxx-/- (Abramowski et al., 2018), Xrcc4-/- (Gao et al., 1998), and Atm-/- (Barlow et al., 1996) mice were maintained in pathogen-free environment. All experiments were performed in compliance with the French Ministry of Agriculture’s regulations for animal experiments (act 87847, 19 October 1987; modified in May 2001).
The Xrcc4M61R allele was generated through CRISPR/cas9 ribonucleoprotein (RNP) complex microinjection in C57BL/6J mouse zygotes pronuclei as described (Ucuncu et al., 2020). Xrcc4 exon 3-specific gRNA (5′-GTACCAGCTGCTCCTGACCC-3′) was designed using Crispor (http://crispor.tefor.net/oligonucleotide). The HR template was the ssODN ([Integrated DNA technologies, IDT] 5′-TCAAGGGAGAAATGCCGAGACTCCTTAGAAAAGAGGAACTTGTATGTACCAGCaGCACCGGAGCCTGGCACCAGTGCCTTTCTCAGCTCATCAATGTATTTTCCTTTCTCCCTAGCCATGTCATCAGCTTCTTGGGAAATCTCCAATTCAGAAACTATGGGAAAGATTAATTAAGGTGAT-3′; see Figure 1—figure supplement 1). F° chimera screen and Xrcc4M61R mice genotyping were obtained upon FspBI digestion of genomic DNA amplified with primers F 5′-AGGAGACGGAGGAAAAAGAGATG-3′ and R 5′-TACCCTCACAGAAACACAACTCA-3′.
Immunoblotting
Whole tissues or MEFs single-cell suspensions were washed in PBS. Cells were lysed in 50–200 μL of 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% TritonX100, 1X phosphatase inhibitor cocktail 2 and 3 (ThermoFisher), and 1X protease inhibitor cocktail EDTA free (Roche) on ice for 45 min. Proteins migrated within Tris-Acrylamide gels and Tris-Glycine-SDS buffer and were transferred on PVDF membranes (Immobilon). Membranes were stained with the following antibodies: β-actin (mouse monoclonal AM4302, ThermoFisher), XRCC4 (goat monoclonal C-20, Santa Cruz), and DNA ligase IV (mouse monoclonal D-8, Santa Cruz). Membranes were revealed with appropriate secondary antibodies conjugated with infrared 700 or 800 dyes (LiCor). Immunoblotting was revealed by LiCor CLx analyzer and analyses were performed on Image Studio Lite software.
X4 transcript analysis
Total RNAs were obtained from thymocytes with Pure Like RNA mini kit (Invitrogen) and used for RT-PCR (SuperScript II reverse transcriptase; Invitrogen). X4 transcripts were amplified with primers F1 5′-TTGGGCGCATCGGTTTATCT-3′ R1 5′-GCTGCTAAGTTGAAAGCCTG-3′ F2 5′-TGCTTCTGAACCCAACGTACC-3′ R2 5′- AGGTGCTCGTTTTTGGCTTG-3′. F1-R1 PCR amplifies full-length transcript from 5′UTR to 3′UTR (1150 bp) and F2-R2 amplifies exon 2 to exon 4 (400 bp).
Flow cytometry analysis of cell populations
Cell phenotyping from 6- to 9-week-old mice was performed on thymus, spleen, and bone marrow using the following antibodies: CD4, CD8, CD25, CD28, CD44, CD69, B220, CD19, CD43, and IgM (all from Sony Biotechnology, using respectively PECy7, FITC, PerCPCy5.5, PE, BV510, APC, PE, PECy7, FITC, APC fluorophores). Intracellular IgM expression in E18.5 FL cells was performed as previously described (Abramowski et al., 2018) using CD19, B220, CD43, IgM for extracellular staining followed by cell fixation and permeabilization (Invitrogen) and intracellular IgM staining (all antibodies from Sony Biotechnology, using respectively PECy7, BV605, PE, FITC, and APC). Cells were recorded by FACS LSR-Fortessa X-20 and analyses were performed with FlowJo 10 software.
