

Abstract
SARS-CoV-2 infection depends on binding its spike (S) protein to angiotensin-converting enzyme 2 (ACE2). The S protein expresses an RGD motif, suggesting that integrins may be co-receptors. Here, we UV-inactivated SARS-CoV-2 and fluorescently labeled the envelope membrane with octadecyl rhodamine B (R18) to explore the role of integrin activation in mediating cell entry and productive infection. We used flow cytometry and confocal microscopy to show that SARS-CoV-2R18 particles engage basal-state integrins. Furthermore, we demonstrate that Mn2+, which induces integrin extension, enhances cell entry of SARS-CoV-2R18. We also show that one class of integrin antagonist, which binds to the αI MIDAS site and stabilizes the inactive, closed conformation, selectively inhibits the engagement of SARS-CoV-2R18 with basal state integrins, but is ineffective against Mn2+-activated integrins. RGD-integrin antagonists inhibited SARS-CoV-2R18 binding regardless of integrin activation status. Integrins transmit signals bidirectionally: 'inside-out' signaling primes the ligand-binding function of integrins via a talin-dependent mechanism, and 'outside-in' signaling occurs downstream of integrin binding to macromolecular ligands. Outside-in signaling is mediated by Gα13. Using cell-permeable peptide inhibitors of talin and Gα13 binding to the cytoplasmic tail of an integrin's β subunit, we demonstrate that talin-mediated signaling is essential for productive infection.
Subject terms: Cell biology, Cell adhesion, Integrins, Diseases, Infectious diseases, Viral infection
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel virus in the Betacoronavirus genus that causes coronavirus disease 2019 (COVID-19)1. SARS-CoV-2 was first reported in Wuhan, China, and currently persists as a global pandemic2,3. SARS-CoV-2 presents similar characteristics with the original SARS-CoV in genome structure, tissue tropism, and viral pathogenesis. However, SARS-CoV-2 is more transmissible than SARS-CoV.
Cellular entry of coronaviruses depends on binding of the viral spike (S) protein to a specific cellular receptor, the angiotensin-converting enzyme 2 (ACE2)4,5, and subsequent S protein priming by cellular protease activity such as Transmembrane Serine Protease 2 (TMPRSS2)6. Interestingly, ACE2 expression across different human tissues7 revealed low expression of ACE2 in the lungs compared to elevated expression in the kidney and heart8,9. Nevertheless, studies have shown that type I and II interferons (IFNs) secreted during viral infection upregulate the transcription and expression of ACE210,11. Unlike its predecessor, SARS-Cov-2 expresses a novel K403R spike protein substitution encoding an Arginine-Glycine-Aspartic acid (RGD) motif12, introducing the potential for interacting with RGD-binding integrins, as likely mediators for viral cell entry and enhanced pathogenicity13. ACE2 contains two integrin-binding domains: an RGD motif at position 204–206 and the sequence RKKKNKAR in the cytoplasmic tail at its C-terminus14. Also, ACE2 binds integrin β1 in the failing human heart14. Correlated increased expressions of β115 and ACE2 have been reported16,17. Others have shown that ACE2 interacts in cis with integrin β1 in a manner that enhances RGD-mediated cell adhesion18.
Integrins are heterodimeric transmembrane adhesion protein receptors composed of α and β subunits whose activation is tightly regulated and bidirectional19. Integrins can exist in three states characterized by their structural conformation and affinity for their ligands (Fig. 1A). The inactive, bent-closed state (BCS) with a closed headpiece has a low affinity for extracellular matrix (ECM) ligands. The bent structure inhibits the receptors from inappropriate signaling due to random binding to extracellular matrix proteins. Integrins exhibit an extended-closed state (ECS) with a closed headpiece and higher ligand binding affinity than BCS when primed. Active and extended-open state (EOS) presents an open headpiece and maximum affinity for ECM ligands20. Integrin function involves coordination with cytoskeletal components whose functions regulate cell adhesion and migration21,22. Changes in integrin conformation can elicit cell-signaling events that increase ligand affinity/avidity, promote cytoskeletal rearrangement, and enable virus internalization. Ligand binding to integrins is mediated by divalent-cations bound at the Metal Ion Dependent Adhesion Site (MIDAS) domain on top of either the αI domain, in I domain-containing integrins, or the βI domain in non-αI integrins23. Physiologically, 1 mM Ca2+ and 1 mM Mg2+ in body fluid stabilize the BCS conformation. Under non-physiological conditions, 1 mM Mn2+ initiates and stabilizes ECS conformation even in the presence of Ca2+.
Figure 1.

Integrin conformational states antagonist targets and SARS-CoV-2 binding. (A) Integrin States: First, the inactive, bent-closed state (BCS), with a closed headpiece and low affinity for extracellular matrix (ECM) ligands. The bent structure inhibits the receptors from inappropriate signaling due to random binding to extracellular matrix proteins. In the BCS form, binding to large ligands is likely limited. Second, when primed, integrins exhibit an extended-closed state (ECS) with a closed headpiece and higher ligand binding affinity than BCS. Third, active and extended-open state (EOS) with an open headpiece and maximum affinity for ECM ligands. Integrin Affinity Regulation: Mn2+ binding to the MIDAS site at the αI and βI domain integrin induces integrin extension. α2β1 integrin antagonist BTT 3033 binds to the α-I domain, and stabilizes the BCS. GLP0187 blocks binding to the RGD ligand-binding domain. EOS binding to a macromolecular ligand or ECM generates a force (F) transmitted through the integrin β subunit. (B) Model of Sars-CoV-2 virion structure (https://www.scientificamerican.com/interactive/inside-the-coronavirus/). SARS-CoV-2 are spherical or ovoid particles of sizes that span the range of 60–140 nm. The SARS-CoV-2 virion consists of a lipid bilayer envelope membrane covering a large nucleoprotein (N)-encapsidated, positive-sense RNA genome. The lipid envelope is decorated with three transmembrane proteins consisting of trimeric spike proteins (S) that project above the lipid bilayer membrane and relatively small membrane (M) and envelope (E) proteins78,79. S proteins bind with high-affinity (1–50 nM)4 to the angiotensin-converting enzyme 2 (ACE2) for productive infection80. (C) Cartoon alignment of the receptor-binding domain (RBD) and RGD sequence on the trimeric spike protein, which favors engagement of activated integrin, adapted from ref.25 The illustrations were generated using Microsoft® PowerPoint Version 16.51 (21071101).
Many viruses use integrin-mediated endocytosis pathways for cell entry5,24. A recent bioinformatics-driven study predicted a model that placed integrins in a central ligating role, whereby SARS-CoV-2 could engage multiple receptors and form a multicomponent receptor complex and functional signaling platform25. Interestingly, ACE2 also has a similar MIDAS motif25. Still, it has not yet been established whether the ACE2 MIDAS domain has a potential role in creating synergy overlap between the ligand-binding profiles and regulation of ACE2 and integrins25. Several in vitro studies have established experimental evidence in support of cognate binding interactions between SARS-CoV-2 spike proteins, integrin β126,27 and integrin β312,28. In addition, the transmembrane glycoprotein neuropilin 1 (NRP1), which is abundantly expressed in the olfactory epithelium and promotes the endocytosis of activated α5β1 integrin29–34, has been recently identified as a receptor for SARS-CoV-2 infection34,35.
In this study, we took a mechanistic approach to examine the role of integrins as effectors of SARS-CoV-2 cell entry and productive infection. First, we tested whether inducing a BCS to ECS integrin conformational change with Mn2+24,36 enhanced cell binding and entry of fluorescently tagged UV-inactivated SARS-CoV-2R18. Conversely, we used integrin extension or RGD-binding inhibitors to determine the inhibitors’ effect on cellular entry. Integrins signal bidirectionally via "inside-out” and “outside-in" signaling22,36–41. Inside-out signaling is initiated by intracellular signaling upstream of talin, and other adaptor proteins binding to the integrin β-subunit cytoplasmic tail (β-CT), which causes integrin extension (ECS) and concomitant increases in high-affinity ligand binding21,22. Integrin engagement with macromolecular ligands stimulates the transient exchange of talin for Gα13’s occupancy of the β-CT42,43 which initiates integrin outside-in signaling. In the context of viral infection, integrin outside-in signaling induces cell spreading, retraction, and internalization of integrin-associated ligands. We used cell-permeable inhibitors of integrin outside-in and inside-out signaling42 to test the role of canonical integrin signaling during cell entry of SARS-CoV-2R18 and infectious SARS-CoV-2. Taken together, our results demonstrate that integrins play a significant role in the infectivity of SARS-CoV-2.
