

Abstract
We report a novel NIPBL-NACC1 gene fusion in a rare primary hepatic neoplasm previously described as the “cholangioblastic variant of intrahepatic cholangiocarcinoma”. The two index cases were identified within our consultation files as morphologically distinctive primary hepatic neoplasms in a 24-year-old female and a 54-year-old male. The neoplasms each demonstrated varied architecture, including trabecular, organoid, microcystic/follicular, and infiltrative glandular patterns, and biphasic cytology with large, polygonal eosinophilic cells and smaller basophilic cells. The neoplasms had a distinctive immunoprofile characterized by diffuse labeling for inhibin, and patchy labeling for neuroendocrine markers (chromogranin and synaptophysin) and biliary marker cytokeratin 19. RNA sequencing of both cases demonstrated an identical fusion of NIBPL exon 8 to NACC1 exon 2, which was further confirmed by break-apart FISH assay for each gene. Review of a tissue microarray including 123 cases originally diagnosed as well-differentiated neuroendocrine neoplasm at one of our hospitals resulted in identification of a third case with similar morphology and immunophenotype in a 52-year-old male, and break-apart FISH probes confirmed rearrangement of both NIPBL and NACC1. Review of The Cancer Genome Atlas (TCGA) sequencing data and digital images from 36 intrahepatic cholangiocarcinomas (www.cbioportal.org) revealed one additional case with the same gene fusion and the same characteristic solid, trabecular, and follicular/microcystic architectures and biphasic cytology as seen in our genetically confirmed cases. The NIPBL-NACC1 fusion represents the third type of gene fusion identified in intrahepatic cholangiocarcinoma, and correlates with a distinctive morphology described herein.
Keywords: Cholangiocarcinoma, Intrahepatic, Translocation
Introduction
The molecular pathology of intrahepatic cholangiocarcinoma correlates well with clinicopathologic subtypes. Intrahepatic cholangiocarcinomas are now generally subdivided into two broad categories, the large duct and small duct types. The large duct subset is more frequently perihilar and characterized by infiltrative glands lined by columnar cells in desmoplastic stroma, similar to the morphology of extrahepatic bile duct adenocarcinomas1–3. Like their extrahepatic counterparts, large duct intrahepatic cholangiocarcinomas frequently harbor mutations in KRAS and TP53. By contrast, the small duct subset is typically peripherally located within the liver parenchyma, often has a pushing border with hypocellular sclerotic stroma, and is composed of glands lined by cuboidal cells. The small duct type of intrahepatic cholangiocarcinoma is frequently associated with mutations in IDH1 or IDH2, or with gene fusions involving the fibroblast growth factor receptor 2 (FGFR2), a transmembrane tyrosine kinase that promotes fibroblast growth factor signaling4–10. Reported FGFR2 fusion partners have included BICC1, AHCYL1, TACC3, TACC1, KIAA1598, MGEA5, and PPHLN. In addition, rare cases have demonstrated a RABGAPLL-NTRK1 gene fusion11.
Over the past 15 years, two small reports have described an apparently distinctive variant of intrahepatic cholangiocarcinoma with an unusual phenotype. In 2005, Vrettou et al. described one case in a 24-year-old female as “hepatic adenocarcinoma expressing inhibin” 12, while in 2017 Braxton et al. described three additional similar cases13. Morphologically, the neoplasms had varied architectural patterns, including trabecular, nested/organoid, and microcystic/follicular, as well as angulated tubules in a fibrous stroma. The cytology was biphasic, including larger epithelioid cells with eosinophilic cytoplasm and smaller cells with minimal cytoplasm. All four of these cases occurred in relatively young females (ages 17–44, median 24.5 years) and all had a distinctive immunoprofile: labeling with biliary cytokeratins cytokeratin 7 and cytokeratin 19 but not Hep-Par1, and surprisingly consistent diffuse immunolabeling for inhibin A. While the initial case was reported to be negative for chromogranin and synaptophysin, the three cases reported by Braxton et al. were positive for chromogranin and synaptophysin. Based upon the resemblance of the small cells to blastema and the relatively young age of patients, Braxton et al. referred to this neoplasm as the “cholangioblastic variant of intrahepatic cholangiocarcinoma”, believing it to represent a malignant counterpart of the ductal plate/biliary tract. Targeted next generation sequencing using a 47-gene solid tumor panel did not reveal any recurring alterations in the three cases studied.
We herein report three cases of this distinctive entity, all of which demonstrate a NIPBL-NACC1 gene fusion.
Materials and Methods
IRB Approval
This study was approved by the Institutional Review Boards at our institutions.
Cases
The three cases reported herein were retrieved from the consultation files of one of the authors (PA) along with the institutional files of The Johns Hopkins Hospital. Case 1 was submitted as a second opinion consultation for tumor classification, while Case 2 was submitted for routine patient care, having been diagnosed at an outside hospital as a well-differentiated neuroendocrine neoplasm of presumed pancreatic origin. Case 3 was identified retrospectively by screening a tissue microarray (TMA) containing an institutional cohort of 123 cases originally classified as well-differentiated neuroendocrine neoplasms, mainly of the gastrointestinal tract, for inhibin immunoreactivity. The sites of origin of the lesions tested on the TMA were pancreas (60 cases), small bowel (51 cases), colon (3 cases), stomach (2 cases), bile duct (2 cases), and lung (1 case). In 4 cases, the tumor sampled was from the liver, but a primary site had not been established. The majority (93%) of cases were negative for inhibin. Seven cases demonstrated immunoreactivity for inhibin, which has previously been described in well-differentiated neuroendocrine neoplasms13, particularly those arising in the pancreas often in association with von-Hippel Lindau syndrome14. These neoplasms had the typical morphologic features of well-differentiated neuroendocrine neoplasms, had known primary sites outside of the liver, and lacked the distinctive morphologic features of the current cases. Thus, they were not studied further. One case, a tumor in a 52-year-old male, demonstrated diffuse immunoreactivity for inhibin on all three cores sampled and was unassociated with a known extrahepatic primary, and was thus selected for further study. This case became our study case 3 after molecular confirmation.
Immunohistochemistry and RNA In-Situ Hybridization
Immunohistochemistry (IHC) was performed as previously described15 for the following markers: chromogranin (Ventana, Tuscon, AZ; LK2H10, predilute), synaptophysin (Novacastra, Buffalo Grove, IL: 27G12, 1:400), inhibin (Bio-Rad, Hercules, CA, MCA951S, R1, 1:25), cytokeratin 7 (Dako/Agilent, Santa Clara, CA, M7018, OV-TL12/30, 1:500), cytokeratin 19 (Dako/Agilent, M0888, RCK108, 1:100), arginase (Sigma St Louis, MO, polyclonal, HPA003595-100UL, 1:500), Hep-Par1 (Cell Marque, St. Louis, MO, PA0801, OCH1E5, Predilute), and INSM1 (Santa Cruz, Dallas, TX, sc-271408, A-8, 1:200). Albumin RNA in situ hybridization was performed with an RNASCOPE Probe (Advanced Cell Diagnostics, Hayward, CA, catalog# RS7752, Predilute). Hybridization was performed on 4 μM sections using a Leica Bond autostainer (Leica Biosystems, Buffalo Grove, IL) according to a standardized automated protocol provided by the manufacturer.
DNA and RNA sequencing
DNA and RNA sequencing were performed by the Genomics and Molecular Pathology Core at UT Southwestern Medical Center16. Briefly, tumor hematoxylin and eosin slides were examined and marked by 2 pathologists (P.A. and D.N.P.) for subsequent macro-dissection, nucleic acid isolation, and molecular testing. Areas enriched with tumor were then scraped from adjacent 5-μm thick formalin-fixed paraffin embedded (FFPE) sections. Adjacent normal tissue was separately isolated and processed for all cases. Extraction and purification were performed using Qiagen Allprep kits (Qiagen, Germantown, MD). Libraries were prepared using KAPA Hyperplus kits (Roche Sequencing and Life Science Kapa Biosystems, Wilmington, MA) with genomic regions of interest captured by custom probes covering all exons of 1,516 cancer-related genes. The libraries were sequenced using Sequencing by Synthesis (SBS) paired-end cluster generation on the Illumina NextSeq 550 platform (Illumina Inc., San Diego, CA. DNA Sequence reads were aligned to reference genome GRCh38. Single nucleotide variants (SNVs), insertions and deletions were called using Strelka2, MuTect2, Freebayes and Platypus. Copy number alterations were called using CNVKit. Fusions were called using the STAR-Fusion algorithm with the RNASeq reads (https://github.com/bcantarel/school). All fusions and variant calls were classified according to the Association for Molecular Pathology/American Society for Clinical Oncology/College of American Pathologists guidelines17 and manually inspected using Integrated Genomics Viewer version 2.3.4 (IGV; Broad Institute, MIT Harvard, Cambridge, MA).
