

Abstract
The molecular chaperone FKBP51 is gaining attention as a meaningful biomarker of metabolic dysfunction. This review examines the emerging contributions of FKBP51 in adipogenesis and lipid metabolism, myogenesis and protein catabolism, and glucocorticoid-induced skin hypoplasia and dermal adipocytes. The FKBP51 signaling mechanisms that may explain these metabolic consequences are discussed. These mechanisms are diverse, with FKBP51 independently and directly regulating phosphorylation cascades and nuclear receptors. We provide a discussion of the newly developed compounds that antagonize FKBP51, which may offer therapeutic advantages for adiposity. These observations suggest we are only beginning to uncover the complex nature of FKBP51 and its molecular chaperoning of metabolism.
Keywords: FKBP5, FKBP4, AKT, obesity, adipose, diabetes
Introduction
Early models of the larger molecular mass FK506 binding proteins (FKBPs) revolved around nuclear receptor (NR) cellular signaling, revealing two major FKBP proteins bound, FKBP51 and FKBP52 [1–3]. The gene nomenclature for these proteins are FKBP5 and FKBP4, respectively, and they are located on independent chromosomes, 6p21.31 and 12p13.33. While their genes are distantly located, the two proteins are closely related, and both have peptidyl-prolyl cis-trans isomerase (PPIase) activity that is inhibited by immunosuppressant ligands, such as FK506 and rapamycin. Hence, they are referred to as ‘immunophilins.’ The immunophilin class contains a larger group of proteins involved in immunoregulation and the cellular processes of protein folding and trafficking. In general, these processes are dependent on their PPIase activity. Interestingly, PPIase action is not required for FKBP regulation of nuclear receptors [4]. However, PPIase activity is likely essential for controlling other proteins and signaling cascades, as discussed below.
FKBP51 and FKBP52 are also classified as tetratricopeptide repeat (TPR) proteins based on the presence of this protein-protein interaction motif, used for binding diverse client proteins. While they both interact with the same nuclear receptor complexes, they exert divergent effects on NR transcriptional activities [1, 2] – a fact that serves as clues to their possible physiological functions. Studies in humans have associated FKBP5 with select diseases, such as major depressive disorder, asthma, obesity, and type II diabetes (for reviews of FKBP51 and mood disorders, see refs: [3, 5, 6]). In contrast, FKBP4 has been linked to male and female infertility [7–9], Parkinson’s disease [10, 11], and adenovirus-associated virus (AAV) optimization in human gene therapy [12]. The tissue distribution of these genes correlates with their diverse functions. FKBP4 is primarily expressed in the testis, kidney, brain, immune system, and liver. In contrast, FKBP5 is highest in adipocytes, skeletal muscle, and the immune system (see below). Based on this profile, it is not surprising that animal and cell culture studies have uncovered new and potentially important roles for FKBP51 in metabolism.
Because of the association of FKBP51 with the glucocorticoid receptor (GR) and peroxisome proliferator-activated receptor-γ (PPARγ), early models of FKBP51 action revolved around NR control of metabolism. We now know that FKBP51 is pleiotropic in its effects and mechanisms [13]. Through its TPR-based scaffolding function, FKBP51 regulates a variety of kinase cascades, most notably insulin signaling via AKT. This review will start with an examination of the major cellular and physiological roles of FKBP51 and conclude with mechanisms that may explain the emerging effects of the chaperone on metabolism.
I. FKBP51 - A Key Regulator of Adipogenesis and Metabolism
A. A Bona Fide Adipocyte Marker
Factors that control gene expression during adipocyte expansion are critical to understanding the mechanisms that influence weight gain and obesity. It is well understood that PPARγ is the primary NR responsible for controlling lipogenesis and lipid accumulation in fat tissues that leads to adipocyte expansion. Cofactors that regulate PPARγ during this process serve as potential therapeutics. One such cofactor is another TPR protein that directly mediates PPARγ phosphorylation, protein phosphatase 5 (PP5). Investigations on PP5 knockout (KO) mouse embryonic fibroblast (MEF) cells showed that PP5 directly dephosphorylates the inhibitory serine 112 (Ser112) of PPARγ to activate transcription [14]. Intriguingly, PP5, like PPAR-γ, can be activated by fatty acids and related compounds, further adding to its importance in metabolism [15–17]. These facts seemingly make PP5 a potential drug target for control of adiposity by indirectly inhibiting PPARγ and adipogenesis [14]. However, therapeutic targeting of PP5 will likely be problematic because it is ubiquitous and regulates numerous client proteins [18, 19], increasing the potential for off-target effects. This may not be the case for FKBP51. The tissue distribution of FKBP5 is much more restricted, with the highest levels in adipose tissue of humans (Figure 1). Murine FKBP51 protein expression significantly increases in 3T3-L1 cells during adipose differentiation [20, 21], while FKBP52 expression is almost completely lost [20]. These facts suggest that an inverse and reciprocal relationship exists between FKBP51 and FKBP52 in the control of adipogenesis. Differentiated FKBP51 KO MEFs exhibited substantially less lipid accumulation (>10-fold lower) and markedly reduced PPARγ transcriptional activity compared to MEFs with intact FKBP51 [20]. Important supporting data was found in FKBP51 null mice, which are resistant to diet-induced obesity [22, 23] and have reduced responses to the PPARγ agonist rosiglitazone [22]. These results posit that FKBP51 is an essential marker of adipocyte differentiation and a mechanism for titrated control of PPAR-γ activity, as complete blockade of PPARγ is known to cause lipodystrophy [24].
Figure 1. FKBP5 expression profile in humans.
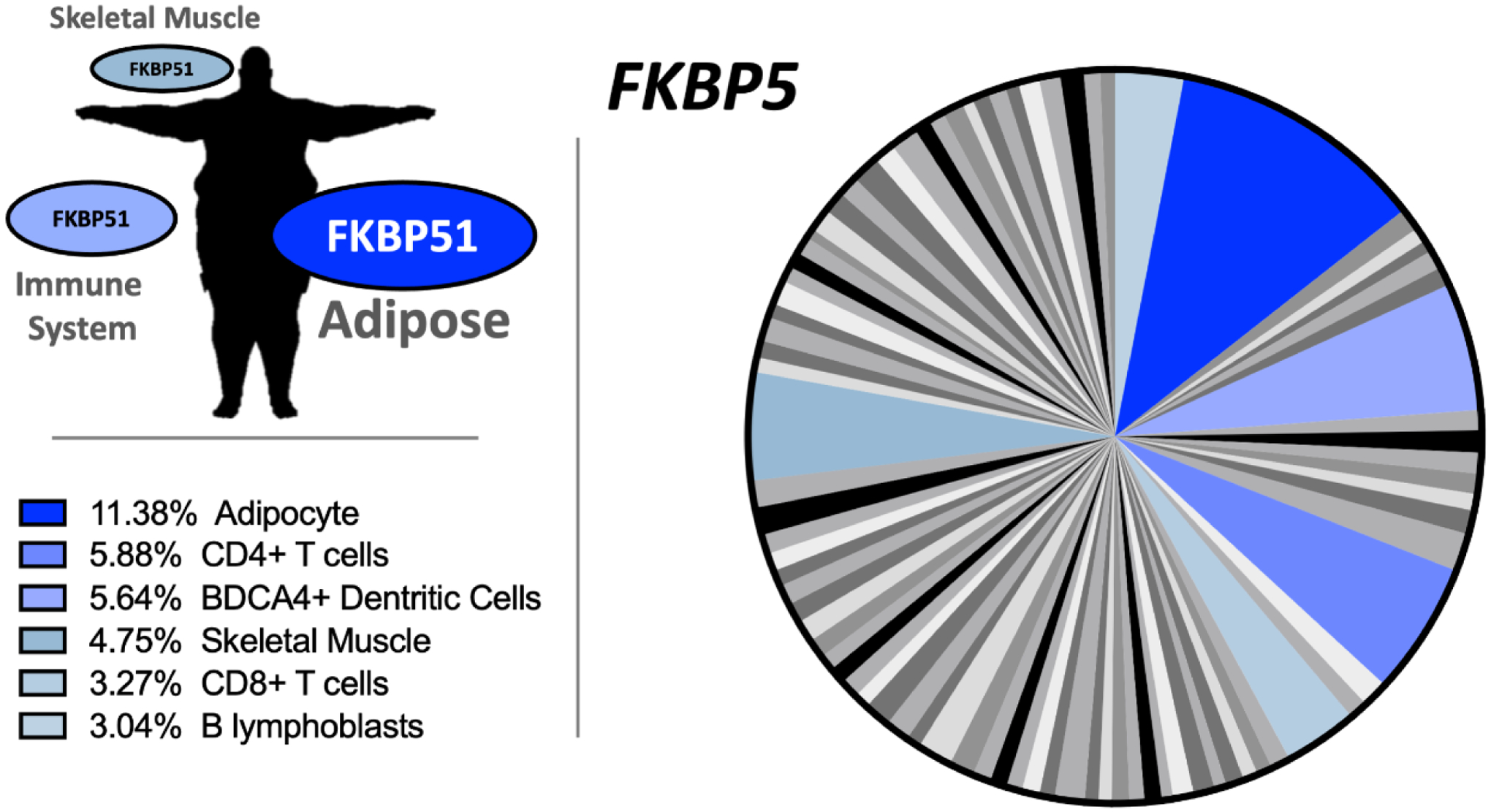
The pie chart with white, gray, black, and blue labeling is a tissue array for FKPB5 expression in 84 human tissues, with blue indicating the highest levels. The color code for the pie chart and percentages are provided in the boxes to the bottom left. The expression of FKBP51 in human tissues is highest in adipose, immune cells, and skeletal muscle. High-throughput gene expression data was downloaded for FKBP5 in human tissues from http://biogps.org [84, 85].
