

Abstract
Neuroinflammation, particularly in the dorsolateral prefrontal cortex, is well-established in a subset of people with schizophrenia, with significant increases in inflammatory markers including several cytokines. Yet the cause(s) of cortical inflammation in schizophrenia remains unknown. Clues as to potential microenvironmental triggers and/or intracellular deficits in immunoregulation may be gleaned from looking further upstream of effector immune molecules to transcription factors that control inflammatory gene expression. Here, we focus on the ‘master immune regulator’ nuclear factor kappa B (NF-κB) and review evidence in support of NF-κB dysregulation causing or contributing to neuroinflammation in patients. We discuss the utility of ‘immune biotyping’ as a tool to analyse immune-related transcripts and proteins in patient tissue, and the insights into cortical NF-κB in schizophrenia revealed by immune biotyping compared to studies treating patients as a single, homogenous group. Though the ubiquitous nature of NF-κB presents several hurdles for drug development, targeting this key immunoregulator with novel or repurposed therapeutics in schizophrenia is a relatively underexplored area that could aid in reducing symptoms of patients with active neuroinflammation.
Subject terms: Molecular neuroscience, Neuroscience
Introduction
Schizophrenia is a severe psychiatric illness that disrupts the normal functioning of the mind and affects roughly 1% of the population worldwide [1]. People with schizophrenia typically suffer from positive symptoms including delusions, hallucinations and disorganised speech, and negative symptoms such as diminished emotional expression and/or lack of motivation [2, 3]. In addition, there is a high prevalence of often profound neurocognitive deficits among patients, most commonly in working memory, attention, problem solving and processing speed [4]. This combination of psychiatric and cognitive symptoms contributes to reduced scholastic and vocational achievements and worse quality of life [5, 6]. As such, the social and economic costs of schizophrenia are substantial and are disproportionate to the disease prevalence, due to the chronic, severe and often treatment-resistant nature of schizophrenia [2, 7, 8]. Better understanding of the aetiology of schizophrenia is needed in order to develop novel treatments that improve clinical and functional outcomes in patients.
One of the most significant insights into the pathophysiology of schizophrenia over the last 10 years has been the identification of inflammation in patients; however, this has not yet led to effective new treatments. The limited success of clinical trials with anti-inflammatories [9] may reflect previous failures to target the correct inflammatory mechanism(s) with available medications. One approach to identifying the correct target(s) at which to aim novel therapies is to start with the known increase in cytokine mRNA and to then examine factors upstream to determine how these are changed. For example, to ask which, how and to what extent molecular switches known to be responsible for turning on these cytokines are changed. This approach may be more informative given that cytokine synthesis is highly regulated at the transcriptional level, and many specific transcription factors controlling cytokine expression have been identified. Further, control over the activity of these transcription factors can be traced back to receptors capable of responding to the external (extracellular) environment. Altered gene expression of one such ‘master’ immune transcription factor, nuclear factor kappa B (NF-κB), co-occurs with increased cytokine mRNA levels in the brains of people with schizophrenia in at least three studies [10–12].
Another reason that anti-inflammatory treatment may not show a high degree of effectiveness or reproducibility is that not everyone with schizophrenia is expected to be inflamed at commencement of anti-inflammatory treatment. Indeed, it is increasingly apparent that some patients are more likely to be in a state of heightened inflammation and to respond to anti-inflammatory treatment than others. Those with more severe symptoms at baseline are more responsive to adjunctive aspirin [9], and one study found that aspirin response in people with schizophrenia differs based on pro- and anti-inflammatory cytokine ratios at baseline [13]. Results from an earlier clinical trial showed that patients who responded to the cyclo-oxygenase 2 inhibitor celecoxib had lower baseline levels of the anti-inflammatory protein sTNFR1 in blood than non-responders [14]. Since few studies into anti-inflammatory adjuvants in schizophrenia have stratified individuals based on their immune status, it may be the case that only a subset of ‘inflamed’ patients respond favourably to anti-inflammatory drugs, and that this accounts for the small overall effect sizes when people with schizophrenia are considered as a single, homogenous patient group [9, 15].