Results
Integrin extension promotes SARS-CoV-2R18 cell entry
To facilitate studies of SARS-CoV-2 host-cell entry outside of BSL-3 containment, we generated UV-inactivated virus particles. Under our experimental conditions, a minimum UV dose of 100 mW s/cm2 was sufficient to completely inactivate 107 virions/ml distributed in 500 µl samples of a twelve well plate (Fig. 2A,B). UV-inactivated virus samples were fluorescently labeled with a lipophilic lipid probe, octadecyl rhodamine B (R18), intercalating the envelope membrane44. Labeled samples were purified and characterized as we have described previously for the Sin Nombre virus45.
Figure 2.

Characterization of UV-inactivated virus for Sars-Cov-2 studies. (A) Duplicate plaque assays of supernatants of Sars-CoV-2 exposed to increasing doses of 254 nm radiation and then tested for viability. The live virus completely lysed the cells at 1:100 dilution relative to UV exposed virions. (B) Graph shows UV dose–response, leading to a significant decrease in plaque-forming units at different doses. For our experiments, a 90 s (450 mW s/cm2) UV dose was used to inactivate the virus before removal from the BSL-3 laboratory. (C) Confocal microscopy imaging of cells after incubation with SARS-CoV-2R18 (magenta) for 15 min, then fixed and labeled for early endosome marker, early endosome antigen 1 or EEA1 (green), an effector protein for Rab5, and nuclei (Hoechst 33,258, blue). SARS-CoV-2R18 vesicles are trafficked to the perinuclear region, and a subset is co-localized with EEA1. Images are maximum projections and have been brightness and contrast-enhanced.
To assess the ability of SARS-CoV-2R18 to enter cells, we used confocal microscopy to image the relative distribution of SARS-CoV-2R18 and EEA1, an effector protein for Rab5 positive early endosomes. Adherent cells were incubated on a coverslip with SARS-CoV-2R18 for 15 min, washed, fixed, and immunolabeled for EEA1 (Fig. 2C). Internalized SARS-CoV-2R18 was frequently found in EEA1 positive early endosomes and perinuclear space, demonstrating that the SARS-CoV-2R18 internalizes and traffics as one might expect29–34. Together, these results show that UV-inactivated SARS-CoV-2R18 is a valuable probe for investigating SARS-CoV-2 entry mechanisms.
We hypothesized that activating integrins by Mn2+24,36, which induces integrin extension and higher ligand affinity, would provide a favorable spatial orientation of the RGD-binding motifs to facilitate SARS-CoV-2R18 binding (Fig. 1A). Therefore, we measured initial rates (< 10 min binding time) of binding in activated cells (Mn2+/Ca2+) relative to resting (1 mM Ca2+ only). SNV-CoV-2R18 bound to the Mn2+ activated samples at 3 times the rate of untreated cells (not shown). However, at equilibrium (> 20 min incubation), the cell occupancy of SARS-CoV-2R18 was only ~ 20% higher in Mn2+-treated samples compared to untreated samples. As expected, the gap increased when the virus was used as a limiting reagent46. We used estimation plots47, to assess the precision of the results from multiple experiments of the integrin function assay that measured the relative binding of SARS-CoV-2R18 to Mn2+-activated cells relative to quiescent cells on different days (Fig. 3A–D). The mean difference between the binding to Mn2+-activated and resting cells was conserved across different samples when we used at least 5000 SARS-CoV-2R18/cell. However, when we used a lower stoichiometric ratio, e.g., 3000 SARS-CoV-2R18/cell, the gap between the site occupancy of Mn2+ activated and resting cells increased to 30%, as discussed below.
Figure 3.

Flow cytometry assays of binding inhibition of SARS-CoV-2R18 show specificity of αI allosteric antagonist BTT 3033, RGD fibronectin synergy domain (F9N) ATN-161, and broad-spectrum RGD antagonist, GLPG0187. Vero E6 suspension cells in 40 µl volumes (1000 cells/µl) were first incubated with 10 µM integrin antagonists or 5× unlabeled Sars-CoV-2 (CoV-2) in ± Mn2+ media for 20 min at 37 °C. Sars-CoV-2R18 was then added to the tubes and incubated for another 20 min. The samples were centrifuged and resuspended in 95 µl HHB buffer and analyzed on a flow cytometer. (A–D) Raw data panels of SARS-CoV-2R18 binding to Mn2+ activated and resting cells. Each data symbol in the left panels shows triplicate measurements of a single functional test experiment measuring the effect of 1 mM Mn2+ on equilibrium binding of SARS-CoV-2R18 to Vero E6 cells. The right panels show differences between the means of Mn2+-replete samples and Mn2+-free samples, and the left panels show 95% confidence intervals (CI). Green arrows are used to link the analysis results of the same experiment. Combined data show negative controls of multiple experiments (n = 6) testing different inhibitors. As demonstrated for GLP, samples treated with the virus as a limiting reagent yielded lower signals for Mn2+ and Ca2+ as shown for GLP). (E) Effect of BTT 3033 on SARS-CoV-2R18 binding to cells. Red vertical bars 1 and 2 denote the difference between mock- and inhibitor-treated samples for Mn2+-replete and Mn2+-free samples. (top) ∆ = 0.2, refers to the relative difference in fluorescence intensity due to SARS-CoV-2R18 in Mn2+-replete samples and Mn2+ free samples; (bottom) ∆ = 0.2, refers to the fractional difference inhibition of SARS-CoV-2R18 binding by BTT 3033 in Mn2+-replete cells and Mn-free samples, indicating that BTT 3033 is a selective inhibitor of quiescent integrins. (F) Effect of ATN-160 on SARS-CoV-2R18 binding to cells indicating that ATN-160 is agnostic of integrin activation status. (G) Effect of GLP0187 on SARS-CoV-2R18 binding to cells. The gap between Mn2+ activated cells, and quiescent is shown to be higher ∆ = 0.3 when the virus was used as a limiting reagent < 5000 SARS-CoV-2R18 /cell as indicated by a lower signal in the raw data (C,D). (H–J) Specific binding of SARS-CoV-2R18 to cells determined by subtraction of non-specific binding represented by 5xCov-2 data points. The data are normalized to mock-treated cells (no inhibitors). Data are representative of at least 3 separate measurements for each inhibitor. The p-values between samples are indicated on the horizontal bar. The figure was produced in GraphPad Prism version 9.2.0.
To further investigate the role of integrins in SARS-CoV-2R18 entry into Vero E6 cells, we used high binding affinity integrin antagonists: (1) BTT 3033, a selective antagonist (EC50 = 130 nM) of integrin α2β1 that binds to a site close to the α2I MIDAS domain and stabilizes the integrin bent conformation state (BCS)48, (2) ATN-161, a non-RGD peptide49 derived from the synergy region of fibronectin50, known to exhibit specific antagonism for α5β1 and αIIbβ3 and also recently shown to inhibit SARS-CoV-2 infectivity27, and (3) GLPG0187, a high-affinity, broad-spectrum (EC50 < 10 nM) integrin receptor antagonist of RGD integrins α5β1, αvβ3, αvβ5, αvβ1, αvβ651. We used a titrated, fivefold excess of unlabeled SARS-CoV-2 relative to fluorescent SARS-CoV-2R18 as a control for competitive inhibition of SARS-CoV-2R18 binding. Paired samples of cell suspensions in Mn2+-replete and Mn2+-free media were treated with the above integrin antagonists. Total viral binding was normalized to Mn2+-treated samples for each experimental condition. The graphs show that Mn2+ treatment increased SARS-CoV-2R18 occupancy of cells by ~ 20% compared to Mn2+-free conditions (Fig. 3E,F). As noted above, using significantly fewer SARS-CoV-2R18 than 5000 increases the gap between binding to the Mn2+-activated and resting as indicated for the GLPG0187 sample (Fig. 3G). We also show in a subsequent experiment that raising SARS-CoV-2R18 to a stoichiometric excess limited the site occupancy gap to 20% (Supplemental Figure 1).