B. Drug Targeting of FKBP51 and Adipogenesis
We have shown that FKBP51 functions as a co-chaperone that reciprocally regulates GR and PPARγ [25]. In brief, FKBP51 acts as a negative transcriptional regulator of GR and a positive regulator of PPARγ signaling. The data also suggested that FKBP51 serves to inhibit GR-induced lipolysis of stored lipids in white adipose tissue (WAT) and to promote PPARγ-induced adipogenesis and lipid storage. For these reasons, compounds that bind FKBP51, such as FK506, rapamycin, and Timcodar (also known as VX-853) [26, 27], may directly influence adipogenesis or lipid storage (Figure 2). Indeed, studies have shown that Timcodar, a specific inhibitor of FKBP51 [28], significantly reduced lipid accumulation and adipogenesis in the 3T3-L1 adipocyte model [26]. Timcodar potentiated GR transcriptional responses in MEF cells treated with dexamethasone [28], a commonly used glucocorticoid. Timcodar and FK506 had different effects on adipogenesis of 3T3-L1 cells, as Timcodar robustly suppressed, while FK506 slightly raised lipid accumulation [26]. FK506 increased the expression of fatty acid-binding protein 4 (FABP4) and stearoyl-CoA desaturase-1 (SCD1), a rate-limiting enzyme in the formation of fatty acids. Timcodar, however, suppressed FABP4 and SCD1, as well as PPARγ protein and mRNA expression while increasing mRNA expression of the glucocorticoid binding isoform, GRα, and its target genes [26]. Timcodar did not affect the inhibitory GR isoform, GRβ [29–31], but did increase the GRα/GRβ ratio and GR target genes [26]. While rapamycin was also a potent suppressor of adipogenesis and has been shown to reduce body weight in animal models of obesity, it induces glucose intolerance, which might occur by its interaction with FKBP12 and mTOR [32]. FK506 also binds FKBP12, which might pose similar complications. These side effects significantly reduce rapamycin’s potential use in the treatment of obesity.
Figure 2. Targeting of FKBP51 in humans to improve metabolic dysfunction.
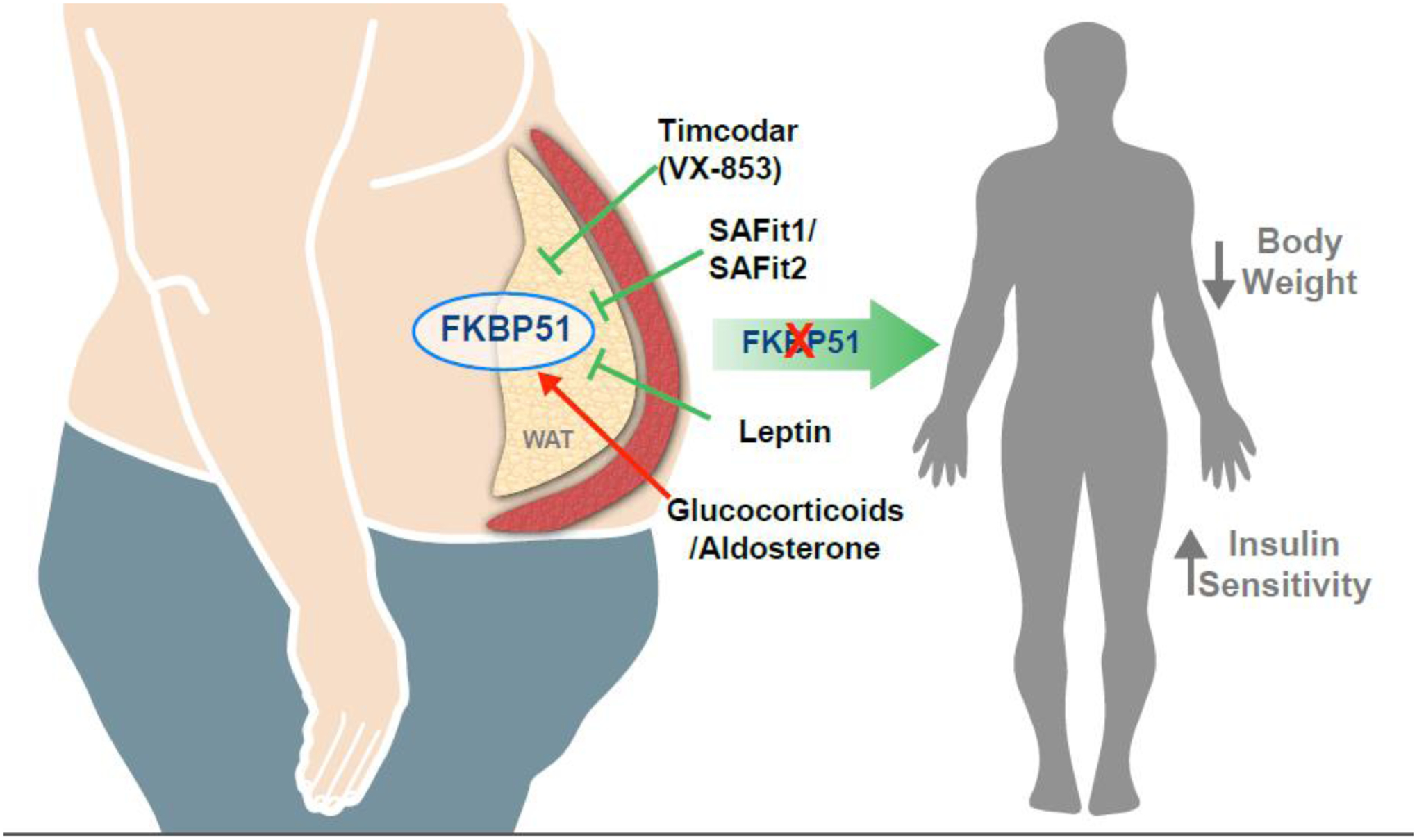
White adipose tissue (WAT) has a high expression of FKBP51, which may occur from stress-related glucocorticoids, aldosterone, weight gain, or metabolic dysfunction. The FKBP51 protein can be inhibited by Timcodar (VX-853), SAFit1/SAFit2, and leptin, which might offer potential therapeutic avenues for metabolic dysfunction. Blockade of FKBP51 is known to reduce body weight and improve insulin sensitivity.
The graphical illustration was developed by Matthew Hazzard at the University of Kentucky College of Medicine.
FK506 and rapamycin both have a macrocyclic linkage that allows them to bind to FKBP12 [26], the smaller FKBP that does not bind to nuclear receptor complexes but regulates immune function [33]. The structure of Timcodar does not contain the macrocyclic linkage, and studies have shown that it targets only the larger FKBP proteins, especially FKBP51 [28]. Others have also generated FKBP51-specific antagonists, such as SAFit1 and SAFit2, which was achieved using an induced-fit mechanism for FKBP51 that is less favorable for FKBP52 [34]. Balsevich et al. showed that a 30-day treatment with SAFit2 in C57/bl6 mice fed a high-fat diet reduced body weight and increased glucose sensitivity [23]. More work on FKBP51 antagonists is needed to reveal their long-term consequences and potential side effects, especially in preclinical animal models of obesity and diabetes (see Outstanding Questions).