Fluorescence in Situ Hybridization (FISH)
FISH on interphase nuclei from paraffin-embedded 4-micron sections was performed by applying custom probes using bacterial artificial chromosomes (BAC), covering and flanking genes that were identified as potential fusion partners in the RNA-seq experiment. BAC clones were chosen according to UCSC genome browser (http://genome.ucsc.edu), see Supplementary Table 1. The BAC clones were obtained from BACPAC sources of Children’s Hospital of Oakland Research Institute (CHORI) (Oakland, CA) (http://bacpac.chori.org) and Life Technologies Corporation (Carlsbad, CA). DNA from individual BACs was isolated according to the manufacturer’s instructions, labeled with different fluorochromes in a nick translation reaction, denatured, and hybridized to pretreated slides. Slides were then incubated, washed, and mounted with DAPI in an antifade solution, as previously described18. The genomic location of each BAC set was verified by hybridizing them to normal metaphase chromosomes. Two hundred successive nuclei were examined using a Zeiss fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany), controlled by Isis 5 software (Metasystems, Newton, MA). A positive score was interpreted when at least 20% of the nuclei showed a break-apart signal. Nuclei with incomplete set of signals were omitted from the score.
Results:
Patient Characteristics
Case 1 was a 24-year-old female who four years earlier had been noted to have a 4 cm liver mass on imaging, which was thought to represent a hepatocellular adenoma. She presented with progressive back pain over one year, and on repeat CT scan the mass was found to have grown to 21 cm (Figure 1) (Table 1). No evidence of extrahepatic spread was apparent at presentation. The patient underwent extended right hepatectomy to remove the tumor. Five months later, the patient developed multifocal local recurrence, with metastasis to the left lobe, epiphrenic/cardiophrenic lymph nodes, and chest wall.
Figure 1 (Case 1):
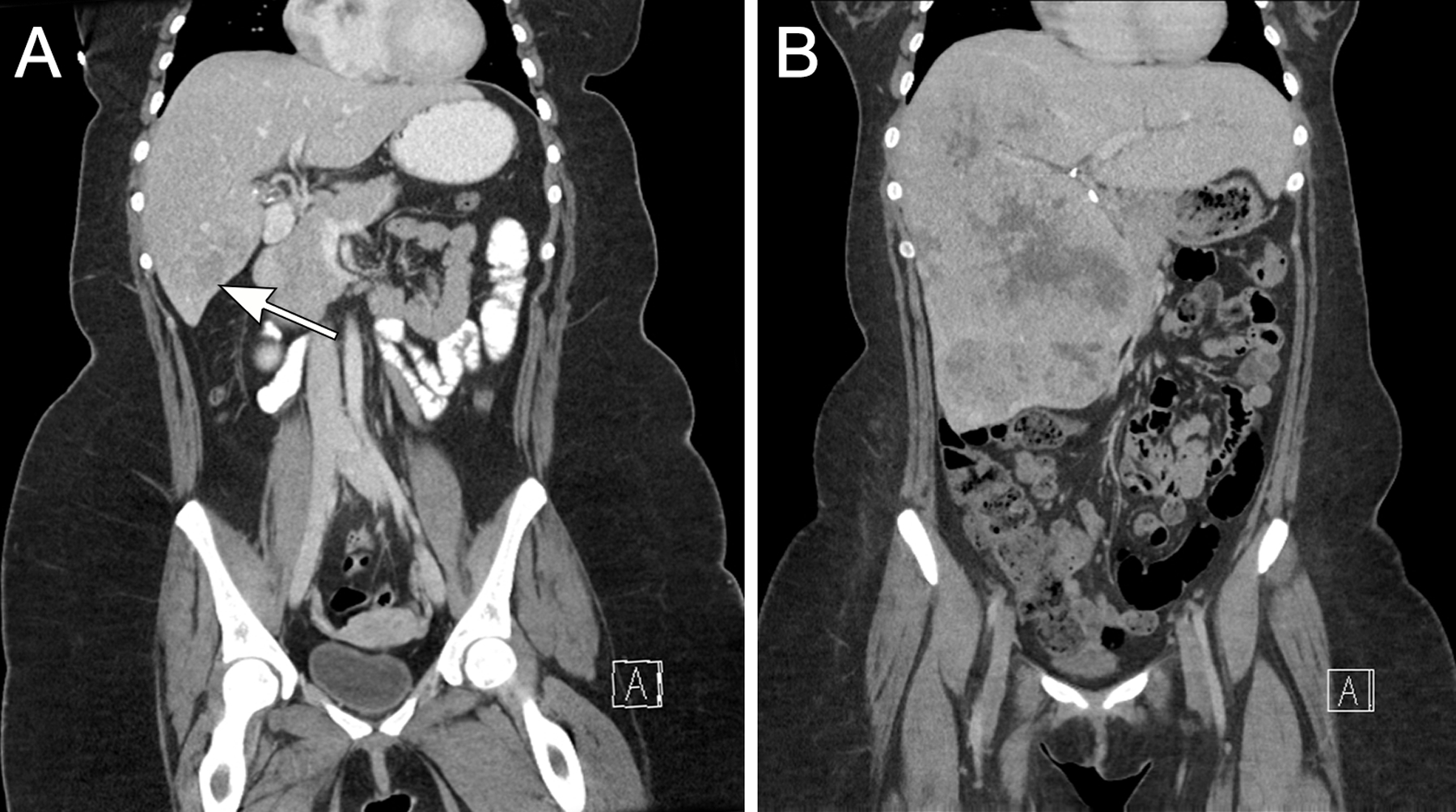
As shown on this coronal CT scan, the neoplasm initially presented as a 4cm fairly well-delineated mass thought to represent a hepatic adenoma (arrow) (A). Four years later, the lesion was 21 cm and poorly delineated (B).
Table 1:
NIPBL-NACC1-Positive Intrahepatic Cholangiocarcinoma in this Study and Similar Cases in the Literature
| Case | Age/Sex | Tumor Size | IHC+ | IHC− | Genetics | Outcome |
|---|---|---|---|---|---|---|
| 1* | 24/F | 25.4cm, multifocal | CK7,CK19, INH, CHR, SYN | Hep-Par1 | NIPBL-NACC1 | Multifocal local recurrence, metastasis to left lobe, epi/cardiophrenic lymph nodes, chest wall at 5 months; AWD at 9 months on Cisplatinum/Gemcitabine |
| 2* | 54/M | 11.5cm | CK7,CK19, INH, CHR, SYN | Hep-Par1 | NIPBL-NACC1 | Multifocal intrahepatic recurrence at 10 months |
| 3* | 51/M | 7.5cm | CK7,CK19, INH, CHR, SYN | Hep-Par1 | NIPBL-NACC1 | NA |
| TCGA-ZH-A8Y6 | 41/F | 3.5cm pT1N0 |
NA | NA | NIPBL-NACC1 | NED 17 months |
| Vrettou et. al. 12 | 24/F | 14cm | CK7,CK19, INH | CHR, SYN, Hep-Par1 | NA | NA |
| Braxton et. al. case 113 | 17/F | 23cm, pT2a | CK7,CK19, INH, CHR, SYN | Hep-Par1 | NA | DOD 41 months |
| Braxton et. al. case 213 | 44/F | 16cm, pT2bNX | CK7,CK19, INH, CHR, SYN | Hep-Par1 | NA | NA |
| Braxton et. al. case 313 | 25/F | 7.5cm, pT3NXM1 | CK7,CK19, INH, CHR, SYN | Hep-Par1 | NA | DOD 30 months |
Cases reported in this study; DOD=dead of disease; NED=no evidence of disease; AWD=alive with disease; CK=cytokeratin; INH=inhibin; CHR=chromogranin; SYN=synaptophysin
Case 2 was a 54-year-old male who developed a urinary tract infection, which lead to a CT scan that showed a liver tumor. A biopsy was performed and interpreted as “carcinoma, consistent with hepatocellular carcinoma” (slides not available for our review). The patient underwent resection of segments 5 and 6 of the liver, which revealed an 11.5 cm tumor that was diagnosed at an outside hospital as a neuroendocrine neoplasm of unknown primary. Ten months later, the patient developed multiple liver nodules that were confirmed by biopsy to represent recurrent disease.
