

Abstract
Rickettsia conorii is a Gram-negative, cytosolic intracellular bacterium that has classically been investigated in terms of endothelial cell infection. However, R. conorii and other human pathogenic Rickettsia species have evolved mechanisms to grow in various cell types, including macrophages, during mammalian infection. During infection of these phagocytes, R. conorii shifts the host cell’s overall metabolism towards an anti-inflammatory M2 response, metabolically defined by an increase in host lipid metabolism and oxidative phosphorylation. Lipid metabolism has more recently been identified as a key regulator of host homeostasis through modulation of immune signaling and metabolism. Intracellular pathogens have adapted mechanisms of hijacking host metabolic pathways including host lipid catabolic pathways for various functions required for growth and survival. In the present study, we hypothesized that alterations of host lipid droplets initiated by lipid catabolic pathways during R. conorii infection is important for bacterial survival in macrophages. Herein, we determined that host lipid droplet modulation is initiated early during R. conorii infection and these alterations rely on active bacteria and lipid catabolic pathways. We also find that these lipid catabolic pathways are essential for efficient bacterial survival. Unlike the mechanisms used by other intracellular pathogens, the catabolism of lipid droplets induced by R. conorii infection is independent of upstream host PPARα signaling. Inhibition of PPARɣ signaling and lipid droplet accumulation in host cells causes a significant decrease in R. conorii survival suggesting a negative correlation with lipid droplet production and R. conorii survival. Together, these results strongly suggest that the modulation of lipid droplets in macrophage cells infected by R. conorii is an important and underappreciated aspect of the infection process.
Keywords: Rickettsia, THP-1 cells, fatty acids, lipid droplets, lipases, β-oxidation
Author Summary
Rickettsia conorii is a human pathogenic rickettsial species that is predominantly transmitted in the Mediterranean basin and Northern Africa with fatality in humans ranging from 3.2%−32%. Pathogenic Rickettsia species do not exclusively grow within endothelial cells and can proliferate within phagocytes in vitro and in vivo. Herein, we demonstrate that initiation of early lipid droplet modulation occurs during R. conorii infection of macrophages. We show that triglyceride-associated lipases and fatty acid β-oxidation regulates the initiation of the identified lipid droplet alterations in a PPAR-independent manner during infection. We have also demonstrated that active R. conorii protein synthesis is required for LD modulation during infection. Studies inhibiting PPARɣ and inducing foam-like cell induction during infection also suggests that LD over-accumulation has a detrimental effect on rickettsial growth. These results indicate that processes required for lipid droplet modulation are potential targets for development of host-directed therapeutics against rickettsial infections.
Introduction
Rickettsia conorii is a Gram-negative obligate intracellular α-proteobacteria that resides within the cytosol of mammalian cells. R. conorii is transmitted predominantly in the Mediterranean basin and Northern Africa by ticks to mammals, with humans being an accidental, dead-end host (1). Infections in humans with pathogenic spotted fever group (SFG) Rickettsia species can cause fatality in 10–40% of untreated cases, with R. conorii fatality rates ranging from 3.2% - 32% (2). It is widely accepted that Rickettsia species can selectively grow in vascular endothelial cells during infections in vivo; therefore, studies to further define the basis underlying Rickettsia-host cell interactions in vitro have focused on using primary endothelial cells and endothelial cell lines (3–8). However, several studies have demonstrated that R. conorii and other pathogenic SFG Rickettsia species are more promiscuous in the cell types they infect during in vivo and in vitro models of rickettsial diseases (8–10). Recent studies have shown that growth by pathogenic SFG Rickettsia species in macrophages is correlated with the ability of species to cause disease in mammals (9, 11–13). Therefore, investigating pathogen-macrophage interactions is critical to further understand the complex interplay between Rickettsia species and target cells in mammals.
To further investigate the consequences of interactions between R. conorii and macrophages in vitro, a non-biased proteomic analysis of uninfected and R. conorii-infected THP-1 macrophages was performed to determine global host protein alterations that are stimulated by R. conorii infection. This study revealed that R. conorii infection leads to an overall transition of infected macrophages to an anti-inflammatory M2 phenotype (12), which is characterized by shifts in host cell energy production and other metabolic pathways, such as lipid metabolic processes, involved in cell homeostasis (11, 12, 14). In addition, host fatty acid synthase (FASN), an enzyme essential for fatty acid production, is required for R. conorii survival during THP-1 macrophage infection, thus highlighting a constraint for the production of host lipids for efficient infection of macrophages (12). Taken together, these data reinforce the importance of better elucidating the modulation of host pathways, such as lipid metabolic processes, that are necessary for shifting macrophage responses to promote survival of R. conorii.
During mammalian cell infection, Rickettsia species quickly escape a newly formed phagosome-like vacuole formed upon entry by lysis and reside within the cytosol of the infected host cell (1). Various other intracellular pathogens, predominantly those that reside within a pathogen containing vacuole, have been shown to interact with and/or alter host lipid pathways for nutrient acquisition and regulation of cellular responses to survive within the host cell (reviewed in (15)). A major host cell components shown to be altered during intracellular infection with these pathogens are lipid droplets (LDs), which act as cellular storage compartments composed of triglycerides and other esterified lipids (16, 17). Dynamic alterations in LD size, number, and composition are involved in regulation of multiple host processes including energy production, immune modulation, membrane integrity, macrophage polarization, and during infection of host cells (reviewed in (15)). Additionally, lipid catabolic pathways such as lipase-driven triglyceride lipolysis and fatty acid β-oxidation (FAO) are well-characterized modulators of LDs that have been associated with infections by various intracellular pathogens (18–22). A shift in host cell metabolism, including increases in metabolic pathways such as lipase-drive triglyceride lipolysis and FAO, is a major driver of the M2 phenotype (14). In addition, lipid catabolic processes promote host cell longevity and anti-inflammation characterized by production of sustainable energy from lipids and other sugar alternative carbon molecules (14, 18). This anti-inflammatory metabolic response, including LD modulation and lipid catabolism, is likely important for development of a hospitable niche for R. conorii during macrophage infection (11, 12). However, the mobilization of LDs and involvement of LD-associated pathways during infections of mammalian cells with R. conorii and other SFG Rickettsia species has not yet been addressed.
Manipulation of LDs and lipid catabolism for maintenance of host cell homeostasis during infection is conducted by various regulatory molecules including host peroxisome proliferator-activated receptors (PPAR) and bacterial mechanisms, such as effector protein interactions with host pathways like with Salmonella typhimurium SseL, and functional RNAs interaction at the post-transcriptional step of host processes like with Mycobacterium tuberculosis miR-33 (23, 24). In macrophages, PPARs are part of a nuclear receptor superfamily that play important roles in transcriptional regulation of proteins required for pathways involved in lipid metabolism and macrophage polarization (25, 26). Specifically, PPARα is a key player in shifting macrophages to an M2 response by regulating energy metabolism through upregulation of FAO-associated and other lipid catabolic processes (26). Alternatively, PPARɣ increases uptake and sequesters gathered lipids into LDs for use upon host cell metabolic shift (26, 27). Interestingly, studies investigating the alteration of PPARα and PPARɣ during intracellular macrophage infection with various pathogens, including Mycobacterium tuberculosis and Chlamydia pneumoniae, determined that the creation of a metabolically favorable niche for sustainable infection requires regulation of expression and function of PPARα and PPARɣ (28–33). For M. tuberculosis and C. pneumoniae, modulation of these signaling molecules promotes development of a foam cell required for bacterial persistence within the infected cell (28, 32, 34, 35). Salmonella typhimurium and M. tuberculosis have also been shown to regulate LDs directly during active bacterial infection suggesting a multitude of mechanisms are potentially involved in modulating lipid metabolism and the M2 macrophage response observed during R. conorii infection of macrophages (23, 24).
In this study, we demonstrate that LD mobilization is initiated early in R. conorii infection of phagocytic cells in vitro. Remarkably, stimulation of the early alterations in LDs requires bacterial de novo protein synthesis suggesting a rickettsial mechanism is involved in modifying host LD composition. Interestingly, lipid catabolic pathways are required for early induction of LD modulation and overall bacterial survival, determined by employing specific pharmacological inhibitors targeting host triglyceride lipases and FAO. Together these data suggest that alterations in host LDs regulated by host lipid catabolism are necessary during R. conorii infection, coinciding with previous studies suggesting a shift to an M2-associated metabolic response. Intriguingly, regulators of these lipid metabolic pathways, PPARα and PPARɣ, are expressed at comparable levels to uninfected cells during R. conorii infection of THP-1 macrophages. In contrast to studies of other intracellular bacteria-host cell interactions (28–31), pharmacological inhibition of PPARα had no effect on R. conorii survival. Inhibition of PPARɣ activity had a positive effect on R. conorii growth, while the induction of a foam cell-like phenotype diminished rickettsial survival. These results give further insight into the significance of host LD and other lipid processes in establishment of a replicative niche for R. conorii within macrophages.
Results
Host lipid droplets are altered during R. conorii infection of THP-1 macrophages
LDs are the center of lipid metabolic pathways and are heavily regulated based on macrophage status (36, 37). To define any changes in host LDs during R. conorii infection of THP-1 macrophages at early (1 hour post infection; hpi) and replicative (24 hpi) time points, Oil Red O was used to target the neutral lipids congregated in LDs. As shown in Figure 1, immunofluorescence microscopy analyses of uninfected and R. conorii-infected THP-1 macrophages demonstrated that the average number of LDs per host cell was increased at 1 hpi (Figure 1A & C) while the average LD area was significantly decreased during infection at both 1 hpi (Figure 1A & C) and 24 hpi (Figure 1B & C). The increase in LDs that are smaller in size early in the infection may suggest a regulation of fission/fusion of LDs or an increase in lipids sequestered into LDs while simultaneously being released during infection. However, the average LD area decreased at 24 hpi without a significant change in LD number, indicating that active release of lipids from LDs is occurring at late stages in R. conorii infection of THP-1 macrophages. To support the observed R. conorii stimulated host LD dynamics, we quantified triglyceride content in uninfected and R. conorii infected THP-1 macrophages at 1 hpi and 24 hpi. As shown in Figure 1D, the triglyceride composition remained unchanged overall at 1 hpi and was significantly decreased at 24 hpi. These results indicate an initiation of alterations in LD phenotypic changes early during infection that is dynamic throughout infection. Interestingly, the intracellular Gram-negative bacteria, Chlamydia trachomatis has been shown to colocalize with and utilize host lipids from LDs during infection (38, 39). To determine whether R. conorii employs a similar strategy to gain access to host lipids, immunofluorescence microscopy analysis on R. conorii-infected THP-1 cells stained with Oil Red O was performed. In contrast to C. trachomatis, R. conorii does not colocalize with host LDs, suggesting an alternative method of LD regulation is employed (Figure S1).
Figure 1.