Before considering these two points, we will review the evidence of inflammation as a potential causal or contributing factor in schizophrenia from three approaches: (1) associations between schizophrenia and immune genes, (2) increased inflammatory markers in postmortem brain tissue from people with schizophrenia and (3) abnormal levels of immune molecules in the blood of living patients. Immune activation assessed in living patients has been linked to more severe positive, negative and cognitive symptoms in patients [16–19], indicating a significant role of inflammation in schizophrenia symptomatology. Heightened or chronic neuroinflammation is not a normal or healthy occurrence, and while the pathogenesis of schizophrenia likely involves several interacting contributors [such as psychosocial stress and/or exposure to recreational drugs [20–23], alleviation of inflammation in people with schizophrenia could bring about therapeutic benefit regardless of its cause.
Inflammation is associated with schizophrenia
Genetic evidence of inflammation in schizophrenia
One of the most consistent signals from genome-wide association studies of schizophrenia is significant association between the disease and genetic variation in the major histocompatibility region of chromosome 6 [24–28] which encodes molecules involved in immunity and inflammation [29]. Most notably, a strong association between schizophrenia and the complement system gene C4 was found in a genome-wide association study of more than 28,000 schizophrenia cases and 35,000 control cases [30]. Genes in the major histocompatibility region often contain NF-κB-binding sequences in their promoter regions, including several that encode potent pro-inflammatory cytokines such as interleukin (IL)-1, IL-6, IL-8 and tumour necrosis factor (TNF) [31–37]. Genetic associations to schizophrenia mark broad chromosomal regions which may contain many DNA changes (SNPs) that combine to increase risk, and DNA changes linked to schizophrenia are known occur in promoter regions that regulate gene expression. Thus, this represents a transcriptional enhancement mechanism by which NF-κB, activated by environmental cues, could potentially interact with genomic regions that then increase risk for developing schizophrenia. In support of this, some studies have found significant links between variation in these regions, most notably in the gene encoding interleukin (IL)-1, and schizophrenia susceptibility [38–44]. However, other studies have failed to find an association between IL-1 polymorphisms and schizophrenia risk [45–49]. Lack of replication of SNPs associated with schizophrenia are common, and there are likely multiple genes that need to interact with particular environmental cues to then trigger inflammation, such that no single gene will sufficiently explain risk in all cohorts. This perspective highlights the importance of studying brain gene expression of immunoregulatory pathways as a way of detecting which microenvironmental drivers of inflammation may precipitate schizophrenia genetic risk.
Postmortem evidence of inflammation in schizophrenia
The study of human postmortem brain tissue allows for the direct measurement of ‘immune’ molecules and markers in neural tissue, and has revealed evidence of immune activation in the dorsolateral prefrontal cortex (PFC), orbital frontal cortex and midbrain of a substantial subset (~40–50%) of people with schizophrenia compared to a significantly smaller proportion (~0–10%) of age-matched non-schizophrenic controls [10, 11, 50–53]. These ‘high neuroinflammation’1 patients have been identified across independent cohorts using two-step recursive clustering of mRNA levels of several pro-inflammatory transcripts such as IL-1β, IL-6, IL-8 and SERPINA3 [50–53]. Given that structural and functional abnormalities in the dorsolateral PFC are hallmarks of schizophrenia [54–60], it is possible that inflammation in this region drives neuropathology—and potentially symptoms—in some patients. Indeed, the high neuroinflammation patient subgroup has worse psychotic symptoms [18], PFC-dependent cognition and neuropathology than patients with ‘normal’ levels of these transcripts, including reduced verbal fluency [19], increased astrogliosis [61], larger reductions in inhibitory interneuron-related transcripts [50, 62] and significant loss of prefrontal grey matter volume [51]. Recent evidence also suggests that this high neuroinflammation schizophrenia subgroup may have altered blood–brain barrier function that facilitates the trafficking of macrophages from blood to brain, evidenced by increased endothelial expression of adhesion molecules that capture white blood cells [63, 64]. Further, Cai et al. [63] and Purves-Tyson et al. [64] found elevated transcript levels of the macrophage marker CD163 mRNA in the PFC and midbrain of high neuroinflammation patients, supporting the contention that macrophages are recruited to the brain in response to cortical and subcortical immune activation. Taken together, these findings suggest that aberrant neuroinflammatory processes play a critical role in causing neuropathology in some patients.