Case 3 was a 52-year-old man who developed increased frequency of bowel movements, after which a CT scan showed a 7.5 cm liver mass that was resected and diagnosed as neuroendocrine carcinoma. The tumor was presumed to represent a metastasis from a pancreatic endocrine neoplasm; however, on clinical workup, no extrahepatic primary site was identified. The patient was disease free after 12 months but was lost to follow-up thereafter.
Pathology
All tumors had a tan cut surface on gross exam. Case 1 presented with multifocal disease, including a dominant 25 cm tumor with 2.1 and 1.0 cm satellite nodules. Case 2 presented as an 11.5 cm unifocal tumor, and Case 3 as a unifocal 7.5 cm tumor (Figure 2). None of the cases showed gross evidence of necrosis, but focal necrosis was noted microscopically in case 1. These three primary liver neoplasms demonstrated similar, distinctive morphology and immunohistochemical profiles, and therefore they are described together (Figures 3–5). All three neoplasms were multinodular, demonstrated an irregular border with the native liver, and showed evidence of vascular invasion. Each demonstrated variable architecture, the most common being trabecular patterns featuring cords of neoplastic cells typically up to 10 cells thick, mimicking hepatocellular carcinoma; nested, organoid patterns mimicking well-differentiated neuroendocrine neoplasms; and microcystic/follicular patterns featuring tubules containing eosinophilic fluid that mimicked thyroid colloid. Focally, each neoplasm contained areas of angulated tubules in fibrous stroma that resembled the architecture of conventional intrahepatic cholangiocarcinoma, though of lower nuclear grade. All three neoplasms had biphasic cytology. The larger epithelioid cells had abundant eosinophilic cytoplasm and round nuclei with evenly dispersed chromatin, and they resembled hepatocytes or raised the possibility of an oncocytic neuroendocrine neoplasm. The second population of smaller cells had scant cytoplasm and were most often located centrally within thick trabeculae or nests of neoplastic cells. Mitotic figures were frequent (Case 1, 10 mitoses per 10 high-power-fields; Cases 2 and 3, 5 mitoses per 10 high power fields). The nuclei were round, with uniform chromatin, lacking significant pleomorphism.
Figure 2 (Case 3):
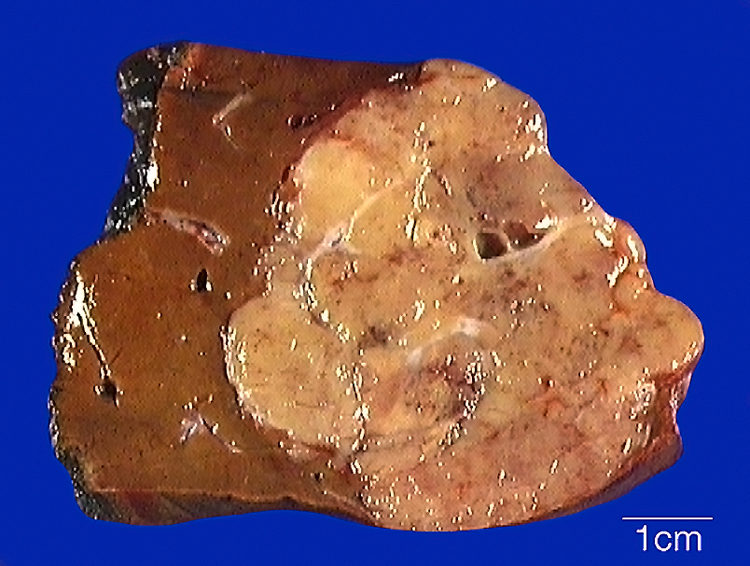
The tumor is tan and multinodular, with a scalloped and subtly infiltrative border in the native liver parenchyma.
Figure 3 (Case 1):
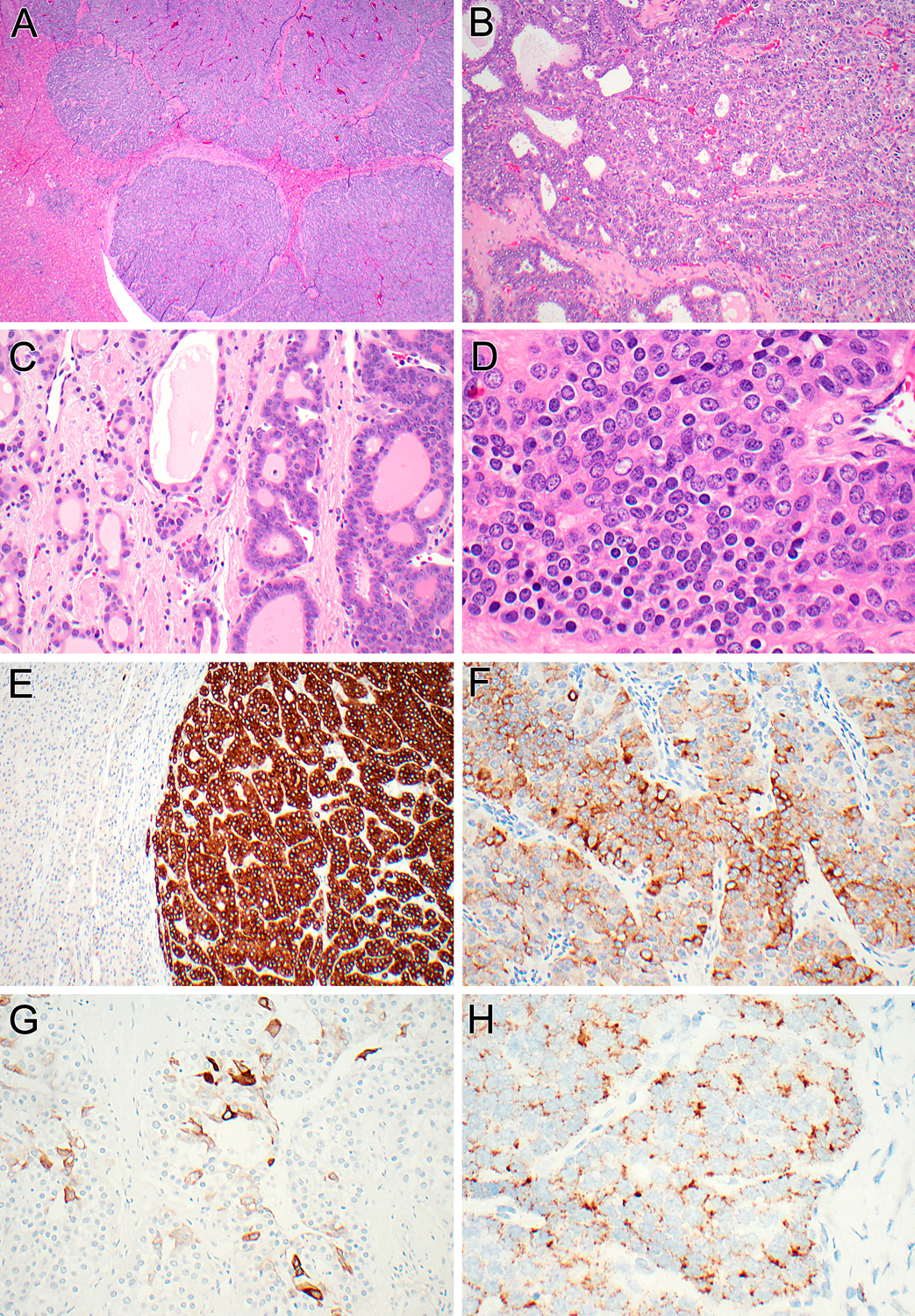
At low power, multinodular and permeative growth within the liver is apparent. Note the entrapped hepatocytes between nodules of tumor (A). At intermediate power, the neoplastic cells lining the follicles and tubules show bland cytology (B). A transition between the microcystic/follicular areas resembling follicular thyroid neoplasia (right) or irregular smaller glands set in fibrotic stroma (left) is noted (C). At higher power, one can appreciate the biphasic phenotype, with larger epithelioid cells with eosinophilic granular cytoplasm (right), and smaller cells with darker round nuclei and scant cytoplasm (left) (D). The neoplasm shows diffuse immunoreactivity for inhibin (E), patchy labeling for synaptophysin (F), patchy labeling for cytokeratin 19 (G), and demonstrates albumin mRNA by in situ hybridization (H).