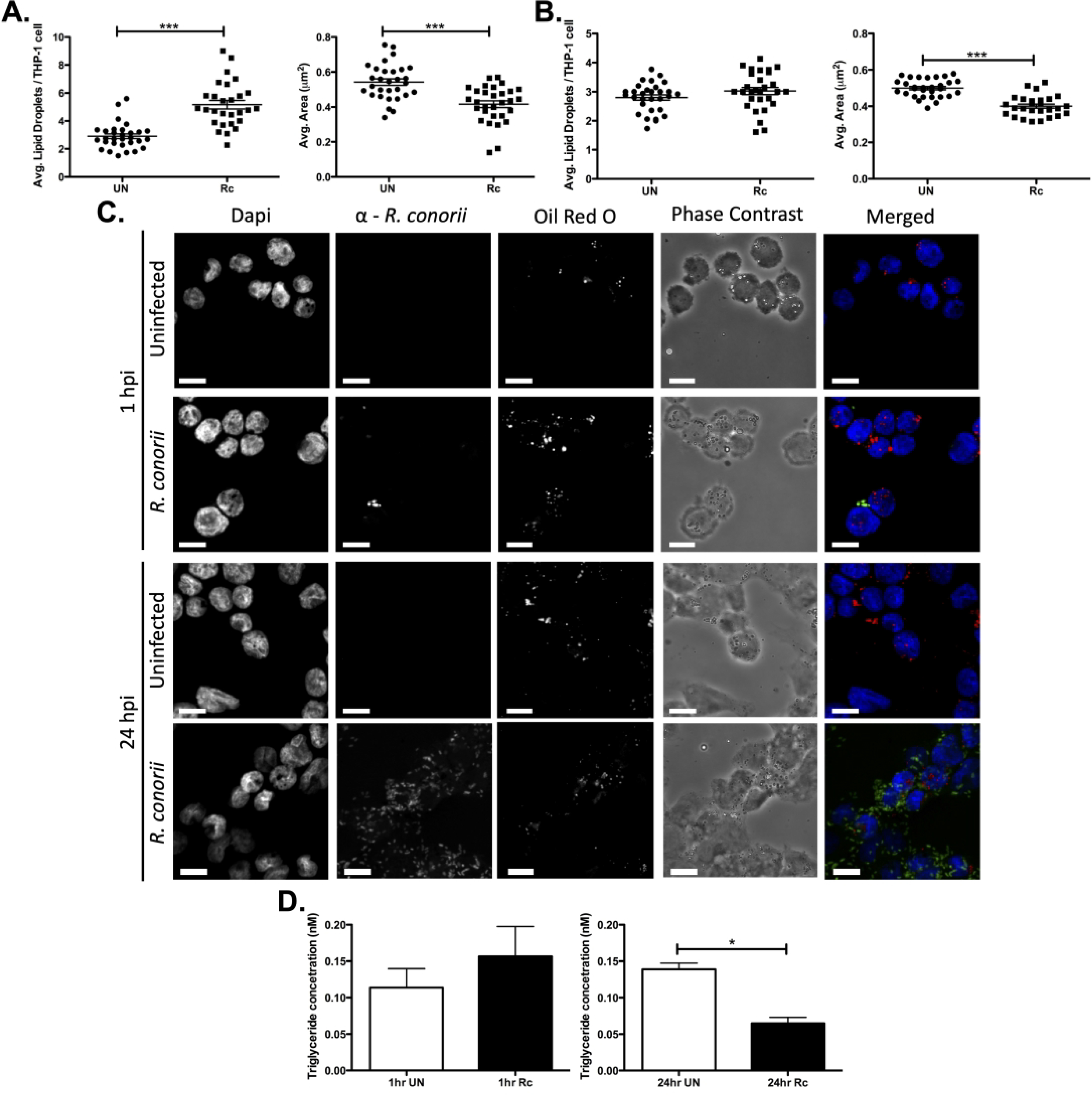
Modulation of host lipid droplets (LDs) during R. conorii THP-1 macrophage infection. THP-1 macrophages were infected with R. conorii at a multiplicity of infection of 2. Oil Red O was used for quantification of average LDs per THP-1 cell and average area (μm2) at 1 hour post infection (hpi; A) and 24 hpi (B). Ten fields of view from three independent experiements with 3–15 cells per field of view were quantified for both uninfected (UN) and R. conorii (Rc)-infected samples using ImageJ software with a constant threshold for all images. (C) A single representative image of fields taken at 100X with oil red O (red) signifying LDs, α-Rickettsia (green) signifying R. conorii, and DAPI (blue) signifying nuclei. White bar is indicative of 10μm. (D) Triglyceride quantification was performed for three independent experiments with three experimental replicates of unifected and R. conorii infected samples at 1 hpi and 24 hpi. Significance is represented by p≤0.05 determined by a one-way Student t-test for each time-point separately. Statistical significance is defined by *p≤0.05, **p≤0.005, or ***p≤0.001.
R. conorii protein synthesis is required for initiation of host LD modulation early in infection
A few bacterial pathogens that reside within a vacuole have demonstrated a requirement for active bacterial mechanisms to alter LD composition during infection (23, 24). Therefore, we determined whether R. conorii de novo protein synthesis is involved in initiating the modulation of LDs during THP-1 macrophage infection. Chloramphenicol is a bacteriostatic antibiotic drug that targets bacterial ribosomes to block de novo protein synthesis and has been previously shown to block rickettsial protein expression (40–42). To determine if rickettsial de novo protein synthesis is involved in initiating the observed LD phenotype in THP-1 macrophages, R. conorii was pre-incubated with chloramphenicol or ethanol as a control 1 hr prior to macrophage infection at a concentration previously utilized for Rickettsia species to inhibit protein synthesis (43). As shown in Figure 2A & C, inhibition of bacterial protein synthesis using chloramphenicol prior to R. conorii infection leads to a significant decrease in the average number of LDs per cell when compared to vehicle treated R. conorii infected cells. In addition, the R. conorii driven decrease in average LD area is blocked by pre-incubation of rickettsial cells with chloramphenicol and results in average LD area comparable to uninfected controls (Figure 2B & C). Taken together, these results demonstrate that active bacterial infection and de novo bacterial protein synthesis is required for initiation of LD modulation in infected THP-1 macrophages.
Figure 2.
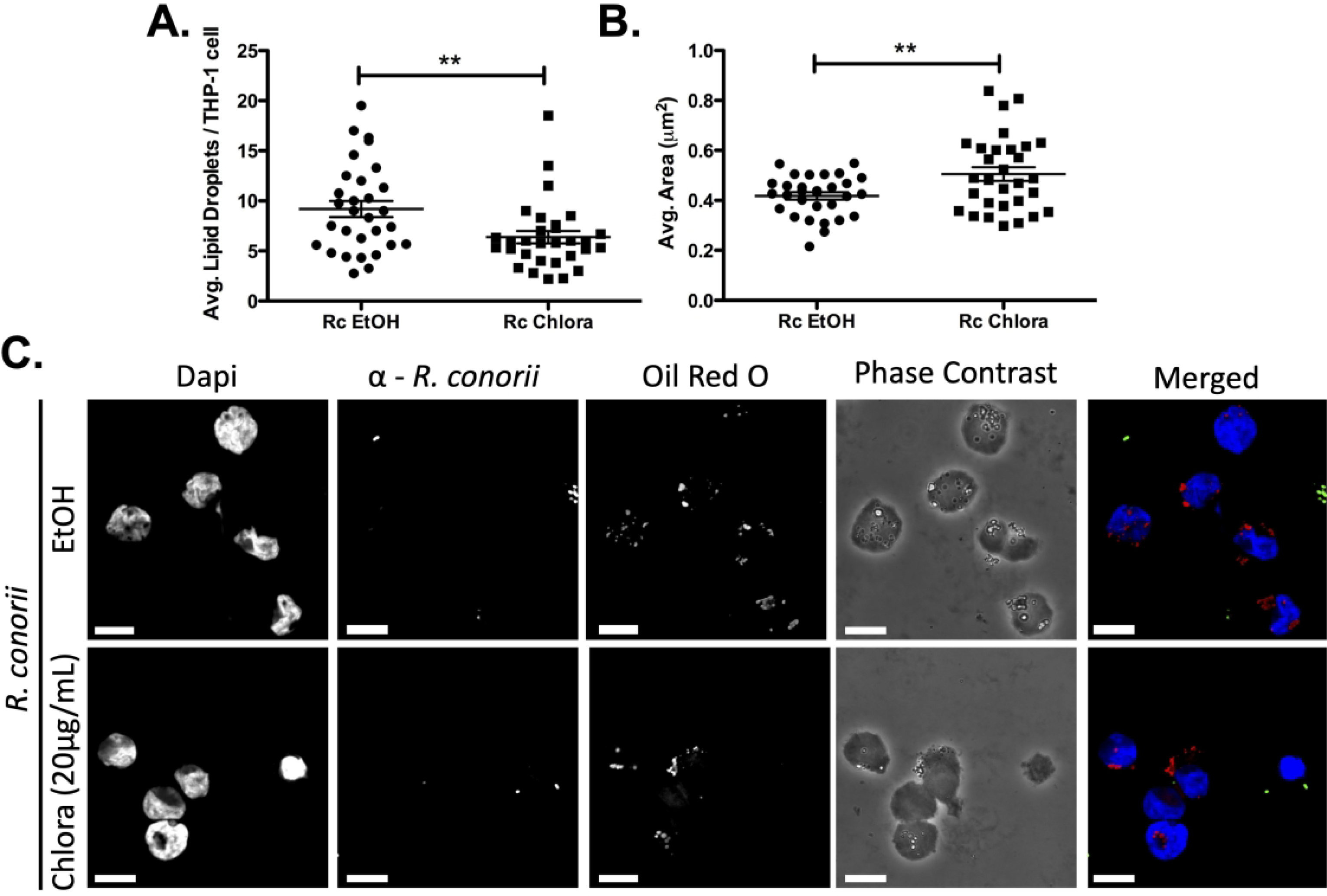
De novo R. conorii protein synthesis is required for stimulation of LD alterations seen during THP-1 macrophage infection. R. conorii was pretreated with Chloramphenicol (Chlora; 20μg/ml) or ethanol (EtOH) prior to THP-1 macrophage infection. Samples were collected at 1 hour post infection (hpi) before being stained with Oil Red O for quantification. Ten fields of view from three independent experiments each with 3–15 cells were quantified for (A) average lipid droplets (LDs) per THP-1 cell and (B) average area (μm2) of all treatment groups using ImageJ software with a constant threshold. (C) A single representative field was taken at 100X for each treatment. Oil red O (red) signifies LDs, α-Rickettsia (green) signifies R. conorii, and DAPI (blue) signifies nuclei. White bar is indicative of 10μm. Significance is represented by p≤0.05 determined by a one-way ANOVA followed by Bonferroni’s correction post hoc test. Statistical significance is defined by *p≤0.05, **p≤0.005, ***p≤0.001.
Initiation of early LD modulation seen during R. conorii infection is blocked by general triglyceride lipase inhibition
Release of lipid from LDs is driven by a multitude of catabolic processes, including lipases both specific to LDs and trafficked to LDs (44). The distinct decrease in average area of LDs during infection suggests that catabolic processes, such as lipase-driven lipid release, are potentially involved in this process. Orlistat is an FDA-approved drug that targets the active domain of a multitude of lipases that are specific for hydrolysis of triglycerides predominantly found within LDs but does not significantly affect the function of phospholipases. Orlistat has been used previously to define host lipid droplet:intracellular pathogen interactions (45, 46) and is currently being investigated for use as an alternative host-directed antiviral treatment (47–49). Additionally, the concentration of Orlistat used herein was previously shown to have minimal “off-target” effects and allowed THP-1 macrophages to remain viable (Figure S2A) (50, 51). To elucidate if active release of triglycerides is involved in initiating the LD phenotype defined at the early stage of infection, Orlistat was added to THP-1 macrophages 24 hrs prior to infection. THP-1 macrophages were infected with R. conorii for 1 hour and processed for immunofluorescence microscopy analysis to determine the average LDs per host cell and the average LD area. Similar to observations seen in Figure 1, R. conorii infection of DMSO-treated THP-1 cells led to an increase in average number of LDs per host cell (Figure 3A & C) and a decrease in the average area of LDs (Figure 3B & C). Interestingly, addition of Orlistat to cells prior to R. conorii infection significantly impacted the early LD changes making the average number of LDs per cell and average LD area in infected cells comparable to uninfected controls (Figure 3A, B, & C). To define the impact perturbing triglyceride lipases with Orlistat has on R. conorii growth in THP-1 macrophages, bacterial survival overtime was monitored. As highlighted in Figure 3D, addition of Orlistat prior to R. conorii infection led to a significant decrease in R. conorii survival in THP-1 macrophages at 3 days post infection (dpi) and 5 dpi, suggesting a requirement for triglyceride catabolism via lipases during R. conorii infection of macrophages. Taken together, these results suggest that lipase-driven triglyceride catabolism is required for the significant early initiation of LD alterations during R. conorii infection and correlates with rickettsial survival in THP-1 macrophages.
Figure 3.