The existence of inflammatory biotypes (subtypes) within schizophrenia may explain why studies measuring immune-related transcripts and/or proteins in the dorsolateral PFC of patients sometimes produce discrepant results. Many studies comparing dorsolateral PFC (and neighbouring PFC) tissue from people with schizophrenia as a single homogenous cohort to tissue from non-schizophrenic controls do find up-regulation of pro-inflammatory cytokines and acute phase proteins (IL-6, IL-8, TNFα, SERPINA3) and cytokine receptors (IL-1 receptor type 1 [IL1R1], TNF receptor 1 [TNFR1]), at the transcriptional level in schizophrenia [10, 50, 65–67]. IL-6 and TNFα have also been shown to be elevated at the protein level in this brain region in patients, along with another TNF family cytokine, lymphotoxin α [65]. Examination of cytokines that serve to dampen the inflammatory response has also revealed that the anti-inflammatory IL-10 transcript and protein are reduced in the dorsolateral PFC in schizophrenia [65], suggestive of a diminished ability to attenuate neuroinflammation in at least some patients. However, data from several postmortem studies dispute the role of neuroinflammation at both the molecular and cellular levels in schizophrenia [68–75]. Surprisingly, though, one such study reported significant enrichment of myeloid leucocyte activation in the DLPFC of people with schizophrenia relative to unaffected controls before concluding that immune activation is not a characteristic of the schizophrenia brain [68]. Notably, studies that fail to find positive associations between inflammatory markers in human postmortem brain tissue and schizophrenia did not investigate the possibility of heterogeneity in regards to immune subtypes. As such, immune ‘biotyping’ is an important research design tool that can be used to uncover previously unrealised neuropathology.
Clinical evidence of inflammation in schizophrenia
Since immune-to-brain communication is bidirectional [76], assessing inflammation in the blood is useful in determining the extent of peripheral immune activation that may contribute to or result from neuroinflammation in schizophrenia. In fact, evidence for altered peripheral immune function predates evidence of increased brain cytokines [77–81]. More recently, elevations in inflammatory markers including pro-inflammatory cytokines, the acute phase protein CRP, the immune regulator S100B and soluble intracellular cell adhesion molecule have been found in the blood of people with schizophrenia [18, 19, 63, 82–97]. However, some studies have found unchanged [98–102] and even decreased [102, 103] levels of these same inflammatory biomarkers in the blood of patients compared to controls, which again may be due to elevations only occurring in a subset of patients. Similar to brain studies, studies of immune biomarkers in patient blood have produced conflicting results, though many strongly support peripheral inflammation in a subset of patients.
