

Abstract
In recent decades, treatments for myocardial infarction (MI), such as stem and progenitor cell therapy, have attracted considerable scientific and clinical attention but failed to improve patient outcomes. These efforts indicate that more rigorous mechanistic and functional testing of potential MI therapies is required. Recent studies have suggested that augmenting post-MI lymphatic growth via VEGF-C administration improves cardiac function. However, the mechanisms underlying this proposed therapeutic approach remain vague and untested. To more rigorously test the role of lymphatic vessel growth after MI, we examined the post-MI cardiac function of mice in which lymphangiogenesis had been blocked genetically by pan-endothelial or lymphatic endothelial loss of the lymphangiogenic receptor VEGFR3 or global loss of the VEGF-C and VEGF-D ligands. The results obtained using all 3 genetic approaches were highly concordant and demonstrated that loss of lymphatic vessel growth did not impair left ventricular ejection fraction 2 weeks after MI in mice. We observed a trend toward excess fluid in the infarcted region of the left ventricle, but immune cell infiltration and clearance were unchanged with loss of expanded lymphatics. These studies refute the hypothesis that lymphangiogenesis contributes significantly to cardiac function after MI, and suggest that any effect of exogenous VEGF-C is likely to be mediated by nonlymphangiogenic mechanisms.
Keywords: Cardiology, Vascular Biology
Keywords: Heart failure, Lymph

Introduction
Myocardial infarction (MI) remains a leading cause of death and disability in the developed world. During MI, luminal coronary thrombosis blocks blood flow to part of the heart, resulting in death of myocardium in that vascular distribution. Since the body’s demand for blood flow is unchanged, the remaining viable myocardium must compensate for this loss, a response that often leads to heart failure after a large MI. Heart failure and death after MI have been shown to correlate tightly with remaining cardiac pump function, typically measured as the fraction of left ventricular (LV) blood volume that is ejected from the heart with each heartbeat (ejection fraction, EF; ref. 1). Thus, preservation of cardiac function through improved myocardial viability has been considered the most important goal of therapy for acute MI, achieved primarily through mechanical and pharmacologic strategies designed to reestablish blood flow to ischemic cardiac muscle. Such therapies have successfully reduced the acute mortality of MI to less than 5% for treated individuals (2), but a consequence of improved mortality is a rising prevalence of long-term complications of MI such as heart failure.
Heart failure after MI is accompanied by ventricular remodeling in which the ventricle enlarges to maintain cardiac output despite reduced overall cardiac function. Established drugs such as angiotensin-converting enzyme inhibitors are believed to improve post-MI cardiac function and remodeling indirectly, e.g., by favorably altering the hemodynamic conditions against which the infarcted heart must pump (3–5). Remodeling of the heart after MI is proposed to be influenced by wound healing in the infarct zone as well as by adaptation of the remaining functional myocardium (6, 7). Therapeutic strategies targeting the wound healing process have also been proposed as a means of improving ventricular function after MI (8). Such approaches, however, require a more complete understanding of the wound healing processes of the infarcted heart and their subsequent impact on cardiac function. Studies in animal models have suggested that there may be distinct phases of wound healing in the infarct zone: an early inflammatory phase in which large numbers of phagocytes remove dead heart tissue and a later reparative phase in which scar tissue is formed and secondary responses such as angiogenesis may be initiated (9, 10). Such studies have led to the hypothesis that altering the wound healing events in the infarct zone (e.g., by reducing the early inflammatory response) could improve cardiac function (9, 11). Translation of these hypotheses to new therapies for MI has been hampered, however, by a lack of studies directly testing whether and how the cellular events that take place in the infarct zone after MI impact function.
After migration of inflammatory cells into the infarcted region of the heart, the immune cells must be removed so that scar formation can occur. In other models of acute or chronic inflammation, in situ death of the inflammatory cells contributes substantially to reduced cell numbers (12, 13). Another mechanism for immune cell exit is through lymphatic vessels, though this hypothesis has not been functionally tested in the infarcted heart. Recent studies have reported extremely robust growth of new lymphatic vessels in the infarct zone after MI in mice (14, 15). Administration of exogenous VEGF-C C156S, a form of the lymphangiogenic factor VEGF-C that acts specifically on VEGFR3 receptors selectively expressed in lymphatic endothelial cells (LECs), has been reported to further augment this response and confer improved heart function after MI (15–19). These studies have led to a paradigm in which lymphangiogenic strategies may be used to improve wound healing and cardiac function in patients who survive large infarcts and are at high risk of subsequent heart failure (20). However, significant weaknesses limit the interpretation and translation of these studies. First, the gain-of-function approaches utilized are global; thus, whether the observed improvements in cardiac EF arise specifically due to effects on cardiac lymphatic vessel growth is not clear. Second, the results have not been consistently reproducible; e.g., administration of VEGF-C protein was found to improve cardiac function after MI in one study (15) but not in another (19). Finally, conflicting findings have recently been reported for gain of VEGF-C and blockade of its receptor VEGFR3 using adeno-associated virus–mediated gene therapy approaches (19). Thus whether, and to what extent, lymphangiogenesis after MI affects subsequent cardiac function remains unclear.
To more rigorously address the role of lymphatic vessel growth after MI, we have used genetic approaches to block the expression of the primary lymphangiogenic receptor VEGFR3 in blood endothelial cells and/or LECs or to block global expression of the required lymphangiogenic factors VEGF-C and VEGF-D in mice that undergo coronary ligation. Each of the 3 complementary genetic approaches successfully reduced the rate of new lymphatic growth in the infarct by greater than 80%, resulting in the formation of myocardial infarcts that were almost entirely devoid of lymphatic vessels. Despite the near absence of lymphangiogenesis in the infarcted heart, we observed no significant change in LV EF 14 days after MI, a time point at which altering lymphatic growth with systemic administration of VEGF-C has previously been reported to improve EF (15). We observed a trend toward excess fluid accumulation in the LV free wall but no difference in immune cell infiltration or clearance. These studies indicate that lymphatic vessel expansion in the infarct zone after MI is not required for maintaining cardiac function after injury, and suggest that lymphangiogenic stimulation as a therapeutic approach to MI should be more carefully examined prior to clinical translation.
Results
LV EF after MI is not altered by loss of lymphangiogenesis conferred by pan-endothelial deletion of VEGFR3.