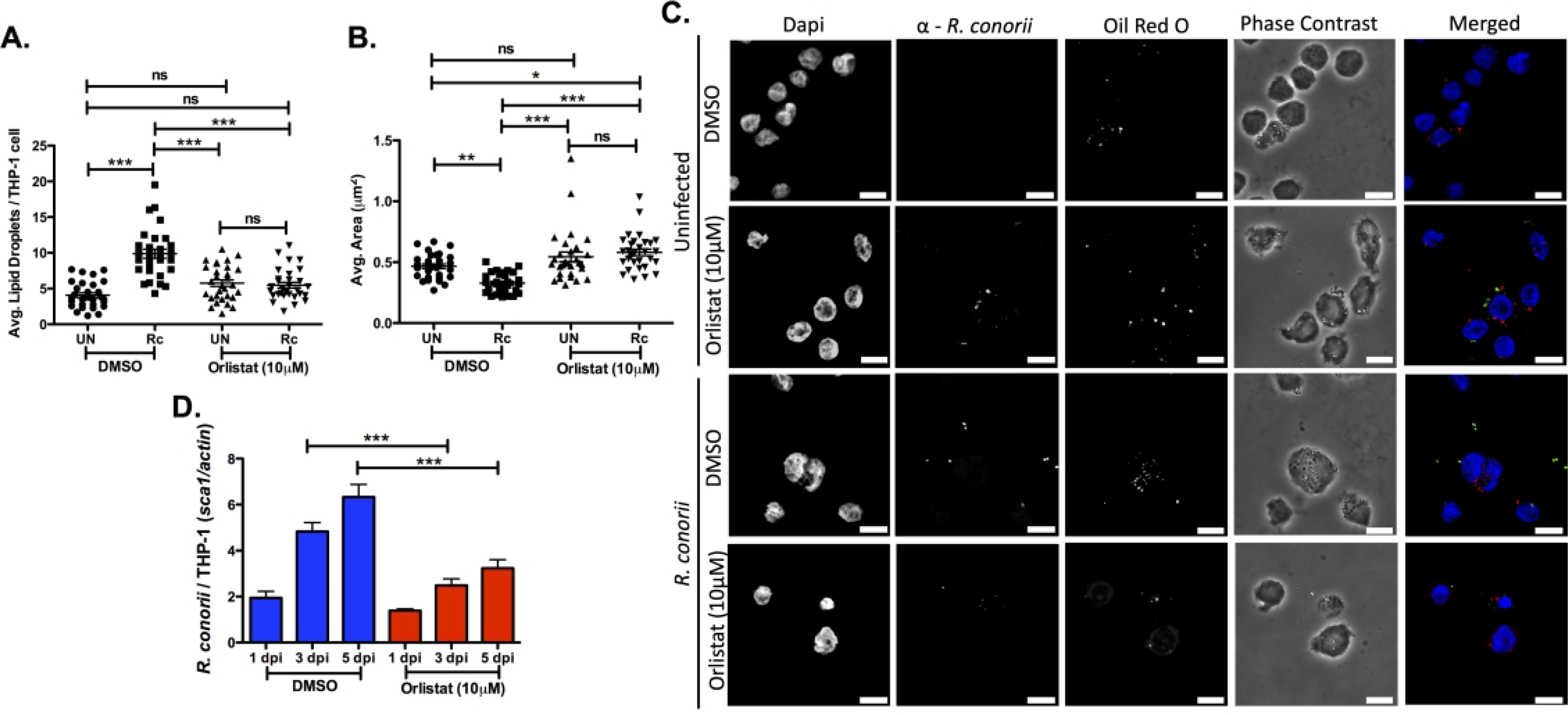
Pharmacological inhibition of triglyceride targeting lipases prevents the initiation of LD modulation early in R. conorii THP-1 macrophage infection. THP-1 macrophages were treated with Orlistat (10 μM) 24 hr prior to infection with R. conorii (MOI of 2). Samples for LD analysis were collected at 1 hour post infection (hpi) before being stained with Oil Red O for visualization of LDs. Ten fields of view from three independent experiements with 3–15 cells per field were quantified to define (A) average lipid droplets (LDs) per THP-1 cell and (B) average area (μm2) for all groups using ImageJ software with a constant threshold. (C) A representative visualization at 100X of one cell within each treatment group is shown. Oil red O (red) signifies LDs, α-Rickettsia (green) signifies R. conorii, and DAPI (blue) signifies nuclei. White bar is indicative of 10μm. (D) Rickettsial survival in the presence of Orlistat or DMSO was quantified by qPCR to analyze R. conorii (sca1) per host cell (actin) at 1 day post infection (dpi), 3 dpi, and 5 dpi. Data is representative of three independent experiments with each condition performed in triplicate. Significance is represented by p≤0.05 determined by a one-way ANOVA followed by Bonferroni’s correction post hoc test. Statistical significance is defined by *p≤0.05, **p≤0.005, ***p≤0.001.
Initiation of early lipid droplet alteration requires active fatty acid β-oxidation during R. conorii infection of THP-1 macrophages
Although lipase-driven lipolysis of triglycerides is important for initiation of the LD modulation seen during R. conorii infection of macrophages, there are other lipid catabolic factors that may also be involved. The recent reports suggesting the promotion of an M2 response during R. conorii infection supports the use of alternative metabolic pathways, such as those involved in lipid catabolism during infection (11, 12). One vital lipid catabolic pathway that utilizes free fatty acids and stored lipids is FAO (52). A key requirement for FAO is the initial conversion of lipids to acylcarnitine esters by carnitine palmitoyltransferase 1a (CPTIa) that can then be transported across the mitochondrial membrane for catabolism (18). To define the role host FAO has on initiation of LD alterations during infection the CPTIa inhibitor, Etomoxir, was used at a concentration previously utilized to elucidate the role of FAO during infection with various intracellular pathogens (45, 53–55). In addition, the dose of Etomoxir was used at a concentration that allowed THP-1 macrophages to remain viable (Figure S2B) and has been shown to exhibit minimal “off-target” effects in macrophages (56). Uninfected and R. conorii-infected THP-1 macrophages treated with Etomoxir (10 μM) were processed to visualize changes in LDs at 1 hpi (Figure 4A, B, & C). As expected, DMSO-treated R. conorii infected THP-1 macrophages showed a significant increase in average LDs per host cell and decrease in average LD area compared to uninfected DMSO treated cells at 1 hpi. Interestingly, treatment with Etomoxir significantly reduced the average number of LDs in R. conorii infected cells comparable to the DMSO infected control (Figure 4A & C) and restored the average area of LDs in R. conorii infected cells to levels seen in uninfected cells (Figure 4B & C). These results suggest that early in the infection process, CPTIa activation and FAO are involved in initiating the observed alterations in host LDs. To determine if CPTIa and FAO are also important for bacterial survival in THP-1 macrophages, host cells were treated with Etomoxir, and rickettsial survival overtime was analyzed by qPCR. Indeed, pharmacological inhibition of CPTIa with Etomoxir prior to R. conorii infection results in a significant decrease in rickettsial survival at 3 dpi and 5 dpi compared to untreated controls (Figure 4D), suggesting a requirement for FAO to develop a sustainable infection in THP-1 macrophages.
Figure 4.
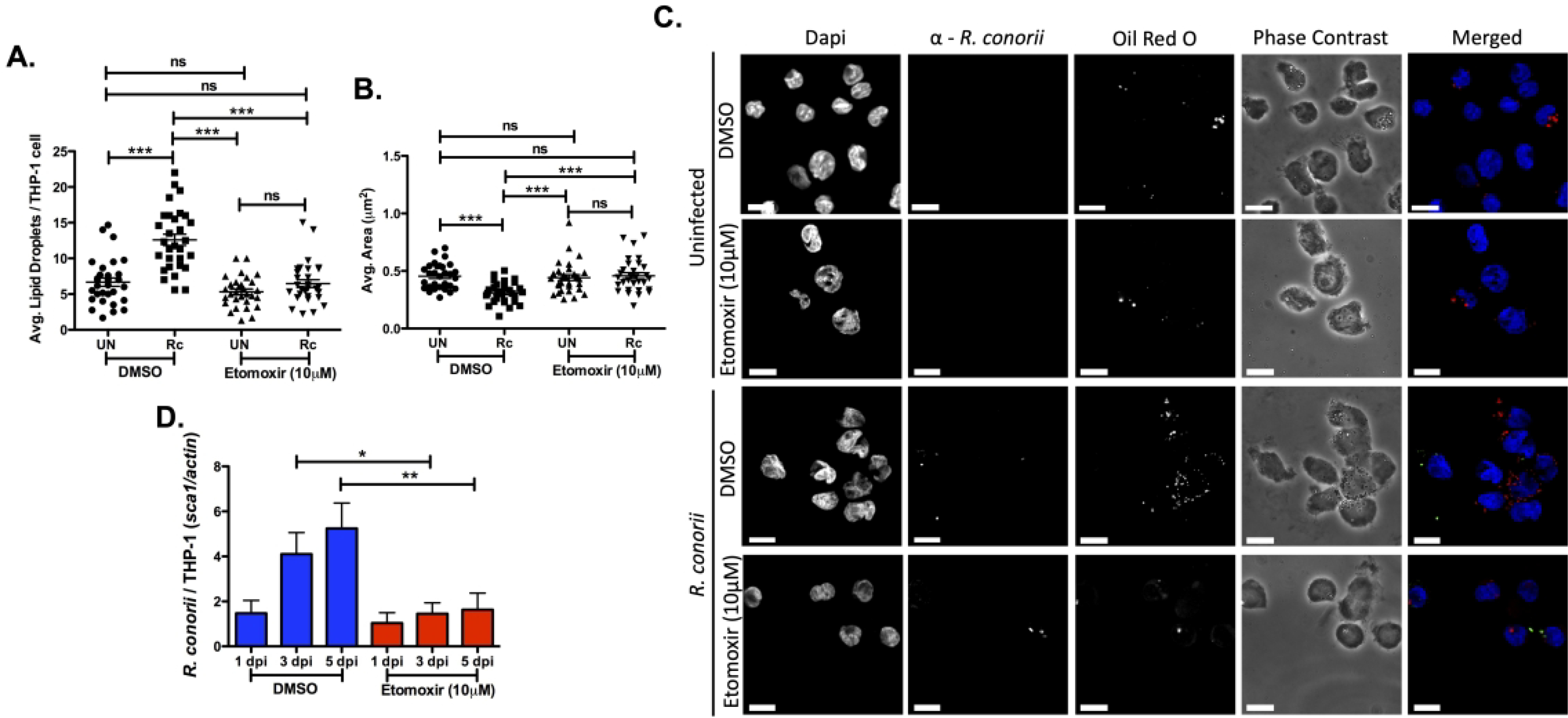
Pharmacological inhibition of fatty acid β-oxidation (FAO) prevents lipid droplet modulation early in R. conorii THP-1 macrophage infection. THP-1 macrophages were treated with Etomoxir (10 μM) 24 hr before infection with R. conorii (MOI of 2). Samples for LD analysis were collected at 1 hour post infection (hpi) before being stained with Oil Red O for visualization of LDs. Ten fields of view from three independent experiements with 3–15 cells per field were quantified to define (A) average lipid droplets (LDs) per THP-1 cell and (B) average area (μm2) for all groups using ImageJ software with a constant threshold. (C) A representative visualization of one cell at 100X within each treatment group. Oil red O (red) signifies LDs, α-Rickettsia (green) signifies R. conorii, and DAPI (blue) signifies nuclei. White bar is indicative of 10μm. (D) Rickettsial survival in the presence of Etomoxir or DMSO was quantified by qPCR to analyze R. conorii (sca1) per host cell (actin) at 1 day post infection (dpi), 3 dpi, and 5 dpi. Data is representative of three independent experiments with each condition performed in triplicate. Significance is represented by p≤0.05 determined by a one-way ANOVA followed by Bonferroni’s correction post hoc test. Statistical significance is defined by *p≤0.05, **p≤0.005, ***p≤0.001.
Host PPARα is not required for initial LD changes or sustainable R. conorii infection of THP-1 macrophages
PPARα is a potent regulator of FAO, LD catabolism, and overall lipid modulation and has been shown to play an important role in the macrophage shift to an M2 metabolic phenotype seen during R. conorii infection of macrophages (26). C. pneumoniae and M. tuberculosis are known modulators of macrophage polarization and specifically LD modulation which have been linked to regulation of PPARα signaling during infection (28, 30). To investigate the role PPARα serves for the initiation of LD modulation early in R. conorii infection, LDs were visualized in the presence of a PPARα inhibitory compound, GW6471 (0.5 μM). GW6471 has been used previously for defining the requirement of FAO and regulation of foam cell formation during C. pneumonia infection of macrophages (28). Pharmacological inhibition of PPARα with GW6471 (0.5 μM) was performed at a concentration previously shown to inhibit PPARα activity (57, 58) and at a concentration that allowed THP-1 macrophages to remain viable (Figure S2C). Interestingly, treatment with GW6471 had no effect on host LD alterations that are stimulated early in infection (Figure 5 A, B, & C), suggesting that PPARα-signaling is not required for early LD modulation during R. conorii infection of THP-1 macrophages.
Figure 5.