Acute vs. chronic inflammation in schizophrenia
As consensus grows that inflammation plays a role in the pathophysiology of schizophrenia, important questions about the nature of this inflammation have arisen: Is inflammation consistent throughout a person with schizophrenia’s life or does it wax and wane? Further, is inflammation only apparent after the development of schizophrenia or can it be detected prior to the onset of symptoms? In terms of neuroinflammation specifically, it is difficult to assess fluctuations over the course of the illness. This is largely due to the inaccessibility of brain tissue and secretions to measure immune markers. As a result, neuroinflammatory status in living patients can only really be inferred from brain imaging studies or cerebrospinal fluid. Brain imaging studies often rely on radioligand-binding to the translocator protein (TSPO) to detect microgliosis and therefore neuroinflammation, and several studies have found increased TSPO binding in high-risk individuals, first episode psychosis and schizophrenia [104–106]. However, others have failed to find this effect in schizophrenia patients [107, 108]. TSPO is not specific for microglia [109–111], and there is inter-individual variability in TSPO binding in brain tissue for certain radioligands [112], complicating the interpretation and use of TSPO imaging. There have also been many reports of increased cytokines, immunoglobulin and antibody abnormalities and altered immune cell populations within the CSF of chronically ill people with schizophrenia as well as individuals with first episode psychosis [113–119]. These findings strongly suggest that neuroinflammation may be detected throughout the course of the illness. However, if schizophrenia was similar to other immune disorders, then we would expect inflammation to fluctuate over time. To the best of our knowledge, no study has directly compared CSF levels of immune markers between acutely and chronically ill patients over time.
Much insight into the course of (peripheral) inflammation in schizophrenia and its relationship to symptoms has been gained via assessment of immune factors in the blood of living patients. Inflammatory markers appear to fluctuate in tandem with symptom severity, peaking during times of acute psychosis and relapse. Acutely-ill patients have higher levels of both pro-inflammatory cytokines and CRP in their blood than those who are relatively stable [18, 120]. Evidence that inflammation actually precedes, rather than simply co-occurs with, the onset of psychosis comes from studies of high-risk individuals, where those who do go on to experience psychosis show elevations in blood inflammatory markers relative to low-risk individuals prior to transitioning [121, 122]. These findings also highlight the likelihood that immune disturbances in schizophrenia are not solely attributable to antipsychotic medications. In fact, several studies have found that some types of antipsychotic drugs can alleviate—but perhaps not fully normalise—inflammation in people with schizophrenia (reviewed in [123]). Despite this, even medicated patients who are chronically ill but clinically stable show evidence of inflammation in their blood [18, 19, 86, 90, 91, 97], albeit to a lesser degree than those experiencing acute psychosis. Overall, the evidence is convincing that inflammation is persistent yet also linked to symptom severity and clinical course in schizophrenia. Whether neuroinflammation in patients is chronic or transient (i.e. during acute psychosis), though, is less clear.
Biotypes: identifying patients with inflammation-associated schizophrenia
Several researchers have attempted to stratify people with schizophrenia based on their immune status in the blood using either pro-inflammatory cytokine transcripts or CRP levels. When white blood cell cytokine mRNAs are used, ~40% of patients are deemed to have high peripheral inflammation [19, 90]. When CRP alone is used, the proportion of patients exhibiting peripheral inflammation varies from ~20–45% [16–18, 97, 124], and much of this variability may be attributable to the use of different cut-off values to define ‘high CRP’ as well as varying clinical status between patient cohorts (stable vs. acutely psychotic).2 Both methods of immune stratification have proven clinically informative: those defined as having high inflammation show more severe positive and negative symptoms, greater cognitive deficits, and more significant reductions in brain volume and cortical thickness [16–19, 97]. People with schizophrenia defined as being in the ‘high cytokine’ group have worse verbal learning and lower cortical grey matter volume in Broca’s area as compared to ‘low cytokine’ patients [19]. People who have both schizophrenia and high circulating CRP have more severe symptoms (psychiatric and cognitive) and reduced PFC thickness compared to low CRP patients [16–18, 97]. Therefore, peripheral inflammation in some people with schizophrenia appears linked to brain pathology (both structural and functional), and this relationship may be overlooked if the existence of inflammatory biotypes within schizophrenia is not considered.