OUTSTANDING QUESTIONS.
Besides glucocorticoid, fatty acid, and insulin signaling, does FKBP51 control additional nuclear receptors or kinase cascades in adipocytes?
What is the precise function of FKBP51 in the regulation of lipogenesis and fat storage?
Do single nucleotide polymorphisms or FKBP51 over-expression contribute to insulin-resistant diabetes?
Will drug antagonism of FKBP51 have a positive effect on obesity or diabetes?
Can glucocorticoid-induced myopathy or skin thinning be ameliorated by inhibition of FKBP51?
C. FKBP51 in Obesity and Diabetes
Human studies have linked the FKBP5 gene with metabolic diseases, suggesting that both rodents and humans may have the same role for FKPB51 in adipogenesis. Sidibeh et al. showed that in humans with Type 2 diabetes, FKBP5 expression was increased in subcutaneous adipose tissue and associated with lipid metabolism and adipogenesis [35]. A study by Pereira et al. found that FKBP5 expression was increased 7.76-fold in subcutaneous and 7.69-fold in omental fat depots of humans with dexamethasone exposure [36], validating GR-mediated regulation of FKBP5 in cell and mouse models. The same study also identified 12 single nucleotide polymorphisms (SNPs) in the FKBP5 gene associated with type II diabetes, triglyceride accumulation, and altered cholesterol blood levels in humans. Others have identified that the rs1360780 SNP in the FKBP5 gene is associated with reduced weight loss after bariatric surgery [37]. The human studies are in line with recent work showing that Fkbp5 null mice have reduced body weight compared to wild-type mice and are resistant to diet-induced obesity [22, 23]. On the contrary, leptin-deficient ob/ob genetically obese mice have a high expression of FKBP51 in WAT that is reduced after 12 days of leptin treatment [38]. The latter suggests that FKBP51 is involved in satiety and hunger, as this is a known role for leptin signaling in the adipose-brain axis. Adeno-associated virus (AAV) overexpression of FKBP51 in the hypothalamus of C57/bl6 mice induced significantly more weight gain on a high-fat diet compared to mCherry AAV control [39]. However, no differences were observed in food intake. Interestingly, adipose-derived leptin is an activator of aldosterone secretion [40] – a mineralocorticoid hormone generated in the adrenal cortex that is elevated in obesity [41]. Aldosterone treatment of adrenalectomized rats increased FKBP51 expression in the colon, kidney, and heart [42]. Interestingly, in aldosterone- or dexamethasone-treated rats, cotreatment with the mineralocorticoid receptor (MR) antagonist, spironolactone, inhibited induction of FKBP51, suggesting that MR, like GR, is an inducer of the Fkbp5 gene. FKBP51 is known to chaperone MR [43], but no investigations have uncovered cellular or physiological roles of the interaction, especially in adipocytes. We speculate that glucocorticoid and mineralocorticoid induction of FKBP51 in adipocytes may be a negative feedback loop, or it may serve to regulate other signaling mechanisms, such as kinase cascades (see below). Since MR is a significant factor in adipocyte expansion [28, 29], future studies on MR-FKBP51 signaling in adipocytes are needed, as are studies to delineate the differences of between GR/FKBP51 and MR/FKBP51 complexes in adipocytes.
Altogether, these studies suggest that stress-induced corticosteroids may prime fat depots to weight gain by a FKBP51 mechanism, which might be reversible by antagonism of FKBP51 (Figure 2). The investigations further posit that FKBP51 is a promising lead candidate in drug targeting for obesity and adipogenesis. More work on FKBP51 antagonists, such as Timcodar, SAFit1, SAFit2, and related variants, is needed to reveal their actions and possible side effects in animal models of obesity and diabetes. Metabolic studies of tissue-specific Fkbp5 knockout mice are needed to delineate its functions in adipose, muscle, and skin (further discussed below).
D. The FKBP51-AKT Metabolic Signaling Axis in Adipocytes
Early studies found that FKBP51 interacts with unliganded NR complexes to control a variety of functions, including hormone-binding affinity, intracellular location, and, ultimately, transcriptional activity [2, 44–46]. More recent work has identified an additional pathway of FKBP51 action via direct regulation of kinases, such as AKT, which leads to phosphorylation-mediated regulation of NRs and, most likely, other regulatory factors. AKT (also known as protein kinase B, PKB) is involved in signaling pathways that control glucose and lipid metabolism, and for some of the AKT isoforms, growth. There are three AKT isoforms, AKT1, AKT2, and AKT3, which have varying functions. AKT1 is widely expressed across tissues and is mainly recognized for its function in cell growth and survival [47, 48]. AKT2 is primarily expressed in metabolic tissues, such as adipose, muscle, and liver, and is the primary component that mediates insulin signaling and glucose sensitivity [48]. Hence, AKT2 is essential to glucose homeostasis [48]. The least studied isoform is AKT3, which most likely functions in the brain, as AKT3 knockout animals have impaired brain development [49]. Where AKT2 and FKBP51 expression coincide, there may be a functional relationship that modulates insulin signaling, glucose uptake, and, possibly, adipogenesis.
AKT1 and AKT2 activity are dependent on the phosphorylation of serine 473 (Ser473), which is regulated by mTOR and the AKT-specific kinases in the PI3 kinase family (PI3K) (Box I) [50]. Phosphatases also exist that dephosphorylate Ser473 to inhibit activity, such as the PH domain and leucine-rich repeat protein phosphatase (PHLPP) [51]. Recent work has shown that FKBP51 serves as a scaffolding protein that recruits PHLPP to AKT to facilitate dephosphorylation of Ser473, leading to inactivation [52]. Activated AKT2 is essential in regulating glucose and lipid metabolism within insulin-responsive tissues [48]. Under insulin stimulation in adipose and muscle tissues, AKT2 phosphorylates the AKT Substrate of 160 kDa (AS160, also known as TBC1D1) [48], which drives the translocation of glucose transporter-4 (GLUT4) to the plasma membrane to import glucose (Box I) [53]. Our studies have demonstrated that the overexpression of FKBP51 reduces the phosphorylation of AKT at Ser473, suppressing activity [25]. FKBP51 knockout animals fed a high-fat diet remain glucose-sensitive, while wild-type counterparts with intact FKPB51 develop glucose intolerance [22]. Obese and insulin-resistant mice treated with the SAFit2 FKBP51 antagonist have improved glucose sensitivity and significantly increased phosphorylation of AS160 and AKT2 [23]. These facts are strong evidence that FKBP51 is a negative regulator of insulin/AKT signaling and possibly an essential factor in adiposity and insulin-resistant diabetes (see Outstanding Questions).