To determine the specific role for lymphangiogenesis after MI, 12-week-old mice were subjected to left anterior descending (LAD) coronary artery ligation immediately distal to the first diagonal branch. Consistent with prior reports (14), lymphatic vessels identified by immunostaining for LYVE1 and PROX1 (membrane and nuclear markers for lymphatic endothelium, respectively) were observed in the infarcted zone of the left ventricle by day 4 after MI (Supplemental Figure 1, A and B; supplemental material available online with this article; https://doi.org/10.1172/JCI147070DS1). These lymphatic vessels were numerous by day 7 after MI (Supplemental Figure 1C) and remained present in the infarct tissue through 14 and 28 days after MI (Supplemental Figure 1, D and E). Importantly, the robust growth of lymphatic vasculature was confined to the infarct zone and arose in the absence of any exogenous lymphangiogenic factor administration.
To test a potential role for lymphatic growth in cardiac function after MI, we first generated Flt4fl/fl; Cdh5-CreERT2 animals in which deletion of both VEGFR3 (gene symbol Flt4, Supplemental Figure 2) alleles was restricted to endothelial cells and temporally controlled by administration of tamoxifen (hereafter called EC VEGFR3–KO animals). Tamoxifen was administered to 8-week-old mice and allowed to wash out for at least 2 weeks prior to LAD ligation. EC VEGFR3–KO and control Flt4fl/fl littermates underwent LAD ligation and were studied using echocardiography prior to sacrifice and histologic analysis of lymphatic vascular density 14 days after MI. Histologic analysis of control animals (where VEGFR3 remains highly expressed in epicardial lymphatics; Figure 1A) revealed robust lymphatic vessel growth in the infarct zone, quantified by measurement of LYVE1+PROX1+ cells/mm2 (Figure 1, B and C). In contrast, EC VEGFR3–KO animals showed loss of VEGFR3 expression in epicardial LYVE1+ lymphatics (Figure 1D) and exhibited infarct areas that were almost completely devoid of LYVE1+PROX1+ lymphatic vessels (Figure 1, E and F). Quantitation revealed a statistically significant 82% reduction in LYVE1+PROX1+ LECs in the infarct zone of EC VEGFR3–KO animals 14 days after MI (Figure 1G). These findings are consistent with prior studies demonstrating that VEGFC/VEGFR3 signaling is required for new lymphatic vessel growth but not for maintenance of existing lymphatic vessels outside of the small intestine (21). Importantly, although existing epicardial lymphatic vessels remained intact after the loss of VEGF signaling, the absolute number of lymphatic cells was decreased due to reduced lymphatic growth.
Figure 1. Pan-endothelial deletion of Flt4 severely reduces lymphangiogenesis in the infarct zone without affecting cardiac function.

(A) Epicardial lymphatics in a Flt4fl/fl control heart were immunostained for LYVE1 (magenta) and VEGFR3 (orange). Myocardium is autofluorescent (green); nuclei are marked with DAPI (blue). Individual LYVE1 and VEGFR3 channels are shown. (B and C) Infarcts from Flt4fl/fl animals 14 days after MI were examined using Masson’s trichrome stain (B) and immunostained for lymphatic endothelial markers LYVE1 and PROX1 (C, representative images are from one animal). (D) Epicardial lymphatics in a Flt4fl/fl; Cdh5-CreERT2 heart were examined for LYVE1 and VEGFR3 expression, with individual LYVE1 and VEGFR3 channels shown. Note the loss of VEGFR3 protein signal. (E and F) Infarcts from Flt4fl/fl; Cdh5-CreERT2 hearts 14 days after MI were examined using Masson’s trichrome stain (E) and immunostained for LYVE1 and PROX1 (F). (G) The number of LYVE1+PROX1+ lymphatic endothelial cells was measured per infarct area (n = 6, 8). (H) Representative 2D (top) and M-mode (bottom) echocardiographic images of the left ventricles of sham-operated Flt4fl/fl animals and Flt4fl/fl and Flt4fl/fl; Cdh5-CreERT2 animals that underwent LAD ligation. Note the dilated left ventricle (dia. denotes diastole and sys. denotes systole in M-mode images). (I) Infarct size 14 days after MI was determined histologically for Flt4fl/fl and Flt4fl/fl; Cdh5-CreERT2 animals (n = 6, 7). (J–L) The cardiac functional parameters ejection fraction (J), end diastolic volume (K), and end systolic volume (L) were measured 14 days after MI (n = 18, 15). Insets in C and F are higher magnification of boxed regions. In B, C, E, and F, a yellow line denotes the infarct border, “epi” denotes epicardium, and “myo” denotes live myocardium. In H, the dashed yellow lines show the locations of M-mode images. Triangles represent female animals in I–L. Bar graphs represent mean ± SEM. Comparisons were made with a 2-tailed t test.
Cardiac function was assessed 14 days after MI, a time point used by prior studies that reported an important functional role for lymphangiogenesis after MI (15), by a trained mouse echocardiographer who was blinded to animal genotypes. Importantly, deletion of Flt4 using the Cdh5-CreERT2 driver by tamoxifen induction did not alter EF at baseline (Supplemental Figure 3). Representative 2D and M-mode echocardiographic images from sham-operated Flt4fl/fl and LAD-ligated Flt4fl/fl and EC VEGFR3–KO animals are shown in Figure 1H. Measurement of infarct size revealed the creation of medium-to-large infarcts that did not differ significantly in size between EC VEGFR3–KO animals and littermate controls (Figure 1I). Consistent with prior studies (22), cardiac function after MI induced by LAD ligation resulted in a substantial but highly variable reduction in EF (mean EF = 31.3%, SEM = 2.6; Figure 1J and Table 1) as well as LV end-diastolic volume (EDV) (Figure 1K) and end-systolic volume (ESV) (Figure 1K). Importantly, in contrast to a previous report that used a dominant negative strategy (23), we did not observe increased mortality after MI following loss of VEGFR3 function (a total of 3 mice across all genotypes died between LAD surgery and 14-day echo time point). Given the high experimental variability in cardiac function after MI, to adequately power the study we analyzed 18 control and 15 EC VEGFR3–KO littermate animals. Blinded analysis of this large cohort of animals revealed no significant difference in EF, EDV, or ESV between EC VEGFR3–KO and control littermate animals by t test (Figure 1, J–L). These studies suggest that blocking lymphatic vessel expansion after MI by pan-endothelial VEGFR3 deletion does not alter cardiac function 14 days later.