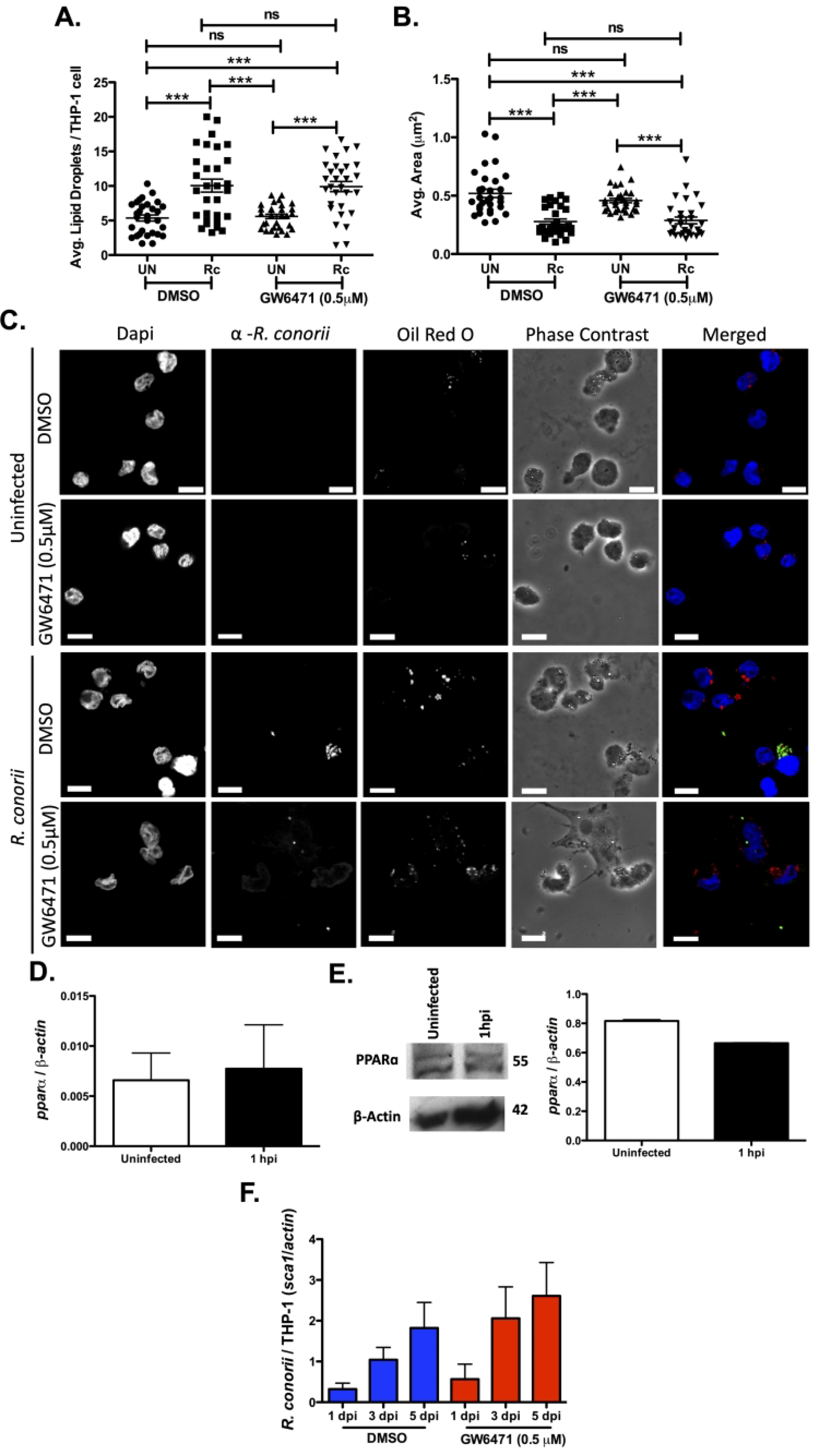
PPARα is not required for R. conorii infection of macrophages. Samples for LD analysis were collected at 1 hour post infection (hpi) before being stained with Oil Red O for visualization of LDs. Ten fields of view from three independent experiements with 3–15 cells per field were quantified to define (A) average lipid droplets (LDs) per THP-1 cell and (B) average area (μm2) for all groups using ImageJ software with a constant threshold. (C) A representative visualization of one cell at 100X within each treatment group. Oil red O (red) signifies LDs, α-Rickettsia (green) signifies R. conorii, and DAPI (blue) signifies nuclei. White bar is indicative of 10μm. (D) qRT-PCR analysis with total RNA from R. conorii infected (MOI of 2) THP-1 macrophages was employed to determine mRNA expression of pparα at 1 hpi. (E) Immunoblotting was conducted with whole cell lysates for protein expression of PPARα at 1 hpi in uninfected or R. conorii infected (MOI of 2) THP-1 macrophages. (F) DMSO or GW6471 qPCR analysis with gDNA from pre-treated THP-1 macrophages infected with R. conorii (MOI = 2) in the presence of DMSO or GW6471 for inhibition of PPARα at 1 days post infection (dpi), 3 dpi and 5 dpi. All data is representative of three independent experiments with each condition performed in triplicate. Significance is represented by p≤0.05 determined by a one-way ANOVA followed by Bonferroni’s correction post hoc test. Statistical significance is defined by *p≤0.05, **p≤0.005, *** p≤0.001.
Other intracellular bacteria that require PPARα signaling have been shown to actively modulate PPARα expression during infection (28–30, 35). To determine if PPARα expression is altered during R. conorii infection of THP-1 macrophages, alterations in pparα mRNA and PPARα protein expression at the initial stage of infection were determined. As shown in Figure 5D & E, R. conorii infection does not stimulate significant changes in pparα mRNA levels and PPARα protein expression at 1 hpi. Similarly, PPARα protein expression remained significantly unaltered at the replicative stage of infection (24 hpi) (Figure S3A). Although mRNA and protein expression levels were unaltered, PPARα activation and function could be modified during infection. To determine if functional PPARα plays a role in R. conorii infection of THP-1 macrophages, GW6471 was utilized to inhibit activation of PPARα. Interestingly inhibition of PPARα did not significantly affect R. conorii survival compared to the vehicle control (Figure 5F) further indicating the absence of a PPARα-signaling requirement during R. conorii infection.
Pharmacological inhibition of host PPARɣ has a positive correlation with R. conorii during THP-1 macrophage infection due to the inhibition of development of a foam cell-like cellular response
PPARɣ is a potent regulator of lipid sequestering and accumulation into LDs and in macrophages is associated with regulation of polarization (26). M. tuberculosis and C. pneumoniae are heavily associated with the development of a foam cell phenotype characterized by an increase in lipid scavenging and accumulation of lipids within LDs. The development of this phenotype during infection requires PPARɣ expression and is important for bacterial sustainability within the host cell (28, 33, 59). To investigate the putative role PPARɣ serves for the initiation of LD modulation early in R. conorii infection, LDs were visualized in the presence of a PPARɣ inhibitory compound, GW9662 (10 μM). This concentration was chosen because it allowed THP-1 macrophages to remain viable (Figure S2D) and has been shown previously to inhibit activation of PPARɣ in macrophages, including in studies with intracellular pathogens such as C. pneumoniae and Brucella abortus (28, 60–62). Intriguingly, treatment with GW9662 had no effect on host LD modulation initiated early in infection (Figure 6A, B, & C). Collectively, these results suggest that PPAR signaling is likely not involved in R. conorii induced LD alterations on THP-1 macrophages.
Figure 6.
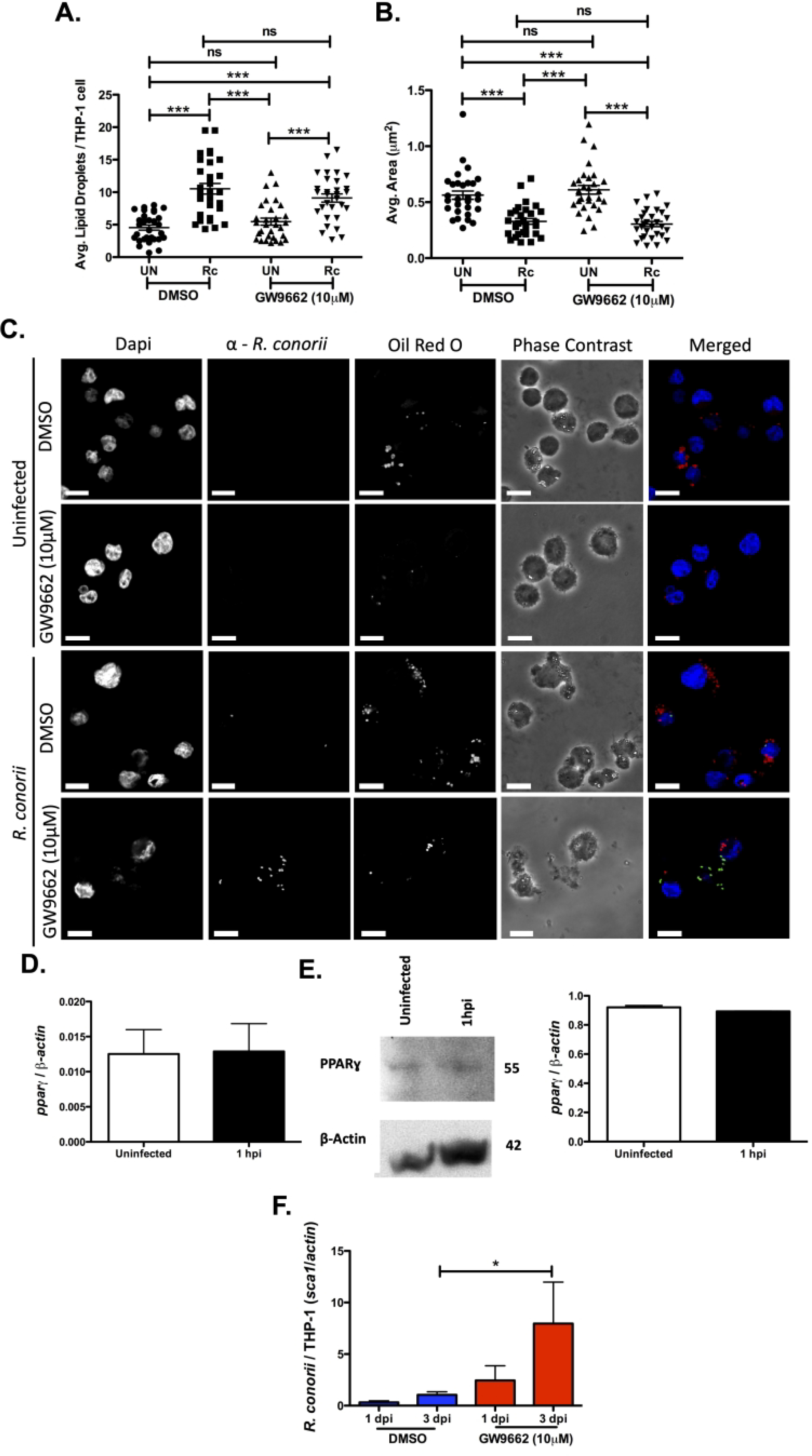
Inhibition of PPARɣ is positively correlated with increased R. conorii infection of macrophages. Samples for LD analysis were collected at 1 hour post infection (hpi) before being stained with Oil Red O for visualization of LDs. Ten fields of view from three independent experiements with 3–15 cells per field were quantified to define (A) average lipid droplets (LDs) per THP-1 cell and (B) average area (μm2) for all groups using ImageJ software with a constant threshold. (C) A representative visualization of one cell at 100X within each treatment group. Oil red O (red) signifies LDs, α-Rickettsia (green) signifies R. conorii, and DAPI (blue) signifies nuclei. White bar is indicative of 10μm. (D) qRT-PCR analysis with total RNA from R. conorii infected (MOI of 2) THP-1 macrophages to determine mRNA expression of pparɣ at 1 hour post infection (hpi). (E) Immunoblotting with whole cell lysates for protein expression of PPARɣ at 1 hpi in uninfected or R. conorii infected (MOI of 2) THP-1 macrophages. (F) qPCR analysis with gDNA from pre-treated THP-1 macrophages infected with R. conorii (MOI of 2) in the presence of DMSO and GW9662 for inhibition of PPARɣ at 1 days post infection (dpi) and 3 dpi. All data is representative of three independent experiments with each condition performed in triplicate. Significance is represented by p≤0.05 determined by a one-way ANOVA followed by Bonferroni’s correction post hoc test. Statistical significance is defined by *p≤0.05, **p≤0.005, *** p≤0.001.
To determine if PPARɣ expression is modulated during R. conorii infection of THP-1 macrophages, alterations in pparɣ mRNA and PPARɣ protein expression at early and replicative stages of infection were analyzed. As shown in Figure 6D & E, R. conorii infection does not stimulate differences in mRNA and protein expression levels at 1 hpi. Interestingly, PPARɣ protein levels at 24 hpi also showed no significant changes (Figure S3B). Inhibition of PPARɣ significantly increased R. conorii growth at 3 dpi (Figure 6F), demonstrating that intracellular survival in THP-1 macrophages is positively impacted by PPARɣ inhibition. At 5 dpi, we observed a significant decrease in cell viability likely caused by continued R. conorii proliferation (data not shown). Together, these results highlight the potential negative impact of PPARɣ and subsequent accumulation of lipids and LDs within infected THP-1 macrophages during R. conorii infection.
To further describe the negative effects a PPARɣ signaling for lipid accumulation may have during R. conorii infection of THP-1 macrophages, a foam cell-like phenotype was induced by addition of oleic acid-albumin. The development and contribution of foam cell development to pathogen intracellular survival has been well characterized for pathogens such as M. tuberculosis and C. pneumoniae (28, 32, 35, 45). Similarly, oleic acid-albumin was used at a concentration of 400μM as done previously during M. tuberculosis infection (34). To confirm a significant increase in LDs present during foam cell induction and to define the LD modulation initiated during infection in foam-like cells, LD visualization and quantification was conducted as done previously. The BSA controls showed similar results as those seen in previous experiments, with a decrease in size of LDs and an increase in the number of LDs present during R. conorii infection of macrophages (Figure 7A, B, & C). In contrast, THP-1 macrophages treated with oleic acid-albumin resulted in a significant increase in LD size in uninfected and infected samples, when compared to BSA controls, with little difference between uninfected and infected oleic acid-albumin treated cells (Figure 7B & C). Interestingly, the number of LDs per host cell in foam-like cells for uninfected and infected samples was comparable to that of the uninfected control (Figure 7A & C). To determine if infection of oleic acid-albumin treated cells is inhibitory for R. conorii infection of macrophages as predicted, R. conorii survival in THP-1 macrophages that present with a foam cell-like phenotype was determined. R. conorii survival was significantly impaired in oleic acid-albumin treated cells after both 3 dpi and 5 dpi. Thus, suggesting that the induction of a foam-like cell phenotype, correlated with a significant increase in LD size, blocks R. conorii-induced early LD modulation and overall survivability in THP-1 macrophages.