Cellular sensitivity to DSBs-inducing agents and thymocyte survival assay
For phleomycin sensitivity assay, 5000 MEF cells were seeded in triplicates and cultivated with increasing doses (0–300 ng/mL) of phleomycin. Living cells were counted by flow analysis with FACS LSR-Fortessa X-20 after 6 days of culture. Radiation sensitivity of T lymphocytes was performed as described (Abramowski et al., 2018). Ex vivo thymocyte survival assay was performed as described (Vera et al., 2013).
Quantitative real-time RT-PCR analysis
TaqMan PCR was performed on triplicates of 8 ng of reverse-transcribed RNA from freshly dissected total thymus as described (Vera et al., 2013).
Analysis of thymic TCR-alpha repertoire
Comprehensive TCR-alpha repertoire analyses were performed by 5′ Rapid amplification of complementary DNA (cDNA) ends (5′RACE) PCR/NGS (switching mechanism at the 5′end of the RNA transcript, SMARTα) from total thymus RNA as described (Abramowski et al., 2018; Roch et al., 2019; Vera et al., 2013). Sequencing data were analyzed with LymAnalyzer (Yu et al., 2016) to retrieve unique CDR3 clonotypes and determine T cell receptor alpha variable (TRAV) and T cell receptor alpha junction (TRAJ) gene segments. Frequencies of TRAV and TRAJ usage were implemented in principal component analysis (PCA) and hierarchical clustering on principal components (HCPC) analyses using the PCA() and HCPC() functions of the FactomineR package, respectively (http://factominer.free.fr/; Le et al., 2008) and graphics were generated using the Factoextra R package (http://www.sthda.com/english/rpkgs/factoextra; Kassambara, 2017).
TCRβ V(D)J recombination analysis
Tcrb rearrangements were analyzed by PCR on genomic DNA from total adult thymocytes or E18.5 fetal thymocytes as described (Abramowski et al., 2018).
E15.5 brain sections immunohistochemistry
Neuronal apoptosis was analyzed as previously described (Abramowski et al., 2018). E15.5 fetal heads were fixed overnight at 4°C by immersion in 4% paraformaldehyde and embedded in paraffin with a Tissu-tek processor (VIP, Leica). 5 μm coronal sections were then obtained using a microtome (Leica RM2125RT) and mounted onto glass slides for histological analyses. After paraffin removal and citrate treatment, the brain sections were permeabilized with 0.5% Triton X-100 in phosphate-buffered saline (PBS) for 15 min and incubated for 2 hr with 7.5% fetal bovine serum and 7.5% goat serum in PBS. The sections were incubated with rabbit anti-CC3 (Cell Signaling 9661) overnight at 4°C. After washing, the sections were incubated with goat anti-rabbit Alexa Fluor 488 or 594 conjugated secondary antibody (ThermoFisher) for 1 hr. After washing, nuclear staining was achieved by incubation with 4′-6-diamidino-2-phenylindole (DAPI) to quantify apoptosis induction by the detection of pyknotic nuclei (Roque et al., 2012). Slides were mounted under Fluoromount (Southern Biotechnologies Associates). Tissues were examined under a fluorescence microscope (50i, Nikon, Japan) with a 10× (NA = 0.3) objective in three channels (appearing red, green, and gray) as separate files. These images were then stacked with Photoshop software (Adobe).
Statistical analysis
Non-parametric Mann–Whitney test or two-tailed Fisher’s exact test was performed with α-risk = 0.05. p-Values were taken to be significant: *significant 0.05≥p>0.01; **very significant 0.01≥p>0.001; ***highly significant 0.001≥p>0.0001; ****highly significant p≤0.0001. Using G*Power software (https://www.psychologie.hhu.de/arbeitsgruppen/allgemeine-psychologie-und-arbeitspsychologie/gpower), we determined that a minimum of 47 newborns was necessary to conclude to an embryonic lethality of the Xrcc4M61R-Nhej1-/- double KO with a power of 80% and a p-value < 0.05.
Acknowledgements
This work was supported by institutional grant from INSERM and Université de Paris, state funding from the Agence National de la Recherche under 'Investissements d’avenir' program (ANR-10-IAHU-01) and grants from Institut National du Cancer (PLBIO 16-280) et Ligue Nationale contre le Cancer (Equipe Labellisée). BR received fellowships from INSERM, ARC, and LNCC.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Jean-Pierre de Villartay, Email: jean-pierre.de-villartay@inserm.fr.