Table 1. Measurement of weight and cardiovascular parameters for cardiac functional experiments.

LV EF after MI is unchanged following LEC-specific loss of VEGFR3.
VEGFR3 is required for LEC proliferation and lymphatic vessel growth (24, 25), but can also be expressed by blood endothelial cells where it is believed to participate in both angiogenic responses and regulation of blood vessel permeability (26, 27). Thus, it is possible that pan-endothelial loss of VEGFR3 could affect both lymphatic and blood vascular responses within the infarcted heart, masking a significant role for VEGFR3-mediated lymphangiogenesis. To more specifically address the requirement for lymphatic vessel growth, we next tested the effect of deleting Flt4 exclusively in lymphatic and not blood endothelium using the Prox1-CreERT2 transgenic driver (ref. 28; Flt4fl/fl; Prox1-CreERT2, hereafter called LEC VEGFR3–KO animals). In control animals, VEGFR3 expression was observed in the epicardial lymphatic vessels (Figure 2A). After infarction, control Flt4fl/fl animals showed a robust lymphangiogenic response in the infarct zone (Figure 2, B and C). LEC VEGFR3–KO animals exhibited loss of VEGFR3 protein expression in large epicardial lymphatic vessels after tamoxifen-induced deletion (Figure 2D), consistent with marked and durable gene deletion, as well as severe reductions in infarct zone lymphatic vasculature 14 days after MI (Figure 2, E and F). Similar to the Cdh5-CreERT2–mediated deletion, examination of the infarct zone 14 days after MI revealed a substantial and statistically significant 86% decrease in LYVE1+PROX1+ lymphatic vessels in LEC VEGFR3–KO animals compared with control littermates (Figure 2G), despite the presence of infarct sizes that were indistinguishable from controls (Figure 2H). Blinded evaluation of LV EF, EDV, and ESV revealed no significant differences between control and LEC VEGFR3–KO animals (Figure 2, I–K). These findings replicate those obtained using EC VEGFR3–KO animals (see summary data in Table 1), and support the conclusion that loss of lymphatic vessel growth due to loss of VEGFR3 function does not significantly impact cardiac function after MI in mice.
Figure 2. Lymphatic endothelial cell–specific deletion of Flt4 severely reduces lymphangiogenesis in the infarct zone without affecting cardiac function.

(A) Epicardial lymphatics in a Flt4fl/fl control heart were immunostained for LYVE1 (magenta) and VEGFR3 (orange). Live myocardium is highly autofluorescent (Auto, green) and cell nuclei are marked with DAPI (blue). Boxed region is shown at higher magnification on the right with individual LYVE1 and VEGFR3 channels. (B and C) Adjacent sections from the same infarct zone in a Flt4fl/fl animal 14 days after MI were examined using Masson’s trichrome stain (B) and immunostained for the lymphatic endothelial markers LYVE1 and PROX1 (C). The inset shows the boxed region in C at higher magnification. (D) Epicardial lymphatics in a Flt4fl/fl; Prox1-CreERT2 heart were immunostained for LYVE1 (magenta) and VEGFR3 (orange). Boxed region is shown at higher magnification on the right with individual LYVE1 and VEGFR3 channels. Note the loss of VEGFR3 protein detection on the LYVE1+ epicardial lymphatic. (E and F) Adjacent sections from the same infarct zone in a Flt4fl/fl; Prox1-CreERT2 heart 14 days after MI were examined using Masson’s trichrome stain (E) and immunostained for the lymphatic endothelial markers LYVE1 and PROX1 (F). The inset shows the boxed region in F at higher magnification. (G) The number of LYVE1+PROX1+ lymphatic endothelial cells was measured per mm2 in the infarct zone of the indicated animals (n = 3, 5). (H) Infarct size 14 days after MI was determined histologically for the Flt4fl/fl and Flt4fl/fl; Prox1-CreERT2 animals (n = 3, 5). (I–K) The cardiac functional parameters ejection fraction (I), end diastolic volume (J), and end systolic volume (K) were measured 14 days after MI in the indicated animals in a fully blinded manner (n = 6, 14). In B, C, E, and F, a dashed yellow line denotes the infarct border, “epi” denotes epicardial surface of the heart, “myo” denotes live myocardium, and “infarct” denotes infarct zone. Triangles represent female animals in H–K. Bar graphs represent mean ± SEM. Statistical comparisons were made with a 2-tailed t test.
Genetic loss of the secreted lymphangiogenic factors VEGF-C and VEGF-D blocks lymphatic outgrowth but does not alter cardiac function after MI.
Prior reports that administration of both the prolymphangiogenic factor VEGF-C and the antilymphangiogenic VEGFR3 ectodomain (designed to sequester the lymphangiogenic factors VEGF-C and VEGF-D) improves cardiac performance after MI are conflicting (15, 19). Since these studies utilized gain-of-function approaches that may compromise specificity, we next assessed the impact of genetic loss of the 2 known lymphangiogenic factors, VEGF-C and VEGF-D, on post-MI cardiac function. Constitutive VEGF-D deficiency is not associated with long-term morbidity or mortality (29); thus, we generated animals lacking both VEGF-C and VEGF-D using global, tamoxifen-inducible deletion of Vegfc in animals that are null for Vegfd (Vegfd–/–; Vegfcfl/fl; R26-CreERT2, hereafter called VEGF-C/D–dKO animals). Since loss of VEGF-D alone did not significantly alter infarct lymphangiogenesis (Supplemental Figure 4) and these compound genetic animals were generated on a mixed strain background, VEGF-D–deficient littermates (Vegfd–/–; Vegfcfl/fl) were used as controls for these experiments. Lymphatic growth after infarction was robust in the Vegfd–/–; Vegfcfl/fl littermates 14 days after MI (Figure 3, A and B). Loss of both VEGF-C and VEGF-D conferred a loss of lymphatic growth in the infarct zone 14 days after MI similar in magnitude to that observed following pan-endothelial or LEC-specific loss of VEGFR3 (86% reduction in VEGF-C/D–dKO animals versus 82% in EC VEGFR3–KO and 86% in LEC VEGFR3–KO animals; Figure 3, C–E). Infarct size was similar between VEGF-C/D–dKO animals and littermate controls (Figure 3F), and VEGF-C/D–dKO animals exhibited LV EF, EDV, and ESV means that were not significantly different from those of Vegfd–/–; Vegfcfl/fl control littermates (Figure 3, G–I). These results are virtually identical to those obtained after loss of VEGFR3 in EC VEGFR3–KO and LEC VEGFR3–KO animals (see summary data in Table 1).