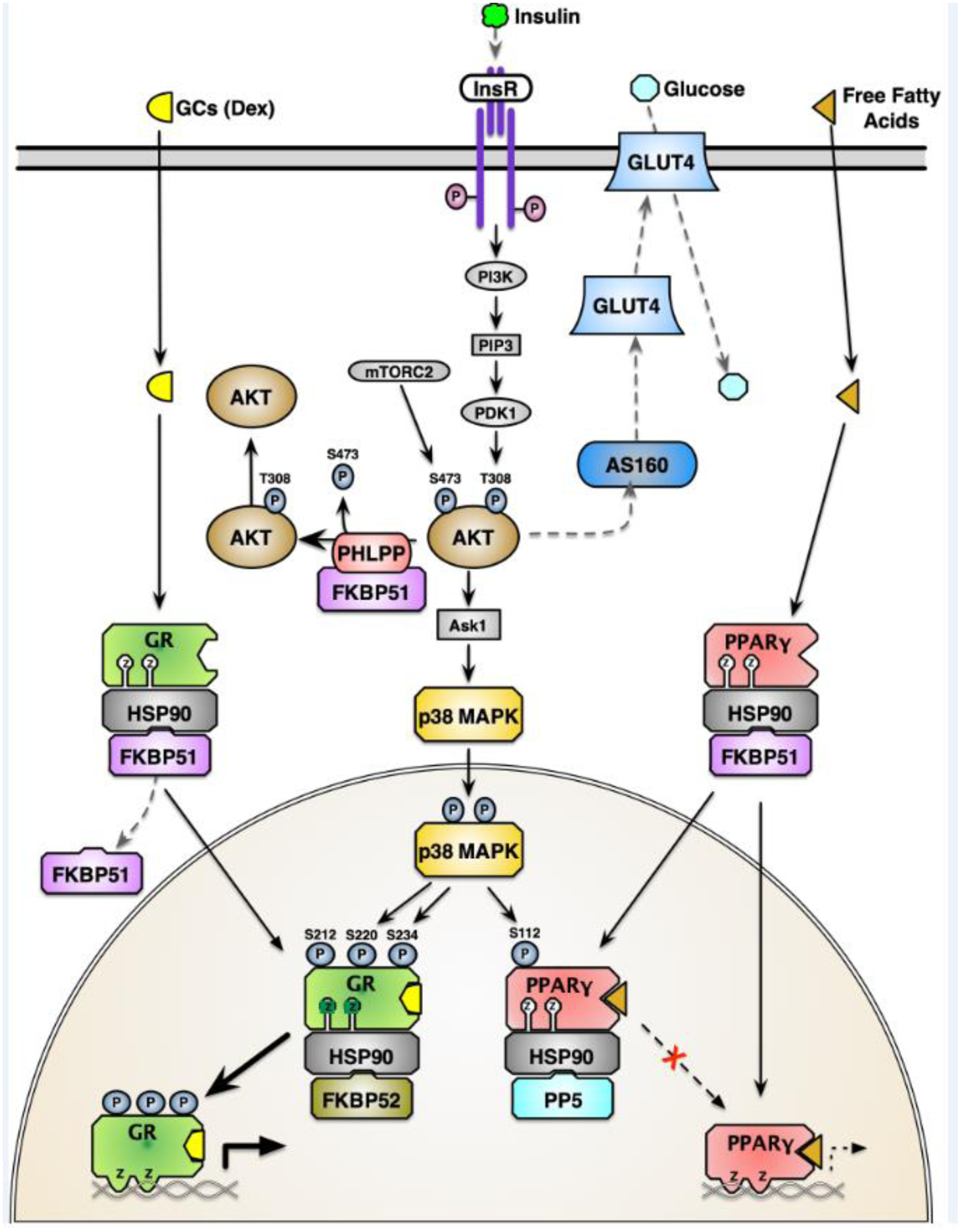
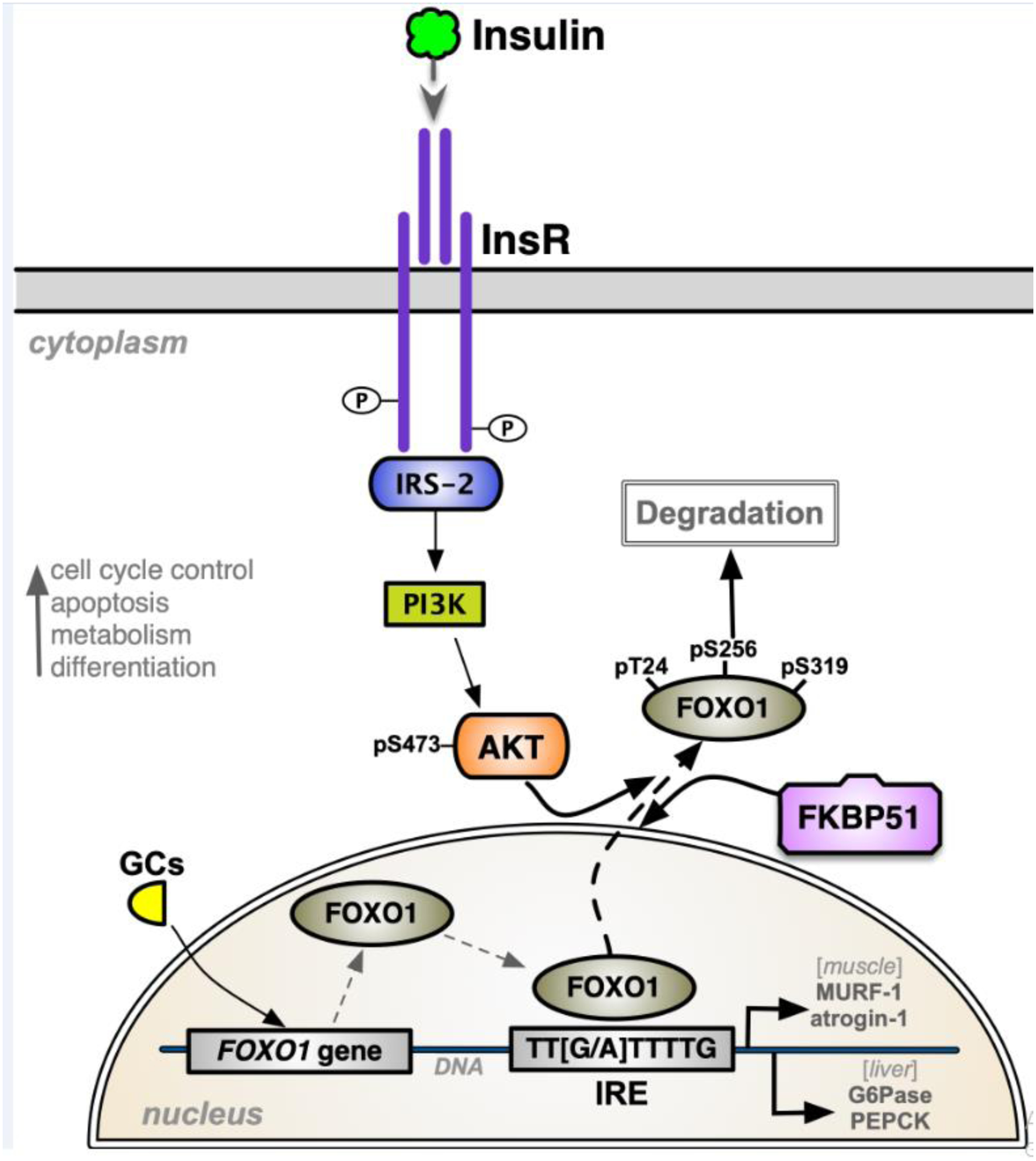
Box I. Converging FKBP51 signaling mechanisms control metabolism.
Insulin stimulation of the insulin receptor (InsR) activates the PI3 kinase (PI3K) pathway, stimulating PDK1 to phosphorylate AKT at threonine 308 (T308). Concomitantly, mTORC2 phosphorylates AKT at serine 473 (S473), increasing kinase activity which can either drive Ask1 phosphorylation or phosphorylate AS160 that activates GLUT4 translocation to the cell membrane to bring in glucose. p38 MAPK regulates phosphorylation of the glucocorticoid receptor (GR) and PPARγ. Independent of kinase cascades, FKBP51 is part of nuclear receptor heterocomplexes containing heat shock protein 90 (HSP90) and serves to sequester GR and PPARγ to the cytoplasm and away from p38 targeting in the nucleus. Upon glucocorticoid (GC) binding, GR dissociates FKBP51 and binds FKBP52, which is required for nuclear translocation. Ligand binding of PPARγ replaces FKBP51 with protein phosphatase 5 (PP5), which dephosphorylates Serine 112 (S112), activating transcriptional activity. Phosphorylation of GR activates transcriptional activity. However, phosphorylation of S112 inhibits transcriptional activity.
The actions of FKBP51 as a negative regulator of GR and a positive regulator of PPARγ may be explained by its role in controlling the AKT/p38 axis (Figure 3) [25]. In the absence of FKBP51, PHLPP activity is reduced, causing increased AKT signaling to the downstream target p38, which in turn increases GRα activity to promote lipolysis and PPARγ-induced adipogenesis [25]. In FKBP51 KO MEF cells, GR phosphorylation at serines 220 and 234 was significantly higher, promoting increased expression of prolipolytic genes [25]. Lentiviral knockdown of FKBP51 in 3T3-L1 pre-adipocytes elevated PPARγ phosphorylation at serine 112, a phospho-residue that inhibits activity [25]. This mechanism for alteration of PPARγ and GR activities is one potential mechanism to explain resistance to adipogenesis in FKBP51 KO cells [20] and suggests that FKBP51 is a vital regulator of the cellular and physiological processes controlled by each receptor.
Figure 3. FKBP51 signaling via nuclear receptors to control lipid metabolism in adipose.