Figure 7.
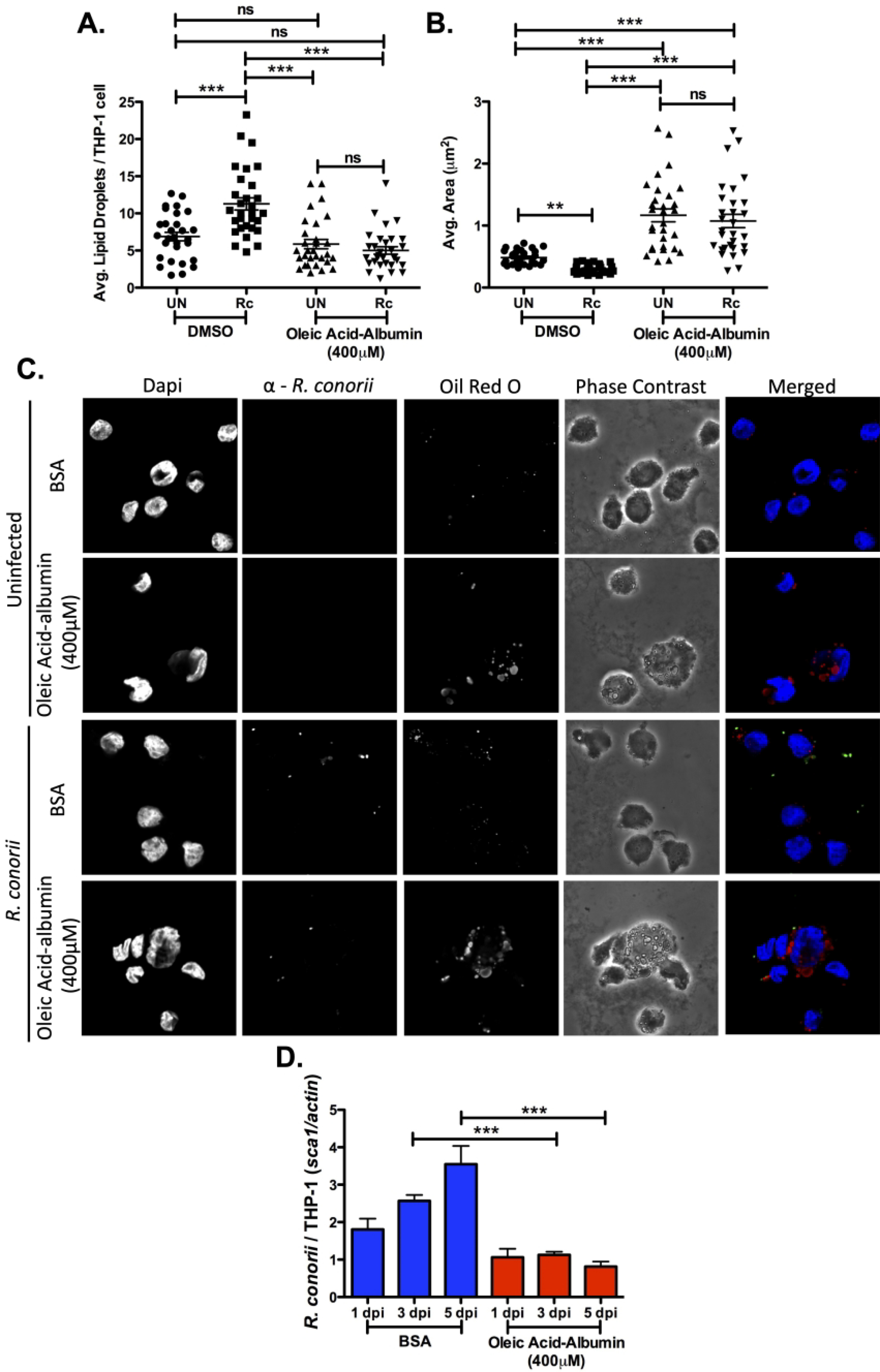
Formation of a foam cell-like phenotype in THP-1 macrophages results in a decrease in R. conorii survival. THP-1 macrophages were treated with oleic acid-albumin (400 μM) 3 hr before infection with R. conorii (MOI of 2). Samples for LD analysis were collected at 1 hour post infection (hpi) before being stained with Oil Red O for visualization of LDs. Ten fields of view from three independent experiements with 3–15 cells per field were quantified to define (A) average lipid droplets (LDs) per THP-1 cell and (B) average area (μm2) for all groups using ImageJ software with a constant threshold. (C) A representative visualization of one cell at 100X within each treatment group. Oil red O (red) signifies LDs, α-Rickettsia (green) signifies R. conorii, and DAPI (blue) signifies nuclei. White bar is indicative of 10μm. (D) Rickettsial survival in the presence of oleic acid-albumin or BSA was quantified by qPCR to analyze R. conorii (sca1) per host cell (actin) at 1 day post infection (dpi), 3 dpi, and 5 dpi. Data is representative of three independent experiments with each condition performed in triplicate. Significance is represented by p≤0.05 determined by a one-way ANOVA followed by Bonferroni’s correction post hoc test. Statistical significance is defined by *p≤0.05, **p≤0.005, ***p≤0.001.
Discussion
Intracellular pathogens rely heavily on host cells to develop a niche required for efficient infection and pathogen growth. Several studies have shed light on host processes important for development of a niche for various vacuolar intracellular bacteria, while neglecting bacteria that live and replicate within the nutrient-poor host cytosol. In macrophages, lipid metabolism has more recently become a known key regulator of host cell homeostasis and polarization, which has been associated with establishment of sustainable intracellular bacterial infections. As previously mentioned, the intracellular bacteria that have defined alterations in host lipid metabolic pathways are predominantly those that reside within a PCV upon infection of the host cell. For example, modulation of various host lipid pathways has been shown to be involved in providing nutrients and regulating host cell signaling to develop a replicative niche, and as structural integrity for pathogens such as Mycobacterium tuberculosis, Chlamydia spp., dengue virus and hepatitis C virus (15, 63–67). During macrophage infection, pathogens, such as M. tuberculosis and C. pneumoniae, also manipulate host lipid processes to initiate polarization of macrophages that is ideal for production of a favorable bacterial niche (68–70). However, R. conorii and other rickettsial species are cytosolic intracellular bacteria that lack the accessibility of associating with host lipids within a pathogen containing vacuole. The requirement of lipid synthesis for R. conorii macrophage infection by FASN suggests a need of lipids for a specific function important for bacterial survival (12); however, the role of host lipid metabolism during intracellular infection by Rickettsia species remains unclear. The current study focuses on further defining the importance of host LDs during R. conorii infection of THP-1 macrophages by elucidating the initiation of LD structural alterations and lipid pathways important for inducing these changes.
There are multiple fates that synthesized lipids during infection can meet including being sequestered into lipid stores and catabolism by lipase-driven lipolysis or FAO. R. conorii infection drives a dynamic shift in host LD phenotype early in infection with changes in both size and number per host cell. However, these alterations are different at replicative stages of infection, implying a temporal dynamic modulation of host LDs and lipid pathways throughout the course of infection (Figure 8A). LDs are also a major source of lipids used for energy production, structural integrity, and immune signaling (44). Indeed, pathogens have evolved mechanisms to sequester lipids from host LDs through lipid catabolic pathways as a mechanism of providing substrates for modulation of signaling, structural lipids, and energy (reviewed in (15)). Early studies with Rickettsia species indicated the presence of membrane phospholipids, including phosphatidylcholine (PC), within the bacterial membrane. Genomic analyses revealed that Rickettsia species lack the machinery to effectively produce certain membrane lipids including PC, suggesting a need to acquire these from host sources (71–73). Rickettsia species have also been shown to stimulate prostaglandin production during infection indicating bacterial immune regulation through lipid metabolites (74). These early studies defining lipid processes modulated during infection coincides with the dynamic nature of LD alterations, including the likely release of lipids from LDs that may contribute to these phenotypes. Intriguingly, at early stages of R. conorii infection of THP-1 macrophages, there is an increase in average LD per host cell with a decrease in the average size of the LDs. This phenotype could be due in part by an infection-induced stimulation of both anabolic and catabolic lipid processes as seen in infections of host cells by other intracellular pathogens, including M. tuberculosis, C. pneumoniae, and dengue virus (45, 53, 55, 63, 64, 75–77). Similarly, Coxiella burnetii infection induced an increase in LDs compared to uninfected controls, while requiring lipid catabolism to regulate LD homeostasis necessary for efficient infection (78). Additionally, this phenotype could also be produced through the stimulation of fission of larger LDs into smaller LDs. Modulation of total lipid content and LD fission has been linked to cell cycle progression with lack of LD catabolism negatively correlating with yeast cell growth (79, 80). However, the mechanisms stimulating LD fission/fusion dynamics in animal cells, and specifically macrophages, remains relatively undefined.
Figure 8.
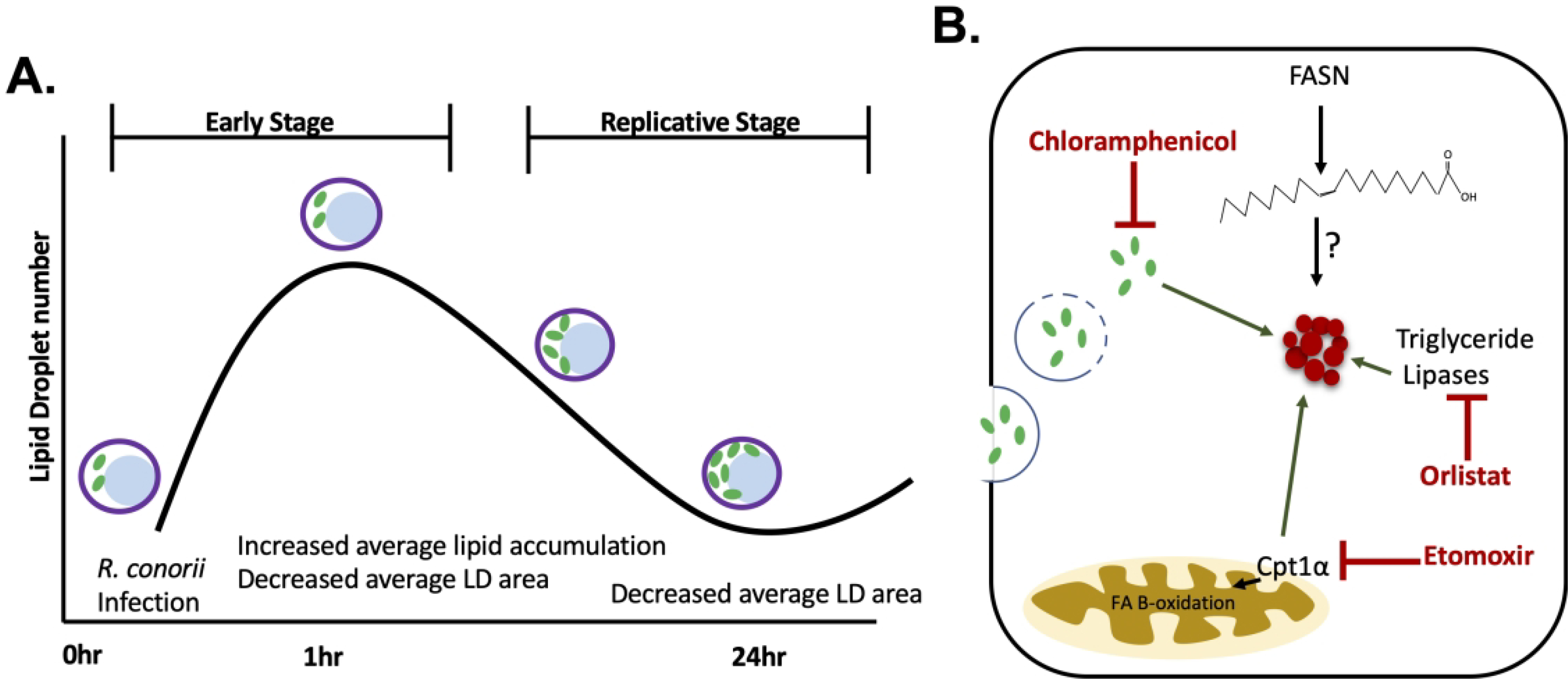
Model of host lipid droplet modulation following R. conorii infection of THP-1 macrophages. (A) Lipid droplet (LD) number and size is dynamic during R. conorii infection, depicted by an increase in average LD production early in infection with a decrease in average LD area throughout infection. Purple circles represent host cells membrane containing nucleus (blue) and R. conorii (green). (B) Host LD (red cluster of circles) alterations are shown to be initated in the early stages of R. conorii infection of THP-1 macrophages. During early stages of infection pharmacological inhibition of lipid catabolic pathways with Orlistat, which targets triglyceride lipases, and Etomoxir, which targets the beginning steps of fatty acid oxiation (FAO) by inhibiting carnitine-palmytol transferase (CPTIa), perturbs the significant LD alterations R. conorii infection induces at 1 hpi (red symbols). This suggests that active FAO and triglyceride lipases are involved in stimulating the LD modulation seen at early stages (dark green arrows). Additionally, fatty acid synthase (FASN) is required for R. conroii infection of THP-1 macrophages (12) and contributes to the production of free fatty acid and in normal states produces lipids for storage in LDs (red cluster of circles); however, the contribution of these free fatty acids to LDs or other pathways during infection remains undetermined. Interestingly, a R. conorii (light green ovals) protein expressed early in infection is necessary for initiation of host LD modulation (dark green arrows).