Figure 3. Deletion of the lymphangiogenic VEGF-C and VEGF-D ligands decreases lymphatic expansion after MI but does not decrease cardiac function.

(A and B) Adjacent sections from the same infarct zone in a Vegfd–/–; Vegfcfl/fl animal 14 days after MI were examined using Masson’s trichrome stain (A) and immunostained for the lymphatic endothelial markers LYVE1 and PROX1 (B). The inset in B shows the boxed region at higher magnification. (C and D) Adjacent sections from the same infarct zone in a Vegfd–/–; Vegfcfl/fl; R26-CreERT2 heart 14 days after MI were examined using Masson’s trichrome stain (C) and immunostained for the lymphatic endothelial markers LYVE1 and PROX1 (D). The inset in D shows the boxed region at higher magnification. (E) The number of LYVE1+PROX1+ lymphatic endothelial cells was measured per mm2 in the infarct zone of the indicated animals (n = 3, 3). (F) The infarct size 14 days after MI was determined histologically (n = 5, 6). (G–I) The cardiac functional parameters ejection fraction (G), end diastolic volume (H), and end systolic volume (I) were measured 14 days after MI in the indicated animals in a fully blinded manner (n = 6, 9). In A–D, a dashed yellow line denotes the infarct border, “epi” denotes epicardial surface of the heart, “myo” denotes live myocardium, and “infarct” denotes infarct zone. Triangles represent female animals in F–I. Bar graphs represent mean ± SEM. Statistical comparisons were made with a 2-tailed t test.
Aggregate data reveal no correlation in cardiac function and lymphatic expansion.
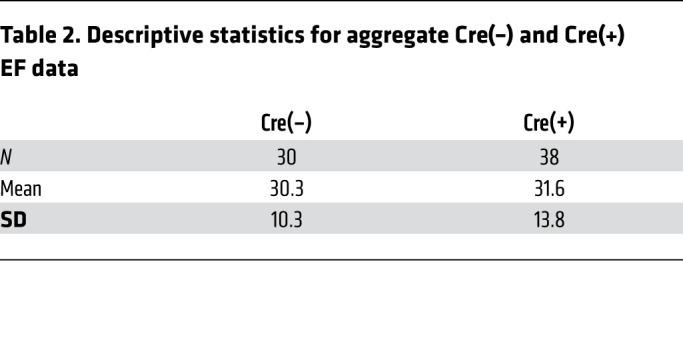
Since our 3 genetic approaches conferred a similar degree of lymphangiogenic blockade (Figures 1G, 2G, and 3E), we estimated an overall effect by combining the 3 experiments using meta-analysis techniques (30). The estimated overall mean difference in EF between animals with and without lymphangiogenic blockade using a random-effects model was 1.64 (SEM = 3.14) (Supplemental Table 1), which is not significantly different from a null effect. The combination of the control genotypes [Cre(–), which exhibit normal lymphatic vascular proliferation, n = 30 individuals] and experimental genotypes [Cre(+), with blunted lymphatic outgrowth, n = 38 individuals] revealed mean EFs that were highly concordant despite large variability of individual values inherent in this technique (Figure 4A and Table 2). Further, when the number of LECs within the infarct was plotted against EF, we observed no significant correlation between the two (Figure 4B). These aggregate data strengthen the conclusion that genetic blockade of new lymphatic growth confers no deleterious effect on cardiac performance in mice.
Figure 4. Aggregate findings demonstrate no correlation between lymphangiogenesis and cardiac performance after MI.

(A) The ejection fraction data for all control animals [2 Flt4fl/fl groups, and Vegfd–/–; Vegfcfl/fl animals, combined as Cre(–)] and animals genetically deficient in lymphangiogenesis [due to loss of pan-endothelial VEGFR3 (Flt4fl/fl; Cdh5-CreERT2), lymphatic endothelial VEGFR3 (Flt4fl/fl; Prox1-CreERT2), or the VEGFC and VEGFD ligands (Vegfd–/–; Vegfcfl/fl; R26-CreERT2), combined as Cre(+)] are shown. Unweighted combination of all ejection fraction experiments for the Cre(–) (n = 30) and Cre(+) (n = 38) groups is shown. (B) The relationship between ejection fraction and the number of lymphatic endothelial cells detected in the infarct zone after MI using data from all studies is shown by dot plot. The overall correlation trendline and R2 are shown. Cre(+) animals clustered to the left of the vertical dotted line, and Cre(–) animals clustered to the right. For A and B, littermates from Flt4fl/fl; Cdh5-CreERT2 experiments are denoted with a purple symbol (EC VEGFR3 cKO); littermates from Flt4fl/fl; Prox1-CreERT2 experiments are denoted with a white symbol (LEC VEGFR3 cKO); and littermates from Vegfd–/–; Vegfcfl/fl; R26-CreERT2 experiments are denoted with a green symbol (VEGF-C cKO; VEGF-D KO). Female animals in A and B are shown as triangles, males are circles. Bar graphs represent mean ± SEM. Statistical comparison in A was made with a 2-tailed t test, and the trendline in B is a linear regression.
Table 2. Descriptive statistics for aggregate Cre(–) and Cre(+) EF data.
Loss of lymphatic expansion after MI: effects on tissue edema and macrophage clearance.