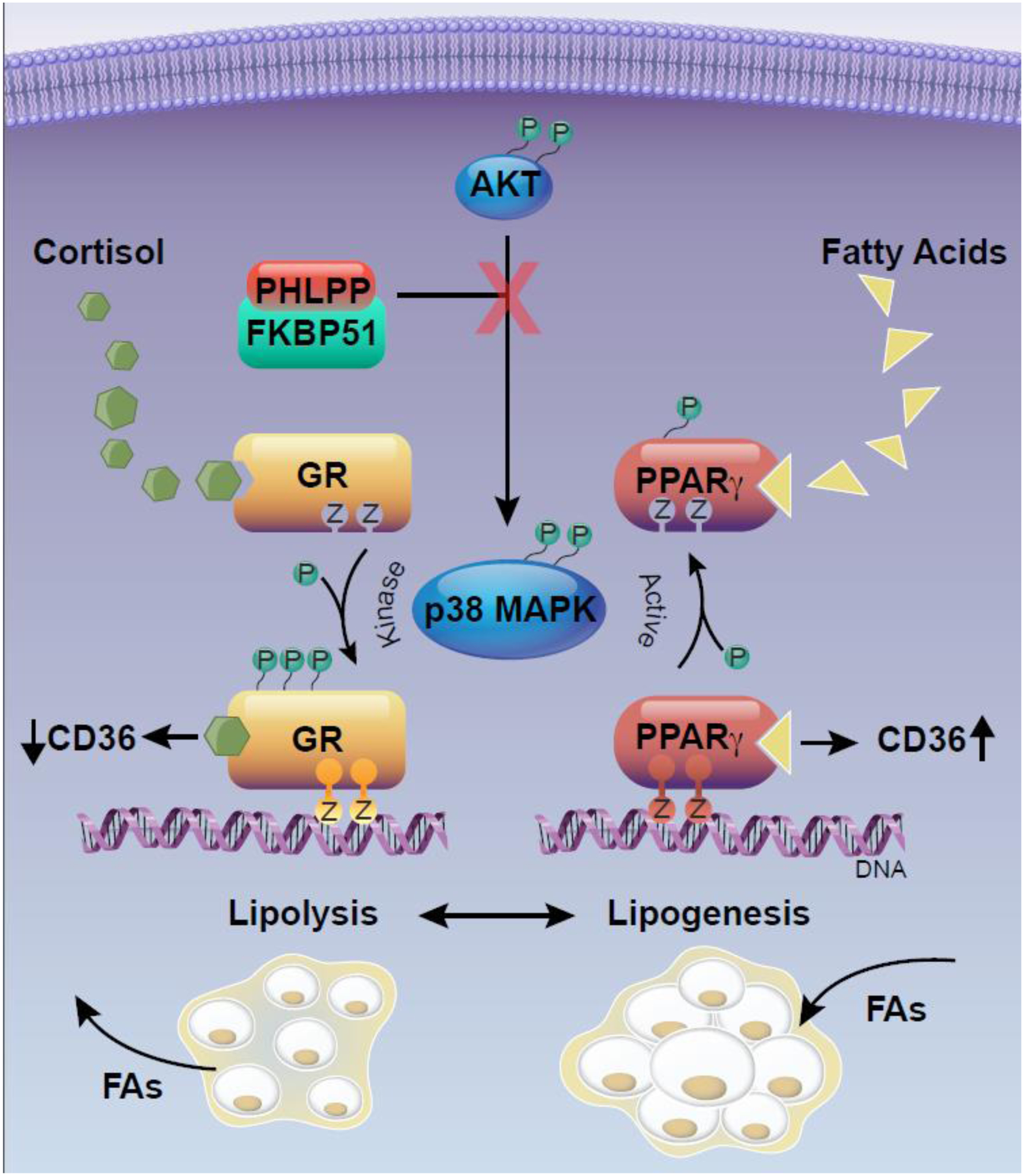
FKBP51 interaction with PHLPP suppresses AKT phosphorylation preventing its kinase activity to phosphorylate p38. The FKBP51-PHLPP-AKT interaction indirectly reduces phosphorylation mediated by p38 on the glucocorticoid receptor (GR), inhibiting cortisol-induced gene activity. In turn, FKBP51-PHLPP-AKT reduces phosphorylation of PPARγ to stimulate fatty acid-induced transcriptional activity and induction of genes for adipogenesis and lipogenesis, such as the fatty acid importer, cluster of differentiation 36 (CD36). The graphical illustration was developed by Matthew Hazzard at the University of Kentucky College of Medicine.
There are other developing facts to consider for future research in this field. The Galigniana laboratory found that FKBP51 acts as a mitochondrial factor where it is highly abundant [54]. The study revealed that at the onset of adipogenesis, FKBP51 translocates from the mitochondria to the nucleus, and over time its expression increases. The mitochondrial shuttling is regulated by cAMP-PKA, where FKBP51 gradually interacts with GRα and restrains its transcription [55]. These findings suggest that mitochondrial-nuclear shuttling of FKBP51 regulated by PKA may be key to transcriptional control of GRα target genes required for the early stages of adipogenesis. These observations pose the interesting questions of whether catecholamine regulation of adipose lipolysis involves FKBP51 and, indeed, whether the adrenergic receptor-CREB pathway controls FKBP51 expression. The Forkhead box-O (FOXO) family of transcription factors are direct targets of AKT (Box II) [56]. Consequently, FOXO1 may be an important target of FKBP51-controlled AKT kinase signaling, as it takes part in cell cycle control, apoptosis, metabolism, and adipocyte differentiation [57]. FKBP51 overexpression in human pancreatic carcinoma cells caused reduced serine 256 (Ser256) phosphorylation of FOXO1, indicating hyperactivity [52]. FOXO1 affects adipocyte function by increasing lipogenesis and regulation of the cell cycle [57]. AKT phosphorylates FOXO1 at Ser256, inhibiting its interaction with the peroxisome proliferator-activated receptor-gamma coactivator-1α (PGC1α) to halt expression of gluconeogenic genes, preventing gluconeogenesis and fatty acid oxidation [58]. Altogether, these observations suggest that FKPB51-AKT mediates insulin signaling in many tissues to control multiple aspects of metabolism.
Box II. FKBP51-FoxO1 signaling mechanisms.
Forkhead box-O-1 (FOXO1) transcription factor expression is induced by glucocorticoids (GCs), elevating its protein levels and target genes. The FOXO1 protein binds to the insulin-response elements (IREs) in promoters of genes to control their expression. The insulin receptor (InsR)-IRS-2-PI3K pathway phosphorylates AKT at pS473 to drive FOXO1 phosphorylation at serine 256 (pSer256), moving it to the cytoplasm where it is degraded, thus inhibiting FOXO1-induced binding to IREs and transcriptional activity. FKBP51 inhibits AKT kinase signaling to reduce pSer256, causing FOXO1 to translocate to the nucleus, where it binds to IREs to manage gene transcription. FOXO1-induced transcriptional activity mediates cell cycle control, apoptosis, metabolism, and differentiation. In muscle, FOXO1 stimulates the expression of atrogenes, such as the Muscle Ring Finger 1 (MURF-1) and atrogin-1, which are muscle-specific E3 ubiquitin-ligases that promote proteasomal degradation. In the liver, FOXO1 activates genes for gluconeogenesis [e.g., glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK)].
II. Role of FKBP51 in Skeletal Muscle and Skin
A. FKBP51/AKT Signaling Pathway in Skeletal Muscle
Skeletal muscle is the most prominent insulin-responsive tissue in the body and is the primary site for insulin-induced glucose disposal, while adipose tissues are the second-highest [59]. Skeletal muscle insulin resistance is fundamental to the metabolic dysregulation associated with obesity and contributes to the development of the metabolic syndrome [60]. In skeletal muscle, glucose transport activity can be activated by an insulin-dependent pathway mediated through AKT [1]. As shown in Figure 1, FKBP51 is highly expressed in adipocytes and skeletal muscle. A recent study by Balsevich et al. showed that antagonism of FKBP51 with SAFit2 increases AKT2 phosphorylation in skeletal muscle of mice [23], indicating that it may play a role in insulin resistance. Moreover, AKT phosphorylates serine 9 of glycogen synthase kinase 3 (GSK3), inhibiting its actions on glycogen synthase [61], which is the rate-limiting enzyme controlling glycogen synthesis. This action increases glycogen content in the skeletal muscle and liver [61, 62]. Investigations using FKBP51 KO mice and rodents treated with the FKBP51 antagonist SAFit2 found that the phosphorylation of AKT2 and its downstream effectors, AS160 and p70S6K, was significantly increased in the skeletal muscle [23]. Genetic ablation and pharmacological inhibition of FKBP51 increased GLUT4 expression at the plasma membrane, causing increased 2-deoxyglucose uptake in primary myocytes [23]. Using co-immunoprecipitation experiments, the study showed that FKBP51 associates with AKT2 and PHLPP and interacts with the downstream molecular target AS160. Altogether, these results implicate FKBP51 in glucose intolerance in obesity and insulin-resistant diabetes, possibly through its signaling with AKT2 in muscle and to a lesser extent in adipose.