The positive control for inhibition (5xCov-2 in data graphs) blocked 80% of SARS-CoV-2R18 and equally inhibited Mn2+ -treated and untreated samples. Reasoning that the residual signal of 5× Cov-2 treated samples was due to non-specific binding to the cell membrane, we subtracted the fluorescence of cells blocked with 5xCov-2 and then normalized the data to mock-treated cells. Finally, we compared the relative efficacy of the inhibitors in Mn2+-replete and -free conditions of the normalized data (Fig. 3H–J). The fraction of Mn2+-activated integrins (20%) were refractory to BTT 3033 treatment (Fig. 3H). BTT 3033 selectively binds to the BCS integrin structure48 and does not bind to Mn2+ activated integrins. In contrast, ATN-161 and GLPG0187 were agnostic to Mn2+ treated cells, as the same baseline was achieved for either condition (Fig. 3I,J). Overall, GLPG0187 (Fig. 3I) appeared to be a better competitive inhibitor of SARS-CoV-2R18 compared to ATN-161 (Fig. 3J). The difference for the latter was potentially due to ATN-161's overall specificity for integrin α5β1. Thus, the expression level of α5β1, in Vero E6 cells, relative to other integrins with which α5β1 would compete for SARS-CoV-2R18 engagement governed its apparent efficacy. Also, ATN-161 is known to exhibit U-shaped dose–response characteristics49 thus, presenting a need to identify an optimally active-dose by titration of ATN-16127 which is beyond the scope of our present study. The mechanistic specificity of integrin inhibition by these antagonists regarding SARS-CoV-2 uptake strongly supports the idea that (1) integrin RGD engagement is an essential co-factor for cell entry and (2) integrin extension is required for cell entry based on BTT 3033's mechanism of action.
Inhibition of integrin activation or binding to SARS-CoV-2R18 blocks intracellular trafficking
We then used live-cell confocal microscopy to visualize Vero E6 cell entry and trafficking of SARS-CoV-2R18 in DMSO (mock)- and BTT 3033-treated cells (Fig. 4A, B). Most of the cells treated with GLPG0187 were de-adhered from the plate and were thus not suitable for imaging. The loss of cells with GLPG0187 was likely due to the loss of integrin-mediated adhesion by the broad-spectrum inhibitor. Cells were imaged at 3-min intervals for 21 min after the addition of ~ 107 SARS-CoV-2R18 particles. In DMSO treated cells (Mock in Fig. 4), SARS-CoV-2R18 particles were visible at cell membranes within 3 min, subsequently developed punctate features at the cell periphery, and trafficked to the perinuclear space. The rate of cell entry (time to perinuclear space ~ 10 min) was comparable to infectious virions52. For the BTT 3033-treated cells, early peripheral membrane localization of SARS-CoV-2R18 showed significant diminution of discernable puncta. It did not undergo retrograde traffic towards the perinuclear region within the timeframe of the experiment. The relative amount of virus binding to the surface was also reduced with BTT 3033 treatment (Fig. 4A,B), consistent with reduced binding observed by flow cytometry measurements (Fig. 2).
Figure 4.
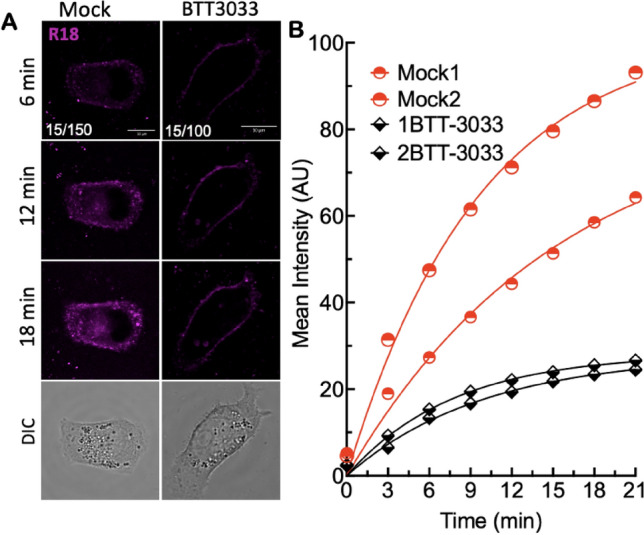
Stabilization of bent closed conformation with aI MIDAS domain binding integrin antagonist inhibits intracellular trafficking of SARS-CoV-2R18. (A) Live cell imaging of SARS-CoV-2R18 (magenta) binding and endocytosis shows perinuclear localization of SARS-CoV-2R18 vesicles. At the same time, the virus is seen to accumulate at the plasma membrane when α2β1 integrins are inhibited by 10 µM BTT 3033. Fluorescence images represent maximum projections of five confocal z slices. Mock and BTT 3033 treated samples are shown with different lookup tables (LUT) since binding in treated cells was lower than untreated cells. LUT lower/upper values are presented in the lower-left corner of 6-min timepoint images. Scale bars, 10 µm. (B) Traces of absolute intensity values of virus binding over time. Two representative cells for each condition are plotted from data acquired on the same day to compare intensity values directly. Data were fit to a non-linear regression function with arbitrary constants for appearance purposes. Imaging data were repeated at least three separate times.
Blocking of integrin signaling significantly inhibits productive infection of cells by SARS-CoV-2
Integrin activation is a complex and well-regulated spatiotemporal process involving the synchronized assembly and disassembly of multiple signaling elements at the integrin’s β-cytoplasmic tail (β-CT)53–55. Various groups have described a network of up to 156 interacting components that comprise the integrin adhesome56–59. Some adhesome components relevant to our study are shown in (Fig. 5A). Most β-CTs contain conserved sequences needed for integrin activity, such as the two β chain NPxY/F sequences, which are sites of competitive binding by adaptor proteins that regulate integrin activation and deactivation41,53, including sorting signals for clathrin-mediated endocytosis60–63. The phosphorylatable tyrosine (Y) residues of NPxY motifs are key regulatory sites of integrin activation on the β-CT. For example, N780PIY783 and N792PKY795 in β1 and N744PLY747 and N756ITY759 in β3 are motifs phosphorylated by Src family kinases (SFK) that may positively or negatively regulate interactions with phosphotyrosine-binding (PTB) domain-containing proteins. During the early stage of integrin activation, inhibitory proteins are displaced from the β-CT in exchange for integrin activators, ending with the recruitment of talin to the integrin tail38. However, the early wave of talin-mediated inside-out signaling is transiently terminated to allow Gα13, the effector of outside-in signaling, to bind to the conserved ExE motif (where x denotes any residue for specific integrins, e.g., EEE for β3-CT and EKE for β1-CT Fig. 5A), which overlaps the talin binding domain42.
Figure 5.