Interestingly, R. conorii de novo protein synthesis in required for initiation of host LD mobilization during infection, suggesting that a bacterial factor(s) is involved in modulating these host lipid shifts important for development of a favorable replicative niche. Although the host metabolic changes required for sustainable intracellular infections are being investigated, the bacterial mechanisms involved in stimulating these responses have not yet been elucidated. To date, very few intracellular bacteria have defined mechanisms that directly regulate host LDs, and those that do are restricted to growth within a pathogen containing vacuole. For example, C. trachomatis uses pathogen containing vacuole-associated bacterial proteins as a mechanism for recruitment of LDs and release of lipids from LDs during intracellular infection (38, 81). S. typhimurium also expresses a secreted effector protein, SseL, that has been associated with direct regulation of host LD modulation to maintain a viable niche for the bacteria (23). Unfortunately, BLAST analysis shows no SseL homologues in the R. conorii genome. In terms of rickettsial infections, the secretome components necessary for development of a desirable niche within the host cell are not well elucidated (82). Rickettsia species including R. conorii encode for phospholipases and putative effector proteins, which have a potential role in modulating host LD responses to infection (82–86). Therefore, further work must be conducted to better characterize the active bacterial mechanism required for modulation of host LDs and other metabolic processes during infection.
M. tuberculosis has been shown to manipulate host lipid catabolic pathways and drive the development of a foam cell phenotype required for bacterial sustainability (45). For M. tuberculosis, the formation of foam cells is necessary for development of chronic infection, and when lipase-driven LD catabolism is inhibited with Orlistat, there is no effect on bacterial growth (87). However, R. conorii infection does not result in the formation of foam cells in macrophages. In addition, the pharmacological inhibition of lipase-driven triglyceride catabolism with Orlistat during R. conorii infection results in a significant decrease in rickettsial survival and early LD modulation during infection (Figure 7B). Similar to M. tuberculosis, R. conorii infection of THP-1 macrophages shifts the host cell towards a M2 macrophage phenotype (12, 88). This shift is typically characterized by a decrease in the glycolytic pathways, suggesting that alternative molecules, such as lipids, are required to compensate (12). The use of lipids as an alternative carbon source relies on the upregulation of lipid catabolic processes such as triglyceride specific lipase-driven lipolysis and FAO (18). We demonstrated that pharmacological inhibition of the host FAO initiation enzyme, CPTIa, causes a significant decrease in rickettsial survival during THP-1 macrophage infection. Comparably, inhibition of FAO by Etomoxir also reduced M. tuberculosis load during replicative stages in macrophages, demonstrating the importance of FAO in establishing M2 macrophage polarization and subsequent sustainable infection (53). Separately, host FAO and LD modulation have been associated with various intracellular infections predominantly for energy production and regulation of the immune response (reviewed in (15)). However, LD regulation as a source of lipids for FAO during the growth of intracellular non-vacuolar bacterial pathogens has been largely unstudied. This study demonstrates that at early stages of infection, inhibition of CPTIa activation leads to a dysfunction in initiation of LD modulation. Together these data suggest a requirement of the lipid catabolic processes, triglyceride lipase driven lipolysis and FAO, for initiation of LD alterations early in infection and bacterial survival in THP-1 macrophages. (Figure 8B).
PPARs are key transcriptional modulators of host homeostasis by regulation of lipid metabolism pathways (27). Previous studies have also identified PPARα and PPARɣ as active regulators of the host immune response making these molecules viable targets for therapeutic intervention (29, 59). Overall, PPARα upregulates lipid catabolism, predominantly through FAO, to maintain cell homeostasis, and PPARɣ is traditionally associated with increases in lipid uptake and LD formation (27). In macrophages, these transcription factors are drivers of cellular metabolism, typically shifting the response to a more M2 macrophage phenotype, like that seen during R. conorii infection of macrophages (12, 26). Many other intracellular pathogens, including M. tuberculosis and C. pneumoniae, regulate PPARα and PPARɣ to mediate host metabolic processes required for establishment a hospitable niche (28, 33, 61). However, the mechanisms for regulation of PPARs throughout infection are largely not understood. Unlike M. tuberculosis and C. pneumoniae, R. conorii infection of THP-1 macrophages does not require PPARα for the lipid metabolic shifts necessary for bacterial survival. As mentioned previously, bacterial protein synthesis is required for initiation of the LD phenotype seen early during infection. There is the potential that instead of hijacking host PPARα to stimulate LD alterations, a R. conorii de novo protein, like SseL in S. typhimurium, bypasses this host signaling pathway to stimulate the desired response (23).
Surprisingly, PPARɣ inhibition significantly enhances R. conorii growth in THP-1 macrophages. A previous report indicated that inhibition of PPARɣ by GW9662 induces an anti-inflammatory environment in macrophages, similar to that seen to be induced during R. conorii infection (11, 12, 60). This would suggest that prior treatment of macrophages with GW9662 likely provides a more favorable niche upon infection with R. conorii that would allow for efficient bacterial growth. PPARɣ is also heavily involved in increasing lipids sequestered into LDs to maintain cellular homeostasis and is required for the foam cell formation observed during infections with M. tuberculosis and Chlamydia pneumoniae (28, 59). Our results during PPARɣ inhibition suggest that the sequestering of lipids into LDs has a negative impact on rickettsial growth and survival in macrophages. Incubation of mammalian cells with GW9662 at relevant concentrations to inhibit PPARɣ have also been shown to reduce lipids sequestered within LDs in macrophage models, including THP-1 macrophages, strengthening this notion (89, 90). In addition, we determine that the stimulation of a foam cell-like phenotype before R. conorii infection negatively impact on bacterial survival and early LD alterations in THP-1 macrophages and correlates with a decrease in R. conorii survival within these cells. Additionally, previous reports define an importance of LDs and subsequent lipid release during C. trachomatis infection of host cells (38, 39, 78, 91, 92). Together, this suggests that an overabundance of LDs developed through PPARɣ-driven lipid accumulation during R. conorii infection of THP-1 macrophages has a detrimental effect on bacterial survival. As mentioned above, PPARɣ signaling and subsequent lipid accumulation to, in some cases, form a foam cell-like phenotype have been associated with activation of host inflammatory responses; therefore, inhibition of PPARɣ before infection could have a positive effect on R. conorii growth by dampening inflammatory signals that Rickettsia would otherwise have to avoid or regulate upon infection to provide the optimum replicative niche (11, 12, 93).
There have been an increasing number of studies addressing the requirement for regulation of host LDs to develop a hospitable niche for survival of various intracellular bacteria (15, 94). Specifically, host lipids derived from LDs are a key source of nutrients, metabolites, and structural and signaling molecules, making it a common target for a variety of intracellular pathogens. We demonstrate that average LDs per host cell are increased early in R. conorii-infected cells with a decrease in average LD area throughout infection. Initiation of early LD alterations require host lipid catabolic processes, such as triglyceride lipases and FAO, as well as de novo bacterial protein synthesis. These catabolic processes are independent of the upstream signaling molecule, PPARα. However, inhibition of PPARɣ positively effects R. conorii growth in THP-1 macrophage. In addition, lipid accumulation to develop a foam cell-like phenotype is detrimental to bacterial survival and LD modulation during THP-1 macrophage infection. Together, the data generated from this study indicate that lipid catabolic processes that regulate host LD modulation are an important understudied aspect of Rickettsia-host cell interactions and may shed further insight into the mechanisms by which non-vacuolar facultative and obligate intracellular bacterial pathogens regulate host cell processes to develop a favorable niche during the infection of mammalian cells.
Materials and Methods
Cell lines, Rickettsia growth and purification
THP-1 (ATCC-TIB-202™) cells were differentiated into macrophage-like cells by addition of 100nM phorbol 12-myristate 12-acetate (PMA; Sigma-Aldrich) 24 hrs prior to start of experiment. PMA-treated THP-1 cells (THP-1 macrophages) were infected with R. conorii Malish7 at a MOI of 2. Cells were grown in RPMI-1640 medium (Gibco) with 10% heat-inactivated fetal bovine serum (FBS; R&D Systems). Vero cells (ATCC-CCL-81™) were used to propagate R. conorii Malish7 as described previously (95, 96). Vero cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) with 10% FBS, 1x non-essential amino acids (Corning), and 0.5 mM sodium pyruvate (Corning). Cells were maintained in 5% CO2 at 34°C.
Lipid droplet visualization and Triglyceride Quantification
THP-1 macrophages were seeded at a density of 5×105-1×106 cells/mL on poly-L-lysine treated in 24 well plates. After 24 hrs, cells were R. conorii infected (MOI of 2) or treated with pharmacological inhibitor as described below. 24 hrs after treatment, R. conorii Malish7 (MOI of 2) was added to cells and bacterial:host cell contact was induced by centrifugation at 300 xg for 5 min. For chloramphenicol-treatment studies, R. conorii required for infection at an MOI of 2 was incubated, at 34°C and 5% CO2, in RPMI with 10% FBS and 20 ug/mL chloramphenicol (43). After 1 hr, chloramphenicol was removed, and THP-1 macrophages were infected with treated R. conorii as described above. Samples were 4% paraformaldehyde (PFA)-fixed at 1-hour post infection (hpi) and 24 hpi for standard infection and 1 hpi for chloramphenicol or inhibitor treatment studies. Oil Red O isopropanol solution (Electron Microscopy Sciences) was utilized according to manufacturer’s protocol. Briefly, after 4% PFA fixation, samples were incubated with 60% isopropanol for 5 min before addition of Oil Red O mixture (6:4 Oil Red O to H2O) for 20 mins. Washed coverslips were probed as described previously with anti-R. conorii antibody RcPFA (97) followed by Goat α-Rabbit Alexa Flour-488 (Invitrogen) and DAPI (Thermo Scientific) before being mounted on slides for visualization with the Olympus Fluoview FV10i confocal microscope (9). Ten fields of view each with 3–15 cells (a total of approximately 100–200 cells) for each treatment group were gathered for determination of average LD area (μm2) and average LD quantification within each field using ImageJ software for analysis of particles with a constant threshold. Fields of view for infected samples contained both uninfected and infected cells with 50–100% infection and all cells within each field of view were included in the analysis. Experiments were repeated three times for a total of 30 fields of view. Colocalization images were analyzed as described previously (9, 13).
1×107 THP-1 macrophage cells were seeded in two wells of a six well plate as described previously. After R. conorii infection (MOI of 2), cells were collected at 1 hpi and 24 hpi and pelleted at 300 xg for 5 min in 1x PBS. Triglycerides were quantified using the Triglyceride Quantification kit (Sigma-Aldrich) following manufacturers protocol. Briefly, triglycerides were released by boiling with 5% Nonidet™ P 40 Substitute and pelleted by centrifugation at high speed for 2 min. Triglyceride was converted to glycerol and fatty acid and quantified by measurement of fluorescent intensity (λex = 535/λem = 590 nm) using the Molecular Devices Spectramax M2 microplate reader.
Pharmacological inhibition of host lipid metabolic processes
THP-1 macrophages were seeded in 96 well plates at 5 × 104 cells per 200μl for each well. Inhibitors were used at concentrations that maintained cell viability and were recommended by supplier. Inhibitors utilized include Etomoxir (10 μM; Sigma-Aldrich), Orlistat (10 μM; Sigma-Aldrich), GW6471 (0.5 μM; BioVision), and GW9662 (10 μM; BioVision). DMSO control cells were treated with equal volumes as inhibitor added. Inhibitors were added 24 hrs prior to R. conorii infection (MOI of 2) as described above. Samples were harvested at 1 day post infection (dpi), 3 dpi, and 5 dpi and processed for total gDNA isolation (Invitrogen) (9). After collection and gDNA processing, qPCR was performed with iTaq Universal Probe Supermix (BioRad) following manufacturers protocol. The PCR reaction was conducted as follows: 50°C for 2 mins, 95°C for 10 mins, followed by 45 cycles of 95°C for 15 secs, 58°C for 1 min. R. conorii sca1 normalized to host β-actin was used to quantify the amount of Rickettsia per host cell present within each sample. Primers and probes used were described previously (10).