Ranjan Sen, National Institute on Aging, United States.
Satyajit Rath, Indian Institute of Science Education and Research (IISER), India.
Funding Information
This paper was supported by the following grants:
Institut National de la Santé et de la Recherche Médicale to Benoit Roch, Vincent Abramowski, Stefania Musilli, Jean-Pierre de Villartay.
Agence Nationale de la Recherche ANR-10-IAHU-01 to Benoit Roch, Vincent Abramowski, Stefania Musilli, Pierre David, Jean-Pierre de Villartay.
Institut National Du Cancer PLBIO 16-280 to Benoit Roch, Vincent Abramowski, Stefania Musilli, Jean-Baptiste Charbonnier, Isabelle Callebaut, Jean-Pierre de Villartay.
Ligue Contre le Cancer Equipe Labellisée to Benoit Roch, Vincent Abramowski, Stefania Musilli, Jean-Pierre de Villartay.
Additional information
Competing interests
No competing interests declared.
No competing interests declared.
Author contributions
Conceptualization, Formal analysis, Investigation, Methodology, Writing - original draft.
Formal analysis, Investigation, Methodology.
Formal analysis, Investigation, Methodology.
Formal analysis, Investigation, Methodology.
Formal analysis, Investigation, Methodology.
Conceptualization.
Conceptualization.
Conceptualization, Data curation, Supervision, Validation, Writing - original draft.
Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Supervision, Validation, Writing - original draft.
Ethics
All experiments were performed in compliance with the French Ministry of Agriculture's regulations for animal experiments (act 87847, 19 October 1987; modified in May 2001) after audit with "Comite' d'Ethique en Expe'rimentation Animale (CEEA) Paris Descartes" (Apafis #25432-2019041516286014 v6).
Additional files
Data availability
The xl file with raw data used in PCA analysis (Fig. 2G) has been deposited on DRYAD https://datadryad.org/stash/share/T4vNOWxXUUGanYkI2H--C39xErpsKK6kSynXqljpxsM.
The following dataset was generated:
Jean-Pierre D, Benoit R, Vincent A, Olivier E, Stefania M, Pierre D, Jean-Baptiste C, Isabelle C, François B. 2021. Data from: A XRCC4 mutant mouse, a model for human X4 syndrome, reveals interplays with Xlf, PAXX, and ATM in lymphoid development. Dryad Digital Repository.
References
- Abramowski V, Etienne O, Elsaid R, Yang J, Berland A, Kermasson L, Roch B, Musilli S, Moussu J-P, Lipson-Ruffert K, Revy P, Cumano A, Boussin FD, de Villartay J-P. PAXX and Xlf interplay revealed by impaired cns development and immunodeficiency of double KO mice. Cell Death and Differentiation. 2018;25:444–452. doi: 10.1038/cdd.2017.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arya R, Bassing CH. V(D)J Recombination Exploits DNA Damage Responses to Promote Immunity. Trends in Genetics. 2017;33:479–489. doi: 10.1016/j.tig.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmus G, Barros AC, Wijnhoven PW, Lescale C, Hasse HL, Boroviak K, Jackson SP. Synthetic lethality between PAXX and XLF in mammalian development. Genes & Development. 2016;30:2152–2157. doi: 10.1101/gad.290510.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Wynshaw-Boris A. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Stamp G, Rosewell I, Denzel A, Lindahl T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Current Biology. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Swat W, Alt FW. The mechanism and regulation of chromosomal V(D)J recombination. Cell. 2002;109 Suppl:S45. doi: 10.1016/s0092-8674(02)00675-x. [DOI] [PubMed] [Google Scholar]
- Beck C, Castaneda-Zegarra S, Huse C, Xing M, Oksenych V. Mediator of DNA Damage Checkpoint Protein 1 Facilitates V(D)J Recombination in Cells Lacking DNA Repair Factor XLF. Biomolecules. 2019;10:E60. doi: 10.3390/biom10010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berland A, Rosain J, Kaltenbach S, Allain V, Mahlaoui N, Melki I. PROMIDISalpha: A T-cell receptor alpha signature associated with immunodeficiencies caused by V(D)J recombination defects. The Journal of Allergy and Clinical Immunology. 2019;143:325–334. doi: 10.1016/j.jaci.2018.05.028. [DOI] [PubMed] [Google Scholar]