Inhibition of integrin activation blocks cell entry of SARS-CoV-2R18, suggesting integrin-mediated signaling is required for productive infection. (A) Aligned sequences of β1 and β3-integrin cytoplasmic tails (β-CT). The NPxY motif tyrosine residues (shown in brown) and the Ser and Thr residues (shown in purple) are important phosphorylation sites required for exchanging adaptor proteins. Srk family kinase-mediated phosphorylation of the NPxY motifs inhibits the binding of talin while promoting the association of inhibitor proteins such as DOK-1. Interaction zones between β-CT and adaptor proteins are denoted by associated horizontal lines. Functional roles of the proteins are indicated in parenthesis. For a detailed description, see refs.41,54. The membrane-permeable peptides mP6 and mP13 were based on the integrin β3 cytoplasmic tail. (B) Model of outside-inside-out signaling for integrin-mediated cell entry. Hypothetical SARS-CoV-2 binding to integrin β1 initiates Gα13 binding to the β1 cytoplasmic tail, which stimulates outside-in signaling in the absence of a known receptor-stimulated GPCR mediated inside-out signaling. mP6 is a specific inhibitor of Gα13 binding to the β1 cytoplasmic tail. The illustrations were generated using Microsoft® PowerPoint Version 16.51 (21071101). (C) Relative fluorescence readings of suspension Vero E6 cells after 30 min incubation with SARS-CoV-2R18 in vehicle- and 100 µM mP6 treated cells. (D) Live cell imaging of SARS-CoV-2R18 (magenta) binding and endocytosis shows cell membrane and perinuclear localization of SARS-CoV-2R18 vesicles. At the same time, the virus is seen to remain at the plasma membrane in cells treated with 50 µM mP6. LUT ranges are shown in the bottom left corner of 6-min timepoint images. Scale bars, 10 µm. (E) Traces of absolute intensity values of virus binding over time. Two representative cells for each condition are plotted from data acquired on the same day to compare intensity values directly. For comparison, mock-treated cell data are the same as in Fig. 4. Data were fit to a non-linear regression function with arbitrary constants for appearance purposes. (F) Inhibition of SARS-CoV-2 productive infection. Suspension Vero E6 cells were preincubated with 250 µM mP6 and 250 µM mP13 for 30 min and followed by infection with 0.01 MOI SARS-CoV-2 for an additional 60 min incubation. Cells were washed twice, transferred to a 12 well plate for 48 h, and assayed for viral RNA by RT-qPCR. Comparisons were performed using Ordinary one-way ANOVA, with Tukey's multiple comparison test using GraphPad Prism version 9.2.0.
Integrin binding to macromolecular ligands, such as SARS-CoV-2, facilitates Gα13-mediated outside-in signaling. Transmission of the tensile force through the integrin to talin stabilizes high-affinity integrin binding (in the EOS) to the ECM promotes the 'second wave' of inside-out signaling (Fig. 5B). The sequential mechanism of inside-out and outside-in signaling was previously established in part by the use of two myristoylated peptides, mP6 (Myr-FEEERA-OH), derived from the Gα13-binding domain and mP13 (Myr-KFEEERARAKWDT-OH) mimicking the β3-CT's talin binding domain42. It is worth noting that the previous mP6 and mP13 related study by Shen et al.42 established that the minimal sequence of EEERA does not interact with talin and is a specific inhibitor of Gα13 association with the β-CT and had no effect on talin-dependent inside-out signaling, or the late phase of outside-in signaling associated with the second wave of talin binding. However, mP13 affects all phases of integrin signaling42. To investigate the relationship between the integrin signaling events and SARS-CoV-2 engagement and cell entry, we treated cells with mP6 peptide, which inhibited cell entry of SARS-CoV-2R18 in flow cytometry and microscopy experiments (Fig. 5C–E). Similarly, mP13 inhibited cell entry in flow cytometry experiments (data not shown). The results for mP6 treated cells suggest that SARS-CoV-2 engagement initiates a Gα13-mediated outside-in integrin activation without a known receptor stimulus which is consistent with the idea that SARS-CoV-2 binding induces integrin activation64, as we previously demonstrated for the Sin Nombre virus24.
Because mP6 and mP13 are membrane-permeable peptides, they were suitable for infectivity experiments while obviating the need to expose cells to DMSO for extended periods. We, therefore, tested the efficacy of mP6 and mP13 at inhibiting cell entry and productive infection in Vero E6 cells with a 0.01 multiplicity of infection (MOI) of SARS-CoV-2. For the productive infection assay, infected cells were plated at confluency (500,000 cells/well in a 12 well plate) to minimize cell growth for 48 h post-infection. We used RT-qPCR to measure viral nucleocapsid RNA in the suspended cells or intact cell monolayers at 48 h post-infection, respectively. At 48 h post-infection, inhibition of productive infection by mP13 was significant relative to mock-treated cells, whereas the effect of mP6 was insignificant (Fig. 5F). The failure of mP6 to inhibit productive infection is consistent with the notion that viral replication52,65 perturbs Ca2+ homeostasis within the infected cells66 and thus dispenses with Gα13 activity in favor of talin-induced outside in-signaling42.
Discussion
This study provides mechanistic evidence for the functionality of extracellular ligand-binding domains of integrin β1 and cytoplasmic tails of integrins in general25,28, which offer possible molecular links between ACE2 and integrins. We show that Mn2+, which induces integrin extension and high-affinity ligand binding, enhances the cell entry of SARS-CoV-2R18. The increased virus binding and entry is consistent with the notion that integrin affinity and/or extension are essential for cell entry. In support of integrin-dependent endocytosis as a pathway of SARS-CoV-2R18 internalization, we used broad-spectrum RGD antagonists such as GLPG0187, which inhibited cell entry regardless of integrin activation status. Our study also suggested integrin specificity. BTT 3033, an αI allosteric antagonist that binds to the bent closed conformation of integrin β1 and stabilizes it, supports the possibility of integrin-dependent endocytosis of SARS-CoV-2R18 upon receptor binding. In a different framework, our data also show that SARS-CoV-2R18 can bind to low affinity and presumptively bent-conformation integrins23, however, in BTT 3033 treated cells, cell entry by SARS-CoV-2R18 is inhibited because integrin activation post- SARS-CoV-2R18 engagement is prevented. Thus, our data contextualize integrin extension as the "sine qua non of integrin cell adhesion function,"23 which in turn is an essential condition for integrin-mediated cell entry by SARS-CoV-2.
Focal adhesion kinase (FAK)67 is a well-established component of the adhesome that potentially bridges the signaling gap between integrin signaling turnover68 and ACE2. FAK is a tyrosine kinase known to direct the recruitment of talin to integrin β1-enriched nascent adhesions60,61. In ailing heart tissues, ACE2 binds integrin α5β1 in an RGD-independent manner. It is known to regulate FAK mediated cell adhesion and integrin signaling18, which terminates with endosomal trafficking30 (of virion-bearing integrins). The binding of macromolecular RGD ligands to resting integrins elicits ligand-induced integrin activation64. We hypothesize that in our in vitro experiments, SARS-CoV-2 binding to inactive integrins triggers a series of spatiotemporally-regulated recruitment of adhesome components, including Gα13 and talin, to the β-CT. Our data show that talin interaction with integrin β-CTs, which causes integrin extension, is indispensable for productive infection (Fig. 5). Talin binding to the β-CT generates the requisite inside-out signal that increases the affinity of the integrin ectodomain for SARS-CoV-2 binding, which in turn increases viral load. Cell entry of SARS-CoV-2 is clathrin-dependent69. Endocytosis of integrins is clathrin-dependent and -independent70 and involves adaptor proteins such as Dab2 and Numb71,72 attached to the β-CTs NPxY/NxxY motifs (Fig. 5A). Alternatively, some integrin α-subunits harbor a common endocytosis motif (Yxxϕ) recognized by the clathrin adaptor protein 2 (AP2)68.
Mészáros et al.25 have used bioinformatics to predict the existence of short amino acid sequences (~ 3–10 residues): short linear motifs (SLiMs), such as NPxY/Nxxy, Yxxϕ in the cytoplasmic tails of ACE2 and integrins that mediate endocytosis and autophagy. Some of their theoretical predictions have been validated by experimental studies. First, Kliche et al.28 confirmed the existence of SLiMs. They extended their findings to establish a potential connection between ACE2 and integrin β3 cytoplasmic tail interactions with scaffolding and adaptor proteins linked to endocytosis and autophagy. Second, SLiM sequences known to bind and activate the transmembrane glycoprotein neuropilin 1 (NRP1) were identified as potential mediators of SARS-CoV-2 endocytosis25. Interestingly, NRP1, which is abundantly expressed in the olfactory epithelium, is now declared as an effector for SARS-CoV-2 infection34,35. NRP1 localizes at adhesion sites and promotes fibronectin-bound, activated α5β1 integrin endocytosis, and directs the cargo to the perinuclear cytoplasm29–34. Studies have shown that the endocytosis of active and inactive integrins to EEA1-containing early endosomes follows distinct mechanisms involving different adaptor proteins. The inactive integrin is promptly recycled back to the plasma membrane via an ARF6‐ and EEA1‐positive compartment in a Rab4 -dependent manner31. We observed that in BTT 3033-treated cells replete with inactive β1 integrins, SARS-CoV-2R18 remained membrane-bound, whereas untreated cells displayed internalization and perinuclear localization of SARS-CoV-2R18. This is consistent with the known trafficking of ligand-bearing integrins, including those directed by NRP1, to the perinuclear space29,30,32.
