

Abstract
Characterisation and diagnosis of idiopathic Parkinson’s disease (iPD) is a current challenge that hampers both clinical assessment and clinical trial development with the potential inclusion of non-PD cases. Here, we used a targeted mass spectrometry approach to quantify 38 metabolites extracted from the serum of 231 individuals. This cohort is currently one of the largest metabolomic studies including iPD patients, drug-naïve iPD, healthy controls and patients with Alzheimer’s disease as a disease-specific control group. We identified six metabolites (3-hydroxykynurenine, aspartate, beta-alanine, homoserine, ornithine (Orn) and tyrosine) that are significantly altered between iPD patients and control participants. A multivariate model to predict iPD from controls had an area under the curve (AUC) of 0.905, with an accuracy of 86.2%. This panel of metabolites may serve as a potential prognostic or diagnostic assay for clinical trial prescreening, or for aiding in diagnosing pathological disease in the clinic.
Subject terms: Diagnostic markers, Metabolomics, Parkinson's disease
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease (AD) and affects around five million people worldwide1. Neuropathological hallmarks of PD include; the loss of catecholaminergic neurons in the substantia nigra, an increase in striatal dopamine deficiency and the presence of ɑ-synuclein aggregate-containing intracellular inclusions2. Moreover, the abundance, structure and function of the striatal N-Methyl-d-Aspartate (NMDA) receptor is altered by the dopamine depletion and pharmacological treatments used in PD3. NMDA receptors are ion channel proteins composed of multiple subunits and allow positively charged ions (like Zn2+, Mg2+ and Ca2+) to pass the cell membrane when activated via glutamate and glycine binding. They have complex regulatory properties and play a central role in synaptic plasticity, learning and memory4. Another potential player in the pathogenesis of PD is the RhoA-ROCK pathway. It plays a critical role in inflammation, and e.g. ROCK inhibitors may provide new protective strategies against PD progression5,6. As is common for diseases where the aetiology is unknown, PD is diagnosed clinically with the presence of7 bradykinesia, supported by the presence of rest tremor, postural instability and rigidity8,9. Non-motor symptoms such as disruption of gastric tract motility (constipation), sleep disturbance and depression are frequently present. The accuracy of clinical diagnosis has been well described10,11 and the clinical phenotype, especially at the onset of the disease, can encompass more than one pathophysiological entity. Since its first description by James Parkinson in 1817, the disease has been split into several different entities, including idiopathic PD (iPD). However, the severity and progression of iPD varies and it is uncertain whether this variation indicates further sub-entities or a broad range of phenotypes of a single entity.
People with PD usually present with symptoms when 50% or more dopaminergic neurons of the substantia nigra are lost12–14. Disease-modifying therapies would be therefore most effective when introduced early. This would be ideally prior to significant neuronal loss and thus well before clinical manifestations were apparent. While there are currently no disease-modifying therapies for PD, early and accurate identification of PD would aid in the discovery of such therapies. Furthermore, research into the understanding of the early pathophysiological event in PD would be aided by presymptomatic recognition.
Currently, no reliable biomarker exist that detect presymptomatic iPD. This underlines the importance of the development of a new diagnostic marker to facilitate both early diagnosis and assessment of new potential treatments. One key phenotype associated with iPD is a pronounced presence of oxidative stress markers including nitration and oxidation15,16. Altered levels of metabolites, including those associated with oxidation, have been measured in a variety of sample types (i.e. brain tissue17,18, cerebrospinal fluid (CSF)19–21, blood serum22,23, blood plasma24,25, red blood cells (RBC)26,27, sebum28 and urine29) from both drug-naïve and l-3,4-dihydroxyphenylalanine (l-DOPA) treated iPD patients with samples taken from healthy, age-matched control groups. Affected metabolic pathways include the tryptophan/kynurenine catabolic pathway (KP)17,19,24,30–32, polyamine pathway27,33,34, glutathione synthesis pathway35–37, lipids and lipid (per)oxidation23,38–42, fatty acid- and beta oxidation43–45, purine pathway22,46–48, energy metabolism20,49 as well as concentration changes of most proteinogenic amino acids25,29,50–53. Metabolites of the kynurenine and polyamine pathways have been found to be neuroprotective54–57 or neurotoxic19,58–60, and small changes to them can substantially disturb pathway equilibrium61. Moreover, both pathways are interconnected as some of their key players have been shown to bind and alter glutaminergic signalling of the NMDA receptor62,63.
In this study, we used a targeted triple quadrupole liquid chromatography-mass spectrometry (QQQ LC/MS) approach to quantify the concentration of 38 iPD-relevant metabolites extracted from the blood serum of 231 individuals to uncover changes solely based on the disease. Included metabolites are 20 proteinogenic amino acids, several metabolites of the kynurenine pathway (KP) including l-Kynurenine (l-KYN), 3-hydroxy-l-kynurenine (3-HK), 3-hydroxyanthranillic acid (3-AA) and the polyamines (PAs) cadaverine-2 (Cad) and putrescine-2 (Put). Metabolites that contain one or two amine groups were derivatized with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC). This reagent is used to increase detection of the amine in the mass spectrometer and enables standard reverse-phase chromatography64. We analyzed the influence of l-DOPA on the tested metabolites as well as the influence of age and sex prior and after confounder adjustment. In addition to this, we investigated changes in the ratios and interactions of all targeted metabolites and used this to identify potential biomarkers. The analysed cohort is one of the largest so far (e.g. see supplement from Stoessel et al. (2018)), containing 103 l-DOPA treated iPD patients, 7 drug-naïve iPD patients (dn-iPD), 93 healthy age-matched controls and 28 patients with AD as a disease-specific control group. Using this cohort, we hypothesised that metabolites could serve as the basis of a diagnostic assay.
Results
Amino acid and metabolite differences between CN, iPD and AD groups
The mean metabolite concentration (in picomoles/µL of serum) and standard deviation (SD) for each metabolite by the group is summarised in Table 1. For the control (CN) vs iPD comparisons, there were 11 significant concentration differences (p < 0.001) prior to adjustment for age and sex. After adjustment, only six remained statistically significant; with 3-HK, l-aspartic acid (Asp), β-alanine (β-ala), homoserine, ornithine (Orn) and tyrosine (Tyr) all being increased in the iPD group as compared with the CN group (Fig. 1). Moreover, there were five metabolites (3-HK, l-cysteine (Cys), l-glutamic acid (Glu), Orn and Tyr) that showed significant differences between the AD and iPD groups after adjustment for both confounders and multiple comparisons, with Cys as the only significantly increased metabolite in AD (Supplementary Table 1). Comparison between CN and AD revealed no significant differences. This is most likely due to the limited power in the AD cohort (n = 28).
Table 1.
Mean metabolite concentrations (pmol µL−1) with standard deviations (SD) and results of pairwise comparison prior and after the adjustment for age and sex.
| Metabolite | Mean (SD); in picomoles/μL | Unadjusted for confounders p values | Adjusted for confounders p values | |||||
|---|---|---|---|---|---|---|---|---|
| CN (n = 93) | naïve-iPD (n = 7) | iPD (n = 103) | AD (n = 28) | iPD vs CN | AD vs CN | iPD vs CN | AD vs CN | |
| 2-Ambut | 22.03 (6.72) | 24.34 (9.77) | 20.7 (5.33) | 20.49 (8.59) | 0.42 | 0.21 | 0.15 | 0.15 |
| 3-AA | 0.04 (0.01) | 0.03 (0.01) | 0.04 (0.02) | 0.03 (0.01) | 0.04 | 0.004 | 0.19 | 0.01 |
| 3-HK | 0.04 (0.02) | 0.05 (0.06) | 0.05 (0.04) | 0.03 (0.01) | 4.00E-05 | 0.002 | 3.96E-04 | 0.01 |
| 4-OH-Pro | 11.37 (5.01) | 13.97 (6.83) | 12.16 (4.94) | 12.41 (5.35) | 0.19 | 0.38 | 0.23 | 0.15 |
| Ala | 414.07 (81.01) | 438.91 (119.44) | 432.91 (90.87) | 407.2 (80.2) | 0.16 | 0.69 | 0.16 | 0.71 |
| Arg | 81.5 (20.04) | 76.93 (24.66) | 73.81 (15.54) | 85.11 (19.45) | 0.04 | 0.36 | 0.34 | 0.50 |
| Asn | 41.65 (6.23) | 42.59 (7.95) | 42.59 (6.61) | 38.65 (4.35) | 0.20 | 0.01 | 0.49 | 0.14 |
| Asp | 9.69 (5.32) | 10.81 (5.71) | 12.57 (4.99) | 9.83 (3.82) | 5.07E-04 | 0.63 | 1.13E-03 | 0.87 |
| β-ala | 4.52 (1.59) | 4.69 (1.96) | 5.19 (1.64) | 4.41 (1.8) | 1.16E-03 | 0.67 | 1.20E-03 | 0.81 |
| Cad | 0.2 (0.09) | 0.13 (0.07) | 0.17 (0.08) | 0.22 (0.1) | 5.92E-04 | 0.45 | 0.02 | 0.74 |
| Citrulline | 34.61 (8.12) | 32.03 (7.7) | 33.06 (7.22) | 37.06 (9.57) | 0.16 | 0.37 | 0.76 | 0.55 |
| Cys | 177.14 (28.59) | 160.55 (29.36) | 157.78 (27.56) | 191.13 (22.99) | 4.00E-06 | 0.01 | 0.06 | 0.04 |
| GABA | 0.32 (0.17) | 0.34 (0.19) | 0.31 (0.17) | 0.35 (0.16) | 0.50 | 0.27 | 0.73 | 0.53 |
| Gln | 710.1 (81.81) | 729.43 (140.56) | 690.75 (80.77) | 711.35 (65.59) | 0.15 | 0.84 | 0.20 | 0.42 |
| Glu | 45.94 (20.46) | 52.99 (13.61) | 54.88 (20.47) | 38.74 (15.61) | 3.05E-04 | 0.09 | 0.002 | 0.12 |
| Gly | 274.57 (67.96) | 268.57 (63.31) | 298.52 (86.63) | 294.61 (61.72) | 0.05 | 0.04 | 0.01 | 0.07 |
| His | 70.47 (12.75) | 75.28 (8.27) | 70.58 (11.31) | 69.25 (11.27) | 0.98 | 0.60 | 0.55 | 0.80 |
| Homoserine | 0.37 (0.14) | 0.42 (0.11) | 0.43 (0.13) | 0.42 (0.15) | 3.48E-04 | 0.11 | 1.39E-03 | 0.11 |
| Ile | 78.89 (14.87) | 81.76 (11.6) | 83.54 (17.3) | 74.58 (19.6) | 0.12 | 0.10 | 0.60 | 0.38 |
| l-KYN | 3.02 (0.78) | 2.45 (0.7) | 2.59 (0.7) | 2.69 (0.94) | 4.70E-05 | 0.04 | 0.02 | 0.01 |
| Leu | 156.23 (28.16) | 167.94 (29.47) | 165.33 (34.58) | 139.5 (25.99) | 0.12 | 0.005 | 0.75 | 0.06 |
| Lys | 229.23 (33.62) | 219.13 (10.11) | 222.4 (36.23) | 214.97 (35.57) | 0.23 | 0.06 | 0.30 | 0.09 |
| Met | 29.94 (4.36) | 30.21 (3.13) | 30.62 (5.23) | 27.73 (4.29) | 0.33 | 0.02 | 0.92 | 0.13 |
| N-acetyl-phenylalanine | 0.03 (0.01) | 0.02 (0) | 0.03 (0.01) | 0.02 (0.01) | 0.01 | 0.01 | 0.11 | 0.06 |
| Orn | 63.82 (16.44) | 66.93 (26.17) | 74.95 (16.14) | 61.41 (12.1) | 3.00E-06 | 0.58 | 1.40E-05 | 0.77 |
| Phe | 79.92 (9.29) | 80.7 (10.2) | 83.44 (11.08) | 75.63 (8.83) | 0.01 | 0.03 | 0.14 | 0.16 |
| Pro | 228.05 (54.83) | 274.65 (98.72) | 246.8 (76.35) | 259.25 (83.25) | 0.08 | 0.09 | 0.26 | 0.01 |
| Put | 0.28 (0.15) | 0.23 (0.07) | 0.24 (0.18) | 0.29 (0.15) | 0.02 | 0.66 | 0.05 | 0.78 |
| Sarco | 1.8 (0.63) | 2.08 (0.89) | 1.83 (0.61) | 1.7 (0.66) | 0.98 | 0.25 | 0.17 | 0.64 |
| Ser | 89.3 (17.26) | 86.46 (16.05) | 92.55 (18.43) | 91.36 (16.96) | 0.14 | 0.53 | 0.15 | 0.44 |
| Serotonin | 0.39 (0.22) | 0.34 (0.21) | 0.43 (0.32) | 0.33 (0.24) | 0.75 | 0.09 | 0.69 | 0.07 |
| Tau | 111.79 (31.34) | 124.97 (35.26) | 124.91 (32.11) | 121.06 (28.68) | 1.22E-03 | 0.20 | 0.01 | 0.37 |
| Thr | 125.32 (23.7) | 122.05 (20.64) | 137.59 (31.11) | 122.77 (26.25) | 0.003 | 0.58 | 0.01 | 0.96 |
| Trp | 75.04 (13.19) | 78.18 (10.4) | 74.46 (12.72) | 65.49 (12.27) | 0.89 | 1.18E-03 | 0.14 | 0.01 |
| Tryptamine | 0.02 (0.01) | 0.02 (0.01) | 0.02 (0.01) | 0.02 (0.01) | 0.58 | 0.02 | 0.95 | 0.03 |
| Tyr | 86.02 (14.66) | 88.82 (7.86) | 102.73 (25.51) | 78.74 (15.1) | <1.00E-4 | 0.03 | 9.00E-06 | 0.05 |
| Val | 276.45 (47.38) | 292.45 (31.63) | 283.03 (53.43) | 248.16 (46.45) | 0.33 | 0.01 | 0.78 | 0.03 |
Significant p values of less than 0.001 are indicated in bold.