FKBP51 has recently been identified as a potential regulator of myoblast differentiation. Ruiz-Estevez et al. revealed that FKBP51 promotes myoblast differentiation and myotube formation by regulating CDK4 signaling in C2C12 cells and FKBP51 KO mice [63], which might transpire through two different mechanisms. One mechanism is that FKBP51 sequesters CDK4 within the HSP90 complex, preventing the formation of the cyclin D1-CDK4 complex, thereby inhibiting myocyte differentiation [63]. Secondly, FKBP51 promotes cis-trans isomerization of the Thr172-Pro173 peptide bond in CDK4 and inhibits phosphorylation of Thr172, which is a critical step for the activation of CDK4 [63]. In support of the in vitro findings, they showed that muscle regeneration is delayed in FKBP51 KO mice. Based on these observations, we now speculate that FKBP51 may also regulate glucocorticoid-induced myopathy in muscle, which we discuss further below.
B. Interactions of FKBP51 and Ubiquitin Ligases Involved in Muscle Atrophy
In the skeletal muscle, GR signaling strongly regulates glucose and protein metabolism. Glucocorticoids antagonize glucose uptake and inhibit glycogen and protein synthesis while increasing protein degradation and preventing the transport of amino acids into muscle. Glucocorticoid-induced atrophy typically occurs in response to starvation to supply the liver with non-hexose substrates such as amino acids that are used for gluconeogenesis [64]. However, muscle atrophy is also commonly seen in individuals with Cushing’s syndrome (excess secretion of adrenal glucocorticoids) and long-term corticosteroid therapy [65]. Blockade of the glucocorticoid-responsive GRα by lentiviral overexpression of GRβ in C2C12 myocytes enhanced myotube fusion [31], suggesting that FKBP51 might have a similar role in myotube formation. Moreover, our studies demonstrate that FKBP51 acts as a negative regulator of GRα by suppressing its phosphorylation through inhibiting the AKT/p38 pathway (Figure 3) [20, 25], suggesting that GRα-induced muscle atrophy may be elevated in the absence of FKBP51. Recent evidence has supported this concept, as Shimoide et al. demonstrated that stable overexpression of FKBP51 in C2C12 murine myocytes enhanced protein synthesis by activating the p70 S6 kinase (S6K) pathway and inhibiting the expression of Muscle Ring Finger 1 (MURF-1) and atrogin-1 [66].
We posit that FKBP51, like GRβ, prevents glucocorticoid-induced muscle atrophy. While GRβ typically inhibits GRα-induced gene activity [31, 67–71], it appears that GRβ does not affect GRα-induction of Fkbp5 [29, 30]. Conversely, FKBP51 regulates the nuclear transport of GRβ and glucocorticoid responsiveness [72], suggesting that there might be a GRβ-FKBP51 axis that is preventive of muscle atrophy. Short-term FKBP51 antagonism with SaFit2 might be helpful for the treatment of persistent pain states that could be induced by the GRβ-FKBP51 axis [73]. However, there is caution for the potential long-term use of FKBP51 antagonists, as they may impede muscle growth, development, or regeneration. Studies in mice have shown that FKBP51 KO animals have increased lean mass [22, 23]. However, treatment with the FKBP51 antagonist SaFit2 for 30 days reduced body weight in high-fat-fed obese and lean animals [23], implying that the weight loss in lean animals might be from reduced muscle mass. Ruiz-Estevez et al. showed that muscle regeneration is delayed in Fkbp5 KO mice [63]. Future studies should determine whether chronic antagonism of FKBP51 promotes complications in muscle atrophy.
Other factors that might contribute to glucocorticoid-induced muscle atrophy are the forkhead transcription factors. Glucocorticoids increase FOXO1 expression and cause the activation of the atrogenes MURF-1 and atrogin-1 (Box II), two muscle-specific E3 ubiquitin-ligases that promote ubiquitination of specific protein substrates that target proteasomal degradation [74]. Interestingly, in muscle and C2C12 myocytes, GRα signaling stimulates myostatin (Mstn) [74, 75], an inhibitor of muscle growth. This most likely occurs via a negative feedback loop, which GRα is well known to activate; FKBP51 and GRβ are examples. Myostatin increases the expression of active FOXO1, causing higher expression of both MURF-1 and atrogin-1 [76]. Gilson et al. showed in vivo that genetic ablation of Mstn caused mice to be resistant to dexamethasone-induced muscle atrophy [77]. Although these data are suggestive, further studies are needed to investigate the involvement of FKBP51 in GRα-induced muscle atrophy and signaling processes that it controls in myocytes and myotube formation.
C. FKBP51/AKT Signaling Pathway in the Skin
Glucocorticoid hormones in the skin are critical regulators of proliferation, differentiation, inflammation, and metabolism and are linked to glucocorticoid-induced skin hypoplasia (atrophy) [78]. Glucocorticoids are highly effective in the treatment of inflammatory skin diseases, including psoriasis and atopic dermatitis. However, chronic treatment with glucocorticoids can lead to adverse effects, partially irreversible, with skin atrophy being the most prominent restriction. Glucocorticoid‐induced skin atrophy involves all skin compartments, including sebaceous glands, hair follicles, and dermal adipose, resulting in thinning of the skin, reducing the size and number of keratinocytes, and inhibition of both dermal fibroblast proliferation and collagen synthesis [79, 80]. Studies in the skin have revealed that FKBP5 is a primary GRα target gene in this tissue, where it was strongly upregulated in keratinocytes in the skin-specific keratin5 (K5)-GR transgenic mice [81] and reduced in GR knockout mice [82]. More recently, it has been shown that robust glucocorticoid-induced expression of FKBP51 was observed in human keratinocytes [83]. Surprisingly, FKBP51 KO mice had larger dermal adipocytes and significantly decreased glucocorticoid-induced skin hypoplasia [83]. Furthermore, the dermal adipose thickness was reduced by glucocorticoid treatment in WT mice with intact FKBP51 by 80%, while FKBP51 KO mice were only reduced by 10% [83]. There were no significant differences for several GR target genes in the epidermis between FKBP51 KO and WT. However, FKBP51 expression was relatively low in the epidermis, and these might have been more dramatically changed in specifically dermal adipocytes, but this was not directly tested. More studies of the role of FKBP51 in the skin are needed, especially the role of FKBP51 in dermal adipocytes. One interesting perspective is that maybe FKBP51 has an important function in regulating dermal adipose and the generation of fat in the skin for protection. Future studies on dermal adipose would be valuable to understanding the function of FKBP51 in the skin.
III. Conclusions
The discussion above suggests a preeminent role for FKBP51 in adipogenesis and possibly myogenesis. The processes controlled by FKBP51 may be through its scaffolding function and interaction with proteins such as PHLPP and AKT, or there may be other kinases involved that are not yet identified. The function of FKBP51 to inhibit GRα and activate PPARγ suggests that targeting the protein might have a beneficial action in adipose and muscle tissues. Given that glucocorticoids, such as cortisol, act via GRα to control carbohydrate, lipid, and protein metabolism, it is not surprising that the evolving literature demonstrates an essential role for FKBP51 in metabolic processes and disorders. There appears to be a clear and vital role for FKBP51 in adipogenesis and adipocyte expansion and possibly in dermal adipocytes. Future work showing FKBP51 signaling in humans is needed to strengthen the data found in cell culture and preclinical animal studies. The recent developments of the FKBP51 antagonists such as Timcodar, SAFit1, and SAFit2 offer opportunities to target the protein as a potential therapeutic for obesity and insulin-resistant diabetes. However, caution is warranted for long-term therapies that antagonize FKBP51, as there appears to be a paramount role of the protein in muscle growth and regeneration. More studies are needed on the role of FKBP51, especially in humans, to better understand its influence on metabolism.
HIGHLIGHTS.
FKBP51 expression is highest in adipose tissue, increases with adipogenesis, and is associated with obesity in rodents and humans.
FKBP51 is a multi-functional protein that regulates several metabolic processes that control adiposity.
FKBP51 directly interacts with the insulin receptor signaling cascade to induce insulin resistance.
The antagonism of FKBP51 is a promising target in the prevention or treatment of obesity and diabetes.