Our study has some limitations. Integrin activation is often initiated by other receptors such as G-protein coupled receptors (GPCRs), growth factors, and other integrins38. Future studies will explore the effect of receptor-mediated inside-out signaling, modeled under inflammatory conditions of COVID-19. In addition, the criteria for selecting specific integrins as co-factors of SARS-CoV-2 infectivity are not known and thus worthy of future investigation. Finally, the study is based on the USA-WA1/2020 SARS-CoV-2 strain. Our present study lays the groundwork for examining the activity of the various emergent SARS-CoV-2 variants.
Although several integrins types12,25–28 are believed to be co-receptors of SARS-CoV-2 infectivity, our study suggests inhibitor specificity for integrin β1. This is consistent with known factors: (1) correlated increased expressions of β115 and ACE2 in relevant tissues16,17, (2) cytoplasmic tail in cis interactions between ACE2 and integrin β114, and (3) synergy between ACE2 and integrin β1 signaling that promotes RGD mediated cell adhesion18. To optimize integrin engagement, our cell-binding assays and primary infection assays were carried out in suspension such that ACE2 and integrins were not segregated by cell polarization73,74. However, our microscopy studies on adherent cells agreed with the flow cytometry results. Thus, our study represents an initial step toward establishing a mechanistic role for SARS-CoV-2-mediated integrin activation required for cell entry and productive infection.
Materials and methods
Materials
USA-WA1/2020 SARS-CoV-2 strain was obtained from BEI Resources (NIAID, NIH). Integrin inhibitors, BTT3033, a selective inhibitor of α2β1, ATN-161 an integrin α5β1 antagonist27, and GLPG0187 a broad-spectrum integrin inhibitor, were purchased as powders from Tocris Bioscience. The EEA1 rabbit monoclonal antibody (clone C45B10) was from Cell Signaling Technologies (CAT# 3288S). Alexa fluor 647 conjugated F(ab')2 fragment goat anti-rabbit IgG was from Invitrogen (CAT# A21246). In addition, myristoylated peptides; mP6 (Myr-FEEERA-OH) and mP13 (Myr-KFEEERARAKWDT-OH) were custom synthesized at Vivitide.
Cell culture
African green monkey kidney cells (Vero E6, ATCC) were maintained in DMEM media from Sigma CAT# D5796. All media contained 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine and were kept at 37 °C in a CO2 water-jacketed incubator of 5% CO2 and 95% air (Forma Scientific, Marietta, OH, USA).
UV inactivation and fluorescent labeling of the envelope membrane of SARS-CoV-2 with octadecyl rhodamine (R18)
USA-WA1/2020 SARS-CoV-2 strain (from BEI Resources, NIAID, NIH) was cultured in Vero E6 cells in a biosafety level 3 (BSL-3) containment under a protocol approved by the University of New Mexico's Institutional Biosafety Committee or IBC (Public Health Service registration number C20041018-0267). First, live SARS-CoV-2 were harvested at peak titers of 107 plaque-forming units/mL (PFU/ml). Next, SARS-CoV-2 was UV inactivated using 254 nm (≈ 5 mW/cm2) U.V. irradiation of a TS-254R Spectroline UV Transilluminator (Spectronics Corp., Westbury, NY) following a similar protocol for inactivating pathogenic orthohantaviruses45,75. Briefly, Vero E6 cells were inoculated with SARS-CoV-2 and maintained at 37 °C for 2–4 days. At 70–75% cell death (due to viral cytopathic effect), the supernatant was harvested and subjected to light centrifugation (1000 rpm, 10 min) to remove cellular debris. For UV inactivation, supernatants were added to a 12 well plate at 500 µl aliquot/well. Then UV -irradiated at 3.8 cm above the sample for 0, 10,15, 20, 25, 30, 60, and 90 s and then tested for viability by a 3-day plaque assay as described elsewhere76,77. The titration of UV irradiation times was used to establish a minimal UV dose for complete inactivation. After UV treatment, the 500 µl fractions were pooled into 15 mL tubes stored in a − 80 °C freezer pending the results of a plaque assay. Under our experimental conditions, we established that a minimum UV irradiation interval of 25 s was required for the complete inactivation of SARS-CoV-2. A 90 s UV dose was approved by the IBC for removal of inactivated SARS-CoV-2 out of the BSL-3 lab after it was established that the virus particles were capable of specific binding to Vero E6 cells.
Crude UV-inactivated SARS-CoV-2 samples were purified by floating 10 ml of SARS-CoV-2 supernatant on a density gradient comprising 2 ml volumes of 1.2 g/ml and 1.0 g/ml CsCl in PBS media in 14 × 89-mm Beckman polyallomer tubes. The samples were centrifuged for 1.5 h at 4 °C using a Beckman SW41Ti rotor at 30,000 pm. A band was collected at the interface and purified by centrifugation in HHB using 100 kDa cutoff Microcon® Centrifugal Filter. The purified SARS-CoV-2 samples were stored in 1.0 ml aliquots at − 80 °C. SARS-CoV-2 particles were fluorescently labeled and calibrated according to the same protocol used for the Sin Nombre virus (SNV)45. The final volume for each labeled batch preparation was limited to 500 µl. The number of SARS-CoV-2R18 particles in each sample preparation was estimated from absorption measurements using the following equation: # of moles of R18 (derived from sample absorbance) × Avogadro’s number (6.02 × 1023 molecules mole−1)/estimated average number of R18 molecules per virion (10,000)45. The yield of SARS-CoV-2R18 particles was typically in the 108/µl range. Batch samples were stored in 20 µl aliquots at − 80 °C.
Flow cytometry binding assays of SARS-CoV-2R18 to vero E6 cells
For flow cytometry assays, cells were cultured in T25 or T75 flasks to 80% confluence. Cells were then treated with 0.25% trypsin and transferred to minimum essential medium (MEM) media. Cell counts and viability were performed using a Life Technologies Countess II FL Automated cell counter (Thermofisher Scientific). Test suspension cell samples were transferred to microfuge tubes in 40 µl-aliquots (1000 cells/µl). SARS-CoV-2R18 was added to tubes at 5000 SARS-CoV-2R18/cell and incubated using a shaker at 500 rpm for 20 min at 37 °C. For blocking assays, cells were incubated with 5 × unlabeled SARS-CoV-2 or 10 µM integrin inhibitors for 20 min before the addition of SARS-CoV-2R18. Samples were centrifuged at 3,000 rpm; the pellet was resuspended in HHB buffer (30 mM HEPES, 110 mM NaCl, 10 mM KCl, 1 mM MgCl2⋅6H2O, and 10 mM glucose, pH 7.4) buffer and read on an Accuri flow cytometer. For kinetic assays, Vero E6 suspension cells in 40 µl volumes (1000 cells/µl) were placed in ± Mn2+ media in duplicate microfuge tubes at 37 °C. Sars-CoV-2R18 was then added (5000 virions/cell) to the tubes and incubated for 1, 3, 5, 7, 9 min. At each time point, the tubes were quenched in an ice bath, then samples were centrifuged and resuspended in 95 µl HHB buffer and analyzed on a flow cytometer.