Fig. 1. Elevation of six metabolites in iPD serum.

A Box plot of top six biomarker iPD vs CN and AD (median values in picomoles/µl serum, data taken from Table 1). *** indicates a p value of <0.001 and ***** of <0.00001. B Multivariate ROC analyses resulted in a linear model being able to separate iPD from CN and AD from CN. In the case of iPD vs CN, a panel of seven metabolites resulted in an AUC value of 0.905. For further details, see Table 2C.
The influence of age and sex on metabolite concentrations
Correlations between metabolites and with age are shown in Supplementary Table 2. Weak to moderate significant correlations were found between age and l-arginine (Arg; R = 0.25, p < 0.001), Cad (R = 0.22, p < 0.001), Citrulline (R = 0.32, p < 0.001), Cys (R = 0.50, p < 0.001), l-leucine (Leu; R = −0.26, p < 0.001) and l-KYN (R = 0.31, p < 0.001). Comparing metabolite concentrations between males and females found that l-asparagine (Asn), β-ala, 4-hydroxyproline (4-OH-Pro), l-isoleucine (Ile), Leu, l-methionine (Met), l-proline (Pro), sarcosine (Sarco), l-tryptophan (Trp) and l-valine (Val) had significantly higher levels in males as compared to females (p < 0.001), while only 3-AA was found to have significantly higher levels in females. There were, however, no significant interactions between either age and CN/iPD or between gender and CN/iPD associated with metabolites (assessed via generalised linear modelling (GLM)).
ROC curve analyses, iPD biomarker performance evaluation and diagnostic tests
We performed receiver operating characteristic (ROC) analyses to determine the AUC (area under the curve) of the top-ranked serum metabolites to predict either AD or iPD participants from healthy controls. For the CN vs iPD comparison, 16 individual metabolites had AUC values ranging between 0.591 and 0.706 with a p value of <0.05 (Table 2A). For the CN group vs AD comparisons, 11 individual metabolites had AUC values ranging between 0.629 and 0.706 with a p value <0.05 (Table 2B).
Table 2.
ROC curve analysis for biomarker performance evaluation and diagnostic test results, including ROC curve with a 95% confidence interval, p value, sensitivity, specificity, positive predicted value (PPV), negative predicted value (NPV) and accuracy (ACC).
| (A) iPD vs CN (top 16) | AUC 95%CI | p value | Sensitivity | Specificity | PPV | NPV | Accuracy |
|---|---|---|---|---|---|---|---|
| Cys | 0.705 (0.63–0.78) | 5.04E-07 | 64.55 | 68.82 | 71 | 62.14 | 66.5 |
| Tyr | 0.698 (0.63–0.77) | 1.16E-06 | 55.45 | 75.27 | 72.62 | 58.82 | 64.53 |
| Orn | 0.689 (0.62–0.76) | 3.40E-06 | 72.73 | 62.37 | 69.57 | 65.91 | 67.98 |
| KYN | 0.664 (0.59–0.74) | 5.67E-05 | 68.18 | 62.37 | 68.18 | 62.37 | 65.52 |
| Asp | 0.662 (0.59–0.74) | 7.08E-05 | 80.91 | 50.54 | 65.93 | 69.12 | 67 |
| 3-OH KYN | 0.641 (0.56–0.72) | 5.62E-04 | 40 | 89.25 | 81.48 | 55.7 | 62.56 |
| Cad | 0.639 (0.56–0.72) | 6.29E-04 | 60 | 61.29 | 64.71 | 56.44 | 60.59 |
| Glu | 0.638 (0.56–0.71) | 6.97E-04 | 64.55 | 58.06 | 64.55 | 58.06 | 61.58 |
| Tau | 0.635 (0.56–0.71) | 9.40E-04 | 76.36 | 52.69 | 65.63 | 65.33 | 65.52 |
| Homoserine | 0.635 (0.56–0.71) | 9.35E-04 | 83.64 | 38.71 | 61.74 | 66.67 | 63.05 |
| beta-Ala | 0.62 (0.54–0.7) | 3.27E-03 | 52.73 | 66.67 | 65.17 | 54.39 | 59.11 |
| Thr | 0.603 (0.53–0.68) | 1.12E-02 | 70 | 49.46 | 62.1 | 58.23 | 60.59 |
| N-Acetyl-phenylalanine | 0.601 (0.52–0.68) | 1.31E-02 | 68.18 | 53.76 | 63.56 | 58.82 | 61.58 |
| Phe | 0.597 (0.52–0.68) | 1.69E-02 | 43.64 | 74.19 | 66.67 | 52.67 | 57.64 |
| Put | 0.591 (0.51–0.67) | 2.58E-02 | 40 | 81.72 | 72.13 | 53.52 | 59.11 |
| Arg | 0.583 (0.5–0.66) | 4.15E-02 | 52.73 | 61.29 | 61.7 | 52.29 | 56.65 |
| (B) AD vs CN (top 11) | |||||||
| Trp | 0.706 (0.6–0.82) | 9.76E-04 | 46.43 | 88.17 | 54.17 | 84.54 | 78.51 |
| 3-OH ANA | 0.686 (0.58–0.79) | 2.99E-03 | 75 | 56.99 | 34.43 | 88.33 | 61.16 |
| Val | 0.672 (0.55–0.79) | 6.07E-03 | 78.57 | 56.99 | 35.48 | 89.83 | 61.98 |
| Leu | 0.662 (0.54–0.78) | 9.76E-03 | 64.29 | 64.52 | 35.29 | 85.71 | 64.46 |
| 3-OH KYN | 0.66 (0.55–0.77) | 1.07E-02 | 92.86 | 38.71 | 31.33 | 94.74 | 51.24 |
| KYN | 0.652 (0.52–0.78) | 1.51E-02 | 67.86 | 61.29 | 34.55 | 86.36 | 62.81 |
| Cys | 0.649 (0.54–0.76) | 1.70E-02 | 89.29 | 39.78 | 30.86 | 92.5 | 51.24 |
| Phe | 0.647 (0.53–0.76) | 1.90E-02 | 78.57 | 55.91 | 34.92 | 89.66 | 61.16 |
| Typtamine | 0.646 (0.53–0.77) | 1.97E-02 | 75 | 59.14 | 35.59 | 88.71 | 62.81 |
| Asn | 0.644 (0.54–0.75) | 2.10E-02 | 82.14 | 53.76 | 34.85 | 90.91 | 60.33 |
| Tyr | 0.629 (0.5–0.76) | 3.92E-02 | 39.29 | 88.17 | 50 | 82.83 | 76.86 |
| (C) Seven-marker model | |||||||
| Cys, 2-Ambut, Tyr, KYN, Arg/3-AA, Asp/KYN, beta-Ala*Orn | 0.905 | <0.0001 | 87.4 | 85.0 | 86.5 | 85.9 | 86.2 |
Results for the binary classification of (A) iPD vs CN (top 16 metabolites), (B) AD vs CN (top 11 metabolites) and (C) the seven-marker model for iPD vs CN is shown.
Ratios and interactions that separate CN from iPD and AD disease groups
In neurodegenerative diseases, the ratio of markers associated with the pathology (e.g. tau, amyloid beta) often serve as more powerful biomarkers than the absolute level of the biomarker alone. One clear example of this is the ratio of cerebral spinal fluid amyloid beta 1–42 peptide to tau protein levels65,66. We hypothesised that this would also be the case for metabolites. To test this, all possible ratios and interactions (the product of two analytes) for the 37 metabolites (excluding l-DOPA) and amino acids were computed providing 1332 markers. Dimension reduction via removal of those ratios/interactions with low variance (SD <0.5) reduced this number to 569. Comparing the mean ratio/interaction levels between the CN and iPD groups, we identified 11 ratios and 23 interactions that were significantly altered (p < 0.00009) between the two groups (Supplementary Table 3). Of these, Asp was involved with the most ratios (7/11) and interactions (9/23). Other metabolites that appeared frequently in the top 34 included l-glutamine (Gln; five interactions), homoserine (four interactions), 3-HK (four interactions and one ratio) and Orn (six interactions and one ratio). Of these 34 markers, seven remained significant post adjusting for age and gender (homoserine*Orn, β-ala*Orn, Asp*Tyr, Gln*Tyr, Asp*Orn, Asp / L-KYN and Gln*Orn).