Established roles of the lymphatic vasculature include drainage of interstitial fluid to prevent tissue edema and a route of egress of immune and inflammatory cells from tissues, functions that could conceivably alter cardiac function after MI through effects on ventricular remodeling. Therefore, we next assessed whether and to what extent genetic blockade of lymphangiogenesis might affect these endpoints in the infarcted heart. To determine whether loss of lymphatic growth in the infarct zone resulted in cardiac edema after MI, we assayed the wet/dry weight ratio of cardiac tissues from Flt4fl/fl; Cdh5-CreERT2 and littermate control animals 14 days after MI. The LV free wall, composed mostly of infarcted tissue, was dissected from the right ventricle and septum (Figure 5A), and each piece was weighed when freshly isolated (“wet”) and after dehydration (“dry”). Although we observed no difference in the wet/dry ratio of the right ventricle and septum, a trend toward a higher wet/dry ratio was observed for the LV free wall that includes the infarcted tissue (Figure 5, B and C). The large variability in wet/dry measurement of the LV free wall likely represents variability in the size of infarct after coronary ligation, as well as variability associated with excision of this small piece of tissue from the remaining heart. These wet/dry findings are consistent with histologic studies demonstrating that new lymphatic vessels in the infarct zone exhibit large, visible lumens consistent with luminal fluid filling (e.g., Supplemental Figure 1), and suggest that new lymphatic growth in the infarct zone may prevent infarct edema without measurably affecting cardiac contractile function. In this regard, it is noteworthy that prior studies have associated edema of the contractile myocardium, not MI, with cardiac performance (31, 32). Future studies will be required to definitively determine whether loss of lymphatic growth results in significant and persistent infarct edema and, if so, whether that has any long-term impact on scar formation or strength after MI.
Figure 5. Infarct lymphangiogenesis effects on cardiac edema and infiltrating macrophages after MI.

(A) The left ventricle (LV) was separated from the right ventricle (RV) and septum and water content was assayed for each tissue piece by gravimetry. Wet-to-dry weight ratios for the right ventricle and septum (B) and the left ventricle including infarcted tissue (C) are presented for Flt4fl/fl and Flt4fl/fl; Cdh5-CreERT2 animals (n = 8, 9). (D–I) Macrophages within the infarct zone were examined by immunohistochemical staining for CD68 (red) on day 3 (D and E), day 7 (F and G), and day 14 (H and I) after MI. Insets in D–I show the boxed region at higher magnification. (J and K) Quantification of the ratio of CD68+ area to infarct area at the 3-, 7-, and 14-day post-MI time points in the Flt4fl/fl (J) and Flt4fl/fl; Cdh5-CreERT2 (K) animals. For the 3-day time point n = 6, 6; for the 7-day time point n = 4, 6; for the 14-day time point n = 5, 6. In D–I, “epi” denotes epicardial surface of the heart and “infarct” denotes infarct zone. Scale bars presented in H and I apply to D–I. Bar graphs represent mean ± SEM. Statistical comparisons were made with a 2-tailed t test.
A second proposed function of lymphatic vessels is to provide a mechanism for egress of some immune cells during resolution of inflammation after injury (33). Histologic analysis revealed that CD68+ macrophages constituted a large population of inflammatory cells in the infarct zone (Figure 5, D–I), consistent with prior reports (34). In contrast, other immune and inflammatory cell types, including B220+ B cells (Supplemental Figure 5), CD3+ T cells, and Ly6G+ lymphocytes were too rare to measure. Temporal analysis revealed that the number of CD68+ macrophages within the infarct zone exhibited a bell-shaped curve, increasing from 3 days after MI to a maximum at 7 days after MI, and declining by 14 days after MI (Figure 5K and ref. 34). An identical rise and fall in CD68+ macrophage numbers was observed in Flt4fl/fl; Cdh5-CreERT2 and littermate control animals (Figure 5K), suggesting that the inflammatory macrophage life cycle in the infarct wound is normal despite severely reduced lymphatic vascular growth. TUNEL staining revealed that few CD68+ cells within the infarct zone were apoptotic in either Flt4fl/fl; Cdh5-CreERT2 or littermate control animals at these time points (Supplemental Figure 6). These findings suggest that new lymphatic growth may be required for fluid drainage but may not contribute substantially to inflammatory cell clearance in a fresh myocardial infarct.
Discussion
We have used 3 complementary genetic strategies to block the robust lymphangiogenic response to MI and test its contribution to subsequent cardiac function in vivo. The results of these 3 approaches are highly concordant and fail to identify a substantial contribution of infarct lymphangiogenesis to cardiac function after MI. However, as for all studies with negative findings, there exist limitations to our work that must be considered during its interpretation.
First, cardiac function after MI is highly variable, even when experimentally induced by the same experienced investigator (Figure 4A). Our studies were performed in a fully blinded manner and no animals with histological evidence of infarction were excluded from analysis, factors that eliminated bias but likely increased the variability of the results. Variability was also likely increased by including males and females (Supplemental Figure 7), especially as sex can affect lymphatic growth (35), and our studies are not powered to detect any sex differences. Thus, confidence in the negative biological result is dictated by the number of animals studied and the reproducibility of the results. We examined a larger number of animals than prior studies (e.g., n > 15 per group in the EC VEGFR3–KO studies vs. n = 8 in each group in prior studies; refs. 15, 19). In studies comparing 3 distinct genetic loss-of-function strategies, we observed no significant difference in means for measured EF related to animal genotype (the 95% confidence intervals for the difference in means were [–9.24, 9.04] in the EC VEGFR3–KO study, [–15.6, 11.6] in the LEC VEGFR3–KO study, and [–16.7, 9.7] in the VEGF-C/D–dKO study). After pooling all individual EF data points into Cre(–) and Cre(+) groups, we observed similar group means (Figure 4A). Additionally, we observed no correlation between infarct lymphatic growth and EF (Figure 4B). Second, since we studied cardiac function 14 days after MI it is possible that loss of lymphatics affects later events in the recovery after MI that we did not observe. However, prior studies that gave rise to the paradigm that lymphatic growth in the infarct zone affects cardiac function were done at this time point (15), refuting the hypothesis that lymphangiogenesis has a major impact on early recovery after MI.
Is there a biological role for infarct lymphangiogenesis and, if so, what might it be? MI is a consequence of coronary atherosclerosis, and not an event that has occurred throughout mammalian or even human history. Thus, there is no a priori reason to assume that the lymphangiogenic response in the infarct zone is a product of evolutionary pressure to improve survival after MI. Instead, it seems more likely that infarct lymphangiogenesis represents a purely pathologic response that, through an as yet undefined mechanism, stimulates robust VEGF-C/VEGFR3 signaling. We observed a trend toward increased LV/infarct edema following loss of infarct lymphangiogenesis that was restricted to the LV free wall, composed largely of infarct tissue. Recent studies have implicated mechanical pressure as a potential driving force for lymphatic growth (36, 37); thus, interstitial fluid accumulation in the infarct zone of the high pressure, contractile ventricle might be responsible for the robust post-MI lymphangiogenic response. In contrast, we observed no effect of lymphangiogenic loss on the numbers of CD68+ macrophages, suggesting that altered inflammatory cell function or life cycle is neither a cause nor a consequence of infarct lymphangiogenesis. Taken together, our studies suggest that the infarct lymphangiogenesis observed after MI most likely represents a pathologic event associated with local wound healing that does not affect the overall contractile function of the remaining myocardium.