Live cell confocal microscopy
Imaging was performed using a Leica TCS SP8 Laser Scanning Confocal Microscope with a 63 × water objective and a Bioptechs objective heater to maintain cells at physiological temperature (~ 36–37 °C). Vero E6 cells were plated in eight-well Lab-Tek (Nunc) chambers at a density of 30,000 cells per well 24 h before imaging. Cells were imaged in Tyrode’s buffer (135 mM NaCl, 10 mM KCl, 0.4 mM MgCl2, 1 mM CaCl2 20 mM glucose, 0.1% BSA, 10 mM HEPES, pH 7.2). For integrin inhibition, cells were treated with 10 μM BTT3033-α2β1 or 50 μM MP6 in Tyrode’s buffer for 30 min before imaging. ~ 1 × 109 SARS-CoV-2R18 particles were added per well, and z-stacks (300 nm thickness) were acquired every 3 min for 21 min to visualize viral cell entry. R18 was excited using 561 nm light, isolated from the white light source. R18 emission and differential interference contrast (DIC) transmitted light were captured with Leica Hybrid detectors (HyD) in a spectral window of 571–636 nm (for R18 emission). Analysis of the accumulation of SARS-CoV-2R18 particles in Vero E6 cells was completed using Matlab. Briefly, regions of interest (ROI) were created around the cell membrane, and the mean SARS-CoV-2R18 intensity was measured within the cell mask at each time point.
Immunofluorescence
Vero E6 cells were plated on 18-mm coverslips overnight in a 6 well plate at a density of 100,000 cells/well. Cells were exposed to ~ 1 × 109 SARS-CoV-2R18 particles/well for 15 min at 37 °C, in the presence or absence of 10 μM BTT 3033. Cells were then washed in phosphate-buffered saline (PBS) and fixed using 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature. Cells were extensively washed with 10 mM Tris (pH 7.4) and PBS and permeabilized with 0.1% Triton. Cells were labeled with anti-EEA1 primary antibody and anti-rabbit Alexa Fluor 647 secondary. Nuclei were stained with Hoechst 33258. Cells were mounted on microscope slides using Prolong Diamond Antifade Mountant (Invitrogen, CAT#P33970). Samples were imaged using a Leica TCS SP8 Laser Scanning Confocal Microscope with a 63× oil objective.
Infection inhibition
Vero E6 cells grown at 80% confluency were trypsinized and divided into microfuge tubes aliquots of 1.5 × 106cells in 750 µl media containing 250 µM mP6, 250 µM mP13, 10 µM BTT 3033, DMSO, and media only. Samples were shaken at 500 rpm for 30 min at 37 °C. After transfer to a BSL-3 laboratory, 0.01 MOI of SARS CoV-2 (lot #P3: 1.2 × 107 pfu) and then incubated for 60 min while shaking. Tubes were spun down (1000 rpm for 3 min), resuspended in fresh media, and spun down again. The cells were then resuspended in 300 µl of media and transferred to a 12 well plate in 100 µl aliquots (500,000 virions/well) for triplicate measurements. An additional 400 µl were added to each well for a final volume of 500 µl. The plate was transferred to an incubator for 48 h to allow the virus to replicate. The cells were then washed with 1xPBS before RNA was extracted with TRIzol™ (Thermofisher, #15596026) according to the manufacturer's protocol:
(https://assets.thermofisher.com/TFS-Assets/LSG/manuals/trizol_reagent.pdf).
The RNA was quantified with a Nanodrop and total cellular cDNA transcribed with the Applied Biosystems™ High-Capacity cDNA Reverse Transcription Kit (Fisher Scientific #43-688-14).
RT-qPCR was performed with the TaqMan Fast Advance Master Mix (Fisher Scientific #4444963) and the following primers ordered from Integrated DNA Technologies: Forward: 5′-CCCTGTGGGTTTTACACTTAA-3′, Reverse: 5′-ACGATTGTGCATCAGCTGA-3′, and probe: 5′-[FAM] CCGTCTGCGGTATGTGGAAAGGTTATGG [BHQ1]-3′. The RT-qPCR was performed using an Applied Biosystems QuantStudio 5 instrument.
Statistical analysis
SARS-CoV-2R18 binding was expressed as the mean channel fluorescence (MCF) output of the flow cytometer. For different batch preparations of SARS-CoV-2R18, we first tested functional binding of SARS-CoV-2R18 to cells by comparing the MCF readings of 1 mM Mn2+- activated cells and resting cells after correcting for autofluorescence. Then, assuming sampling from a Gaussian distribution, the two groups were compared using estimation plots47 with unpaired two-tailed t-tests to check for consistency between batch preparations of SARS-CoV-2R18. For integrin activation inhibitor tests involving three or more groups, comparisons were performed using Ordinary one-way ANOVA with Tukey's multiple comparison test. Data analysis was done with GraphPad Prism software version 9.2.0. Statistical significance was defined as p < 0.05.
Supplementary Information
Acknowledgements
This project is supported by an award from the National Center for Advancing Translational Sciences, National Institutes of Health under grant number UL1TR001449, NIH R35GM-126934 (DSL). Fluorescence microscopy was performed in the University of New Mexico Comprehensive Cancer Center fluorescence microscopy shared resource (NIH P30CA118100).
Author contributions
T.B. conceived and designed the project. T.B., D.S.L. conceived the flow cytometry and microscopy experiments, respectively; A.M.K. and S.B. provided essential materials; P.S., T.B., V.B., D.A.R., A.M.K., S.B., and D.S.L. carried out the experiments; all authors reviewed the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-99893-7.
References
- 1.WHO https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (2020).
- 2.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, Guan L, Wei Y, Li H, Wu X, Xu J, Tu S, Zhang Y, Chen H, Cao B. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet. 2020;395:1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia J, Yu T, Zhang X, Zhang L. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet. 2020;395:507–513. doi: 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stewart PL, Nemerow GR. Cell integrins: Commonly used receptors for diverse viral pathogens. Trends Microbiol. 2007;15:500–507. doi: 10.1016/j.tim.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Muller MA, Drosten C, Pohlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280.e278. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sungnak W, Huang N, Becavin C, Berg M, Queen R, Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Sampaziotis F, Worlock KB, Yoshida M, Barnes JL, Network, H.C.A.L.B SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li MY, Li L, Zhang Y, Wang XS. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty. 2020;9:45. doi: 10.1186/s40249-020-00662-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hikmet F, Mear L, Edvinsson A, Micke P, Uhlen M, Lindskog C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020;16:e9610. doi: 10.15252/msb.20209610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrison AG, Lin T, Wang P. Mechanisms of SARS-CoV-2 transmission and pathogenesis. Trends Immunol. 2020;41:1100–1115. doi: 10.1016/j.it.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, Cao Y, Yousif AS, Bals J, Hauser BM, Feldman J, Muus C, Wadsworth MH, 2nd, Kazer SW, Hughes TK, Doran B, Gatter GJ, Vukovic M, Taliaferro F, Mead BE, Guo Z, Wang JP, Gras D, Plaisant M, Ansari M, Angelidis I, Adler H, Sucre JMS, Taylor CJ, Lin B, Waghray A, Mitsialis V, Dwyer DF, Buchheit KM, Boyce JA, Barrett NA, Laidlaw TM, Carroll SL, Colonna L, Tkachev V, Peterson CW, Yu A, Zheng HB, Gideon HP, Winchell CG, Lin PL, Bingle CD, Snapper SB, Kropski JA, Theis FJ, Schiller HB, Zaragosi LE, Barbry P, Leslie A, Kiem HP, Flynn JL, Fortune SM, Berger B, Finberg RW, Kean LS, Garber M, Schmidt AG, Lingwood D, Shalek AK, Ordovas-Montanes J, lung-network@humancellatlas.org. H.C.A.L.B.N.E.a. & Network, H.C.A.L.B SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181:1016–1035.e1019. doi: 10.1016/j.cell.2020.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nader D, Fletcher N, Curley GF, Kerrigan SW. SARS-CoV-2 uses major endothelial integrin alphavbeta3 to cause vascular dysregulation in-vitro during COVID-19. PLoS ONE. 2021;16:e0253347. doi: 10.1371/journal.pone.0253347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makowski L, Olson-Sidford W, Weisel JW. Biological and clinical consequences of integrin binding via a rogue RGD motif in the SARS CoV-2 spike protein. Viruses. 2021;13:146. doi: 10.3390/v13020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin Q, Keller RS, Weaver B, Zisman LS. Interaction of ACE2 and integrin beta1 in failing human heart. Biochem. Biophys. Acta. 2004;1689:175–178. doi: 10.1016/j.bbadis.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Sun M, Opavsky MA, Stewart DJ, Rabinovitch M, Dawood F, Wen WH, Liu PP. Temporal response and localization of integrins beta1 and beta3 in the heart after myocardial infarction: Regulation by cytokines. Circulation. 2003;107:1046–1052. doi: 10.1161/01.CIR.0000051363.86009.3C. [DOI] [PubMed] [Google Scholar]