Multivariate analyses of metabolites for iPD and AD
Following up the univariate assessment of metabolites, we conducted a multivariate analysis including both individual analytes and ratio’s/interactions to see if a panel of markers together could provide better discrimination between CN and disease groups. Using a combination of feature selection (LASSO) and model selection via Akaike information criterion (AIC) reduction, seven markers were selected in a linear model to separate CN from iPD participants (Cys [p = 0.008], 2-aminobutyric acid (2-Ambut) [p = 0.0002], Tyr [p = 0.0005], l-KYN [p = 0.0003], ratio of Arg/3-AA [p = 0.004], ratio of Asp/l-KYN [p = 0.007] and product of β-ala*Orn [p < 0.0001]). These seven metabolites resulted in an AUC value of 0.905 with an accuracy of 86.2% (sensitivity: 87.4%, specificity: 85.0%, positive predicted value (PPV): 86.5% and negative predicted value (NPV): 85.9%, Table 2C and Fig. 1B). For the comparison between CN and AD participants, using the same method to define a multivariate set of analytes, we identified a set of six markers (Asp [p = 0.019], Cys [p = 0.0008], Tryp [p = 0.022], Homoserine/N-Acetyl-phenylalanine [p = 0.055], Pro/3-HK [p = 0.002] and Gln*Typtamine [p = 0.063]) that worked together to separate AD from CN participants. Here, ROC analyses calculated an AUC of 0.884 to predict AD from CN with 79.3% accuracy (sensitivity: 89.3%, specificity: 76.3%, PPV: 53.2% and NPV: 95.9%).
Discussion
While proteins are recognised as playing important roles in iPD and its progression67, there is increasing recognition of the importance of metabolites in the disease phenotype68. In the current study, we quantified 37 iPD-relevant metabolites (- l-DOPA) from the blood serum of 231 individuals with the goal to find potential biomarkers to separate CN from the disease. l-DOPA was removed from the statistics, as it results in the most prominent and expected change. After adjustment for age and sex, a multivariate analysis followed by ROC predictions defined a biomarker panel of four metabolites (Cys, 2-Ambut, Tyr, l-KYN), two ratios (Arg/3-AA, Asp/l-KYN) and one interaction (β-ala*Orn) to separate iPD from CN with an AUC of 0.91 and an accuracy of 86.2%. The high accuracy of this biomarker panel indicates that there may be a metabolite signature that could be used to assist in the diagnosis of iPD cases. Importantly, using samples from the AIBL study on AD, we demonstrated that the metabolite panel for iPD was specific for iPD compared to AD with the only overlapping metabolite being Cys. In AD, a panel of three metabolites (Asp, Cys, Trp), two ratios (Homoserine/N-Acetyl-phenylalanine, Pro/3-HK) and one interaction (Glu*Tryptamine) were able to separate AD from CN with an AUC of 0.88 and an accuracy of 79.3%. Future studies will be needed to validate the potential clinical impact of these diagnostic markers for iPD and AD.
Looking at individual analyte concentrations, ratios and interactions, the most significant changes were observed for three metabolites of the KP (3-HK, l-KYN, 3-AA), and the amines of Asp, Orn, β-ala, Gln, Tyr, Homoserine and Cys. In the case of 3-HK, Orn and Tyr, all are increased in iPD compared to CN and AD and are therefore disease-specific. Asp was significantly increased in the serum of iPD patients (mean: 12.57 pmol µL−1) and slightly in naïve-iPD patients (mean: 10.81 pmol µL−1), when compared to CN group (mean: 9.69 pmol µL−1). In previous studies, changes in plasma Asp levels were inconsistent in iPD patients, as probably a reaction to the treatment25,69. However, as Asp is also involved in two interactions (Asp*Tyr, Asp*Orn), one ratio (Asp/l-KYN) and is also part of the iPD/CN separation model, it may have an important role in iPD. Further, Asp is converted into Homoserine by a two-reduction step of the terminal carboxyl group. Not surprisingly, both metabolites were increased in PD patients in this study (Fig. 2). The Asp-Homoserine intermediate is the branching point for the lysine pathway, and Homoserine itself is the metabolic branching point of threonine. Tyr was significantly increased in l-DOPA treated iPD patients (mean: 102.73 pmol µL−1) compared to CN (mean: 86.02 pmol µL−1). In drug-naïve patients, Tyr-increase was not significantly changed validating the lack of l-DOPA treatment and that the changes in Tyr are associated with the treatment (mean: 88.82 pmol µL−1). Tyr is converted to l-DOPA via tyrosine hydroxylases (TH1-4) and l-DOPA is the precursor of dopamine (Fig. 3)70. TH enzymes can also catalyze the hydroxylation of phenylalanine to Tyr71. TH enzymes are mainly expressed in dopaminergic neurons. In iPD, however, most dopaminergic neurons are dead due to dopamine deficiency. The reason for the Tyr increase in l-DOPA treated iPD patients is unclear and could be related to the use of peripheral decarboxylase inhibitors contained in Levodopa treatment and/or the metabolism of the individual’s gut microbiome72. Cys was decreased ca. 10% in both iPD and naïve-iPD participants as compared with CN participants, while it was increased ca. 20% in AD participants (Table 1). Some studies report an increase of cysteine in serum after l-DOPA intake23, others report a decrease in plasma73,74. A decrease of cysteine is thought to be a reaction to l-DOPA intake and an indication of increased glutathione (GSH) synthesis due to oxidative stress73–75. GSH itself acts as a redox buffer and antioxidant defence, and its homoeostasis dysregulation is believed to contribute to the progression of neurodegenerative diseases76. However, this does not explain why iPD participants have a lower cysteine concentration when compared to untreated participants.
Fig. 2. Polyamine pathway and Cadaverine pathway.

A Polyamine pathway and B Cadaverine pathway. The metabolites highlighted in bold have been targeted and detected in this study. l-Ornithine, l-Glutamine and l-Aspartate are all significantly increased in the iPD cohort (arrows and text written in green). ARG arginase, ODC ornithine decarboxylase, SRM spermidine synthase, PAO polyamine oxidases, SMS spermine synthase, SPMO spermidine oxidase, SSAT1 and 2 diamine acetyltransferase 1 and 2, APAO acetylated polyamine oxidase, LDC lysine decarboxylase, OAT ornithine aminotransferase, ALDH4A1 delta-1-pyrroline-5-carboxylate dehydrogenase), ASNS asparagine synthetase [glutamine-hydrolysing]).
Fig. 3. Dopamine-catecholaminergic pathway.
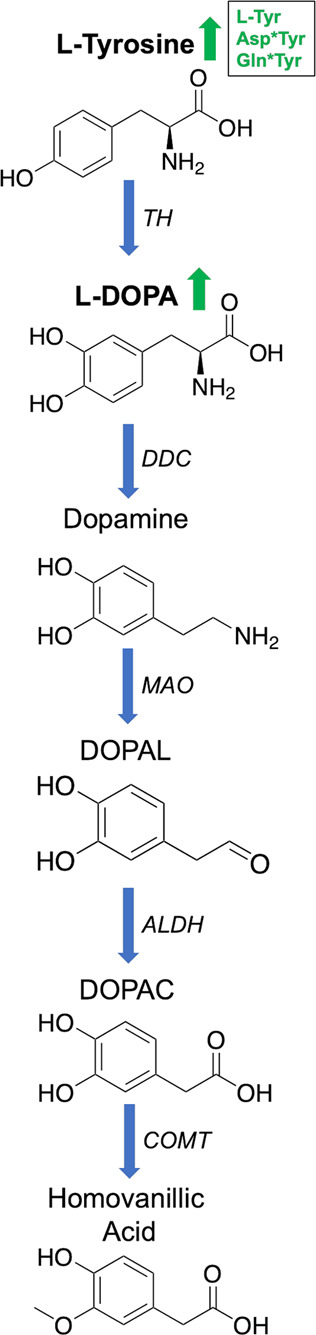
The metabolites highlighted in bold have been targeted and detected in this study. Both Tyr and l-DOPA are significantly increased in iPD patients (arrows and text in green). TH tyrosine hydroxylases, DDC dopa decarboxylase, MOA monoamine oxidase, DOPAL 3,4-dihydroxyphenylacetaldehyde; toxic intermediate, ALDH aldehyde dehydrogenase, DOPAC 3,4-dihydroxyphenylacetic acid and COMT catechol-O-methyltransferase.
The KP is the central route in the Trp metabolism, ~95% of Trp is catabolized via the KP, leading to the formation of nicotinamide adenine dinucleotide and its phosphate (NAD + /NADP; Fig. 4)77,78. The remainder forms a substrate for serotonin and melatonin synthesis. Trp and KP metabolites have been studied since the early 1930s79,80. Altered KP metabolism is involved in a number of neurodegenerative diseases (e.g. Epilepsy81, Huntington’s disease82, Multiple sclerosis77,83, Amyotrophic lateral sclerosis51 and AD84). Their role in iPD has been known since the early 1990s17. This pathway is highly regulated, with small changes substantially disturbing its equilibrium61. l-KYN is the central metabolite of this pathway and is either degraded into kynurenic acid (KYNA), 3-HK or anthranilic acid (AA) (Fig. 4)19. In the central nervous system, ~ 40 % of l-KYN is locally produced, whereas the other 60 % are absorbed from blood85. It can be transported across the blood–brain barrier (BBB) by a neutral amino acid carrier86, which is thought to be modulated by l-valine in metabolic disorders. KYNA acts as a neuroprotectant and could therefore have therapeutic effects in neurological disorders like iPD54,55, but its use is restricted due to its very limited ability to cross the BBB86. 3-HK and AA have been shown to cause neuronal damage, as they generate free radicals and elevate oxidative stress19,58. 3-AA, synthesised from 3-HK and/or AA, exhibits an increased level in iPD patients26. Another metabolite of the KP pathway is quinolinic acid (QUIN), acting as an excitotoxic agonist of the NMDA receptor87. In contrast, KYNA has been shown to protect rat neurons against the damage caused by QUIN88,89. NAD + /NADP is one of the final products of the KP, produced by the catabolism of QUIN. Monocytes can be activated by high levels of inflammatory cytokines, which upregulate the expression of KP enzymes, favouring the production and secretion of QUIN90. Furthermore, QUIN has been shown to induce damage to dendrites and axons, when present in high/toxic levels, leading to cytoskeleton destabilization by the phosphorylation of structural proteins91,92. In this study, l-KYN was significantly decreased in the serum of iPD patients (iPD 2.59 vs CN 3.02 pmol µL−1, mean values), whereas 3-HK was increased (iPD 0.05 vs CN 0.04 pmol µL−1, mean values) (Table 1). Our results are in line with other studies19,22,93,94. Further, we observed a decrease in the ratios of l-KYN/Trp (CN: 0.04; iPD: 0.035), l-KYN/3-HK (CN: 75.5; iPD: 51.8) and Arg/3-AA (CN: 2037.5; iPD: 1845.3) in iPD patients, and an increase of Asp/l-KYN (CN: 3.21; iPD: 4.85) (Table 1). Importantly, we show that the changes were also detected in naïve-iPD patients (l-KYN/Trp: 0.031; KYN/3-HK: 49; Asp/l-KYN: 4.41), indicating that the concentration changes of l-KYN and 3-HK are based on iPD and not on l-DOPA treatment. A decrease of l-KYN/3-HK is associated with an increased kynurenine 3-monooxygenase (KMO) activity32. The KMO enzyme catalyses the hydroxylation of l-KYN to form 3-HK. Inhibition of the KMO enzyme has been shown to reduce LID in Parkinson’s like disease in monkeys95. Several reviews about the KP pathway and its role in the central nervous system are available54,96–98. It is highly recommended that future studies should simultaneously analyse all relevant KP metabolites (Trp, l-KYN, 3-HK, xanthurenic acid, AA, 3-AA, QUIN, KYNA, 2-picolinic acid and NAD + /NADP; Fig. 4) as their function can be either neuroprotective or neurotoxic. It is unclear at this point, if the metabolite changes found in serum are originating from the brain, or if they influence brain homoeostasis.