Viewed more broadly, there are parallels between our study and prior studies addressing recent paradigms for treatment of MI that lacked transparent cellular and molecular mechanisms. Following the emergence of stem cell treatment for MI, a recent study demonstrated that the effect of injected stem cells was not due to renewal of cardiomyocytes, as had been proposed, but was instead the result of secondary inflammatory responses to the injection (38). Similarly, if the beneficial effects of administered VEGF-C prove to be reproducible, it is likely that they would be mediated by mechanisms other than infarct lymphangiogenesis, e.g., effects on cardiac afterload (39). Our genetic studies suggest that further efforts to treat MI by targeting cardiac lymphangiogenesis are likely to end in failure like those for cardiac stem cell therapy.
Methods
Study design.
The overall objective of our study was to determine if lymphatic vascular expansion is required for recovery from myocardial infarction. Within the animal studies, experimental and control genotypes were littermates and all received tamoxifen. The same skilled surgeon performed all LAD ligations blinded to the genotypes of the mice. At the experimental endpoint of 14 days, a mouse echocardiographer (the same echocardiographer was used for all animals), again blinded to mouse genotypes, analyzed cardiac performance. Mice were excluded from analysis if there was no histological evidence of MI after tissue harvest (this occurred with 2 mice across all groups).
A priori power analysis was used to determine group sizes necessary for appropriate statistical significance. For the most robust genetic experiment, Cdh5-CreERT2–mediated knockout of Flt4, experimental numbers were estimated based on previous results for an effect size of 1.1004 and an α of 0.05. Group sample sizes of 16 and 16 achieve 85.4% power to reject the null hypothesis of zero effect size when the population effect size is 1.1004 and the significance level (α) is 0.050 using a 2-sided, 2-sample, equal-variance t test. Thus, we aimed for 16 animals/group in that study, and used other genetic studies (Prox1-Cre deletion and VEGF-C/D–dKO) as confirmatory results with lower animal numbers.
Mouse models.
A conditional Vegfr3 allele (Flt4fl/fl) was generated by our laboratory using established mouse embryonic stem cell targeting techniques. Details of the targeting are shown in Supplemental Figure 2. Genotyping primers for the conditional allele are FWD 5′-GAATACGGGGCCTCACACAG-3′ and REV 5′-GAAGGGAGTGGTACCGAGC-3′ for band sizes of 334 bp (loxP-flanked allele) and 266 bp (WT allele). The Cdh5(PAC)-CreERT2 (a gift from R. Adams, Max Planck Institute, Muenster, Germany) has been described previously (40). The Prox1-CreERT2 (stock 022075) and R26-CreERT2 (stock 008463; ref. 41) mice were purchased from The Jackson Laboratory. Global Vegfd-deficient mice have been described previously (29), and were bred with our laboratory’s conditional Vegfcfl/fl allele (42).
All mice were maintained on a mixed genetic background. Tamoxifen administration (described below) was begun at 8 weeks of age and was allowed to wash out for 2 weeks before experimentation. All mice underwent LAD ligation surgery between 12 and 16 weeks of age.
Tamoxifen administration.
To induce conditional allele recombination in adult mice, tamoxifen (Sigma-Aldrich) was administered to adult mice via intragastric gavage (200 μL of 25 mg/mL for 5 days) beginning at 8 weeks of age. After the gavage schedule was completed, mice were maintained on tamoxifen chow (Envigo) for 2 weeks. Before the experiment, mice were returned to normal chow for at least 2 weeks.
MI and echocardiographic analysis.
LAD coronary artery ligation was used to induce MI, as described previously (43). Briefly, MI was induced by permanent ligation with an (8-0 silk suture) of the LAD after the first diagonal branch by intracostal thoracotomy. Mice were administered analgesics and allowed to recover for up to 14 days after MI. On day 14, echocardiography was performed by a trained technician to determine LV ESV and EDV. Ultrasound examination of the left ventricle was performed using a Fujifilm VisualSonics Ultrasound System (VisualSonics Inc.) and using an MS400 (18–38 MHz) transducer. Mice were lightly anesthetized with an i.p. injection of 0.005 mL/g of 2% Avertin (2,2,2-tribromoethanol, Sigma-Aldrich). Hair was removed from the anterior chest using chemical hair remover (Nair), and the animals were placed on a warming pad in a left lateral decubitus position to maintain normothermia (37°C), monitored by a rectal thermometer. Ultrasound gel was applied to the chest, and care was taken to maintain adequate contact while avoiding excessive pressure on the chest. LV systolic function: 2-dimensional long-axis, short-axis M-mode images were obtained. After completion of the imaging studies, mice were allowed to recover from anesthesia and returned to their cages. 2D long-axis images were analyzed for heart rate, EDV, ESV, and EF using Vevo Lab software (Visual Sonics Inc).
Histology and immunofluorescence imaging.
After sacrifice, mouse hearts were isolated and fixed overnight in 4% paraformaldehyde in PBS (v/v) and 4°C, prior to paraffin embedding. Thin sections of hearts in cross section (6 μm) were placed on charged glass slides for Masson’s trichrome staining or immunofluorescence imaging.
For immunofluorescence, citrate antigen retrieval buffer, pH 6.0 (Sigma-Aldrich, C9999), was used to enhance antibody-epitope binding; the blocking solution was PBS with 10% donkey serum and 1% bovine serum albumin. Primary antibodies used were against LYVE1 (Abcam, ab14917; 1:200), VEGFR3 (R&D Systems, AF743; 1:100), PROX1 (R&D Systems, AF2727; 1:100), CD68 (clone SR-D1, Novus, MAB101141; 1:100), and B220/CD45R (clone RA3-6B2, R&D Systems, MAB1217; 1:100). Secondary antibodies were from Invitrogen (donkey, Alexa Fluor 568, 594, or 647, all used at 1:400 dilution). TUNEL staining was performed using the In Situ Cell Death Detection Kit, Fluorescein (Roche).
Images were obtained with an Olympus BX53 widefield fluorescence microscope, equipped with a DP80 camera using 10× (NA 0.4) and 20× (NA 0.75) air objectives and CellSense Dimension software. Raw.vsi files were processed with FIJI (44) for pseudocoloring and analysis.