- 16.Krishnamurthy P, Subramanian V, Singh M, Singh K. Deficiency of beta1 integrins results in increased myocardial dysfunction after myocardial infarction. Heart. 2006;92:1309–1315. doi: 10.1136/hrt.2005.071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM. Trilogy of ACE2: A peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010;128:119–128. doi: 10.1016/j.pharmthera.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clarke NE, Fisher MJ, Porter KE, Lambert DW, Turner AJ. Angiotensin converting enzyme (ACE) and ACE2 bind integrins and ACE2 regulates integrin signalling. PLoS ONE. 2012;7:e34747. doi: 10.1371/journal.pone.0034747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo BH, Springer TA. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006;18:579–586. doi: 10.1016/j.ceb.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu. Rev. Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nordenfelt P, Elliott HL, Springer TA. Coordinated integrin activation by actin-dependent force during T-cell migration. Nat. Commun. 2016;7:13119. doi: 10.1038/ncomms13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schurpf T, Springer TA. Regulation of integrin affinity on cell surfaces. EMBO J. 2011;30:4712–4727. doi: 10.1038/emboj.2011.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore TI, Aaron J, Chew TL, Springer TA. Measuring integrin conformational change on the cell surface with super-resolution microscopy. Cell Rep. 2018;22:1903–1912. doi: 10.1016/j.celrep.2018.01.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bondu V, Wu C, Cao W, Simons PC, Gillette J, Zhu J, Erb L, Zhang XF, Buranda T. Low-affinity binding in cis to P2Y2R mediates force-dependent integrin activation during hantavirus infection. Mol. Biol. Cell. 2017;28:2887–2903. doi: 10.1091/mbc.e17-01-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mészáros B, et al. Short linear motif candidates in the cell entry system used by SARS-CoV-2 and their potential therapeutic implications. Sci. Signal. 2021;14:eabd0334. doi: 10.1126/scisignal.abd0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park EJ, Myint PK, Appiah MG, Darkwah S, Caidengbate S, Ito A, Matsuo E, Kawamoto E, Gaowa A, Shimaoka M. The spike glycoprotein of SARS-CoV-2 binds to beta1 integrins expressed on the surface of lung epithelial cells. Viruses. 2021;13:645. doi: 10.3390/v13040645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beddingfield BJ, Iwanaga N, Chapagain PP, Zheng W, Roy CJ, Hu TY, Kolls JK, Bix GJ. The integrin binding peptide, ATN-161, as a novel therapy for SARS-CoV-2 infection. JACC Basic Transl. Sci. 2021;6:1–8. doi: 10.1016/j.jacbts.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kliche J, Kuss H, Ali MA, Ivarsson Y. Cytoplasmic short linear motifs in ACE2 and integrin β3 link SARS-CoV-2 host cell receptors to mediators of endocytosis and autophagy. Sci. Signal. 2021;14:eabf1117. doi: 10.1126/scisignal.abf1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valdembri D, Caswell PT, Anderson KI, Schwarz JP, Konig I, Astanina E, Caccavari F, Norman JC, Humphries MJ, Bussolino F, Serini G. Neuropilin-1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol. 2009;7:e25. doi: 10.1371/journal.pbio.1000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mana G, Valdembri D, Serini G. Conformationally active integrin endocytosis and traffic: Why, where, when and how? Biochem. Soc. Trans. 2020;48:83–93. doi: 10.1042/BST20190309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arjonen A, Alanko J, Veltel S, Ivaska J. Distinct recycling of active and inactive beta1 integrins. Traffic. 2012;13:610–625. doi: 10.1111/j.1600-0854.2012.01327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Franceschi N, Hamidi H, Alanko J, Sahgal P, Ivaska J. Integrin traffic—The update. J. Cell Sci. 2015;128:839–852. doi: 10.1242/jcs.161653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lobert VH, Stenmark H. The ESCRT machinery mediates polarization of fibroblasts through regulation of myosin light chain. J. Cell Sci. 2012;125:29–36. doi: 10.1242/jcs.088310. [DOI] [PubMed] [Google Scholar]
- 34.Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, van der Meer F, Kallio K, Kaya T, Anastasina M, Smura T, Levanov L, Szirovicza L, Tobi A, Kallio-Kokko H, Osterlund P, Joensuu M, Meunier FA, Butcher SJ, Winkler MS, Mollenhauer B, Helenius A, Gokce O, Teesalu T, Hepojoki J, Vapalahti O, Stadelmann C, Balistreri G, Simons M. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370:856–860. doi: 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Anton-Plagaro C, Shoemark DK, Simon-Gracia L, Bauer M, Hollandi R, Greber UF, Horvath P, Sessions RB, Helenius A, Hiscox JA, Teesalu T, Matthews DA, Davidson AD, Collins BM, Cullen PJ, Yamauchi Y. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. 2020;370:861–865. doi: 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye F, Kim C, Ginsberg MH. Reconstruction of integrin activation. Blood. 2012;119:26–33. doi: 10.1182/blood-2011-04-292128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banno A, Ginsberg MH. Integrin activation. Biochem. Soc. Trans. 2008;36:229–234. doi: 10.1042/BST0360229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu. Rev. Cell Dev. Biol. 2011;27:321–345. doi: 10.1146/annurev-cellbio-100109-104104. [DOI] [PubMed] [Google Scholar]
- 39.Zhu J, Zhu J, Springer TA. Complete integrin headpiece opening in eight steps. J. Cell Biol. 2013;201:1053–1068. doi: 10.1083/jcb.201212037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen B, Delaney MK, Du X. Inside-out, outside-in, and inside-outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Curr. Opin. Cell Biol. 2012;24:600–606. doi: 10.1016/j.ceb.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gahmberg CG, Fagerholm SC, Nurmi SM, Chavakis T, Marchesan S, Gronholm M. Regulation of integrin activity and signalling. Biochem. Biophys. Acta. 2009;1790:431–444. doi: 10.1016/j.bbagen.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen B, Zhao X, O'Brien KA, Stojanovic-Terpo A, Delaney MK, Kim K, Cho J, Lam SC, Du X. A directional switch of integrin signalling and a new anti-thrombotic strategy. Nature. 2013;503:131–135. doi: 10.1038/nature12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong H, Shen B, Flevaris P, Chow C, Lam SC, Voyno-Yasenetskaya TA, Kozasa T, Du X. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin "outside-in" signaling. Science. 2010;327:340–343. doi: 10.1126/science.1174779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buranda T, Wu Y, Perez D, Chigaev A, Sklar LA. Real-time partitioning of octadecyl rhodamine B into bead-supported lipid bilayer membranes revealing quantitative differences in saturable binding sites in DOPC and 1:1:1 DOPC/SM/cholesterol membranes. J. Phys. Chem. 2010;114:1336–1349. doi: 10.1021/jp906648q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buranda T, Wu Y, Perez D, Jett SD, BonduHawkins V, Ye C, Edwards B, Hall P, Larson RS, Lopez GP, Sklar LA, Hjelle B. Recognition of decay accelerating factor and alpha(v)beta(3) by inactivated hantaviruses: Toward the development of high-throughput screening flow cytometry assays. Anal. Biochem. 2010;402:151–160. doi: 10.1016/j.ab.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benson SW. Foundations of Chemical Kinetics. McGraw-Hill; 1960. [Google Scholar]