Fig. 4. Tryptophan/kynurenine pathway.
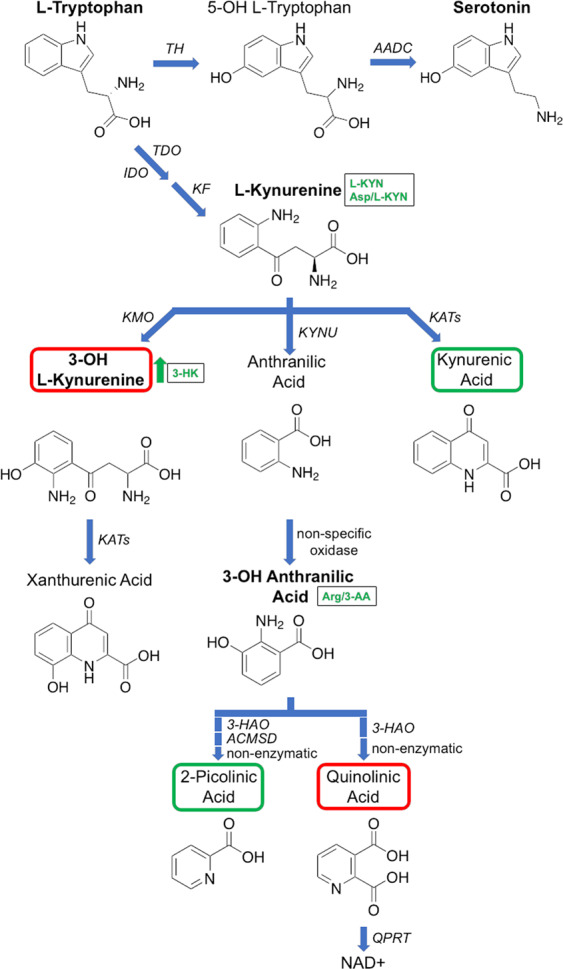
The main end product of this pathway is NAD+ (nicotinamide adenine dinucleotide). Trp can also be converted to serotonin. The metabolites highlighted in bold have been targeted and detected in this study. Metabolites surrounded by red boxes are neurotoxic and by green boxes are neuroprotective. l-Kynurenine, 3-OH l-Kynurenine and 3-OH anthranilic acid are all significantly changed in the iPD cohort (arrow and text written in green). IDO indoleamine 2,3-dioxygenase), TDO tryptophan 2,3-dioxygenase), KF kynurenine formamidase, KMO kynurenine 3-monooxygenase, KYNU kynureninase, KAT kynurenine aminotransferase, 3-HAO 3-hydroxyanthranilic acid dioxygenase, ACMSD 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase, QPRT nicotinate-nucleotide pyrophosphorylase, TH tryptophan hydroxylase and AADC aromatic amino acid decarboxylase.
PAs are small aliphatic polycations that are derived from the amino acids Met, Orn, Arg and Lys99. The most common PA are spermine, spermidine, Put and Cad (Fig. 2). When the PA metabolism is disturbed, multiple cellular processes are influenced (e.g. gene expression, protein translation, autophagy and membrane function59,99). PA have been associated with neurodegenerative diseases (e.g. AD100, Amyotrophic lateral sclerosis27 and iPD101). In iPD patients, the metabolites Cad and Put were increased21,102, as well as Orn103 and the Put/Orn ratio104. PA were also found to be increased in astroglial cells105. Cellular PA also promotes the aggregation and fibrillization of α-synuclein, which is the major protein component of Lewy bodies in iPD106. However, it is thought, albeit controversially, that PA may be neuroprotective as they can induce autophagy as a way to protect cells from stress56,57 and have been shown to be elevated in neurodegenerative diseases27,100. Additionally, they have been found to be cytotoxic, with their increase leading to an elevated concentration of toxic metabolites such as aldehydes and hydrogen peroxide59,60. PA can also have several opposing effects on NMDA receptors, including a glycine-dependent potentiation, a voltage-dependent inhibition and a voltage- and glycine-independent potentiation3,107. For example, spermine can bind to NMDA receptor and potentiate agonist-induced currents108. Finally, the increase of PA in iPD and AD brain could also be based on the level of their enzymes increasing as a reaction to proteasomal impairment109. Although we did not find any significant changes in Cad, Put and Put/Orn ratio after confounder adjustment, its precursor Orn was significantly increased in iPD patients (iPD 74.95 vs CN 63.82 pmol µL−1, mean values), involved in four interactions (Homoserine*Orn, β-ala*Orn, Asp*Orn and Gln*Orn) and also part of the iPD/CN separation model. Reiterating the above, future studies should aim to include all possible PAs, in order to gain a better understanding of the role of this highly relevant and regulated pathway.
Both kynurenines and PAs can bind to the NMDA receptors62, indicating that this ion channel complex is a potential therapeutic target. In particular, spermine was shown to attenuate or prevent QUIN-induced damages in rat striatum through NMDA receptor interaction and/or its antioxidant function110. Spermidine was also shown to be neuroprotective against QUIN-induced excitotoxic cell death due to its NMDA receptor antagonistic properties111. Therefore, a common denominator of both pathways are NMDA receptors. Moreover, it seems to be crucial that both pathways need to be in perfect balance to guarantee normal cellular function. Neither spermine, spermidine nor Quin were part of this study but should be included in future studies due to their significant biological importance.
To get an overview of already known metabolic changes in iPD patients in regard to proteinogenic amino acids and metabolites of the kynurenine and polyamine pathways, we performed a literature review in PubMed (details outlined in method section). In total, we identified 32 cohort studies, comparing iPD with CN and/or AD, restless leg syndrome (RLS), traumatic brain injury (TBI), multiple system atrophy (MSA), Amyotrophic lateral sclerosis, Huntington’s Disease and progressive supranuclear palsy (PSP). From that 32 studies, 12 analysed metabolites of the KP (Table 3A)17,19,22–24,26,29,31,32,93,112,113, 18 analysed amino acids (Table 3B)20,23,25,29,43,50–53,69,73,112,114–119 and five PA (Table 3C)18,21,27,102,120. Like in this study, eight studies also analysed the blood serum of iPD patients, whereof five covered amino acids and another five metabolites of the KP. In the remaining studies, metabolites were extracted and analysed from the brain, plasma, CSF, urine and RBC. Only the main and significant changes between iPD and CN are shown. A red arrow (↓) indicates a decrease in the metabolite concentration in iPD, a green arrow (↑) an increase, a yellow one (⟷) no changes and an empty box indicates nonsignificant changes/non-analysed metabolites. Moreover, two stoichiometric/pathway enrichment analyses were also included. The pathway enrichment analysis was performed by Kori M. et al. (2016) and 54 metabolite biomarkers were proposed for iPD, including many proteinogenic amino acids119. However, many studies led to controversial results where the same bio-fluid was analysed; and where the same metabolites had discorded results. A possible explanation is the sample size of the analysed cohorts, with large variance within biomarkers across groups resulting in nonsignificant increases or decreases in the same biomarkers. In 43 %, the cohort size was ≤50 participants and 73% of all studies had ≤100 participants. Only three studies have a cohort size of >200, including ours.
Table 3.
Literature review of metabolite changes in PD patients compared to control, including (A) metabolites of the kynurenine pathway, (B) proteinogenic amino acids and (C) metabolites of the polyamine pathway.
| No | Study | Research focus | Biomaterial | Cohort size (n) | Screening technique | Metabolites | Metabolite ratios | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trp | KYN | 3-HK | AA | KYNA | QUIN | 3-HK/ KYNA | KYN/Trp | KYN/3-HK | KYN/KYNA | KYNA/QUIN | ||||||
| (A) Kynurenine pathway metabolites | ||||||||||||||||
| 0 | This study | PD vs. CN/AD | Serum | 231 | LC-MS | ↓ | ↑ | ↓ | ↓ | |||||||
| 1 | Widner et al., 200293 | PD vs. CN | Serum + CSF | 33 | HPLC | ↓ | ↓ | ↑ | ||||||||
| 2 | Schulte et al., 201623 | PD vs. RLS/CN | Serum | 1449 | LC-MS + GC-MS | ⟷ | ⟷ | |||||||||
| 3 | Hatano et al., 201631 | PD vs. CN | Serum | 50 | LC-MS + GC-MS | ↓ | ||||||||||
| 4 | Han et al., 201722 | PD vs. CN | Serum | 85 | LC-MS | ↓ | ↓ | ↑ | ||||||||
| 5 | Sorgdrager et al., 2019112 | PD vs. AD/CN | Serum & CSF | 105 | LC-MS | ↓ | ↓ | ↓ | ⟷ | ↓ | ||||||
| 6 | Oxenkrug et al., 201724 | PD vs. AD/CN | Plasma | 62 | LC-MS | ↓ | ↑ | ↑ | ↑ | ↑ | ||||||
| 7 | LeWitt et al., 201319 | PD vs. CN | CSF | 105 | LC-MS + GC-MS | ↑ | ↑ | |||||||||
| 8 | Iwaoka et al., 2020113 | PD vs. CN | CSF | 33 | HPLC ECD | ↑ | ↑ | |||||||||
| 9 | Havelund et al., 201768 | PD vs. CN | Plasma & CSF | 40 | LC-MS | ↓ | ↑ | ↓ | ||||||||
| 10 | Hartai et al., 200583 | PD vs. CN | Plasma & RBC | 36 | HPLC | ↓ plasma ↑ RBC | ||||||||||
| 11 | Ogawa et al., 199217 | PD vs. CN | Brain regions | −85 | HPLC ECD | ↓ | ↑ | ↓ | ⟷ | ↓ | ⟷ | |||||
| 12 | Luan et al., 201547 | PD vs. CN | Urine | 157 | LC-MS + GC-MS | ↑ | ↑ | ↑ | ||||||||
| No | Study | Research focus | Biomaterial | Cohort size (n) | Screening technique | Arg | His | Lys | Asp | Glu | Ser | Thr | Asn | Gln | Cys | Sec | Gly | Pro | Ala | Val | Ile | Leu | Met | Phe | Tyr | Trp |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (B) Amino acids | ||||||||||||||||||||||||||
| 0 | This study | PD vs. CN/AD | Serum | 231 | LC-MS | ↑ | ↑ | ↑ | ||||||||||||||||||
| 1 | Hirayama M. et al., 201653 | PD vs. CN | Serum & sweat | 58 & 107 | LC-MS + HPLC fluorescence | ↑ | ↓ | |||||||||||||||||||
| 2 | Schulte et al., 201623 | PD vs. CN/RLS | Serum | 1449 | LC-MS + GC-MS | ↓ | ↓ | ↓ | ↓ | ↑ | ↑ | ↑ | ↑ | ↑ | ↓ | ↓ | ↑ | ⟷ | ||||||||
| 3 | Fiandaca M. S. et al., 2018114 | PD vs. CN/TBI | Serum | 155 | LC-MS | ↓ | ||||||||||||||||||||
| 4 | Figura M. et al., 201852 | ePD, lPD + /− LID (no CN) | Serum | 73 | HPLC fluorescence | ↓ | ↑ | ↓ | ↓ | |||||||||||||||||
| 5 | Sorgdrager et al., 2019112 | PD vs. CN/AD | Serum & CSF | 105 | LC-MS | ↓ | ↓ | ↓ | ↓ | ↓ | ||||||||||||||||
| 6 | Iwasaki Y. et al., 199225 | PD vs. CN | Plasma | 40 | Ion exchange | ↑ | ↑ | ↑ | ||||||||||||||||||
| 7 | Mueller T. et al., 201273 | l-Dopa & PD (no CN) | Plasma | 13 | HPLC ECD | ↓ | ||||||||||||||||||||