Figure 5 (Case 3):
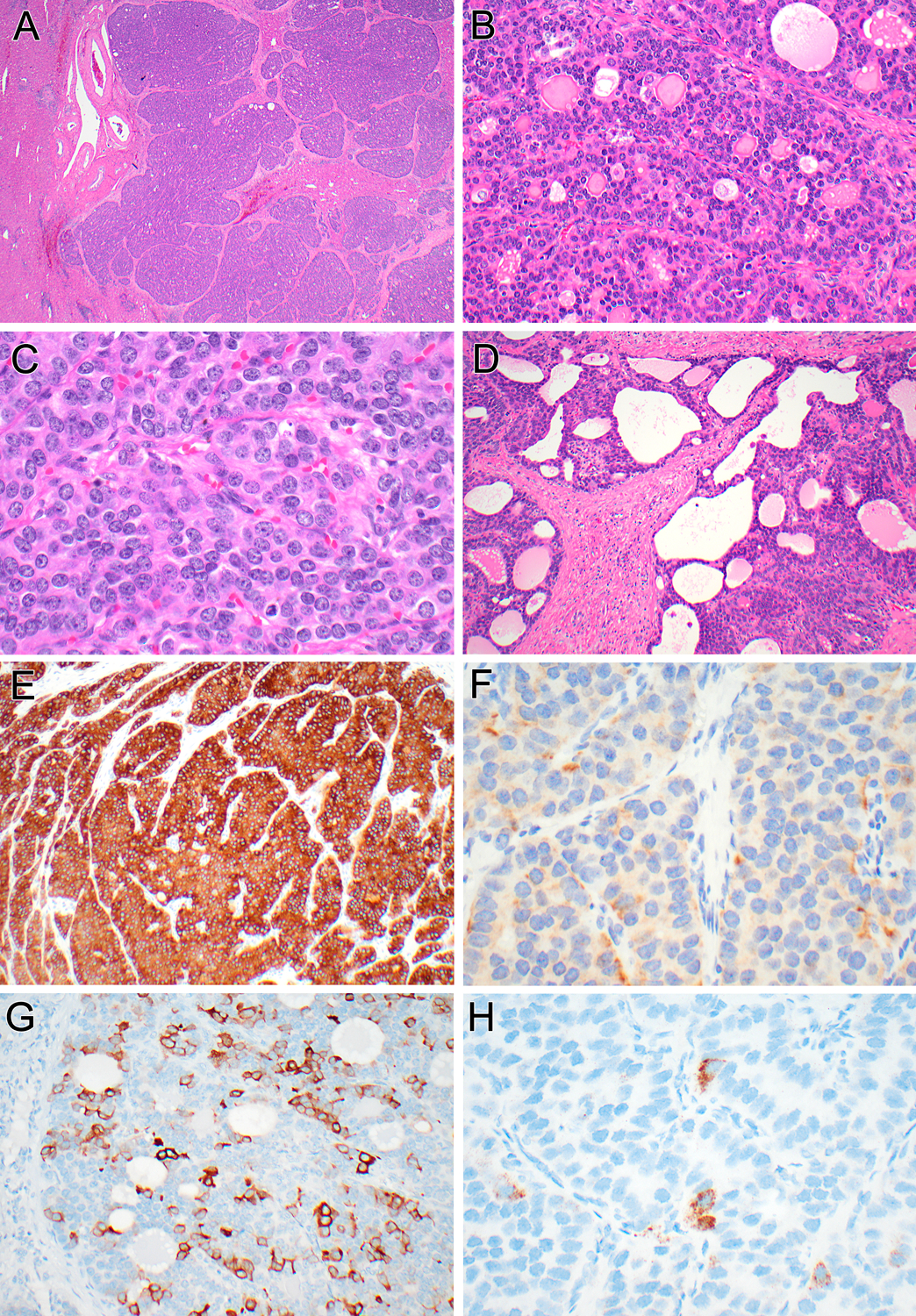
While this neoplasm presented as a solitary liver mass, one can appreciate the multinodular appearance and entrapped hepatocytes within the neoplasm (A). At higher magnification, the solid trabecular areas with microcystic/follicular patterns and the biphasic cytology are evident (B). High power shows solid nested areas resembling well-differentiated neuroendocrine neoplasm (C). More fibrotic areas demonstrate more angulated glands merging with the microfollicular areas (D). The neoplasm is diffusely immunoreactive for inhibin (E), demonstrates patchy labeling for synaptophysin (F), and multifocal patchy labeling for cytokeratin 19 (G), and focally contains albumin mRNA transcripts (H).
By immunohistochemistry, the three neoplasms had an identical phenotype: all labeled for cytokeratin 7 and cytokeratin 19 (Figures 3G, 4G, 5G), but not Hep-Par1. Strikingly, all demonstrated diffuse strong immunolabeling for inhibin A (Figure 3E, 4E, 5E). Additionally, all showed patchy immunoreactivity for synaptophysin (ranging from 30–70% of neoplastic cells) (Figures 3F, 4F, 5F) and focal labeling for chromogranin (ranging from 5–20% of neoplastic cells), but no labeling for INSM1. Albumin transcripts were detected by in situ hybridization (Figure 3H, 4H, 5H). The background liver demonstrated no pathologic abnormalities in all three cases.