MTT Assay for analysis of THP-1 macrophages viability in the presence of inhibitors
THP-1 macrophages were seeded in 96 well plates at 5 × 104 cells per 200μl for each well. Varying concentrations of Etomoxir, Orlistat, GW6471, or GW9662 were added. At 6 days post treatment, the Vybrant MTT Cell Proliferation Assay kit (Invitrogen) was used according to the manufactures protocol. Briefly, the media was replaced with 100μL of fresh culture media and 10μL of 12mM MTT stock solution per well before incubation at 34°C. After 2 hrs, 100μL SDS-HCl solution was added to each well and incubated for 2 hrs. Absorbance was read at 570nm using the Molecular Devices Spectramax M2 microplate reader.
PPARα and PPARɣ mRNA and protein expression
THP-1 macrophages were seeded in 6 well plates at 5×105-1×106 cells/mL with 4mL per well. Infections were carried out as described above at an MOI of 2. Total RNA was collected from uninfected control and R. conorii infected THP-1 macrophages at 1 hpi and 24 hpi for pparα and pparɣ mRNA expression using the PureLink RNA Mini Kit (Invitrogen). Total RNA from DMSO-, GW6471-, and GW9662-treated cells was collected. Immediately after extraction, RNA was DNase treated using the TURBO DNA-free kit (Invitrogen) before SYBR-based qRT-PCR was performed. Quantitative RT-PCR was performed using qScript One-Step SYBR Green RT-qPCR (QuantaBio) following the manufactures protocol with the PCR reaction protocol as: 10 mins at 48°C followed by 5 mins at 95°C, then 50 cycles of 10 secs at 95°C, 20 secs at 53°C, and 30 secs at 68°C, ending with a melt curve analysis from 40°C to 95°C. mRNA expression was normalized to β-actin for quantification. Primers used for pparα and pparɣ were published previously (28). The sequences for β-actin primers are forward primer: 5’-CCTGTATGCCTCTGGTCGTA-3’ and reverse primer: 5’-CCATCTCCTGCTCGAAGTCT-3’.
Whole cell lysates (WCL) were collected from uninfected control and R. conorii infected THP-1 macrophages at 1 hpi and 24 hpi. WCLs were quantified and processed as described previously (98). The membranes were blocked with 1X TBST and 2% BSA before incubation with primary antibodies: anti-PPARα (1:250; Santa Cruz Biotechnology), anti-PPARɣ (1:250; Santa Cruz Biotechnology) and anti-β-actin (1:1,000; Santa Cruz Biotechnology). After washing, the membranes were incubated with anti-Mouse horseradish peroxidase (1:25,000; Sigma-Aldrich). The membranes were visualized using chemiluminescence horseradish peroxidase substrates for film exposure. Protein detected by western immunoblot was quantified with ImageJ.
Macrophage lipid loading with oleic acid-albumin
Oleic acid (Enzo Life Sciences) was associated with albumin as previously described (99) before addition to THP-1 macrophages. THP-1 macrophages were seeded in a 24 well plate with coverslips or a 96 well plate at 5 ×105-1×106 cells per mL with 1 mL per well or 200 μL per well, respectively. After 24 hrs, oleic acid-albumin or BSA (Thermo-Scientific) was added to the corresponding cells at a concentration of 400 μM as used previously to induce a foam cell-like phenotype in THP-1 macrophages (34). Oleic acid-albumin was incubated with THP-1 cells for 3 hrs before infection with R. conorii at an MOI of 2. Rickettsial survival overtime, and LD visualization and analysis at 1 hpi was conducted as described above.
Data analysis and Statistics
Quantitative PCR analysis were analyzed by one-way ANOVA followed by Bonferroni correction post-hoc and qRT-PCR and western immunoblot results were analyzed by an one-way Student’s t-test. Significance of average LDs per host cell, average LD area, and triglyceride quantification was determined by one-way Student’s t-test for comparison of samples in each time-point independently. Significance for average LDs per host cell and average LD area in the presence of pharmacological treatments or chloramphenicol were determined by one-way ANOVA followed by Bonferroni correction post-hoc. MTT assay data was analyzed using the non-linear regression model. Statistics were performed using GraphPad Prism Version 5.0b. Significance is defined by p value *≤ 0.05, **≤ 0.05, ***≤0.001.
Supplementary Material
Take Aways.
Host lipid droplets are differentially altered in early and replicative stages of THP-1 macrophage infection with Rickettsia conorii.
Lipid droplet alterations are initiated in a bacterial-dependent manner and do not require host peroxisome proliferator-activated receptors α or ɣ activation.
Pharmacological inhibition of host lipid catabolic processes during R. conorii infection indicates a requirement of lipid catabolism for bacterial survival and initiation of lipid droplet modulation.
A significant increase in host lipid droplets during infection has a negative impact on R. conorii survival in THP-1 macrophages.
Acknowledgments
We would like to thank members of Paige Allen’s dissertation committee (Dr. Kevin Macaluso and Dr. Ronald Thune) and current (M. Nathan Kristof and Lane Yutzy) and former (Dr. Sean P. Riley [University of Maryland]) members of the Martinez lab for helpful discussions. This work was supported by an award by the National Institutes of Health, National Institute of Allergy and Infectious Diseases (grant AI072606) to JJM.
Footnotes
Competing Interest Statement: The authors declare no competing interests.
References
- 1.Hackstadt T The Biology of Rickettsiae. Infectious Agents and Disease. 1996;5(3):127–43. [PubMed] [Google Scholar]
- 2.Nicholson WL, Paddock CD. Rickettsial (spotted and typhus fevers) and related infections, including Anaplasmosis and Ehrlichiosis Center of Disease Control and Prevention; 2017 [Google Scholar]
- 3.Silverman DJ, Bond SB. Infection of Human Vascular Endothelial Cells by Rickettsia rickettsii. The journal of Infectious Diseases. 1984;149(2). [DOI] [PubMed] [Google Scholar]
- 4.Silverman DJ. Rickettsia rickettsii-Induced Cellular Injury of Human Vascular Enothelium in vitro. Infection and Immunity. 1984;44(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker DH, Ismail N. Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nat Rev Microbiol. 2008;6(5):375–86. [DOI] [PubMed] [Google Scholar]
- 6.Walker DH. Endothelial-target rickettsial infection. Lab Anim Sci. 1997;47. [PubMed] [Google Scholar]
- 7.Walker DH, Popov V, Wen J, Feng HM. Rickettsia conorii infection of C3H/HeN mice. A model of enothelial-target rickettsiosis. Lab Invest. 1994;70. [PubMed] [Google Scholar]
- 8.Walker DH. Rickettsiae and rickettsial infections: the current state of knowledge. Clin Infect Dis. 2007;45 Suppl 1:S39–44. [DOI] [PubMed] [Google Scholar]
- 9.Curto P, Simoes I, Riley SP, Martinez JJ. Differences in Intracellular Fate of Two Spotted Fever Group Rickettsia in Macrophage-Like Cells. Front Cell Infect Microbiol. 2016;6:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riley SP, Fish AI, Garza DA, Banajee KH, Harris EK, del Piero F, et al. Nonselective Persistence of a Rickettsia conorii Extrachromosomal Plasmid during Mammalian Infection. Infect Immun. 2016;84(3):790–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curto P, Riley SP, Simoes I, Martinez JJ. Macrophages Infected by a Pathogen and a Non-pathogen Spotted Fever Group Rickettsia Reveal Differential Reprogramming Signatures Early in Infection. Front Cell Infect Microbiol. 2019;9:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Curto P, Santa C, Allen P, Manadas B, Simoes I, Martinez JJ. A Pathogen and a Non-pathogen Spotted Fever Group Rickettsia Trigger Differential Proteome Signatures in Macrophages. Front Cell Infect Microbiol. 2019;9:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kristof MN, Allen PE, Yutzy LD, Thibodaux B, Paddock CD, Martinez JJ. Significant Growth by Rickettsia Species within Human Macrophage-Like Cells Is a Phenotype Correlated with the Ability to Cause Disease in Mammals. Pathogens. 2021;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price JV, Vance RE. The macrophage paradox. Immunity. 2014;41(5):685–93. [DOI] [PubMed] [Google Scholar]
- 15.Allen PE, Martinez JJ. Modulation of Host Lipid Pathways by Pathogenic Intracellular Bacteria. Pathogens. 2020;9(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Libbing CL, McDevitt AR, Azcueta RP, Ahila A, Mulye M. Lipid Droplets: A Significant but Understudied Contributor of Host(−)Bacterial Interactions. Cells. 2019;8(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nature Reveiw Molecular Cell Biology. 2018;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lodish H, Berk A, Zipursky SL, al E. Oxidation of Glucose and Fatty Acids to CO2. Molecular Cell Biology 4hth Edition. New York: W.H. Freeman; 2000. [Google Scholar]
- 19.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sathyanarayan A, Mashek MT, Mashek DG. ATGL Promotes Autophagy/Lipophagy via SIRT1 to Control Hepatic Lipid Droplet Catabolism. Cell Rep. 2017;19(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med. 2011;17(9):1076–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fozo EM, Rucks EA. The Making and Taking of Lipids: The Role of Bacterial Lipid Synthesis and the Harnessing of Host Lipids in Bacterial Pathogenesis. Adv Microb Physiol. 2016;69:51–155. [DOI] [PubMed] [Google Scholar]
- 23.Arena ET, Auweter SD, Antunes LC, Vogl AW, Han J, Guttman JA, et al. The deubiquitinase activity of the Salmonella pathogenicity island 2 effector, SseL, prevents accumulation of cellular lipid droplets. Infect Immun. 2011;79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ouimet M, Koster S, Sakowski E, Ramkhelawon B, van Solingen C, Oldebeken S, et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. 2016;17(6):677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou D, Cheng H, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell signalling. 2014;26(2):192–7. [DOI] [PubMed] [Google Scholar]
- 26.Chawla A Control of macrophage activation and function by PPARs. Circ Res. 2010;106(10):1559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong F, Pan S, Guo Y, Xu P, Zhai Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules. 2019;24(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mei C, He P, Cheng B, Liu W, Wang YF, Wan JJ. Chlamydia pneumoniae induces macrophage-derived foam cell formation via PPAR alpha and PPAR gamma-dependent pathway. Cell Biol Int. 2009;33. [DOI] [PubMed] [Google Scholar]
- 29.Crane DD, Ireland R, Alinger JB, Small P, Bosio CM. Lipids derived from virulent Francisella tularensis broadly inhibit pulmonary inflammation via toll-like receptor 2 and peroxisome proliferator-activated receptor alpha. Clin Vaccine Immunol. 2013;20(10):1531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim YS, Lee HM, Kim JK, Yang CS, Kim TS, Jung M, et al. PPAR-alpha Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J Immunol. 2017;198(8):3283–95. [DOI] [PubMed] [Google Scholar]
- 31.Cheng B, Wu X, Sun S, Wu Q, Mei C, Xu Q, et al. MAPK-PPARalpha/gamma signal transduction pathways are involved in Chlamydia pneumoniae induced macrophage-derived foam cell formation. Microb Pathog. 2014. [DOI] [PubMed] [Google Scholar]
- 32.Reza JZ, Doosti M, Salehipour M, Packnejad M, Mojarrad M, Heidari M. Modulation peroxisome proliferators activated receptor alpha (PPAR alpha) and acyl coenzyme A: cholesterol acyltransferase1 (ACAT1) gene expression by fatty acids in foam cell. Lipids Health Dis. 2009;8:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arnett E, Weaver AM, Woodyard KC, Montoya MJ, Li M, Hoang KV, et al. PPARgamma is critical for Mycobacterium tuberculosis induction of Mcl-1 and limitation of human macrophage apoptosis. PLoS Pathog. 2018;14(6):e1007100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agarwal P, Combes TW, Shojaee-Moradie F, Fielding B, Gordon S, Mizrahi V, et al. Foam Cells Control Mycobacterium tuberculosis Infection. Front Microbiol. 2020;11:1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng B, Wu X, Sun S, Wu Q, Mei C, Xu Q, et al. MAPK-PPARalpha/gamma signal transduction pathways are involved in Chlamydia pneumoniae-induced macrophage-derived foam cell formation. Microb Pathog. 2014;69–70:1–8. [DOI] [PubMed] [Google Scholar]
- 36.Rosas-Ballina M, Guan XL, Schmidt A, Bumann D. Classical Activation of Macrophages Leads to Lipid Droplet Formation Without de novo Fatty Acid Synthesis. Front Immunol. 2020;11:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu H, Han Y, Rodriguez Sillke Y, Deng H, Siddiqui S, Treese C, et al. Lipid droplet-dependent fatty acid metabolism controls the immune suppressive phenotype of tumor-associated macrophages. EMBO Mol Med. 2019;11(11):e10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cocchiaro JL, Kumar Y, Fischer ER, Hackstadt T, Valdivia RH. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc Natl Acad Sci U S A. 2008;105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol. 2006;16(16):1646–51. [DOI] [PubMed] [Google Scholar]