| 8 | Kuiper M.A. et al., 200050 | PD vs. CN/AD/MSA | CSF | 156 | HPLC fluorescence | ↑ | ||||||||||||||||||||
| 9 | Engelborghs S. et al., 2003115 | PD vs. CN | CSF | 54 | HPLC ECD | - No significant changes detected (Asn,Cys, Sec, Pro and Trp not included) - | ||||||||||||||||||||
| 10 | Oehman A. et al., 201520 | PD vs. CN | CSF | 20 | 1H-NMR spectr. | ↓ | ↓ | |||||||||||||||||||
| 11 | Jimenez-Jimenez F. J. et al., 199669 | PD vs. CN | CSF & plasma | 76 | Ion exchange | ↓ | ↑ | ↑ | ↑ | |||||||||||||||||
| 12 | Molina J.A. et al., 1997116 | PD vs. CN | CSF & plasma | 76 | Ion exchange | ↑ | ↓ | ↓ | ↑ | ↑ | ↓ | |||||||||||||||
| 13 | Trupp et al., 201443 | PD vs. CN | CSF & plasma | 40 | GC-MS | ↑ | ↑ | ↑ | ↓ | |||||||||||||||||
| 14 | Wuolikainen et al., 201651 | PD vs. CN/ALS | CSF & plasma | 72 | LC-MS + GC-MS | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | |||||||||||||||
| 15 | Mally J., et al., 1997117 | PD vs. CN | CSF & serum | 20 | HPLC fluorescence | ↓ | ↑ | |||||||||||||||||||
| 16 | Luan et al., 201547 | PD vs. CN | Urine | 157 | LC-MS + GC-MS | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ||||||||||||
| 17 | Sertbas et al., 2014118 | PD vs. CN | / | / | Stoichiometric model | (+) | (+) | (+) | (+) | (+) | (+) | (+) | ||||||||||||||
| 18 | Kori M. et al., 2016119 | PD vs. AD/ALS | / | / | Pathway enrichment | (+) | (+) | (+) | (+) | (+) | (+) | |||||||||||||||
| No | Study | Research focus | Biomaterial | Cohort Size (n) | Screening technique | Cad | Put | Spd | Spm |
|---|---|---|---|---|---|---|---|---|---|
| (C) Polyamines | |||||||||
| 0 | This study | PD vs. CN/AD | Serum | 231 | LC-MS | ||||
| 1 | Saiki et al., 2019120 | PD vs. CN/AD | Plasma | 467 | LC-MS + CE-MS | ↑ | ↓ | ||
| 2 | Paik M-J. et al., 201021 | PD vs. CN/MSA | CSF | 42 | GC-MS | ↑ | ↑ | ↓ | |
| 3 | Gomes-Trolin C. et al., 200227 | PD vs. CN/ALS | RBC | 60 | HPLC fluorescence | ↓ | ↑ | ↑ | |
| 4 | Betancourt L. et al., 2018102 | PD vs. CN | RBC | 24 | LC-MS | ↑ | |||
| 5 | Vivo M. et al., 200118 | PD vs. CN/HD/PSP | Brain | 48 | HPLC fluorescence | no difference to CN | |||
The following parameters are shown: Study origin, research focus, source of biomaterial, total cohort size, screening technique, metabolite changes (↓ decrease, ↑ increase, ⟷ no change) and metabolite-ratio changes. The gaps in the table refer to non-analyzed or nonsignificantly changed metabolites.
iPD and its progression seem to lead to global metabolic changes not only in the brain but also in peripheral body fluids. Overall, our study adds to the body of evidence that amino acids and metabolites of the KP are changed in patients with iPD. Most above-mentioned studies, including our own, did not collect information about the diet of the participants (e.g. western diet, ketogenic diet, vegan, etc.). However, it is well known that the diet and the gut microflora have a significant impact on the metabolome121–124. In particular, the microbiome has been documented to alter the metabolic profile and activity in humans125. Therefore, the impact of the diet on the results cannot be excluded, and this is a limitation of this study.
Taking all these changes into consideration, the detailed molecular mechanism of iPD is still poorly defined. Therefore, a panel of biomarker is urgently needed to increase iPD diagnosis and treatment success. Nevertheless, serum metabolomics is a powerful tool for the discovery and development of a blood-based small-molecule biomarker for neurodegenerative diseases like PD.
Methods
Patient recruitment and cohort details
In the present study, a targeted metabolite screen was performed on provided blood sera from 231 clinically assessed individuals, collected as previously described126. All samples were processed and stored under identical conditions; at −178 °C in liquid nitrogen dewars as previously described127. Eighty-eight serum samples were taken from the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL; 60 healthy age-matched controls with 29 males and 31 females and 28 patients with diagnosed AD including 8 males and 20 females). About 143 samples were taken from the Australian Parkinson’s Disease Registry (APDR; 33 healthy age-matched controls with 20 males and 13 females, 103 patients with l-DOPA treated iPD including 65 males and 38 females and 7 drug-naïve iPD patients with 6 males and 1 female). The demographic and clinical features of the analysed cohort are summarised in Table 4.
Table 4.
(A) Demographic and (B) clinical features of an analyzed cohort.
| iPD (n = 103) | drug-naïve iPD (n = 7) | Control (CN) (n = 93) | AD (n = 28) | n (Sum) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | Male | Female | ||
| (A) Demographic features of analysed cohort | |||||||||
| n | 65 | 38 | 6 | 1 | 49 | 44 | 8 | 20 | 231 |
| Median age years (SD) | 65 (9.7) | 67.5 (7.9) | 66.5 (7.8) | 48 (−) | 76 (9.5) | 74 (8.6) | 75.5 (7.9) | 73 (8.5) | NA |
| Age at assessment | Age at onset of PD symptoms | Duration of disease | Total levodopa equivalent | MDS-UPRDS I | MDS-UPDRS II | MDS-UPRDS III | MDS-Part IV | MDS-UPDRS total | Schwab & England | Hohn & Yahr | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (B) Clinical features of analysed cohort | |||||||||||
| Number of values | 104 | 104 | 104 | 103 | 103 | 103 | 104 | 104 | 104 | 104 | 104 |
| Minimum | 41 | 29 | 0 | 0 | 1 | 1 | 0 | 0 | 11 | 0 | 0 |
| 25% Percentile | 60 | 52 | 3 | 450 | 7 | 5 | 12 | 0 | 31 | 80 | 2 |
| Median | 66 | 58 | 6 | 750 | 10 | 8 | 20 | 3 | 43.5 | 90 | 2 |
| 75% Percentile | 71 | 63.8 | 10 | 1178 | 15 | 14 | 32.8 | 6 | 61 | 90 | 2 |
| Maximum | 87 | 80 | 31 | 3351 | 31 | 31 | 68 | 17 | 136 | 100 | 5 |
| Range | 46 | 51 | 31 | 3351 | 30 | 30 | 68 | 17 | 125 | 100 | 5 |
| 10% Percentile | 54 | 43 | 2 | 40 | 4.4 | 3 | 7 | 0 | 22 | 75 | 1 |
| 90% Percentile | 75 | 71.5 | 13 | 1500 | 18 | 19 | 43 | 9.5 | 80.5 | 100 | 3 |
| 95% CI of median | |||||||||||
| Lower confidence limit | 63 | 56 | 5 | 675 | 9 | 7 | 16 | 2 | 37 | 90 | 2 |
| Upper confidence limit | 68 | 61 | 8 | 900 | 13 | 10 | 24 | 4 | 52 | 90 | 2 |
| Mean | 65 | 57.9 | 7.1 | 868.1 | 11.3 | 10.1 | 22.9 | 3.7 | 47.8 | 84.6 | 2 |
| Std. deviation | 8.9 | 10.1 | 5.2 | 632.9 | 5.8 | 6.4 | 14.2 | 3.8 | 23.7 | 14.1 | 0.8 |
| Std. errror of mean | 0.9 | 1 | 0.5 | 62.4 | 0.6 | 0.6 | 1.4 | 0.4 | 2.3 | 1.4 | 0.1 |
| Lower 95% CI of mean | 63.3 | 55.9 | 6.1 | 744.4 | 10.2 | 8.8 | 20.2 | 3 | 43.2 | 81.9 | 1.8 |
| Upper 95% CI of mean | 66.7 | 59.9 | 8.1 | 991.8 | 12.4 | 11.3 | 25.7 | 4.5 | 52.4 | 87.4 | 2.1 |
Ethics statement
For both cohorts, experiments were conducted under The University of Melbourne human ethics committee approval ID1136882. All participants provided written informed consent prior to enrolment.
Chemicals
The following chemicals were used in this study: Acetonitrile (AcN) (Sigma A955-4), boric acid (Sigma B0394-500G), 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC, Synchem UG & Co KG S041), formic acid (FA) (Sigma 27001-500ML-R). Amino acid standards were purchased from Sigma Aldrich at a minimum purity of ≥99%.
Sample preparation and derivatization
Metabolites were extracted and derivatized as previously reported64. In detail, 50 μL of human serum was transferred into a 1.5 mL Eppendorf tube. Next, 150 μL of ice-cold methanol containing an internal standard (ISTD, 13C5,15N-l-Valine, Sigma Aldrich, 25 µM) was added. All samples were vortexed and cooled on wet ice for 30 min. Samples were then centrifuged at 15,000 × g (maximum speed) for 10 min to precipitate protein, and the supernatant was transferred to a clean, 1.5 mL Eppendorf tube. Two separate sample concentrations were prepared, an undiluted sample and a 5× concentrated sample. The undiluted sample, 10 µL of the supernatant was derivatized directly as described below. For the concentrated sample, 50 µL of each supernatant was dried down and reconstituted in 10 µL of 75% methanol (MeOH), 0.1% FA to form a fivefold concentration ready for derivatization. In addition to this, a pooled biological quality control (PBQC) was prepared to monitor the performance of the 6410 and 6490 QQQ LC/MS instruments. For derivatization, 2.85 mg of AQC was dissolved in 1 mL anhydrous AcN. Next, 70 μL of borate buffer (pH 8.8) was added to 10 μL of each sample. The samples were vortexed and centrifuged at 15,000xg for 1 min. Next, 20 μL of AQC solution was added and the samples were vortexed and centrifuged (1 min, maximum speed) again, followed by a 10 min incubation step at 55 ˚C. The samples were then vortexed and centrifuged at maximum speed for 10 min. Finally, 20 μL of each sample was transferred to a glass vial for analysis on the 6410 and 6490 QQQ LC/MS instruments.