- 47.Ho J, Tumkaya T, Aryal S, Choi H, Claridge-Chang A. Moving beyond P values: Data analysis with estimation graphics. Nat. Methods. 2019;16:565–566. doi: 10.1038/s41592-019-0470-3. [DOI] [PubMed] [Google Scholar]
- 48.Nissinen L, Koivunen J, Kapyla J, Salmela M, Nieminen J, Jokinen J, Sipila K, Pihlavisto M, Pentikainen OT, Marjamaki A, Heino J. Novel alpha2beta1 integrin inhibitors reveal that integrin binding to collagen under shear stress conditions does not require receptor preactivation. J. Biol. Chem. 2012;287:44694–44702. doi: 10.1074/jbc.M111.309450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Donate F, Parry GC, Shaked Y, Hensley H, Guan X, Beck I, Tel-Tsur Z, Plunkett ML, Manuia M, Shaw DE, Kerbel RS, Mazar AP. Pharmacology of the novel antiangiogenic peptide ATN-161 (Ac-PHSCN-NH2): Observation of a U-shaped dose-response curve in several preclinical models of angiogenesis and tumor growth. Clin. Cancer Res. 2008;14:2137–2144. doi: 10.1158/1078-0432.CCR-07-4530. [DOI] [PubMed] [Google Scholar]
- 50.Benito-Jardon M, Klapproth S, Gimeno LI, Petzold T, Bharadwaj M, Muller DJ, Zuchtriegel G, Reichel CA, Costell M. The fibronectin synergy site re-enforces cell adhesion and mediates a crosstalk between integrin classes. Elife. 2017;6:e22264. doi: 10.7554/eLife.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cirkel GA, Kerklaan BM, Vanhoutte F, Van der Aa A, Lorenzon G, Namour F, Pujuguet P, Darquenne S, de Vos FY, Snijders TJ, Voest EE, Schellens JH, Lolkema MP. A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. Investig. New Drugs. 2016;34:184–192. doi: 10.1007/s10637-015-0320-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bar-On YM, Flamholz A, Phillips R, Milo R. SARS-CoV-2 (COVID-19) by the numbers. Elife. 2020;9:e57309. doi: 10.7554/eLife.57309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goult BT, Yan J, Schwartz MA. Talin as a mechanosensitive signaling hub. J. Cell Biol. 2018;217:3776–3784. doi: 10.1083/jcb.201808061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morse EM, Brahme NN, Calderwood DA. Integrin cytoplasmic tail interactions. Biochemistry. 2014;53:810–820. doi: 10.1021/bi401596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anthis NJ, Campbell ID. The tail of integrin activation. Trends Biochem. Sci. 2011;36:191–198. doi: 10.1016/j.tibs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zaidel-Bar R, Geiger B. The switchable integrin adhesome. J. Cell Sci. 2010;123:1385–1388. doi: 10.1242/jcs.066183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Geiger T, Zaidel-Bar R. Opening the floodgates: Proteomics and the integrin adhesome. Curr. Opin. Cell Biol. 2012;24:562–568. doi: 10.1016/j.ceb.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 58.Ye F, Lagarrigue F, Ginsberg MH. SnapShot: Talin and the modular nature of the integrin adhesome. Cell. 2014;156:1340.e1341. doi: 10.1016/j.cell.2014.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horton ER, Humphries JD, James J, Jones MC, Askari JA, Humphries MJ. The integrin adhesome network at a glance. J. Cell Sci. 2016;129:4159–4163. doi: 10.1242/jcs.192054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lawson C, Lim ST, Uryu S, Chen XL, Calderwood DA, Schlaepfer DD. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J. Cell Biol. 2012;196:223–232. doi: 10.1083/jcb.201108078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nader GP, Ezratty EJ, Gundersen GG. FAK, talin and PIPKIgamma regulate endocytosed integrin activation to polarize focal adhesion assembly. Nat. Cell Biol. 2016;18:491–503. doi: 10.1038/ncb3333. [DOI] [PubMed] [Google Scholar]
- 62.Ohno H, Stewart J, Fournier MC, Bosshart H, Rhee I, Miyatake S, Saito T, Gallusser A, Kirchhausen T, Bonifacino JS. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science. 1995;269:1872–1875. doi: 10.1126/science.7569928. [DOI] [PubMed] [Google Scholar]
- 63.Maginnis MS, Mainou BA, Derdowski A, Johnson EM, Zent R, Dermody TS. NPXY motifs in the beta1 integrin cytoplasmic tail are required for functional reovirus entry. J. Virol. 2008;82:3181–3191. doi: 10.1128/JVI.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Du XP, Plow EF, Frelinger AL, 3rd, O'Toole TE, Loftus JC, Ginsberg MH. Ligands "activate" integrin alpha IIb beta 3 (platelet GPIIb-IIIa) Cell. 1991;65:409–416. doi: 10.1016/0092-8674(91)90458-B. [DOI] [PubMed] [Google Scholar]
- 65.Ogando NS, Dalebout TJ, Zevenhoven-Dobbe JC, Limpens R, van der Meer Y, Caly L, Druce J, de Vries JJC, Kikkert M, Barcena M, Sidorov I, Snijder EJ. SARS-coronavirus-2 replication in Vero E6 cells: Replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 2020;101:925–940. doi: 10.1099/jgv.0.001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Y, Frey TK, Yang JJ. Viral calciomics: Interplays between Ca2+ and virus. Cell Calcium. 2009;46:1–17. doi: 10.1016/j.ceca.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 68.Moreno-Layseca P, Icha J, Hamidi H, Ivaska J. Integrin trafficking in cells and tissues. Nat. Cell Biol. 2019;21:122–132. doi: 10.1038/s41556-018-0223-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bayati A, Kumar R, Francis V, McPherson PS. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021;296:100306. doi: 10.1016/j.jbc.2021.100306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ezratty EJ, Bertaux C, Marcantonio EE, Gundersen GG. Clathrin mediates integrin endocytosis for focal adhesion disassembly in migrating cells. J. Cell Biol. 2009;187:733–747. doi: 10.1083/jcb.200904054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishimura T, Kaibuchi K. Numb controls integrin endocytosis for directional cell migration with aPKC and PAR-3. Dev. Cell. 2007;13:15–28. doi: 10.1016/j.devcel.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 72.Calderwood DA, Fujioka Y, de Pereda JM, Garcia-Alvarez B, Nakamoto T, Margolis B, McGlade CJ, Liddington RC, Ginsberg MH. Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: A structural prototype for diversity in integrin signaling. Proc. Natl. Acad. Sci. USA. 2003;100:2272–2277. doi: 10.1073/pnas.262791999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ren X, Glende J, Al-Falah M, de Vries V, Schwegmann-Wessels C, Qu X, Tan L, Tschernig T, Deng H, Naim HY, Herrler G. Analysis of ACE2 in polarized epithelial cells: Surface expression and function as receptor for severe acute respiratory syndrome-associated coronavirus. J. Gen. Virol. 2006;87:1691–1695. doi: 10.1099/vir.0.81749-0. [DOI] [PubMed] [Google Scholar]
- 74.Drubin DG, Nelson WJ. Origins of cell polarity. Cell. 1996;84:335–344. doi: 10.1016/S0092-8674(00)81278-7. [DOI] [PubMed] [Google Scholar]
- 75.Buranda T, Swanson S, Bondu V, Schaefer L, Maclean J, Mo ZZ, Wycoff K, Belle A, Hjelle B. Equilibrium and kinetics of sin nombre hantavirus binding at DAF/CD55 functionalized bead surfaces. Viruses. 2014;6:1091–1111. doi: 10.3390/v6031091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Darnell ME, Subbarao K, Feinstone SM, Taylor DR. Inactivation of the coronavirus that induces severe acute respiratory syndrome, SARS-CoV. J. Virol. Methods. 2004;121:85–91. doi: 10.1016/j.jviromet.2004.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Darnell ME, Taylor DR. Evaluation of inactivation methods for severe acute respiratory syndrome coronavirus in noncellular blood products. Transfusion. 2006;46:1770–1777. doi: 10.1111/j.1537-2995.2006.00976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ke Z, Oton J, Qu K, Cortese M, Zila V, McKeane L, Nakane T, Zivanov J, Neufeldt CJ, Cerikan B, Lu JM, Peukes J, Xiong X, Krausslich HG, Scheres SHW, Bartenschlager R, Briggs JAG. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature. 2020;588:498–502. doi: 10.1038/s41586-020-2665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Neuman BW, Buchmeier MJ. Supramolecular architecture of the coronavirus particle. Adv. Virus Res. 2016;96:1–27. doi: 10.1016/bs.aivir.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;183:1735. doi: 10.1016/j.cell.2020.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.