In sum, examination of patient brain tissue and blood has revealed that inflammation is prevalent both within and outside of the brain in some people with schizophrenia, and that symptoms and morphological changes to the PFC may be more severe in this high inflammation subset of patients. Together, such findings support the contention that heightened inflammation exists in a significant number of people with schizophrenia and that the initiating pro-inflammatory signal could be body-derived or brain-derived. However, it has not yet been conclusively determined which markers of inflammation produce the most information about clinical course or severity, or which patient subgroups would be more likely to respond to which anti-inflammatory treatment. Thus, despite these recent advances in our molecular and cellular understanding of inflammation in the pathophysiology of schizophrenia, the expression of cytokines and acute phase proteins tells us little about the cause of inflammation in patients. Here, we consider that it may be more aetiologically informative to analyse inflammatory regulators upstream of cytokine and acute phase protein synthesis in patient subgroups, thereby identifying specific aspects of immunoregulation instead of inflammatory endpoints. In the next section, we will discuss evidence suggesting that NF-κB, one of the most significant immune regulators in the human body, is itself dysregulated in schizophrenia.
Nuclear factor kappa B: a hub for immune regulation
Since many different physiological events can cause increased cytokines in brain (brain infection, neurodegenerative disease/tissue damage, ageing, neuronal stress), it is useful to determine if there are convergent or divergent changes in the molecular machinery controlling pro-inflammatory gene expression in the brain in schizophrenia. Further, there has long been consensus that schizophrenia has a strong genetic component and is a highly heterogenous disease, with only ~40% of patients showing evidence of neuroinflammation. Thus, we should consider potential pivotal points at which immune dysregulation and genetics may converge to ‘prime’ certain people for the development of inflammation-associated schizophrenia. Transcription factors are key molecules serving as genetic switches that change the phenotype and function of the cells in which they are expressed, including the control of cytokine gene expression. NF-κB is a family of five transcription factors that controls the expression of genes involved in the initiation, maintenance and termination of immune responses [125, 126] and is therefore considered a master regulator of inflammation. The overall level of NF-κB activity is regulated at the mRNA level [127–130], thus NF-κB is a transcription factor that is itself controlled by transcriptional cues within the cell.
Recently, dysregulation of the NF-κB pathway has been linked to schizophrenia [11, 12, 131], making NF-κB an attractive candidate for investigations into the cause of neuroimmune dysregulation in schizophrenia. The first study to examine NF-κB in the postmortem brains of people with schizophrenia without apparent neuroinflammation found that the entire NF-κB system was downregulated in patients in several brain regions, most notably in the temporal cortex [132]. However, more recently, overactivity of NF-κB in the dorsolateral PFC specifically has been linked to schizophrenia [11, 131] and we found that prefrontal cortical NF-κB dysregulation in a subset of patients appears to drive neuroinflammation in this region in these individuals [12]. These later findings align with overexpression of immune biomarkers that are under the control of NF-κB—such as IL-6, IL-1β, IL-8 TNFα and SERPINA3—in the brain and blood of patients [31–37, 50, 52, 53, 66, 133, 134]. Determining which specific aspects of NF-κB induction and/or inhibition are disrupted in people with inflammation-associated schizophrenia at the mRNA level may help us to better understand the cause of inflammation in these people. To appreciate the many ways in which NF-κB signalling may be disrupted by altered transcription of NF-κB-related mRNAs, it is necessary to first understand the structure and regulation of NF-κB.