Quantification of lymphatic cells in the infarct.
Low-power (×10 magnification) images of hearts stained for LYVE1 and PROX1 were used for quantification of LEC density in the infarct. Each heart used for quantification was imaged on at least 4 slides that captured sections every approximately 60 μm from the apex to the top of the infarct to sample the entire infarct across the length of the ventricle. On each slide, one section contained a full view of the infarct, and the infarct was imaged at least 3 times per section (thus, at least 12 images from locations spanning the X, Y, and Z range of each infarct were used for lymphatic cell quantification). In each image, the infarct area was measured (in FIJI, using the myocardial autofluorescence as the infarct border) and the number of LYVE1+PROX1+ cells per image was normalized to the infarct area. Only cells within the infarct border were counted.
Immune cell quantification in the infarct zone.
Quantification of the CD68+ area in the infarct was done by threshold area measurement in FIJI. Briefly, the infarct area was measured (using autofluorescence as the infarct boundary), and the fluorescence channel with CD68 staining was thresholded and measured within the area to include only CD68+ signals. The selected area was measured in FIJI.
Infarct size determination.
Infarct size was determined based on the midline length protocol described in Takagawa et al. (45). Images used for quantification were taken every 100 μm across the infarct from the apex toward the atria. The length of the LV midline where the infarct covered greater than 50% of the wall thickness was measured.
Cardiac gravimetric analysis after MI.
Hearts were removed after euthanasia by CO2 and immediately placed into ice-cold PBS. After gross dissection, the atria were removed and discarded. The left ventricle was cut away from the right ventricle and septum. After thoroughly drying excess liquid on a square of gauze, the wet weights of the left ventricle and right ventricle plus septum were recorded and tissues were put into open tubes in a 65°C heat block for 96 hours. Weights were recorded at 48, 72, and 96 hours to ensure that tissue weights were completely equilibrated and dry weights were stable for a 24-hour period. The ratio presented is (wet weight – dry weight)/(dry weight).
Statistics.
A 2-sample, 2-tailed t test was used to evaluate differences between means of the cardiac functional measurements and means of the lymphatic cell quantification between littermate groups. P values of less than 0.05 were considered significant. In all cases, no statistical outliers were observed, and all animals with a histologically observable infarct were included in the functional analyses. The mean differences for the 3 experiments were analyzed using a random-effects meta-analysis (30).
Study approval.
All animal studies and protocols used in this manuscript have been approved by the University of Pennsylvania IACUC.
Author contributions
TCSK, LL, AAG, and MLK designed and performed experiments. SVS and KM performed echocardiography and mouse MI surgery. LL, ATT, CW, SS, and NAL generated and characterized the Flt4 conditional allele. SVS, IMP, and MSC assisted with generation and analysis of echocardiography data. PAG assisted with statistical analysis. TCSK, LL, and MLK interpreted data and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank members of the Kahn laboratory for thoughtful discussions during the course of these studies. We thank the UPenn Cardiovascular Institute Mouse Cardiovascular Phenotyping Core for their contributions to data analysis and interpretation. This work was supported by NIH grants R01 HL121650 and HL120846 (to MLK), NIH training grant T32HL007971 and American Heart Association Postdoctoral Fellowship number 836238 (to TCSK), and by the Agency for Science, Technology and Research (Singapore) (to LL).
Version 1. 08/17/2021
In-Press Preview
Version 2. 10/15/2021
Electronic publication
Footnotes
AAG’s present address is: Department of Biology, Brandeis University, Waltham, Massachusetts, USA.
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(20):e147070.https://doi.org/10.1172/JCI147070.
See the related Commentary at Lymphatics in the broken heart.
Contributor Information
T.C. Stevenson Keller, IV, Email: kellerth@pennmedicine.upenn.edu.
Lillian Lim, Email: lillianlyx@gmail.com.
Swapnil V. Shewale, Email: vsw@pennmedicine.upenn.edu.
Kendra McDaid, Email: Kendra.McDaid@pennmedicine.upenn.edu.
Alan T. Tang, Email: tangalan@pennmedicine.upenn.edu.
Carl Wittig, Email: carlwitt2@yahoo.com.
Andrea A. Guerrero, Email: andreag@brandeis.edu.
Stephanie Sterling, Email: sterlin@vet.upenn.edu.
N. Adrian Leu, Email: nleu@vet.upenn.edu.
Marielle Scherrer-Crosbie, Email: Marielle.Scherrer-Crosbie@pennmedicine.upenn.edu.
Phyllis A. Gimotty, Email: pgimotty@pennmedicine.upenn.edu.
Mark L. Kahn, Email: markkahn@mail.med.upenn.edu.