Figure 4 (Case 2):
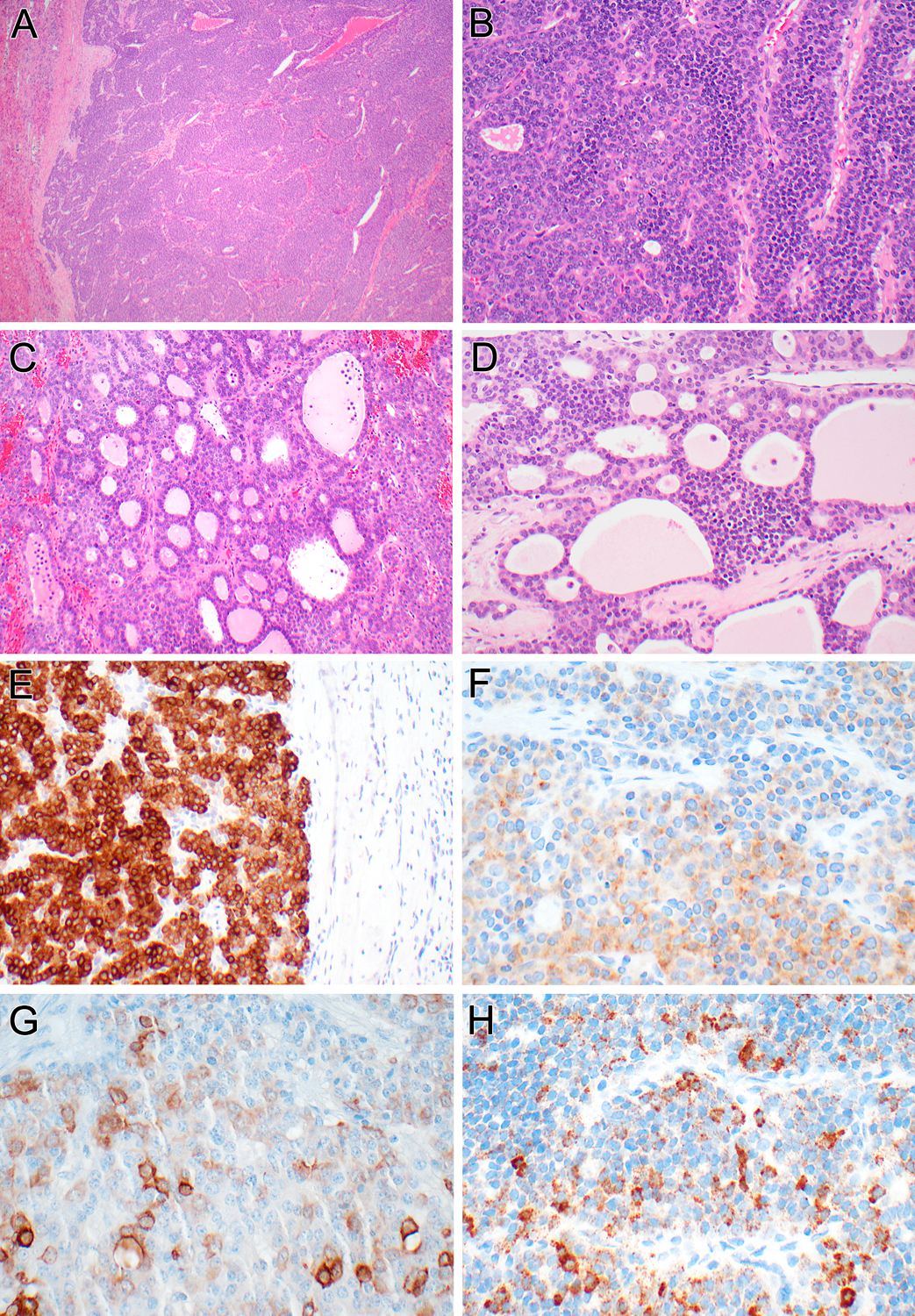
At low power, the multinodular neoplasm has an irregular border with the liver at the left (A). Intermediate power of the trabecular areas reveals biphasic morphology, with larger epithelioid cells and smaller basophilic cells (B). At intermediate power, the follicular pattern area is apparent (C). The biphasic cytology is also evident in the more trabecular cribriform/microfollicular areas (D). The neoplasm is diffusely immunoreactive for inhibin (E), demonstrates patchy labeling for synaptophysin (F), and cytokeratin 19 (G), and demonstrates albumin mRNA by in situ hybridization (H).
Molecular Pathology
RNA sequencing revealed identical NIPBL-NACC1 gene fusions in the two index cases (cases 1 and 2) involving exon 8 of NIPBL and exon 2 of NACC1 (Figure 6). Break-apart FISH confirmed rearrangement of both NIPBL and NACC1 in both neoplasms (Case 2 illustrated in Figure 7). Additional somatic point mutations and copy number alterations were noted (Table 2) with both cases harboring point mutations in LAMA5 (laminin subunit alpha 5) (case 1, p.Arg2561Gln; case 2; p.His749Tyr) and showing overlapping losses in chromosome 1p at the TGFBR3 (transforming growth factor beta-receptor 3) locus and segments of chromosome 16q. RNA sequencing was attempted on the paraffin-embedded material from Case 3; however, RNA quality was suboptimal and a NIPBL-NACC1 fusion was not identified. Given the concern that low quality RNA from the 15-year-old FFPE block could have contributed to a false negative result, break-apart FISH for NIPBL and NACC1 was performed, and both genes demonstrated rearrangements (Figure 7). With the caveat that DNA quality was low, LAMA5 mutations were not detected, and losses at 1p and 16q as seen in cases 1 and 2 were not identified.
Figure 6:
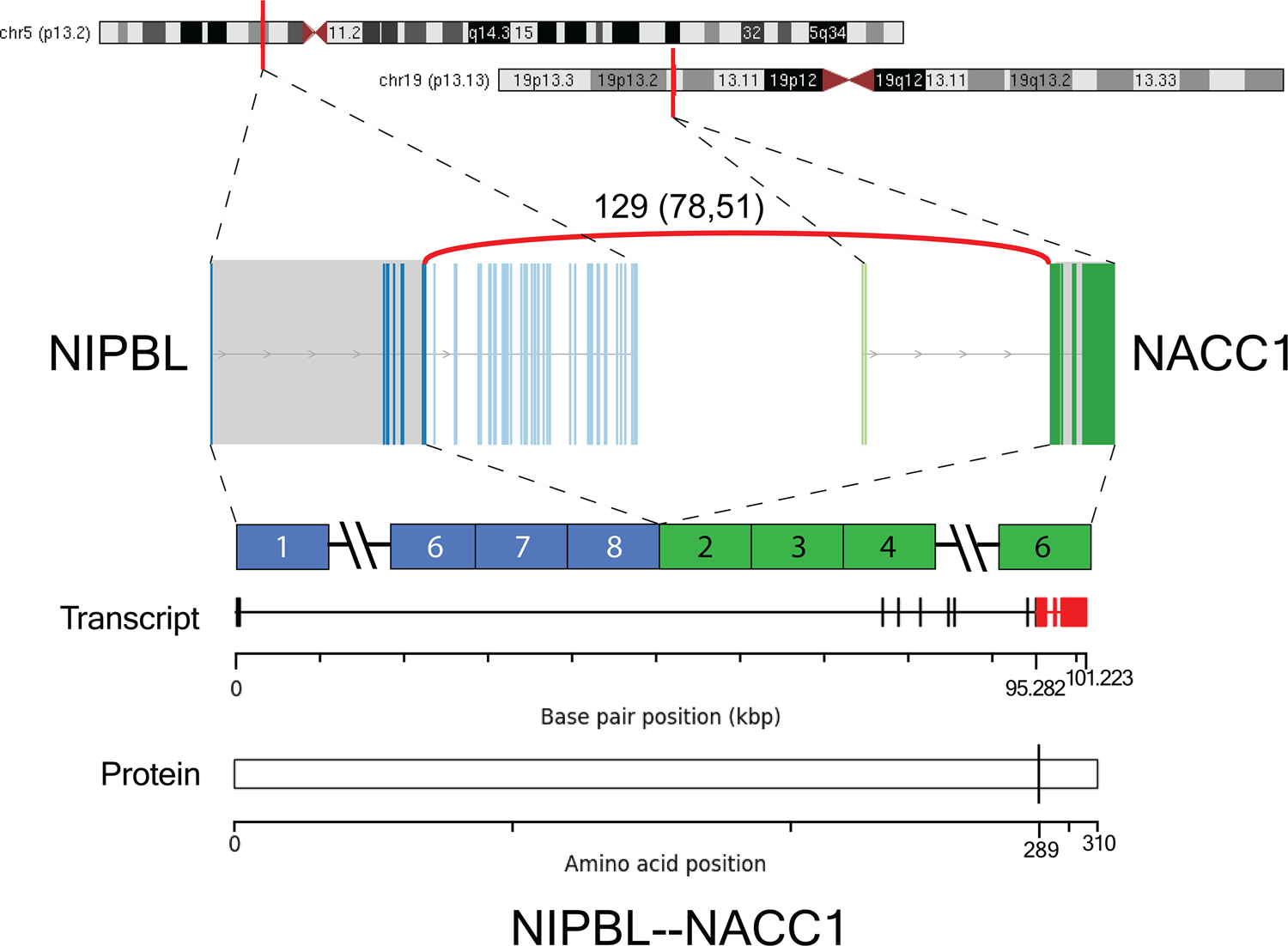
Schematic representation of NIPBL-NACC1 fusion. Gene loci of NIPBL and NACC1 are shown within chromosomes 5 and 19, respectively. RNA-seq revealed fusion transcripts consisting of exon 8 of NIPBL (chr 5p13.2; ENST00000282516) and exon 2 of NACC1 (chr 19p13.13; ENST00000292431). The number of discordant (split and spanning) reads supporting the breakpoint of Case 1 is shown above the curved red line. All fusion-supporting RNA-seq reads contained the identical fusion junction.