Acknowledgments:
This work was supported by the National Institutes of Health NIDDK grants R01DK121797 (T.D.H.J.) and R01DK121017 (E.R.S). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We would like to thank Matthew Hazzard at the University of Kentucky College of Medicine for the development of the graphical illustrations in Figures 2 and 3.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Sanchez ER, Chaperoning steroidal physiology: lessons from mouse genetic models of Hsp90 and its cochaperones. Biochim Biophys Acta, 2012. 1823(3): p. 722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zgajnar NR, et al. , Biological Actions of the Hsp90-binding Immunophilins FKBP51 and FKBP52. Biomolecules, 2019. 9(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahle A, et al. , The Many Faces of FKBP51. Biomolecules, 2019. 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riggs DL, et al. , Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol Cell Biol, 2007. 27(24): p. 8658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zannas AS, et al. , Gene-Stress-Epigenetic Regulation of FKBP5: Clinical and Translational Implications. Neuropsychopharmacology, 2016. 41(1): p. 261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matosin N, Halldorsdottir T, and Binder EB, Understanding the Molecular Mechanisms Underpinning Gene by Environment Interactions in Psychiatric Disorders: The FKBP5 Model. Biol Psychiatry, 2018. 83(10): p. 821–830. [DOI] [PubMed] [Google Scholar]
- 7.Tranguch S, et al. , Cochaperone immunophilin FKBP52 is critical to uterine receptivity for embryo implantation. Proc Natl Acad Sci U S A, 2005. 102(40): p. 14326–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Z, et al. , FK506-binding protein 52 is essential to uterine reproductive physiology controlled by the progesterone receptor A isoform. Mol Endocrinol, 2006. 20(11): p. 2682–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yong W, et al. , Essential role for Co-chaperone Fkbp52 but not Fkbp51 in androgen receptor-mediated signaling and physiology. J Biol Chem, 2007. 282(7): p. 5026–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chambraud B, et al. , A role for FKBP52 in Tau protein function. Proc Natl Acad Sci U S A, 2010. 107(6): p. 2658–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gold BG and Nutt JG, Neuroimmunophilin ligands in the treatment of Parkinson’s disease. Curr Opin Pharmacol, 2002. 2(1): p. 82–6. [DOI] [PubMed] [Google Scholar]
- 12.Zhao W, et al. , Role of cellular FKBP52 protein in intracellular trafficking of recombinant adeno-associated virus 2 vectors. Virology, 2006. 353(2): p. 283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hausl AS, et al. , Focus on FKBP51: A molecular link between stress and metabolic disorders. Mol Metab, 2019. 29: p. 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinds TD Jr., et al. , Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-gamma (PPARgamma). J Biol Chem, 2011. 286(50): p. 42911–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen MX and Cohen PT, Activation of protein phosphatase 5 by limited proteolysis or the binding of polyunsaturated fatty acids to the TPR domain. FEBS Lett, 1997. 400(1): p. 136–40. [DOI] [PubMed] [Google Scholar]
- 16.Ramsey AJ and Chinkers M, Identification of potential physiological activators of protein phosphatase 5. Biochemistry, 2002. 41(17): p. 5625–32. [DOI] [PubMed] [Google Scholar]
- 17.Cher C, et al. , Identification of chaulmoogric acid as a small molecule activator of protein phosphatase 5. Appl Biochem Biotechnol, 2010. 160(5): p. 1450–9. [DOI] [PubMed] [Google Scholar]
- 18.Hinds TD Jr. and Sanchez ER, Protein phosphatase 5. Int J Biochem Cell Biol, 2008. 40(11): p. 2358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu M, et al. , Deciphering the Roles of Thiazolidinediones and PPARgamma in Bladder Cancer. PPAR Res, 2017. 2017: p. 4810672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stechschulte LA, et al. , FKBP51 Controls Cellular Adipogenesis through p38 Kinase-Mediated Phosphorylation of GRα and PPARγ. Molecular Endocrinology, 2014. 28(8): p. 1265–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeh WC, et al. , Identification and characterization of an immunophilin expressed during the clonal expansion phase of adipocyte differentiation. Proc Natl Acad Sci U S A, 1995. 92(24): p. 11081–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stechschulte LA, et al. , FKBP51 Null Mice Are Resistant to Diet-Induced Obesity and the PPARgamma Agonist Rosiglitazone. Endocrinology, 2016: p. en20151996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balsevich G, et al. , Stress-responsive FKBP51 regulates AKT2-AS160 signaling and metabolic function. Nature Communications, 2017. 8: p. 1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ludtke A, et al. , New PPARG mutation leads to lipodystrophy and loss of protein function that is partially restored by a synthetic ligand. J Med Genet, 2007. 44(9): p. e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stechschulte LA, et al. , FKBP51 Reciprocally Regulates GRα and PPARγ Activation via the Akt-p38 Pathway. Molecular Endocrinology, 2014. 28(8): p. 1254–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinds TD, et al. , Timcodar (VX-853) Is a Non-FKBP12 Binding Macrolide Derivative That Inhibits PPARγand Suppresses Adipogenesis. PPAR Research, 2016. 2016: p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Periyasamy S, et al. , FKBP51 and Cyp40 are positive regulators of androgen-dependent prostate cancer cell growth and the targets of FK506 and cyclosporin A. Oncogene, 2010. 29(11): p. 1691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hinds TD, et al. , Analysis of FK506, timcodar (VX-853) and FKBP51 and FKBP52 chaperones in control of glucocorticoid receptor activity and phosphorylation. Pharmacol Res Perspect, 2014. 2(6): p. e00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stechschulte LA, et al. , Glucocorticoid receptor beta stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem, 2014. 289(25): p. 17885–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hinds TD Jr., et al. , Discovery of glucocorticoid receptor-beta in mice with a role in metabolism. Mol Endocrinol, 2010. 24(9): p. 1715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marino JS, et al. , Glucocorticoid receptor beta induces hepatic steatosis by augmenting inflammation and inhibition of the peroxisome proliferator-activated receptor (PPAR) alpha. J Biol Chem, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang GR, et al. , Long-term administration of rapamycin reduces adiposity, but impairs glucose tolerance in high-fat diet-fed KK/HlJ mice. Basic Clin Pharmacol Toxicol, 2009. 105(3): p. 188–98. [DOI] [PubMed] [Google Scholar]
- 33.Harding MW, Immunophilins, mTOR, and pharmacodynamic strategies for a targeted cancer therapy. Clin Cancer Res, 2003. 9(8): p. 2882–6. [PubMed] [Google Scholar]
- 34.Gaali S, et al. , Selective inhibitors of the FK506-binding protein 51 by induced fit. Nat Chem Biol, 2015. 11(1): p. 33–7. [DOI] [PubMed] [Google Scholar]
- 35.Sidibeh CO, et al. , FKBP5 expression in human adipose tissue: potential role in glucose and lipid metabolism, adipogenesis and type 2 diabetes. Endocrine, 2018. 62(1): p. 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pereira MJ, et al. , FKBP5 expression in human adipose tissue increases following dexamethasone exposure and is associated with insulin resistance. Metabolism, 2014. 63(9): p. 1198–1208. [DOI] [PubMed] [Google Scholar]
- 37.Hartmann IB, et al. , The FKBP5 polymorphism rs1360780 is associated with lower weight loss after bariatric surgery: 26 months of follow-up. Surgery for Obesity and Related Diseases, 2016. 12(8): p. 1554–1560. [DOI] [PubMed] [Google Scholar]
- 38.Soukas A, et al. , Leptin-specific patterns of gene expression in white adipose tissue. Genes Dev, 2000. 14(8): p. 963–80. [PMC free article] [PubMed] [Google Scholar]
- 39.Yang L, et al. , Hypothalamic Fkbp51 is induced by fasting, and elevated hypothalamic expression promotes obese phenotypes. Am J Physiol Endocrinol Metab, 2012. 302(8): p. E987–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huby AC, et al. , Adipocyte-Derived Hormone Leptin Is a Direct Regulator of Aldosterone Secretion, Which Promotes Endothelial Dysfunction and Cardiac Fibrosis. Circulation, 2015. 132(22): p. 2134–45. [DOI] [PubMed] [Google Scholar]
- 41.Calhoun DA and Sharma K, The role of aldosteronism in causing obesity-related cardiovascular risk. Cardiol Clin, 2010. 28(3): p. 517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petrovich E, Asher C, and Garty H, Induction of FKBP51 by aldosterone in intestinal epithelium. J Steroid Biochem Mol Biol, 2014. 139: p. 78–87. [DOI] [PubMed] [Google Scholar]