Mass spectrometry instrumentation
Serum extracts were separated on an Agilent 1200 LC-system using an Agilent ZORBAX Eclipse PLUS C18 column (2.1 mm × 50 mm, 1.8 µM, Product number 959757-902). Elution was carried out with a water/AcN mobile phase binary solvent system. Mobile phase A consisted of 100% water/0.1% FA; mobile phase B consisted of 100% AcN/0.1% FA. The samples were analyzed by Agilent 6410 and 6490 ESI-QQQ-MS instruments (Santa Clara, CA) in dynamic multiple reaction monitoring (dMRM) positive ionisation mode using the same instrument settings and method as previously described64.
Selected metabolites
In total, we included 38 metabolites in our targeted screen. Each metabolite was identified based on a standard, which were all purchased from Sigma Aldrich. They represent several different pathways, including the cysteine pathway, the phenylalanine/tyrosine/l-DOPA pathway, the polyamine pathway and the tryptophan/kynurenine catabolic pathway. The following 20 proteinogenic amino acids were included: l-Arginine (Arg), l-Histidine (His), l-Lysine (Lys), l-Aspartic Acid (Asp), l-Glutamic Acid (Glu), l-Serine (Ser), l-Threonine (Thr), l-Asparagine (Asn), l-Glutamine (Gln), l-Cysteine (Cys), Glycine (Gly), l-Proline (Pro), l-Alanine (Ala), l-Valine (Val), l-Isoleucine (Ile), l-Leucine (Leu), l-Methionine (Met), l-Phenylalanine (Phe), l-Tyrosine (Tyr) and l-Tryptophan (Trp). The other metabolites included were: the two PAs of cadaverine-2 (Cad) and putrescine-2 (Put) and l-3,4-dihydroxyphenylalanine (l-DOPA), serotonin (sero), l-KYN, 3-HK, 3-AA, 4-OH-Pro, homoserine, β-ala, N-acetyl-phenylalanine, tryptamine, Orn, citrulline, Sarco, γ-aminobutyric acid (GABA), 2-Ambut and taurine (Tau). Quantitation was conducted by constructing an external standard curve as described previously64. The chemical structures of the metabolites have been drawn with ChemDraw JS online (https://chemdrawdirect.perkinelmer.cloud/js/sample/index.html#).
Statistical analyses
Metabolite and amino acid biomarker data were cleaned via interquartile range filtering and log-transformed prior to analyses. Means and SD are presented post interquartile range filtering (Table 1). Age and gender effects were tested via Pearson’s correlation and independent samples t-test (Supplementary Table 2). Interactions between disease state (Control (CN) vs iPD participants) and age/gender were tested for each metabolite/amino acid via GLM to determine whether either confounder had a significant effect on metabolite via disease state (Supplementary Table 2).
Statistical analyses of metabolites and amino acids was set in two separate hypotheses; (1) targeted assessment of 37 known biomarkers (excluding l-DOPA) using nominal significance (Table 1) and (2) discovery of ratio’s and interactions between all possible metabolites and amino acids using a Bonferroni adjusted alpha (α = 0.05/K ratio’s and interactions, Supplementary Tables 3 and 4). For both hypotheses, independent samples t-test was used to test mean analyte levels between all groups (CN vs iPD, CN vs drug-naïve iPD and CN vs AD), and where the sample size was large enough (i.e. not including the drug-naïve participants) a GLM was used to account for age and gender. Disease specificity was tested across the 37 analytes between iPD and AD groups (targeted assessment using independent samples t-test, Table 1 and discovery using GLM, Supplementary Tables 3 and 4). The final number of ratios and interactions was reduced via the removal of those with an SD of less than 0.5.
Multivariate modelling to find an optimal set of metabolites associated with outcome was performed using both the least absolute shrinkage and selection operator (LASSO) followed by model selection to reduce the possibility of over fitting. Both individual metabolite biomarkers and selected sets of biomarkers from the multivariate testing were then tested using ROC analyses. Multivariate and ROC analyses were performed using the R statistical environment128 (https://www.R-project.org/). Biomarker significance was retained using Bonferroni correction to account for multiple testing.
Literature review
The 32 cohort studies presented in Table 3 were selected the following way: We searched PubMed for publications with the terms ‘PD’ AND ‘Kynurenines’ or ‘Amino Acids’ or ‘PAs’. Only significant changes for PD are shown.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We would like to acknowledge the Australian Imaging and Biomarker, Lifestyle (AIBL) research team (a complete list of researchers can be found at www.aibl.csiro.au) and the volunteers and their families. Further, we would like to thank The Victorian Government’s Operational Infrastructure Support Programme, NHMRC Dementia Leadership Fellowship (B.R.R., APP1138673), The Cooperative Research Centre (CRC) for Mental Health, The Florey Neuroproteomics Facility and NHMRC project grant (B.R.R., APP1164692). B.R.R., S.K. and C.L.M. receive support from the Michael J. Fox Foundation for Parkinson’s Research (MJFF-008030).
Author contributions
B.R.R., A.R., M.H. and B.A.B. designed the experiments and performed the analytical run on the mass spectrometer. S.K., J.D.D., B.A.B., M.H. and B.R.R. analysed the data. M.H. and C.L.M. oversaw the collection of clinical samples. M.H. conducted clinical assessments. S.K., J.D.D. and B.R.R. wrote the manuscript. All authors critically reviewed and edited the manuscript and approved the completed version.
Data availability
Anonymized data will be shared on request from any qualified investigator for purposes of replicating procedures and results.
Competing interests
B.R.R. is an inventor on PCT application AU2014/000849. B.R.R. receives research support from Agilent Technologies (VIC, Australia) and from eMSion Inc. (OR, USA). The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41531-021-00239-x.
References
- 1.Dorsey ER, et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- 2.Poewe W, et al. Parkinson disease. Nat. Rev. Dis. Prim. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 3.Hallett PJ, Standaert DG. Rationale for and use of NMDA receptor antagonists in Parkinson’s disease. Pharm. Ther. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Hansen KB, et al. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018;150:1081–1105. doi: 10.1085/jgp.201812032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villar-Cheda B, et al. Involvement of microglial RhoA/Rho-kinase pathway activation in the dopaminergic neuron death. Role of angiotensin via angiotensin type 1 receptors. Neurobiol. Dis. 2012;47:268–279. doi: 10.1016/j.nbd.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Iyer M, et al. Role of RhoA-ROCK signaling in Parkinson’s disease. Eur. J. Pharm. 2021;894:173815. doi: 10.1016/j.ejphar.2020.173815. [DOI] [PubMed] [Google Scholar]
- 7.National Collaborating Centre for Chronic Conditions. Parkinson’s Disease: National Clinical Guideline for Diagnosis and Management in Primary and Secondary Care (Royal College of Physicians, 2006). [PubMed]
- 8.Xia R, Mao ZH. Progression of motor symptoms in Parkinson’s disease. Neurosci. Bull. 2012;28:39–48. doi: 10.1007/s12264-012-1050-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Postuma RB, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015;30:1591–1601. doi: 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- 10.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125:861–870. doi: 10.1093/brain/awf080. [DOI] [PubMed] [Google Scholar]
- 12.Agid Y. Parkinson’s disease: pathophysiology. Lancet. 1991;337:1321–1324. doi: 10.1016/0140-6736(91)92989-F. [DOI] [PubMed] [Google Scholar]
- 13.Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- 14.Greffard S, et al. Motor score of the unified Parkinson disease rating scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch. Neurol. 2006;63:584–588. doi: 10.1001/archneur.63.4.584. [DOI] [PubMed] [Google Scholar]
- 15.Danielson SR, Andersen JK. Oxidative and nitrative protein modifications in Parkinson’s disease. Free Radic. Biol. Med. 2008;44:1787–1794. doi: 10.1016/j.freeradbiomed.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He Y, Yu Z, Chen S. Alpha-synuclein nitration and its implications in Parkinson’s disease. ACS Chem. Neurosci. 2019;10:777–782. doi: 10.1021/acschemneuro.8b00288. [DOI] [PubMed] [Google Scholar]
- 17.Ogawa T, et al. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology. 1992;42:1702–1706. doi: 10.1212/WNL.42.9.1702. [DOI] [PubMed] [Google Scholar]
- 18.Vivo M, et al. Polyamines in the basal ganglia of human brain. Influence of aging and degenerative movement disorders. Neurosci. Lett. 2001;304:107–111. doi: 10.1016/S0304-3940(01)01776-1. [DOI] [PubMed] [Google Scholar]
- 19.Lewitt PA, et al. 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov. Disord. 2013;28:1653–1660. doi: 10.1002/mds.25555. [DOI] [PubMed] [Google Scholar]
- 20.Ohman A, Forsgren L. NMR metabonomics of cerebrospinal fluid distinguishes between Parkinson’s disease and controls. Neurosci. Lett. 2015;594:36–39. doi: 10.1016/j.neulet.2015.03.051. [DOI] [PubMed] [Google Scholar]
- 21.Paik MJ, et al. Polyamine patterns in the cerebrospinal fluid of patients with Parkinson’s disease and multiple system atrophy. Clin. Chim. Acta. 2010;411:1532–1535. doi: 10.1016/j.cca.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 22.Han W, et al. Profiling novel metabolic biomarkers for Parkinson’s disease using in-depth metabolomic analysis. Mov. Disord. 2017;32:1720–1728. doi: 10.1002/mds.27173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulte EC, et al. Alterations in lipid and inositol metabolisms in two dopaminergic disorders. PLoS ONE. 2016;11:e0147129. doi: 10.1371/journal.pone.0147129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oxenkrug, G., van der Hart, M., Roeser, J. & Summergrad, P. Peripheral tryptophan - Kynurenine metabolism associated with metabolic syndrome is different in Parkinson’s and Alzheimer’s diseases. Endocrinol. Diabetes Metab. J. 1, http://researchopenworld.com/wp-content/uploads/2017/11/EDMJ-2017-113-Gregory-F-Oxenkrug-USA.pdf (2017). [PMC free article] [PubMed]
- 25.Iwasaki Y, Ikeda K, Shiojima T, Kinoshita M. Increased plasma concentrations of aspartate, glutamate and glycine in Parkinson’s disease. Neurosci. Lett. 1992;145:175–177. doi: 10.1016/0304-3940(92)90015-Y. [DOI] [PubMed] [Google Scholar]
- 26.Hartai Z, et al. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J. Neurol. Sci. 2005;239:31–35. doi: 10.1016/j.jns.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 27.Gomes-Trolin C, Nygren I, Aquilonius SM, Askmark H. Increased red blood cell polyamines in ALS and Parkinson’s disease. Exp. Neurol. 2002;177:515–520. doi: 10.1006/exnr.2002.7952. [DOI] [PubMed] [Google Scholar]