- Betermier M, Borde V, de Villartay JP. Coupling DNA Damage and Repair: an Essential Safeguard during Programmed DNA Double-Strand Breaks. Trends in Cell Biology. 2020;30:87–96. doi: 10.1016/j.tcb.2019.11.005. [DOI] [PubMed] [Google Scholar]
- Brouwer I, Sitters G, Candelli A, Heerema SJ, Heller I, de Melo AJ, Wuite GJ. Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA. Nature. 2016;535:566–569. doi: 10.1038/nature18643. [DOI] [PubMed] [Google Scholar]
- Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, Revy P. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Callebaut I, Malivert L, Fischer A, Mornon JP, Revy P, de Villartay JP. Cernunnos interacts with the XRCC4 x DNA-ligase IV complex and is homologous to the yeast nonhomologous end-joining factor Nej1. The Journal of Biological Chemistry. 2006;281:13857–13860. doi: 10.1074/jbc.C500473200. [DOI] [PubMed] [Google Scholar]
- Chang HHY, Pannunzio NR, Adachi N, Lieber MR. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nature Reviews. Molecular Cell Biology. 2017;18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplin AK, Hardwick SW, Liang S, Kefala Stavridi A, Hnizda A, Cooper LR, Blundell TL. Dimers of DNA-PK create a stage for DNA double-strand break repair. Nature Structural & Molecular Biology. 2021a;28:13–19. doi: 10.1038/s41594-020-00517-x. [DOI] [PubMed] [Google Scholar]
- Chaplin AK, Hardwick SW, Stavridi AK, Buehl CJ, Goff NJ, Ropars V, Liang S, De Oliveira TM, Chirgadze DY, Meek K, Charbonnier JB, Blundell TL. Cryo-em of NHEJ supercomplexes provides insights into dna repair. Molecular Cell. 2021b;81:3400–3409. doi: 10.1016/j.molcel.2021.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Elos MT, Viboolsittiseri SS, Gowan K, Leach SM, Rice M, Wang JH. Combined deletion of Xrcc4 and Trp53 in mouse germinal center B cells leads to novel B cell lymphomas with clonal heterogeneity. Journal of Hematology & Oncology. 2016;9:2. doi: 10.1186/s13045-015-0230-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BR, Quinet A, Byrum AK, Jackson J, Berti M, Thangavel S, Sleckman BP. XLF and H2AX function in series to promote replication fork stability. The Journal of Cell Biology. 2019;218:2113–2123. doi: 10.1083/jcb.201808134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair. 2007;6:712–722. doi: 10.1016/j.dnarep.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Craxton A, Somers J, Munnur D, Jukes-Jones R, Cain K, Malewicz M. XLS (c9orf142) is a new component of mammalian DNA double-stranded break repair. Cell Death and Differentiation. 2015;22:890–897. doi: 10.1038/cdd.2015.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Villartay JP, Fischer A, Durandy A. The mechanisms of immune diversification and their disorders. Nature Reviews. Immunology. 2003;3:962–972. doi: 10.1038/nri1247. [DOI] [PubMed] [Google Scholar]
- de Villartay JP. When natural mutants do not fit our expectations: the intriguing case of patients with XRCC4 mutations revealed by whole-exome sequencing. EMBO Molecular Medicine. 2015;7:862–864. doi: 10.15252/emmm.201505307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng HL, Alt FW. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396:173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- Gao Y, Sun Y, Frank KM, Dikkes P, Fujiwara Y, Seidl KJ, Alt FW. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891–902. doi: 10.1016/s0092-8674(00)81714-6. [DOI] [PubMed] [Google Scholar]
- Gao Y, Ferguson DO, Xie W, Manis JP, Sekiguchi J, Frank KM, Alt FW. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature. 2000;404:897–900. doi: 10.1038/35009138. [DOI] [PubMed] [Google Scholar]
- Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, Mann M, Lieber MR. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388:492–495. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- Hung PJ, Johnson B, Chen BR, Byrum AK, Bredemeyer AL, Yewdell WT, Sleckman BP. MRI Is a DNA Damage Response Adaptor during Classical Non-homologous End Joining. Molecular Cell. 2018;71:332–342. doi: 10.1016/j.molcel.2018.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annual Review of Immunology. 2006;24:541–570. doi: 10.1146/annurev.immunol.23.021704.115830. [DOI] [PubMed] [Google Scholar]
- Kajikhina K, Tsuneto M, Melchers F. B-Lymphopoiesis in Fetal Liver, Guided by Chemokines. Advances in Immunology. 2016;132:71–89. doi: 10.1016/bs.ai.2016.07.002. [DOI] [PubMed] [Google Scholar]
- Kassambara A. Practical guide to cluster analysis in r. STHDA. 2017 https://xsliulab.github.io/Workshop/week10/r-cluster-book.pdf
- Le S, Josse J, Husson F. FactoMineR: An R Package for Multivariate Analysis. Journal of Statistical Software. 2008;25:1–18. doi: 10.18637/jss.v025.i01. [DOI] [Google Scholar]
- Lescale C, Abramowski V, Bedora-Faure M, Murigneux V, Vera G, Roth DB, Deriano L. RAG2 and XLF/Cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nature Communications. 2016a;7:10529. doi: 10.1038/ncomms10529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescale C, Lenden Hasse H, Blackford AN, Balmus G, Bianchi JJ, Yu W, Deriano L. Specific Roles of XRCC4 Paralogs PAXX and XLF during V(D)J Recombination. Cell Reports. 2016b;16:2967–2979. doi: 10.1016/j.celrep.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Alt FW, Cheng HL, Brush JW, Goff PH, Murphy MM, Zha S. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Molecular Cell. 2008;31:631–640. doi: 10.1016/j.molcel.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Shao Z, Jiang W, Lee BJ, Zha S. PAXX promotes KU accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nature Communications. 2017;8:13816. doi: 10.1038/ncomms13816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musilli S, Abramowski V, Roch B, de Villartay JP. An in vivo study of the impact of deficiency in the DNA repair proteins PAXX and XLF on development and maturation of the hemolymphoid system. The Journal of Biological Chemistry. 2020;295:2398–2406. doi: 10.1074/jbc.AC119.010924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, Tamura N, Jackson SP. DNA repair PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science. 2015;347:185–188. doi: 10.1126/science.1261971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenych V, Alt FW, Kumar V, Schwer B, Wesemann DR, Hansen E, Guo C. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. PNAS. 2012;109:2455–2460. doi: 10.1073/pnas.1121458109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, Concannon P. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Molecular Cell. 2001;8:1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- Ramond C, Berthault C, Burlen-Defranoux O, de Sousa AP, Guy-Grand D, Vieira P, Cumano A. Two waves of distinct hematopoietic progenitor cells colonize the fetal thymus. Nature Immunology. 2014;15:27–35. doi: 10.1038/ni.2782. [DOI] [PubMed] [Google Scholar]
- Recio MJ, Dominguez-Pinilla N, Perrig MS, Rodriguez Vigil-Iturrate C, Salmon-Rodriguez N, Martinez Faci C, Gonzalez-Granado L. Extreme Phenotypes With Identical Mutations: Two Patients With Same Non-sense NHEJ1 Homozygous Mutation. Frontiers in Immunology. 2018;9:2959. doi: 10.3389/fimmu.2018.02959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid DA, Keegan S, Leo-Macias A, Watanabe G, Strande NT, Chang HH, Rothenberg E. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. PNAS. 2015;112:E2575. doi: 10.1073/pnas.1420115112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roch B, Abramowski V, Chaumeil J, de Villartay JP. Cernunnos/Xlf Deficiency Results in Suboptimal V(D)J Recombination and Impaired Lymphoid Development in Mice. Frontiers in Immunology. 2019;10:443. doi: 10.3389/fimmu.2019.00443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropars V, Drevet P, Legrand P, Baconnais S, Amram J, Faure G, Charbonnier JB. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. PNAS. 2011;108:12663–12668. doi: 10.1073/pnas.1100758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque T, Haton C, Etienne O, Chicheportiche A, Rousseau L, Martin L, Boussin FD. Lack of a p21waf1/cip -dependent G1/S checkpoint in neural stem and progenitor cells after DNA damage in vivo. Stem Cells. 2012;30:537–547. doi: 10.1002/stem.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Kurosawa A, Adachi N. Mutations in XRCC4 cause primordial dwarfism without causing immunodeficiency. Journal of Human Genetics. 2016;61:679–685. doi: 10.1038/jhg.2016.46. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Swanson PC. V(D)J recombination: mechanisms of initiation. Annual Review of Genetics. 2011;45:167–202. doi: 10.1146/annurev-genet-110410-132552. [DOI] [PubMed] [Google Scholar]
- Shull ER, Lee Y, Nakane H, Stracker TH, Zhao J, Russell HR, McKinnon PJ. Differential DNA damage signaling accounts for distinct neural apoptotic responses in ATLD and NBS. Genes & Development. 2009;23:171–180. doi: 10.1101/gad.1746609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibanda BL, Chirgadze DY, Ascher DB, Blundell TL. DNA-PKcs structure suggests an allosteric mechanism modulating DNA double-strand break repair. Science. 2017;355:520–524. doi: 10.1126/science.aak9654. [DOI] [PubMed] [Google Scholar]
- Staines Boone AT, Chinn IK, Alaez-Versón C, Yamazaki-Nakashimada MA, Carrillo-Sánchez K, García-Cruz M, Poli MC, González Serrano ME, Medina Torres EA, Muzquiz Zermeño D, Forbes LR, Espinosa-Rosales FJ, Espinosa-Padilla SE, Orange JS, Lugo Reyes SO. Failing to Make Ends Meet: The Broad Clinical Spectrum of DNA Ligase IV Deficiency Case Series and Review of the Literature. Frontiers in Pediatrics. 2018;6:426. doi: 10.3389/fped.2018.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ucuncu E, Rajamani K, Wilson MSC, Medina-Cano D, Altin N, David P, Cantagrel V. MINPP1 prevents intracellular accumulation of the chelator inositol hexakisphosphate and is mutated in Pontocerebellar Hypoplasia. Nature Communications. 2020;11:6087. doi: 10.1038/s41467-020-19919-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg M, Gennery AR. Educational paper The expanding clinical and immunological spectrum of severe combined immunodeficiency. European Journal of Pediatrics. 2011;170:561–571. doi: 10.1007/s00431-011-1452-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera G, Rivera-Munoz P, Abramowski V, Malivert L, Lim A, Bole-Feysot C, de Villartay JP. Cernunnos deficiency reduces thymocyte life span and alters the T cell repertoire in mice and humans. Molecular and Cellular Biology. 2013;33:701–711. doi: 10.1128/MCB.01057-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Yang M, Huo W, Feng F, Wei L, Jiang W, Xu D. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nature Communications. 2015;6:6233. doi: 10.1038/ncomms7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Bjoras M, Daniel JA, Alt FW, Oksenych V. Synthetic lethality between murine DNA repair factors XLF and DNA-PKcs is rescued by inactivation of Ku70. DNA Repair. 2017;57:133–138. doi: 10.1016/j.dnarep.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Ceredig R, Seoighe C. LymAnalyzer: a tool for comprehensive analysis of next generation sequencing data of T cell receptors and immunoglobulins. Nucleic Acids Research. 2016;44:e31. doi: 10.1093/nar/gkv1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Guo C, Boboila C, Oksenych V, Cheng HL, Zhang Y, Alt FW. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 2011;469:250–254. doi: 10.1038/nature09604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Rothenberg E, Ramsden DA, Lieber MR. The molecular basis and disease relevance of non-homologous DNA end joining. Nature Reviews. Molecular Cell Biology. 2020;21:765–781. doi: 10.1038/s41580-020-00297-8. [DOI] [PMC free article] [PubMed] [Google Scholar]