- 43.Galigniana MD, et al. , The hsp90-FKBP52 complex links the mineralocorticoid receptor to motor proteins and persists bound to the receptor in early nuclear events. Mol Cell Biol, 2010. 30(5): p. 1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davies TH, Ning YM, and Sanchez ER, A new first step in activation of steroid receptors: hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem, 2002. 277(7): p. 4597–600. [DOI] [PubMed] [Google Scholar]
- 45.Davies TH, Ning YM, and Sanchez ER, Differential control of glucocorticoid receptor hormone-binding function by tetratricopeptide repeat (TPR) proteins and the immunosuppressive ligand FK506. Biochemistry, 2005. 44(6): p. 2030–8. [DOI] [PubMed] [Google Scholar]
- 46.Vandevyver S, Dejager L, and Libert C, On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic, 2012. 13(3): p. 364–74. [DOI] [PubMed] [Google Scholar]
- 47.Häusl AS, et al. , Focus on FKBP51: A molecular link between stress and metabolic disorders. Molecular metabolism, 2019. 29: p. 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez E and McGraw TE, The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle, 2009. 8(16): p. 2502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tschopp O, et al. , Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development, 2005. 132(13): p. 2943–54. [DOI] [PubMed] [Google Scholar]
- 50.O’Brien L, et al. , Biliverdin reductase isozymes in metabolism. Trends Endocrinol Metab, 2015. 26(4): p. 212–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gao T, Furnari F, and Newton AC, PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell, 2005. 18(1): p. 13–24. [DOI] [PubMed] [Google Scholar]
- 52.Pei H, et al. , FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer cell, 2009. 16(3): p. 259–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakamoto K and Holman GD, Emerging role for AS160/TBC1D4 and TBC1D1 in the regulation of GLUT4 traffic. American journal of physiology. Endocrinology and metabolism, 2008. 295(1): p. E29–E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gallo LI, et al. , The 90-kDa Heat-shock Protein (Hsp90)-binding Immunophilin FKBP51 Is a Mitochondrial Protein That Translocates to the Nucleus to Protect Cells against Oxidative Stress. Journal of Biological Chemistry, 2011. 286(34): p. 30152–30160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toneatto J, et al. , Dynamic mitochondrial–nuclear redistribution of the immunophilin FKBP51 is regulated by the PKA signaling pathway to control gene expression during adipocyte differentiation. Journal of Cell Science, 2013. 126(23): p. 5357–5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wuescher L, et al. , Insulin regulates menin expression, cytoplasmic localization, and interaction with FOXO1. Am J Physiol Endocrinol Metab, 2011. 301(3): p. E474–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Junye C, et al. , Molecular mechanisms of FOXO1 in adipocyte differentiation. Journal of Molecular Endocrinology, 2019. 62(3): p. R239–R253. [DOI] [PubMed] [Google Scholar]
- 58.Puigserver P, et al. , Insulin-regulated hepatic gluconeogenesis through FOXO1–PGC-1α interaction. Nature, 2003. 423(6939): p. 550–555. [DOI] [PubMed] [Google Scholar]
- 59.Lambadiari V, Triantafyllou K, and Dimitriadis GD, Insulin action in muscle and adipose tissue in type 2 diabetes: The significance of blood flow. World J Diabetes, 2015. 6(4): p. 626–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stump CS, et al. , The metabolic syndrome: Role of skeletal muscle metabolism. Annals of Medicine, 2006. 38(6): p. 389–402. [DOI] [PubMed] [Google Scholar]
- 61.Hinds TD Jr., et al. , Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3beta phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) alpha. J Biol Chem, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedrichsen M, et al. , Akt2 influences glycogen synthase activity in human skeletal muscle through regulation of NH₂-terminal (sites 2 + 2a) phosphorylation. American journal of physiology. Endocrinology and metabolism, 2013. 304(6): p. E631–E639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruiz-Estevez M, et al. , Promotion of Myoblast Differentiation by Fkbp5 via Cdk4 Isomerization. Cell Reports, 2018. 25(9): p. 2537–2551.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.John K, et al. , The glucocorticoid receptor: cause of or cure for obesity? Am J Physiol Endocrinol Metab, 2016. 310(4): p. E249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vegiopoulos A and Herzig S, Glucocorticoids, metabolism and metabolic diseases. Molecular and Cellular Endocrinology, 2007. 275(1): p. 43–61. [DOI] [PubMed] [Google Scholar]
- 66.Shimoide T, et al. , Novel roles of FKBP5 in muscle alteration induced by gravity change in mice. Biochemical and Biophysical Research Communications, 2016. 479(3): p. 602–606. [DOI] [PubMed] [Google Scholar]
- 67.Nwaneri AC, McBeth L, and Hinds TD Jr., Sweet-P inhibition of glucocorticoid receptor beta as a potential cancer therapy. Cancer Cell Microenviron, 2016. 3(3). [PMC free article] [PubMed] [Google Scholar]
- 68.McBeth L, et al. , Glucocorticoid receptor beta increases migration of human bladder cancer cells. Oncotarget, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hinds TD, et al. , Overexpression of Glucocorticoid Receptor beta Enhances Myogenesis and Reduces Catabolic Gene Expression. Int J Mol Sci, 2016. 17(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McBeth L, et al. , Involvement of the Androgen and Glucocorticoid Receptors in Bladder Cancer. Int J Endocrinol, 2015. 2015: p. 384860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.John K, et al. , The Glucocorticoid Receptor: Cause or Cure for Obesity? Am J Physiol Endocrinol Metab, 2015: p. ajpendo 00478 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang X, Clark AF, and Yorio T, FK506-binding protein 51 regulates nuclear transport of the glucocorticoid receptor beta and glucocorticoid responsiveness. Invest Ophthalmol Vis Sci, 2008. 49(3): p. 1037–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maiaru M, et al. , The stress regulator FKBP51: a novel and promising druggable target for the treatment of persistent pain states across sexes. Pain, 2018. 159(7): p. 1224–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma K, et al. , Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. American Journal of Physiology-Endocrinology and Metabolism, 2003. 285(2): p. E363–E371. [DOI] [PubMed] [Google Scholar]
- 75.Artaza JN, et al. , Endogenous expression and localization of myostatin and its relation to myosin heavy chain distribution in C2C12 skeletal muscle cells. Journal of Cellular Physiology, 2002. 190(2): p. 170–179. [DOI] [PubMed] [Google Scholar]
- 76.McFarlane C, et al. , Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-κB-independent, FoxO1-dependent mechanism. Journal of Cellular Physiology, 2006. 209(2): p. 501–514. [DOI] [PubMed] [Google Scholar]
- 77.Gilson H, et al. , Myostatin Gene Deletion Prevents Glucocorticoid-Induced Muscle Atrophy. Endocrinology, 2007. 148(1): p. 452–460. [DOI] [PubMed] [Google Scholar]
- 78.Agarwal S, et al. , PI3K inhibitors protect against glucocorticoid-induced skin atrophy. EBioMedicine, 2019. 41: p. 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schoepe S, Schäcke H, and Asadullah K, Test systems for the determination of glucocorticoid receptor ligand induced skin atrophy. Dermato-endocrinology, 2011. 3(3): p. 175–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schoepe S, et al. , Glucocorticoid therapy-induced skin atrophy. Experimental Dermatology, 2006. 15(6): p. 406–420. [DOI] [PubMed] [Google Scholar]
- 81.Chebotaev D, et al. , The tumor suppressor effect of the glucocorticoid receptor in skin is mediated via its effect on follicular epithelial stem cells. Oncogene, 2007. 26(21): p. 3060–3068. [DOI] [PubMed] [Google Scholar]
- 82.Sevilla LM, et al. , Glucocorticoid receptor regulates overlapping and differential gene subsets in developing and adult skin. Molecular endocrinology (Baltimore, Md.), 2010. 24(11): p. 2166–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baida G, et al. , Deletion of the glucocorticoid receptor chaperone FKBP51 prevents glucocorticoid-induced skin atrophy. Oncotarget, 2018. 9(78): p. 34772–34783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu C, et al. , BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol, 2009. 10(11): p. R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu C, Macleod I, and Su AI, BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res, 2013. 41(Database issue): p. D561–5. [DOI] [PMC free article] [PubMed] [Google Scholar]