Figure 7:
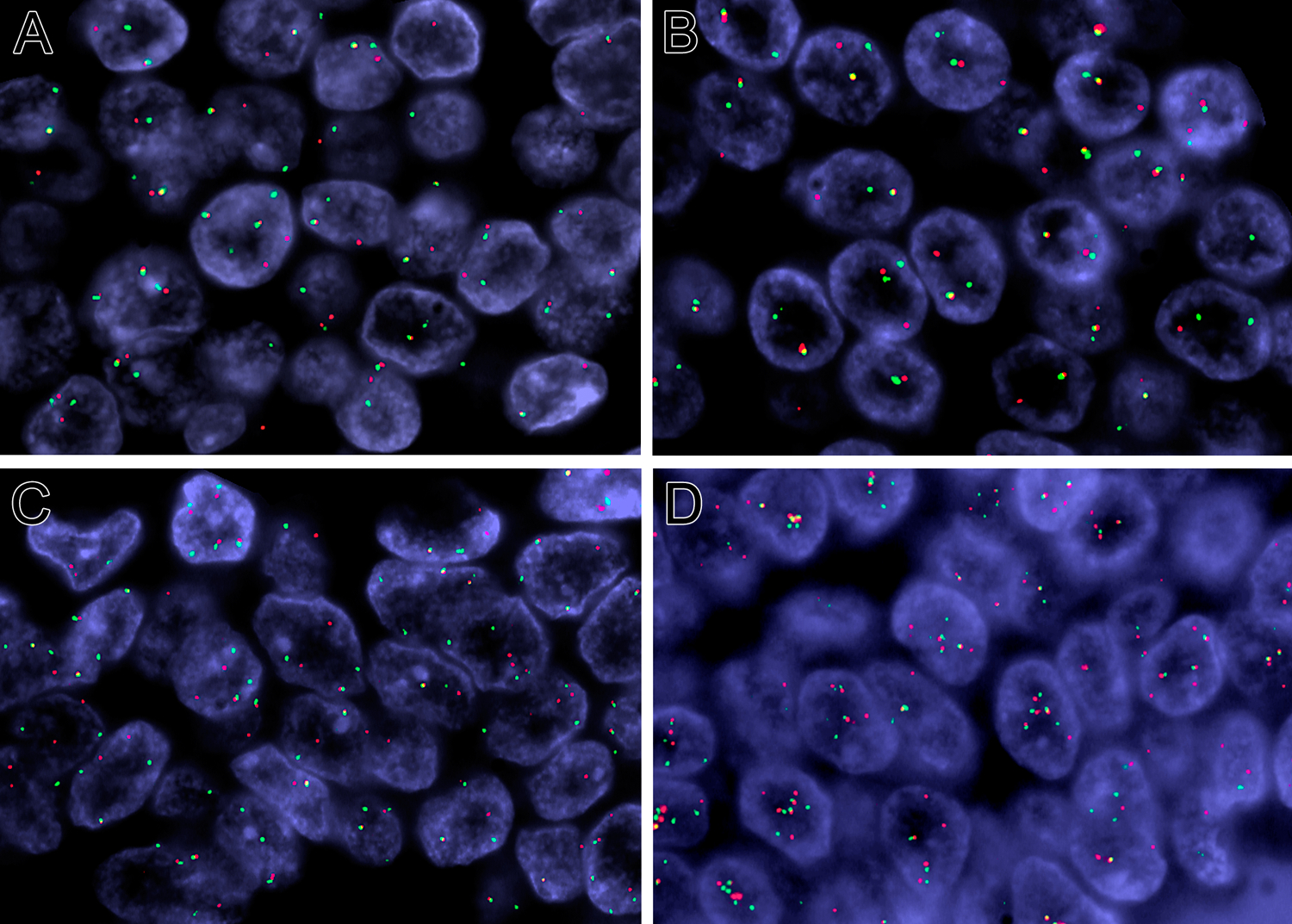
FISH analysis for NIPBL and NACC1 gene rearrangements (red centromeric, green telomeric). NIPBL break apart (A) and NACC1 break apart (case 2) (B). NIPBL break apart and (C) NACC1 break apart (case 3) (D).
Table 2.
Genetic Alterations Found in Index cases of Cholangioblastic Variant of Intrahepatic Cholangiocarcinoma.
| Genetic Alteration | Case 1 | Case 2 |
|---|---|---|
| Point Mutations |
STAT5B p.Glu315* APC p.Glu1891* LAMA5 p.Arg2561Gln PRKDC p.Arg782Lys BRAF p.Leu711Phe FLNC p.Glu2171Lys PPARG p.Asp490Asn BRINP3 p.Phe107Leu KAT6A p.Glu986Lys TP63 p.Glu248Gln ROBO2 p.Asp197Tyr RBM15 p.Met45Ile CHD4 p.Pro468Ala LRRC37B p.Pro748Ser EPHA3 p.Val58Leu CACNA2D3 p.Leu169Val CENPF p.Ser2741Leu RYR3 p.Asp4008Asn FHL2 p.Trp181Arg GPS2 p.Leu128Val BRWD3 p.Ser222Cys INPP4A c.1363–1G>C NOS3 c.2897–1G>A PTK2 c.2603–1G>A |
LAMA5 p.His749Tyr CTNNB1 p.Ser348Tyr |
| Copy Number Alterations | Aneuploidy Loss of 1p36.33-q11 including TGFBR3 Loss of 6p25.3-q27 Loss of 11p15.5-q25 Loss of 16p11.1-q24.3 Gain of 5p15.33-p13.2 Gain of 7p22.1-q36.3 Gain of 9p24.3-q34.3 Gain of 19p13.3-q13.43 |
Loss of 1p22.2-p22.1 involving TGFBR3 Loss of 13q32.3-q33.3 Loss of 16q11.2-q24.3 |
| Gene Fusions | NIPBL-NACC1 | NIPBL-NACC1 |
Review of TCGA Cholangiocarcinoma Data Identifies an Additional NIPBL-NACC1 Fusion-Positive Intrahepatic Cholangiocarcinoma with Similar Morphology
To determine the prevalence of the NIPBL-NACC1 fusion in intrahepatic cholangiocarcinoma, we reviewed the published online data from the TCGA cohort (www.cbioportal.org). Of the 36 intrahepatic cholangiocarcinomas reported, one case (TCGA-ZH-A8Y6) demonstrated the NIPBL-NACC1 fusion. This neoplasm was a 3.5 cm unifocal pT1N0 tumor in a 41-year-old female who was disease free after 17-month follow-up. Review of the online-published H&E images from this case demonstrates the characteristic multinodular pattern, as well as areas with the microcystic thyroid follicular-like architecture and biphasic cytology, as seen in the three genetically confirmed cases reported above (Figure 8). Of note, LAMA5 mutations were not detected, and losses at 1p and 16q as seen in cases 1 and 2 were not identified.