- 40.Choi J, Marks J, Zhang J, Chen DH, Wang J, Vazquez-Laslop N, et al. Dynamics of the context-specific translation arrest by chloramphenicol and linezolid. Nat Chem Biol. 2020;16(3):310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raoult D, Roussellier P, Vestris G, Tamalet J. In vitro antibiotic susceptibility of Rickettsia rickettsii and Rickettsia conorii: plaque assay and microplaque colorimetric assay. Jounral of Infectious Disease. 1987;155. [DOI] [PubMed] [Google Scholar]
- 42.Rolain J, Maurin M, Vestris G, Raoult D. In Vitro Susceptibilities of 27 Rickettsiae to 13 Antimicrobials. Antimicrobial Agents and Chemotherapy. 1998;42(7):1537–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Heinzen RA, Hayes SF, Peacock MG, Hackstadt T. Directional Actin Polymerization Associated with Spotted Fever Group Rickettsia Infection of Vero Cells. Infect Immun. 1993;61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20(3):137–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Genoula M, Marin Franco JL, Maio M, Dolotowicz B, Ferreyra M, Milillo MA, et al. Fatty acid oxidation of alternatively activated macrophages prevents foam cell formation, but Mycobacterium tuberculosis counteracts this process via HIF-1alpha activation. PLoS Pathog. 2020;16(10):e1008929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tongluan N, Ramphan S, Wintachai P, Jaresitthikunchai J, Khongwichit S, Wikan N, et al. Involvement of fatty acid synthase in dengue virus infection. Virol J. 2017;14(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hitakarun A, Khongwichit S, Wikan N, Roytrakul S, Yoksan S, Rajakam S, et al. Evaluation of the antiviral activity of orlistat (tetrahydrolipstatin) against dengue virus, Japanese encephalitis virus, Zika virus and chikungunya virus. Sci Rep. 2020;10(1):1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esser K, Lucifora J, Wettengel J, Singethan K, GLinzer A, Zernecke A, et al. Lipase inhibitor orlistat prevents hepatitis B virus infection by targeting an early step in the virus life cycle. Antiviral Research. 2018;151. [DOI] [PubMed] [Google Scholar]
- 49.Ammer E, Nietzsche S, Rien C, Kuhnl A, Mader T, Heller R, et al. The anti-obesity drug orlistat reveals anti-viral activity. Med Microbiol Immunol. 2015;204(6):635–45. [DOI] [PubMed] [Google Scholar]
- 50.Xiong Q, Lin M, Huang W, Rikihisa Y. Infection by Anaplasma phagocytophilum Requires Recruitment of Low-Density Lipoprotein Cholesterol by Flotillins. mBio. 2019;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hack AM, Yanovaki JA, Calis KA. Orlistat, a New Lipase Inhibitor for the Management of Obesity. Pharmacotherapy. 2000;20(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schlaepfer IR, Rider L, Rodrigues LU, Gijon MA, Pac CT, Romero L, et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol Cancer Ther. 2014;13(10):2361–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chandra P, He L, Zimmerman M, Yang G, Koster S, Ouimet M, et al. Inhibition of Fatty Acid Oxidation Promotes Macrophage Control of Mycobacterium tuberculosis. mBio. 2020;11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eisele NA, Ruby T, Jacobson A, Manzanillo PS, Cox JS, Lam L, et al. Salmonella require the fatty acid regulator PPARdelta for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe. 2013;14(2):171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jordan TX, Randall G. Dengue Virus Activates the AMP Kinase-mTOR Axis To Stimulate a Proviral Lipophagy. J Virol. 2017;91(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018;28(3):490–503 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu X, Li X, Zhao G, Xiao J, Mo Z, Yin K, et al. OxLDL up-regulates Niemann-Pick type C1 expression through ERK1/2/COX-2/PPARalpha-signaling pathway in macrophages. Acta Biochim Biophys Sin (Shanghai). 2012;44(2):119–28. [DOI] [PubMed] [Google Scholar]
- 58.Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415. [DOI] [PubMed] [Google Scholar]
- 59.Leopold Wager CM, Arnett E, Schlesinger LS. Mycobacterium tuberculosis and macrophage nuclear receptors: What we do and don’t know. Tuberculosis (Edinb). 2019;116S:S98–S106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zizzo G, Cohen PL. The PPAR-gamma antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas6 axis: a key role for PPAR-gamma in human macrophage polarization. J Inflamm (Lond). 2015;12:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xavier MN, Winter MG, Spees AM, den Hartigh AB, Nguyen K, Roux CM, et al. PPARgamma-mediated increase in glucose availability sustains chronic Brucella abortus infection in alternatively activated macrophages. Cell Host Microbe. 2013;14(2):159–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han S, Sidell N. Peroxisome-proliferator-activated-receptor gamma (PPARc) independent induction of CD36 in THP-1 monocytes by retinoic acid. Immunology. 2002;106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heaton NS, Perera R, Berger KL, Khadka S, Lacount DJ, Kuhn RJ, et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc Natl Acad Sci U S A. 2010;107(40):17345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8(5):422–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang W, Hood BL, Chadwick SL, Liu S, Watkins SC, Luo G, et al. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology. 2008;48(5):1396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miyanari Y, Atsuzawa K, Usuda N, Watachi K, Hishiki T, Zayas M, et al. The lipid droplet is an important organelle for hepatitis C virus production. Nature Cell Biology. 2007;9. [DOI] [PubMed] [Google Scholar]
- 67.Zhang J, Zhang Z, Chukkapalli V, Nchoutmboube JA, Li J, Randall G, et al. Positive-strand RNA viruses stimulate host phosphatidylcholine synthesis at viral replication sites. Proc Natl Acad Sci U S A. 2016;113(8):E1064–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cao F, Castrillo A, Tontonoz P, Re F, Byrne GI. Chlamydia pneumoniae—Induced macrophage foam cell formation is mediated by Toll-like receptor 2. Infect Immun. 2007;75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buchacher T, Ohradanova-Repic A, Stockinger H, Fischer MB, Weber V. M2 Polarization of Human Macrophages Favors Survival of the Intracellular Pathogen Chlamydia pneumoniae. PLoS One. 2015;10(11):e0143593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toledo A, Benach JL. Hijacking and Use of Host Lipids by Intracellular Pathogens. Microbiol Spectr. 2015;3(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Winkler H, Miller E. Phospholipid Composition of Rickettsia prowazeki Grown in Chicken Embryo Yolk Sacs. Journal Bacteriology. 1978;136(1):175–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zezerov E, Loginov V, Berezneva A. Polypeptide and phospholipid composition of the Rickettsia prowazekii membrane and its immunogenic properties. Zhurnal Mikrobiologii, Epidemiologii, i Immunobiologii. 1985;6:6–13. [PubMed] [Google Scholar]
- 73.Driscoll TP, Verhoeve VI, Guillotte ML, Lehman SS, Rennoll SA, Beier-Sexton M, et al. Wholly Rickettsia! Reconstructed Metabolic Profile of the Quintessential Bacterial Parasite of Eukaryotic Cells. MBio. 2017;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rydkina E, Sahni A, Baggs RB, Silverman DJ, Sahni SK. Infection of human endothelial cells with spotted Fever group rickettsiae stimulates cyclooxygenase 2 expression and release of vasoactive prostaglandins. Infect Immun. 2006;74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 2011;7(6):e1002093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barisch C, Soldati T. Breaking fat! How mycobacteria and other intracellular pathogens manipulate host lipid droplets. Biochimie. 2017;141:54–61. [DOI] [PubMed] [Google Scholar]
- 77.Walenna NF, Kurihara Y, Chou B, Ishii K, Soejima T, Itoh R, et al. Chlamydia pneumoniae exploits adipocyte lipid chaperone FABP4 to facilitate fat mobilization and intracellular growth in murine adipocytes. Biochem Biophys Res Commun. 2018;495(1):353–9. [DOI] [PubMed] [Google Scholar]
- 78.Mulye M, Zapata B, Gilk SD. Altering lipid droplet homeostasis affects Coxiella burnetii intracellular growth. PLoS One. 2018;13(2):e0192215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Long AP, Manneschmidt AK, VerBrugge B, Dortch MR, Minkin SC, Prater KE, et al. Lipid droplet de novo formation and fission are linked to the cell cycle in fission yeast. Traffic. 2012;13(5):705–14. [DOI] [PubMed] [Google Scholar]
- 80.Kwok AC, Wong JT. Lipid biosynthesis and its coordination with cell cycle progression. Plant Cell Physiol. 2005;46(12):1973–86. [DOI] [PubMed] [Google Scholar]
- 81.Soupene E, Wang D, Kuypers FA. Remodeling of host phosphatidylcholine by Chlamydia acyltransferase is regulated by acyl-CoA binding protein ACBD6 associated with lipid droplets. Microbiologyopen. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gillespie JJ, Kaur SJ, Rahman MS, Rennoll-Bankert K, Sears KT, Beier-Sexton M, et al. Secretome of obligate intracellular Rickettsia. FEMS Microbiol Rev. 2015;39(1):47–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Driskell LO, Yu XJ, Zhang L, Liu Y, Popov VL, Walker DH, et al. Directed mutagenesis of the Rickettsia prowazekii pld gene encoding phospholipase D. Infect Immun. 2009;77(8):3244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rahman MS, Ammerman NC, Sears KT, Ceraul SM, Azad AF. Functional characterization of a phospholipase A(2) homolog from Rickettsia typhi. J Bacteriol. 2010;192(13):3294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Renesto P, Dehoux P, Gouin E, Touqui L, Cossart P, Raoult D. Identification and Characterization of a Phospholipase D–Superfamily Gene in Rickettsiae. [DOI] [PubMed] [Google Scholar]
- 86.Walker DH, Feng HM, Popov VL. Rickettsial Phospholipase A2 as a Pathogenic Mechanism in a Model of Cell Injury by Typhus and Spotted Fever Group Rickettsiae. The American Journal of Tropical Medicine and Hygeine. 2001;65(6):936–42. [DOI] [PubMed] [Google Scholar]
- 87.Jaisinghani N, Dawa S, Singh K, Nandy A, Menon D, Bhandari PD, et al. Necrosis Driven Triglyceride Synthesis Primes Macrophages for Inflammation During Mycobacterium tuberculosis Infection. Front Immunol. 2018;9:1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mege JL, Mehraj V, Capo C. Macrophage polarization and bacterial infections. Curr Opin Infect Dis. 2011;24(3):230–4. [DOI] [PubMed] [Google Scholar]
- 89.Yang T, Chen J, Gao L, Huang Y, Liao G, Cao Y. Induction of lipid droplets in THP-1 macrophages by multi-walled carbon nanotubes in a diameter-dependent manner: A transcriptomic study. Toxicology Letters. 2020;332(10). [DOI] [PubMed] [Google Scholar]
- 90.Souza-Moreira L, Soares VC, Dias S, Bozza PT. Adipose-derived Mesenchymal Stromal Cells Modulate Lipid Metabolism and Lipid Droplet Biogenesis via AKT/mTOR -PPARgamma Signalling in Macrophages. Sci Rep. 2019;9(1):20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saka HA, Thompson JW, Chen YS, Dubois LG, Haas JT, Moseley A, et al. Chlamydia trachomatis Infection Leads to Defined Alterations to the Lipid Droplet Proteome in Epithelial Cells. PLoS One. 2015;10(4):e0124630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sharma M, Recuero-Checa MA, Fan FY, Dean D. Chlamydia trachomatis regulates growth and development in response to host cell fatty acid availability in the absence of lipid droplets. Cell Microbiol. 2018;20(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11. [DOI] [PubMed] [Google Scholar]
- 94.Walpole GFW, Grinstein S, Westman J. The role of lipids in host-pathogen interactions. IUBMB Life. 2018;70(5):384–92. [DOI] [PubMed] [Google Scholar]
- 95.Chan YG, Cardwell MM, Hermanas TM, Uchiyama T, Martinez JJ. Rickettsial outer-membrane protein B (rOmpB) mediates bacterial invasion through Ku70 in an actin, c-Cbl, clathrin and caveolin 2-dependent manner. Cell Microbiol. 2009;11(4):629–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ammerman NC, Beier-Sexton M, Azad AF. Laboratory maintenance of Rickettsia rickettsii. Curr Protoc Microbiol. 2008;Chapter 3:Unit 3A 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chan YG, Riley SP, Chen E, Martinez JJ. Molecular basis of immunity to rickettsial infection conferred through outer membrane protein B. Infect Immun. 2011;79(6):2303–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Martinez JJ, Cossart P. Early signaling events involved in the entry of Rickettsia conorii into mammalian cells. J Cell Sci. 2004;117(Pt 21):5097–106. [DOI] [PubMed] [Google Scholar]
- 99.Brasaemle DL, Wolins NE. Isolation of Lipid Droplets from Cells by Density Gradient Centrifugation. Curr Protoc Cell Biol. 2016;72:3 15 1–3 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.