References
- 1.Chew DS, et al. Change in left ventricular ejection fraction following first myocardial infarction and outcome. JACC Clin Electrophysiol. 2018;4(5):672–682. doi: 10.1016/j.jacep.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 2.Roe MT, et al. Treatments, trends, and outcomes of acute myocardial infarction and percutaneous coronary intervention. J Am Coll Cardiol. 2010;56(4):254–263. doi: 10.1016/j.jacc.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Domanski MJ, et al. Effect of angiotensin converting enzyme inhibition on sudden cardiac death in patients following acute myocardial infarction. A meta-analysis of randomized clinical trials. J Am Coll Cardiol. 1999;33(3):598–604. doi: 10.1016/S0735-1097(98)00609-3. [DOI] [PubMed] [Google Scholar]
- 4.Pfeffer MA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med. 1992;327(10):669–677. doi: 10.1056/NEJM199209033271001. [DOI] [PubMed] [Google Scholar]
- 5.Pfeffer JM, et al. Angiotensin-converting enzyme inhibition and ventricular remodeling after myocardial infarction. Annu Rev Physiol. 1995;57:805–826. doi: 10.1146/annurev.ph.57.030195.004105. [DOI] [PubMed] [Google Scholar]
- 6.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81(4):1161–1172. doi: 10.1161/01.CIR.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 7.Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101(25):2981–2988. doi: 10.1161/01.CIR.101.25.2981. [DOI] [PubMed] [Google Scholar]
- 8.Seropian IM, et al. Anti-inflammatory strategies for ventricular remodeling following ST-segment elevation acute myocardial infarction. J Am Coll Cardiol. 2014;63(16):1593–1603. doi: 10.1016/j.jacc.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Nahrendorf M, et al. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121(22):2437–2445. doi: 10.1161/CIRCULATIONAHA.109.916346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dutta P, Nahrendorf M. Monocytes in myocardial infarction. Arterioscler Thromb Vasc Biol. 2015;35(5):1066–1070. doi: 10.1161/ATVBAHA.114.304652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Amerongen MJ, et al. Macrophage depletion impairs wound healing and increases left ventricular remodeling after myocardial injury in mice. Am J Pathol. 2007;170(3):818–829. doi: 10.2353/ajpath.2007.060547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gautier EL, et al. Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood. 2013;122(15):2714–2722. doi: 10.1182/blood-2013-01-478206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Potteaux S, et al. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe-/- mice during disease regression. J Clin Invest. 2011;121(5):2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishikawa Y, et al. Lymphangiogenesis in myocardial remodelling after infarction. Histopathology. 2007;51(3):345–353. doi: 10.1111/j.1365-2559.2007.02785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klotz L, et al. Cardiac lymphatics are heterogeneous in origin and respond to injury. Nature. 2015;522(7554):62–67. doi: 10.1038/nature14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henri O, et al. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation. 2016;133(15):1484–1497. doi: 10.1161/CIRCULATIONAHA.115.020143. [DOI] [PubMed] [Google Scholar]
- 17.Vieira JM, et al. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J Clin Invest. 2018;128(8):3402–3412. doi: 10.1172/JCI97192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang HF, et al. Enhancement of cardiac lymphangiogenesis by transplantation of CD34+VEGFR-3+ endothelial progenitor cells and sustained release of VEGF-C. Basic Res Cardiol. 2019;114(6):43. doi: 10.1007/s00395-019-0752-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houssari M, et al. Lymphatic and immune cell cross-talk regulates cardiac recovery after experimental myocardial infarction. Arterioscler Thromb Vasc Biol. 2020;40(7):1722–1737. doi: 10.1161/ATVBAHA.120.314370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brakenhielm E, Alitalo K. Cardiac lymphatics in health and disease. Nat Rev Cardiol. 2019;16(1):56–68. doi: 10.1038/s41569-018-0087-8. [DOI] [PubMed] [Google Scholar]
- 21.Nurmi H, et al. VEGF-C is required for intestinal lymphatic vessel maintenance and lipid absorption. EMBO Mol Med. 2015;7(11):1418–1425. doi: 10.15252/emmm.201505731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, et al. Variability in coronary artery anatomy affects consistency of cardiac damage after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2017;313(2):H275–H282. doi: 10.1152/ajpheart.00127.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vuorio T, et al. Downregulation of VEGFR3 signaling alters cardiac lymphatic vessel organization and leads to a higher mortality after acute myocardial infarction. Sci Rep. 2018;8(1):16709. doi: 10.1038/s41598-018-34770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliver G. Lymphatic vasculature development. Nat Rev Immunol. 2004;4(1):35–45. doi: 10.1038/nri1258. [DOI] [PubMed] [Google Scholar]
- 25.Makinen T, et al. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. 2001;7(2):199–205. doi: 10.1038/84651. [DOI] [PubMed] [Google Scholar]
- 26.Tammela T, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008;454(7204):656–660. doi: 10.1038/nature07083. [DOI] [PubMed] [Google Scholar]
- 27.Tammela T, et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nat Cell Biol. 2011;13(10):1202–1213. doi: 10.1038/ncb2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srinivasan RS, et al. Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev. 2007;21(19):2422–2432. doi: 10.1101/gad.1588407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baldwin ME, et al. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol Cell Biol. 2005;25(6):2441–2449. doi: 10.1128/MCB.25.6.2441-2449.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Borenstein M, et al. A basic introduction to fixed-effect and random-effects models for meta-analysis. Res Synth Methods. 2010;1(2):97–111. doi: 10.1002/jrsm.12. [DOI] [PubMed] [Google Scholar]
- 31.Laine GA, Allen SJ. Left ventricular myocardial edema. Lymph flow, interstitial fibrosis, and cardiac function. Circ Res. 1991;68(6):1713–1721. doi: 10.1161/01.RES.68.6.1713. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen NR, et al. A murine model of increased coronary sinus pressure induces myocardial edema with cardiac lymphatic dilation and fibrosis. Am J Physiol Heart Circ Physiol. 2020;318(4):H895–H907. doi: 10.1152/ajpheart.00436.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliver G, et al. The lymphatic vasculature in the 21(st) century: novel functional roles in homeostasis and disease. Cell. 2020;182(2):270–296. doi: 10.1016/j.cell.2020.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan X, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. doi: 10.1016/j.yjmcc.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 35.Trincot CE, Caron KM. Lymphatic function and dysfunction in the context of sex differences. ACS Pharmacol Transl Sci. 2019;2(5):311–324. doi: 10.1021/acsptsci.9b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Planas-Paz L, et al. Mechanoinduction of lymph vessel expansion. EMBO J. 2012;31(4):788–804. doi: 10.1038/emboj.2011.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Urner S, et al. Identification of ILK as a critical regulator of VEGFR3 signalling and lymphatic vascular growth. EMBO J. 2019;38(2):e99322. doi: 10.15252/embj.201899322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vagnozzi RJ, et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature. 2020;577(7790):405–409. doi: 10.1038/s41586-019-1802-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song L, et al. Lymphangiogenic therapy prevents cardiac dysfunction by ameliorating inflammation and hypertension. Elife. 2020;9:e58376. doi: 10.7554/eLife.58376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465(7297):483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 41.Ventura A, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 42.Lim L, et al. Hemostasis stimulates lymphangiogenesis through release and activation of VEGFC. Blood. 2019;134(20):1764–1775. doi: 10.1182/blood.2019001736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salto-Tellez M, et al. Myocardial infarction in the C57BL/6J mouse: a quantifiable and highly reproducible experimental model. Cardiovasc Pathol. 2004;13(2):91–97. doi: 10.1016/S1054-8807(03)00129-7. [DOI] [PubMed] [Google Scholar]
- 44.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Meth. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takagawa J, et al. Myocardial infarct size measurement in the mouse chronic infarction model: comparison of area- and length-based approaches. J Appl Physiol. 2007;102(6):2104–2111. doi: 10.1152/japplphysiol.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.