Figure 8:
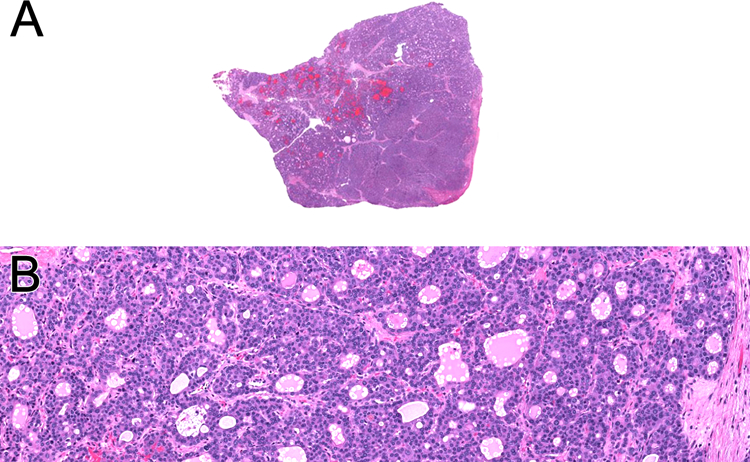
Online images of TCGA-ZH-A8Y6, an intrahepatic cholangiocarcinoma in a 41-year-old female that demonstrated NIPBL-NACC1 gene fusion. The neoplasm has a solid, multinodular appearance (A). A solid pattern and follicular architecture are evident (B)
Discussion
We report that a novel NIPBL-NACC1 gene fusion is characteristic of a rare primary hepatic neoplasm previously referred to as the cholangioblastic variant of intrahepatic cholangiocarcinoma. This neoplasm features distinctive architecture (trabecular, nested, microcystic/follicular, and glandular patterns), cytology (biphasic with large eosinophilic and smaller basophilic cells), and immunoprofile (diffuse labeling for inhibin; typically patchy labeling for biliary markers cytokeratin 7 and 19 and for neuroendocrine markers chromogranin and synaptophysin). Given its presentation in patients of young age, as well its biphasic cytology and absence of significant nuclear pleomorphism, it is not surprising that this neoplasm is characterized genetically by a recurrent chromosomal translocation, akin to translocation-associated sarcomas or MiT family translocation renal cell carcinoma19.
Our cases generally fit the previously reported profile of these distinctive but rare neoplasms. Our patients were significantly younger (mean 42.5 years) than those with typical intrahepatic cholangiocarcinoma (mean 70 years) (https://www.cancer.net/cancer-types/bile-duct-cancer-cholangiocarcinoma/statistics). Two of our four cases occurred in males, which expands the clinicopathologic features of this neoplasm that was previously reported only in females. While we have limited follow-up for our cases, the presence of vascular invasion in all cases, multifocality and multifocal recurrence/metastasis in case 1, and multifocal recurrence in case 2 is consistent with the aggressive clinical behavior reported in the literature.
The main clinicopathologic differential diagnosis for these neoplasms is that of well-differentiated neuroendocrine neoplasms metastatic to the liver. Indeed, three of the four previously reported cases of this neoplasm were initially misdiagnosed as metastatic well-differentiated neuroendocrine neoplasms, as were two of three cases in our cohort. It is possible that this neoplasm accounts for some cases previously considered well-differentiated neuroendocrine neoplasms in the liver for which a primary site outside of the liver was not found. Aside from the distinctive microcystic/follicular architecture (may not be evident in all sections), a clue to the diagnosis could be the only patchy immunoreactivity for synaptophysin and chromogranin in these neoplasms, coupled with absence of INSM1 labeling. Typically, a well-differentiated neuroendocrine neoplasm with abundant eosinophilic cytoplasm would demonstrate diffuse labeling for chromogranin and/or synaptophysin and label with INSM1, and its absence in these cases should prompt additional studies. Screening with inhibin immunochemistry is the logical next step to identify candidate cases (as suggested by Braxton et al.)13. Other considerations given the usual absence of diffuse chromogranin/synaptophysin labeling in these neoplasms include metastatic breast carcinoma with neuroendocrine differentiation20 and metastatic acinar cell carcinoma of the pancreas21, both of which may show patchy labeling for neuroendocrine markers and be mistaken for a well-differentiated neuroendocrine neoplasm.
The NIPBL-NACC1 gene fusion has not previously been reported in any other distinct neoplasm. However, a literature search revealed one recent report of NIPBL-NACC1 fusions in two of 41 patient samples with advanced cancer from “non-colorectal and non-pancreas gastrointestinal primaries”22. Interestingly, these two cases share the same junctional breakpoint (exon 8 of NIPBL and exon 2 of NACC1) as our cases, but were not illustrated and further clinicopathologic features were not provided.
Both genes involved in this novel gene fusion have previously been associated with cancer. NIPBL (Nipped-b homolog), located at chromosome 5p13, encodes for delangin, a 188-kilo Dalton chromatin cohesion regulator23, and is the most commonly mutated gene in the Cornelia de Lange developmental syndrome (approximately 50% of cases24,25). Delangin protein functions to load cohesin onto chromosomes, which regulates sister chromatid cohesion during mitosis/meiosis, DNA damage repair, and gene expression. Fusions involving NIPBL have been rarely reported in other cancer types: NIPBL-ERG in tenosynovial giant cell tumor26, and NIPBL-HOXB927, NIPBL-HOXA928, and NIPBL-ETV629 in acute megakaryoblastic leukemia. In all these cases, only the noncoding sequence of exon 1 of NIPBL is retained, whereas the NIPBL-NACC1 fusion reported herein also retains the coding sequence of exons 2–8. The known DNA binding and cohesion and HDAC interacting domains, which are part of the C-terminal part of the protein, are notably lost in all fusions.
NAC1 (nucleus accumbens-associated protein 1), encoded by the NACC1 gene on 19p13, is a transcriptional coregulator that lacks a DNA binding domain but complexes with other DNA binding cofactors. It is a member of the broad complex tramtrack bric-a-brac/pox virus and zinc finger (BTB/POZ) family of transcription factors. It has been reported to be overexpressed in multiple gynecological cancers, including ovarian, uterine, and cervical cancers30–32, as well as melanoma33, where it is predicted to increase cellular motility, cellular proliferation, resistance to chemotherapy, and autophagy. NAC1 is a 527 amino acid protein that contains a BTB/POZ domain which mediates homodimerization and heterodimerization (amino acids 30–124), a nuclear localization signal (amino acids 180–207), and a C-terminal BEN (BANP, ESR or NAC1) domain (amino acids 374–471) which mediates DNA-protein and protein-protein interactions with the Myc-interacting zinc finger protein 1 (Miz1). The NIPBL-NACC1 fusion retains exons 2–6 of NACC1, which includes the N-terminal BTB/POZ and C-terminal BEN domains.
While the biological function of the NIPBL-NACC1 fusion at present is not known at present due to a lack of mechanistic studies, a possible consequence of the NIPBL-NACC1 gene fusion could be dysregulated expression of a largely intact NAC1 protein, promoting neoplastic growth as is seen in melanoma and gynecologic cancers30–33. Unfortunately, we were unable to assess for NAC1 expression in these cases using our sequencing platform, so further studies will be needed to address this hypothesis. As both NIPBL and NAC1 proteins play a role in binding to regulatory regions of genes, there may also be cooperativity between these proteins in regulating other genetic loci, such as c-Myc.
In summary, the NIPBL-NACC1 gene fusion is characteristic of a distinctive primary hepatic neoplasm previously described as the cholangioblastic variant of intrahepatic cholangiocarcinoma. While it may mimic metastatic well-differentiated neuroendocrine neoplasms, this neoplasm can be identified prospectively by its distinctive morphology (follicular architecture) and immunohistochemical profile (typically only patchy labeling for synaptophysin/chromogranin, negativity for INSM1, diffuse labeling for inhibin).
Supplementary Material
Acknowledgement:
We thank Norman Barker MA, MS, RBP for expert photographic assistance.