- 28.Sinclair E, et al. Metabolomics of sebum reveals lipid dysregulation in Parkinson’s disease. Nat. Commun. 2021;12:1592. doi: 10.1038/s41467-021-21669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luan H, et al. Comprehensive urinary metabolomic profiling and identification of potential noninvasive marker for idiopathic Parkinson’s disease. Sci. Rep. 2015;5:13888. doi: 10.1038/srep13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiarugi A, Meli E, Moroni F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J. Neurochem. 2001;77:1310–1318. doi: 10.1046/j.1471-4159.2001.00335.x. [DOI] [PubMed] [Google Scholar]
- 31.Hatano T, et al. Identification of novel biomarkers for Parkinson’s disease by metabolomic technologies. J. Neurol. Neurosurg. Psychiatry. 2016;87:295–301. doi: 10.1136/jnnp-2014-309676. [DOI] [PubMed] [Google Scholar]
- 32.Havelund JF, et al. Changes in kynurenine pathway metabolism in Parkinson patients with L-DOPA-induced dyskinesia. J. Neurochem. 2017;142:756–766. doi: 10.1111/jnc.14104. [DOI] [PubMed] [Google Scholar]
- 33.Roede JR, et al. Serum metabolomics of slow vs. rapid motor progression Parkinson’s disease: a pilot study. PLoS ONE. 2013;8:e77629. doi: 10.1371/journal.pone.0077629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krasnoslobodtsev AV, et al. Effect of spermidine on misfolding and interactions of alpha-synuclein. PLoS ONE. 2012;7:e38099. doi: 10.1371/journal.pone.0038099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sian J, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994;36:348–355. doi: 10.1002/ana.410360305. [DOI] [PubMed] [Google Scholar]
- 36.Bogdanov M, et al. Metabolomic profiling to develop blood biomarkers for Parkinson’s disease. Brain. 2008;131:389–396. doi: 10.1093/brain/awm304. [DOI] [PubMed] [Google Scholar]
- 37.Smeyne M, Smeyne RJ. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013;62:13–25. doi: 10.1016/j.freeradbiomed.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fessel JP, et al. Isofurans, but not F2-isoprostanes, are increased in the substantia nigra of patients with Parkinson’s disease and with dementia with Lewy body disease. J. Neurochem. 2003;85:645–650. doi: 10.1046/j.1471-4159.2003.01709.x. [DOI] [PubMed] [Google Scholar]
- 39.Baillet A, et al. The role of oxidative stress in amyotrophic lateral sclerosis and Parkinson’s disease. Neurochem. Res. 2010;35:1530–1537. doi: 10.1007/s11064-010-0212-5. [DOI] [PubMed] [Google Scholar]
- 40.Chan RB, et al. Elevated GM3 plasma concentration in idiopathic Parkinson’s disease: a lipidomic analysis. PLoS ONE. 2017;12:e0172348. doi: 10.1371/journal.pone.0172348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Sanz P, et al. N370S-GBA1 mutation causes lysosomal cholesterol accumulation in Parkinson’s disease. Mov. Disord. 2017;32:1409–1422. doi: 10.1002/mds.27119. [DOI] [PubMed] [Google Scholar]
- 42.Stoessel D, et al. Promising metabolite profiles in the plasma and CSF of early clinical Parkinson’s disease. Front. Aging Neurosci. 2018;10:51. doi: 10.3389/fnagi.2018.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trupp M, et al. Metabolite and peptide levels in plasma and CSF differentiating healthy controls from patients with newly diagnosed Parkinson’s disease. J. Parkinsons Dis. 2014;4:549–560. doi: 10.3233/JPD-140389. [DOI] [PubMed] [Google Scholar]
- 44.Burte F, et al. Metabolic profiling of Parkinson’s disease and mild cognitive impairment. Mov. Disord. 2017;32:927–932. doi: 10.1002/mds.26992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saiki S, et al. Decreased long-chain acylcarnitines from insufficient beta-oxidation as potential early diagnostic markers for Parkinson’s disease. Sci. Rep. 2017;7:7328. doi: 10.1038/s41598-017-06767-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johansen KK, et al. Metabolomic profiling in LRRK2-related Parkinson’s disease. PLoS ONE. 2009;4:e7551. doi: 10.1371/journal.pone.0007551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luan H, et al. LC-MS-based urinary metabolite signatures in idiopathic Parkinson’s disease. J. Proteome Res. 2015;14:467–478. doi: 10.1021/pr500807t. [DOI] [PubMed] [Google Scholar]
- 48.LeWitt PA, et al. Metabolomic biomarkers as strong correlates of Parkinson disease progression. Neurology. 2017;88:862–869. doi: 10.1212/WNL.0000000000003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmed SS, Santosh W, Kumar S, Christlet HT. Metabolic profiling of Parkinson’s disease: evidence of biomarker from gene expression analysis and rapid neural network detection. J. Biomed. Sci. 2009;16:63. doi: 10.1186/1423-0127-16-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuiper MA, et al. L-glutamate, L-arginine and L-citrulline levels in cerebrospinal fluid of Parkinson’s disease, multiple system atrophy, and Alzheimer’s disease patients. J. Neural Transm. 2000;107:183–189. doi: 10.1007/s007020050016. [DOI] [PubMed] [Google Scholar]
- 51.Wuolikainen A, et al. Multi-platform mass spectrometry analysis of the CSF and plasma metabolomes of rigorously matched amyotrophic lateral sclerosis, Parkinson’s disease and control subjects. Mol. Biosyst. 2016;12:1287–1298. doi: 10.1039/C5MB00711A. [DOI] [PubMed] [Google Scholar]
- 52.Figura M, et al. Serum amino acid profile in patients with Parkinson’s disease. PLoS ONE. 2018;13:e0191670. doi: 10.1371/journal.pone.0191670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirayama M, et al. Serum tyrosine-to-phenylalanine ratio is low in Parkinson’s disease. J. Parkinsons Dis. 2016;6:423–431. doi: 10.3233/JPD-150736. [DOI] [PubMed] [Google Scholar]
- 54.Stone TW. Kynurenines in the CNS: from endogenous obscurity to therapeutic importance. Prog. Neurobiol. 2001;64:185–218. doi: 10.1016/S0301-0082(00)00032-0. [DOI] [PubMed] [Google Scholar]
- 55.Stone TW, et al. Tryptophan metabolites and brain disorders. Clin. Chem. Lab Med. 2003;41:852–859. doi: 10.1515/CCLM.2003.129. [DOI] [PubMed] [Google Scholar]
- 56.Eisenberg T, et al. Induction of autophagy by spermidine promotes longevity. Nat. Cell Biol. 2009;11:1305–1314. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 57.Madeo F, et al. Spermidine: a novel autophagy inducer and longevity elixir. Autophagy. 2010;6:160–162. doi: 10.4161/auto.6.1.10600. [DOI] [PubMed] [Google Scholar]
- 58.Eastman CL, Guilarte TR. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem. Res. 1990;15:1101–1107. doi: 10.1007/BF01101711. [DOI] [PubMed] [Google Scholar]
- 59.Pegg AE. Toxicity of polyamines and their metabolic products. Chem. Res. Toxicol. 2013;26:1782–1800. doi: 10.1021/tx400316s. [DOI] [PubMed] [Google Scholar]
- 60.Wood PL, Khan MA, Moskal JR. The concept of “aldehyde load” in neurodegenerative mechanisms: cytotoxicity of the polyamine degradation products hydrogen peroxide, acrolein, 3-aminopropanal, 3-acetamidopropanal and 4-aminobutanal in a retinal ganglion cell line. Brain Res. 2007;1145:150–156. doi: 10.1016/j.brainres.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 61.Nemeth, H., Toldi, J. & Vécsei, L. Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J. Neural Transm. Suppl. 285–304 10.1007/978-3-211-45295-0_45 (2006). [DOI] [PubMed]
- 62.Ghasemi M, Schachter SC. The NMDA receptor complex as a therapeutic target in epilepsy: a review. Epilepsy Behav. 2011;22:617–640. doi: 10.1016/j.yebeh.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 63.Sirrieh RE, MacLean DM, Jayaraman V. Subtype-dependent N-methyl-D-aspartate receptor amino-terminal domain conformations and modulation by spermine. J. Biol. Chem. 2015;290:12812–12820. doi: 10.1074/jbc.M115.649723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boughton BA, et al. Comprehensive profiling and quantitation of amine group containing metabolites. Anal. Chem. 2011;83:7523–7530. doi: 10.1021/ac201610x. [DOI] [PubMed] [Google Scholar]
- 65.Motter R, et al. Reduction of beta-amyloid peptide42 in the cerebrospinal fluid of patients with Alzheimer’s disease. Ann. Neurol. 1995;38:643–648. doi: 10.1002/ana.410380413. [DOI] [PubMed] [Google Scholar]
- 66.Hansson O, et al. Advantages and disadvantages of the use of the CSF Amyloid beta (Abeta) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimers Res. Ther. 2019;11:34. doi: 10.1186/s13195-019-0485-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Halbgebauer S, et al. Protein biomarkers in Parkinson’s disease: focus on cerebrospinal fluid markers and synaptic proteins. Mov. Disord. 2016;31:848–860. doi: 10.1002/mds.26635. [DOI] [PubMed] [Google Scholar]
- 68.Havelund, J. F., Heegaard, N. H. H., Faergeman, N. J. K. & Gramsbergen, J. B. Biomarker research in Parkinson’s disease using metabolite profiling. Metabolites. 7, 42 (2017). [DOI] [PMC free article] [PubMed]
- 69.Jimenez-Jimenez FJ, et al. Neurotransmitter amino acids in cerebrospinal fluid of patients with Parkinson’s disease. J. Neurol. Sci. 1996;141:39–44. doi: 10.1016/0022-510X(96)00115-3. [DOI] [PubMed] [Google Scholar]
- 70.Waloen K, Kleppe R, Martinez A, Haavik J. Tyrosine and tryptophan hydroxylases as therapeutic targets in human disease. Expert Opin. Ther. Targets. 2017;21:167–180. doi: 10.1080/14728222.2017.1272581. [DOI] [PubMed] [Google Scholar]
- 71.Fossbakk A, et al. Functional studies of tyrosine hydroxylase missense variants reveal distinct patterns of molecular defects in Dopa-responsive dystonia. Hum. Mutat. 2014;35:880–890. doi: 10.1002/humu.22565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Kessel SP, et al. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat. Commun. 2019;10:310. doi: 10.1038/s41467-019-08294-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Muller T, Muhlack S. Cysteine decrease following acute Levodopa intake in patients with Parkinson’s disease. Neurosci. Lett. 2012;521:37–39. doi: 10.1016/j.neulet.2012.05.054. [DOI] [PubMed] [Google Scholar]
- 74.Muller T, Trommer I, Muhlack S, Mueller BK. Levodopa increases oxidative stress and repulsive guidance molecule A levels: a pilot study in patients with Parkinson’s disease. J. Neural Transm. 2016;123:401–406. doi: 10.1007/s00702-016-1519-4. [DOI] [PubMed] [Google Scholar]
- 75.Muller T, Muhlack S. Cysteinyl-glycine reduction as marker for levodopa-induced oxidative stress in Parkinson’s disease patients. Mov. Disord. 2011;26:543–546. doi: 10.1002/mds.23384. [DOI] [PubMed] [Google Scholar]
- 76.Johnson WM, Wilson-Delfosse AL, Mieyal JJ. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients. 2012;4:1399–1440. doi: 10.3390/nu4101399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lovelace MD, et al. Current evidence for a role of the kynurenine pathway of tryptophan metabolism in multiple sclerosis. Front. Immunol. 2016;7:246. doi: 10.3389/fimmu.2016.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vamos E, et al. The role of kynurenines in disorders of the central nervous system: possibilities for neuroprotection. J. Neurol. Sci. 2009;283:21–27. doi: 10.1016/j.jns.2009.02.326. [DOI] [PubMed] [Google Scholar]