Footnotes
Disclosures: Supported in part by: Biliary Cancer Research Fund at Johns Hopkins (PA)
References:
- 1.Banales JM, Marin JJ, Lamarca A, Rodrigues PM. Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol 2020; 17:557–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakanuma Y, Kakuda Y. Pathologic classification of cholangiocarcinoma: New concepts. Best Pract Res Clin Gastroenterol 2015; 29:277–93. [DOI] [PubMed] [Google Scholar]
- 3.Krasinskas AM. Cholangiocarcinoma Surg Pathol Clin. 2018;11:403–429. [DOI] [PubMed] [Google Scholar]
- 4.Liau JY, Tsai JH, Yuan RH, Chang CN, Lee HJ, Jeng YM. Morphological subclassification of intrahepatic cholangiocarcinoma: etiological, clinicopathological, and molecular features Mod Pathol. 2014. ;27:1163–73. [DOI] [PubMed] [Google Scholar]
- 5.Valle JW, Lamarca A, Goyal L, Barriuso J, Zhu AX. New horizons for precision medicine in biliary tract cancers. Cancer Discov 2017; 7:943–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arai Y, Totoki Y, Hosoda F, Shirota T, Hama N, Nakamura H, Ojima H, Furuta K, Shimada K, Okusaka T, Kosuge T, Shibata T. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014; 59:1427–1434. [DOI] [PubMed] [Google Scholar]
- 7.Louis C, Papoutsoglou P, Coulouarn C. Molecular classification of cholangiocarcinoma. Curr Opin Gastroenterol 2020; 36:57–62. [DOI] [PubMed] [Google Scholar]
- 8.Graham RP, Barr Fritcher EG, Pestova E, Schulz J, Sitailo LA, Vasmatzis G, Murphy SJ, McWilliams RR, Hart SN, Halling KC, Roberts LR, Gores GJ, Couch FJ, Zhang L, Borad MT, Kipp BR. Fiboblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum Pathol 2014; 45:1630–1638. [DOI] [PubMed] [Google Scholar]
- 9.Ang C Role of the fibroblast growth factor receptor axis in cholangiocarcinoma. J Gastroenterol Hepatol 2015; 30:1116–22. [DOI] [PubMed] [Google Scholar]
- 10.Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas Nat Genet. 2013;45:1470–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross JS, Wang K, Gay L, Al-Rohil R, Rand JV, Jones DM, Lee HJ, Sheehan CE, Otto GA, Palmer G, Yelensky R, Lipson D, Morosini D, Hawryluk M, Catenacci DV, Miller VA, Churi C, Ali S, Stephens PJ. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist. 2014;19:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vrettou E, Hytiroglou P, Sikas N, Soultoyannis I, Goodman ZD. Hepatic adenocarcinoma expressing inhibin in a young patient on oral contraceptives. Virchows Arch. 2005;446:560–5. [DOI] [PubMed] [Google Scholar]
- 13.Braxton DR, Saxe D, Damjanov N, Stashek K, Shroff S, Morrissette JD, Tondon R, Furth EE. Molecular and cytogenomic profiling of hepatic adenocarcinoma expressing inhibin A, a mimicker of neuroendocrine tumors: proposal to reclassify as “cholangioblastic variant of intrahepatic cholangiocarcinoma. Hum Pathol 2017; 62:232–241. [DOI] [PubMed] [Google Scholar]
- 14.Sinkre PA, Murakata L, Rabin L, Hoang MP, Albores-Saavedra J. Clear cell carcinoid tumor of the gallbladder: another distinctive manifestation of von Hippel-Lindau disease Am J Surg Pathol. 2001;25:1334–9 [DOI] [PubMed] [Google Scholar]
- 15.Argani P, Hicks J, De Marzo AM, Albadine R, Illei PB, Ladanyi M, Reuter VE, Netto GJ. Xp11 translocation renal cell carcinoma (RCC): extended immunohistochemical profile emphasizing novel RCC markers Am J Surg Pathol. 2010;34:1295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Argani P, Reuter VE, Eble JN, Vlatkovic L, Yaskiv O, Swanson D, Dickson BC, Antonescu CR, Matoso A, Gagan J, Palsgrove DN. Biphasic Hyalinizing Psammomatous Renal Cell Carcinoma (BHP RCC): A Distinctive Neoplasm Associated With Somatic NF2 Mutations Am J Surg Pathol. 2020;44:901–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Antonescu CR, Zhang L, Chang NE, et al. EWSR1-POU5F1 fusion in soft tissue myoepithelial tumors. A molecular analysis of sixty-six cases, including soft tissue, bone, and visceral lesions, showing common involvement of the EWSR1 gene. Genes Chromosomes Cancer. 2010; 49:1114–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Argani P, Laé M, Hutchinson B, Reuter VE, Collins MH, Perentesis J, Tomaszewski JE, Brooks JS, Acs G, Bridge JA, Vargas SO, Davis IJ, Fisher DE, Ladanyi M. Renal carcinomas with the t(6;11)(p21;q12): clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol. 2005. ;29:230–40. [DOI] [PubMed] [Google Scholar]
- 20.Cloutier J, Thompson ED, Cimino-Mathews A, Rooper LM, Matoso A, Argani P. Metastatic breast cancer simulating well-differentiated neuroendocrine neoplasms of visceral organs Hum Pathol. 2018. ;82:76–86. [DOI] [PubMed] [Google Scholar]
- 21.Stelow EB, Bardales RH, Shami VM, Woon C, Presley A, Mallery S, Lai R, Stanley MW. Cytology of pancreatic acinar cell carcinoma. Diagn Cytopathol. 2006; 34:367–72. [DOI] [PubMed] [Google Scholar]
- 22.Tsang ES, Grisdale CJ, Pleasance E, Topham JT, Mungall K, Reisle C, et al. Uncovering Clinically Relevant Gene Fusions with Integrated Genomic and Transcriptomic Profiling of Metastatic Cancers. Clin Cancer Res. 2021;27:522–531. [DOI] [PubMed] [Google Scholar]
- 23.Gao D, Zhu B, Cao X, Zhang M, Wang X. Roles of NIPBL in maintenance of genome stability. Seminars Cell Dev Biol 2019; 90:181–186. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Baynam G. Cornelia de Lange Syndrome. Adv Exp Med Biol 2010; 685:111–23. [PubMed] [Google Scholar]
- 25.Pienkowski VM, Kucharcyzk M, Mlynek M, Szczaluba K, Rydzanicz M, Poszewiecka B, et al. Mapping of breakpoints in balanced chromosomal translocations by shallow whole-genome sequencing points to EFNA5, BAHD1 and PPP2R5E as novel candidates for genes causing human Mendelian disorders. J Med Genet 2019; 56:104–112. [DOI] [PubMed] [Google Scholar]
- 26.Vougiouklakis T Shen G, Feng X, Hoda ST, Jour G. Molecular profiling of atypical tenosynovial giant cell tumors reveals novel non-CSF1 fusions. Cancers 2019; 12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dang J, Nance S, Ma J, Cheng J, Walsh MP, Vogel P, Easton J, Song G, Rusch M, Gedman A, Koss C, Downing JR, Gruber TA. AMKL chimeric transcription factors are potent inducers of leukemia. Leukemia 2017; 31:2228–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Braekeleer E, Auffret R, Gonzalez Garcia JR, Padilla JM, Fletes CC, Morel F, Douet-Guilbert N, De Braekeleer M. Identification of NIPBL, a new ETV6 partner gene in t(5;12) (p13;p13)-associated acute megakaryoblastic leukemia. Leuk Lymphoma 2013; 54:423–4. [DOI] [PubMed] [Google Scholar]
- 29.Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia Cancer Cell. 2012;22:683–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatemichi Y, Shibazaki M, Yasuhira S, Kasai S, Tada H, Oikawa H, Suzuki Y, Takikawa Y, Masuda T, Maesawa C. Nucleus accumbens associated 1 is recruited within the promyelocytic leukemia nuclear body through SUMO modification. Cancer Sci 2015; 106:848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakayama K, Rahman MT, Rahman M, Yeasmin S, Ishikawa M, Katagiri A, Iida K, Nakayama N, Miyazaki K. Biological role and prognostic significance of NAC1 in ovarian cancer Gynecol Oncol. 2010. ;119:469–78. [DOI] [PubMed] [Google Scholar]
- 32.Yeasmin S, Nakayama K, Ishibashi M, Katagiri A, Iida K, Purwana IN, Nakayama N, Miyazaki K. Expression of the bric-a-brac tramtrack broad complex protein NAC-1 in cervical carcinomas seems to correlate with poorer prognosis. Clin Cancer Res. 2008; 15;14:1686–91. [DOI] [PubMed] [Google Scholar]
- 33.Tsunoda K, Oikawa H, Tada H, Tatemichi Y, Muraoka S, Miura S, Shibazaki M, Maeda F, Takahashi K, Akasaka T, Masuda T, Maesawa C. Nucleus accumbens-associated 1 contributes to cortactin deacetylation and augments the migration of melanoma cells. J Invest Dermatol. 2011;131:1710–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.