- 79.Kotake Y. Studies on the intermediary metabolism of tryptophane. On the mode of the formation of kynurenic acid in the organism. Hoppe-Seyler’s Z. fur physiologische Chem. 1931;195:158–166. [Google Scholar]
- 80.Kotake Y, Iwao J. Studies on the intermediatary metabolism of tryptophan. I. Kynurenine, an intermediary metabolic product of tryptophan. Z. Physiol. Chem. 1931;195:139–147. doi: 10.1515/bchm2.1931.195.3-6.139. [DOI] [Google Scholar]
- 81.Young SN, Joseph MH, Gauthier S. Studies on kynurenine in human cerebrospinal fluid: lowered levels in epilepsy. J. Neural Transm. 1983;58:193–204. doi: 10.1007/BF01252805. [DOI] [PubMed] [Google Scholar]
- 82.Beal MF, et al. Kynurenine pathway measurements in Huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J. Neurochem. 1990;55:1327–1339. doi: 10.1111/j.1471-4159.1990.tb03143.x. [DOI] [PubMed] [Google Scholar]
- 83.Hartai Z, et al. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005;112:93–96. doi: 10.1111/j.1600-0404.2005.00442.x. [DOI] [PubMed] [Google Scholar]
- 84.Gulaj E, Pawlak K, Bien B, Pawlak D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010;55:204–211. doi: 10.2478/v10039-010-0023-6. [DOI] [PubMed] [Google Scholar]
- 85.Gal EM, Sherman AD. Synthesis and metabolism of L-kynurenine in rat brain. J. Neurochem. 1978;30:607–613. doi: 10.1111/j.1471-4159.1978.tb07815.x. [DOI] [PubMed] [Google Scholar]
- 86.Fukui S, et al. Blood-brain barrier transport of kynurenines: implications for brain synthesis and metabolism. J. Neurochem. 1991;56:2007–2017. doi: 10.1111/j.1471-4159.1991.tb03460.x. [DOI] [PubMed] [Google Scholar]
- 87.Klivenyi P, Toldi J, Vecsei L. Kynurenines in neurodegenerative disorders: therapeutic consideration. Adv. Exp. Med. Biol. 2004;541:169–183. doi: 10.1007/978-1-4419-8969-7_10. [DOI] [PubMed] [Google Scholar]
- 88.Ferreira FS, et al. Kynurenic acid restores Nrf2 levels and prevents quinolinic acid-induced toxicity in rat striatal slices. Mol. Neurobiol. 2018;55:8538–8549. doi: 10.1007/s12035-018-1003-2. [DOI] [PubMed] [Google Scholar]
- 89.Pierozan P, et al. Kynurenic acid prevents cytoskeletal disorganization induced by quinolinic acid in mixed cultures of rat striatum. Mol. Neurobiol. 2018;55:5111–5124. doi: 10.1007/s12035-017-0749-2. [DOI] [PubMed] [Google Scholar]
- 90.Jones SP, et al. Expression of the kynurenine pathway in human peripheral blood mononuclear cells: implications for inflammatory and neurodegenerative disease. PLoS ONE. 2015;10:e0131389. doi: 10.1371/journal.pone.0131389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pierozan P, et al. Acute intrastriatal administration of quinolinic acid provokes hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons of rats. Exp. Neurol. 2010;224:188–196. doi: 10.1016/j.expneurol.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 92.Rahman A, et al. The excitotoxin quinolinic acid induces tau phosphorylation in human neurons. PLoS ONE. 2009;4:e6344. doi: 10.1371/journal.pone.0006344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Widner B, Leblhuber F, Fuchs D. Increased neopterin production and tryptophan degradation in advanced Parkinson’s disease. J. Neural Transm. 2002;109:181–189. doi: 10.1007/s007020200014. [DOI] [PubMed] [Google Scholar]
- 94.Heilman PL, et al. Tryptophan metabolites are associated with symptoms and nigral pathology in Parkinson’s disease. Mov. Disord. 2020;35:2028–2037. doi: 10.1002/mds.28202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gregoire L, et al. Prolonged kynurenine 3-hydroxylase inhibition reduces development of levodopa-induced dyskinesias in parkinsonian monkeys. Behav. Brain Res. 2008;186:161–167. doi: 10.1016/j.bbr.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 96.Vecsei L, Szalardy L, Fulop F, Toldi J. Kynurenines in the CNS: recent advances and new questions. Nat. Rev. Drug Disco. 2013;12:64–82. doi: 10.1038/nrd3793. [DOI] [PubMed] [Google Scholar]
- 97.Venkatesan D, et al. Kynurenine pathway in Parkinson’s disease-An update. eNeurologicalSci. 2020;21:100270. doi: 10.1016/j.ensci.2020.100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Torok, N., Tanaka, M. & Vecsei, L. Searching for peripheral biomarkers in neurodegenerative diseases: the tryptophan-kynurenine metabolic pathway. Int. J. Mol. Sci. 21, 9338 (2020). [DOI] [PMC free article] [PubMed]
- 99.Miller-Fleming L, Olin-Sandoval V, Campbell K, Ralser M. Remaining mysteries of molecular biology: the role of polyamines in the cell. J. Mol. Biol. 2015;427:3389–3406. doi: 10.1016/j.jmb.2015.06.020. [DOI] [PubMed] [Google Scholar]
- 100.Inoue K, et al. Metabolic profiling of Alzheimer’s disease brains. Sci. Rep. 2013;3:2364. doi: 10.1038/srep02364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lewandowski NM, et al. Polyamine pathway contributes to the pathogenesis of Parkinson disease. Proc. Natl Acad. Sci. USA. 2010;107:16970–16975. doi: 10.1073/pnas.1011751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Betancourt L, et al. Micellar electrokinetic chromatography with laser induced fluorescence detection shows increase of putrescine in erythrocytes of Parkinson’s disease patients. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018;1081–1082:51–57. doi: 10.1016/j.jchromb.2018.02.015. [DOI] [PubMed] [Google Scholar]
- 103.Picca A, et al. Circulating amino acid signature in older people with Parkinson’s disease: a metabolic complement to the EXosomes in PArkiNson Disease (EXPAND) study. Exp. Gerontol. 2019;128:110766. doi: 10.1016/j.exger.2019.110766. [DOI] [PubMed] [Google Scholar]
- 104.Chang KH, et al. Alternations of metabolic profile and kynurenine metabolism in the plasma of Parkinson’s disease. Mol. Neurobiol. 2018;55:6319–6328. doi: 10.1007/s12035-017-0845-3. [DOI] [PubMed] [Google Scholar]
- 105.Sonninen TM, et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020;10:14474. doi: 10.1038/s41598-020-71329-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Antony T, et al. Cellular polyamines promote the aggregation of alpha-synuclein. J. Biol. Chem. 2003;278:3235–3240. doi: 10.1074/jbc.M208249200. [DOI] [PubMed] [Google Scholar]
- 107.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharm. Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 108.Mony L, Zhu S, Carvalho S, Paoletti P. Molecular basis of positive allosteric modulation of GluN2B NMDA receptors by polyamines. EMBO J. 2011;30:3134–3146. doi: 10.1038/emboj.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dantuma NP, Bott LC. The ubiquitin-proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014;7:70. doi: 10.3389/fnmol.2014.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Velloso NA, et al. Spermine attenuates behavioral and biochemical alterations induced by quinolinic acid in the striatum of rats. Brain Res. 2008;1198:107–114. doi: 10.1016/j.brainres.2007.12.056. [DOI] [PubMed] [Google Scholar]
- 111.Jamwal S, Singh S, Kaur N, Kumar P. Protective effect of spermidine against excitotoxic neuronal death induced by quinolinic acid in rats: possible neurotransmitters and neuroinflammatory mechanism. Neurotox. Res. 2015;28:171–184. doi: 10.1007/s12640-015-9535-y. [DOI] [PubMed] [Google Scholar]
- 112.Sorgdrager FJH, et al. Age- and disease-specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J. Neurochem. 2019;151:656–668. doi: 10.1111/jnc.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Iwaoka K, et al. Impaired metabolism of kynurenine and its metabolites in CSF of parkinson’s disease. Neurosci. Lett. 2020;714:134576. doi: 10.1016/j.neulet.2019.134576. [DOI] [PubMed] [Google Scholar]
- 114.Fiandaca, M. S. et al. Potential metabolomic linkage in blood between Parkinson’s disease and traumatic brain injury. Metabolites8, 50 (2018). [DOI] [PMC free article] [PubMed]
- 115.Engelborghs S, Marescau B, De Deyn PP. Amino acids and biogenic amines in cerebrospinal fluid of patients with Parkinson’s disease. Neurochem. Res. 2003;28:1145–1150. doi: 10.1023/A:1024255208563. [DOI] [PubMed] [Google Scholar]
- 116.Molina JA, et al. Decreased cerebrospinal fluid levels of neutral and basic amino acids in patients with Parkinson’s disease. J. Neurol. Sci. 1997;150:123–127. doi: 10.1016/S0022-510X(97)00069-5. [DOI] [PubMed] [Google Scholar]
- 117.Mally J, Szalai G, Stone TW. Changes in the concentration of amino acids in serum and cerebrospinal fluid of patients with Parkinson’s disease. J. Neurol. Sci. 1997;151:159–162. doi: 10.1016/S0022-510X(97)00119-6. [DOI] [PubMed] [Google Scholar]
- 118.Sertbas M, Ulgen K, Cakir T. Systematic analysis of transcription-level effects of neurodegenerative diseases on human brain metabolism by a newly reconstructed brain-specific metabolic network. FEBS Open Bio. 2014;4:542–553. doi: 10.1016/j.fob.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kori M, et al. Metabolic biomarkers and neurodegeneration: a pathway enrichment analysis of Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. OMICS. 2016;20:645–661. doi: 10.1089/omi.2016.0106. [DOI] [PubMed] [Google Scholar]
- 120.Saiki S, et al. A metabolic profile of polyamines in parkinson disease: a promising biomarker. Ann. Neurol. 2019;86:251–263. doi: 10.1002/ana.25516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sheard JM, et al. Malnutrition in a sample of community-dwelling people with Parkinson’s disease. PLoS ONE. 2013;8:e53290. doi: 10.1371/journal.pone.0053290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mulak A, Bonaz B. Brain-gut-microbiota axis in Parkinson’s disease. World J. Gastroenterol. 2015;21:10609–10620. doi: 10.3748/wjg.v21.i37.10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Seidl SE, Santiago JA, Bilyk H, Potashkin JA. The emerging role of nutrition in Parkinson’s disease. Front. Aging Neurosci. 2014;6:36. doi: 10.3389/fnagi.2014.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wikoff WR, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl Acad. Sci. USA. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wilmanski T, et al. Blood metabolome predicts gut microbiome alpha-diversity in humans. Nat. Biotechnol. 2019;37:1217–1228. doi: 10.1038/s41587-019-0233-9. [DOI] [PubMed] [Google Scholar]
- 126.Cardoso BR, et al. Selenium levels in serum, red blood cells, and cerebrospinal fluid of Alzheimer’s disease patients: a report from the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL) J. Alzheimers Dis. 2017;57:183–193. doi: 10.3233/JAD-160622. [DOI] [PubMed] [Google Scholar]
- 127.Watt AD, et al. Variability in blood-based amyloid-beta assays: the need for consensus on pre-analytical processing. J. Alzheimers Dis. 2012;30:323–336. doi: 10.3233/JAD-2012-120058. [DOI] [PubMed] [Google Scholar]
- 128.R Core Team. R: a language and environment for statistical computing (R Foundation for Statistical Computing, 2018).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Anonymized data will be shared on request from any qualified investigator for purposes of replicating procedures and results.