The structure of NF-κB dimers and their changed expression in the schizophrenia cortex
NF-κB exists as homo- or hetero-dimers3 which are mostly sequestered in the cytoplasm until activated by pro-inflammatory stimuli. NF-κB dimers may be made up of any combination of five transcription factors (subunits) including RelA, RelB, cRel, NF-κB1 and NF-κB2, which readily dimerise in the cytoplasm. For an NF-κB dimer to be transcriptionally active, it must contain at least one of the Rel subunits, since these contain transactivation domains that allow the dimer to initiate transcription on upstream DNA regions of target genes [135]. In addition, NF-κB1 and NF-κB2 are synthesised as precursors and must be processed into mature subunits (p50 and p52, respectively) before translocating to the nucleus (Fig. 1). However, even p50 and p52 are transcriptionally inactive (and may even repress transcription of target genes) if bound to each other or themselves. Dimers containing RelA and cRel, most commonly bound to processed NF-κB1 (p50), are induced by the canonical pathway of NF-κB activation, while the dimer formed by RelB and processed NF-κB2 (p52) is induced by the non-canonical pathway of NF-κB activation. Though the DNA-binding affinities of dimers are largely overlapping [136], canonical NF-κB activation is rapid and typically transient, whereas activation of the non-canonical NF-κB pathway is characteristically slower and more persistent [137]. While the non-canonical NF-κB pathway is mainly involved in B-cell development and lymphoid organogenesis as opposed to acute immune responses orchestrated by the canonical pathway in immune cells [138], distinct functions of the two pathways are not well understood in the brain. However, the most abundant NF-κB dimers in mature glia contain RelA, suggesting the prime importance of NF-κB activation through the canonical pathway in microglia and astrocytes [139]. Though it has long been believed that neurons exhibit high basal levels of NF-κB reflective of a role for NF-κB in memory formation [140–145], more recent evidence points to very low NF-κB activity in cortical neurons both basally and after stimulation, but very high inducibility of NF-κB in cortical glia in inflammatory contexts [146, 147]. Thus, it appears that while neurons also possess the NF-κB ‘machinery’ to participate in neuroinflammation, they do so to a much lesser degree than glia.
Fig. 1. NF-κB dimers.

NF-κB dimers may be made up of any two NF-κB subunits, however, unprocessed NF-κB1 and NF-κB2 retain Rel subunits in the cytoplasm.
NF-κB dimers binding to κB-binding sites on DNA promoters is the final step of NF-κB activation, and higher levels of subunit mRNA could logically lead to a higher rate of NF-κB dimer formation in the cytoplasm and hence a higher rate of pro-inflammatory gene expression. When considered as one homogenous group, people with schizophrenia have ~20–30% higher levels of RelA, cRel and NF-κB1 transcript and >80% higher levels of NF-κB2 transcript in the dorsolateral PFC compared to non-schizophrenic controls, while levels of RelB mRNA do not differ between the two groups [11]. However, we found that cRel was unchanged in people with schizophrenia, inflamed or not, despite being upregulated in high inflammation controls relative to low inflammation controls [12] (for comparison between the findings of Volk et al. and Murphy et al. refer to Fig. 2). Further, increases in RelA and NF-κB1 mRNAs may not occur in all patients but uniquely in the high inflammation patient subgroup [12]. In fact, it appears that RelA and NF-κB1 mRNAs are also upregulated in non-schizophrenic controls with increased inflammation and surprisingly, to an even greater degree than in high inflammation patients. By contrast, NF-κB2 mRNA is increased in people with inflammation to the same degree regardless of diagnosis (control or schizophrenia). This perhaps explains the greater magnitude of increase in NF-κB2 mRNA found when all people with schizophrenia are grouped together [11] as compared to RelA and NF-κB1 mRNA increases that would be less, on average, considering that non-inflamed people with schizophrenia make about 60% of the cases [50–52]. Taken together, these findings suggest that NF-κB activation (at least through the canonical pathway that activates the RelA/NF-κB1 heterodimer) may actually be blunted in people with schizophrenia compared to people without schizophrenia who have brain inflammation. Thus, this presents the intriguing possibility that there could actually be an inadequate neuroimmune response to physiological stress or tissue damage in people with schizophrenia. This is particularly interesting given the findings of Roussos et al. [132] that people with schizophrenia who do not appear to be in a state of neuroinflammation (the authors reported no change in several cytokine transcripts) show decreased expression of NF-κB pathway genes in the brain, particularly those molecules involved in the translocation of RelA. Lower basal NF-κB activity and/or a deficit in NF-κB activation may limit the capacity for inflammation to be resolved and explain the increases in pro-inflammatory cytokines observed by us and others. Therefore, it is possible that chronic low-grade inflammation in the dorsolateral PFC of people with schizophrenia continues unchecked due to inadequate NF-κB activation to meet a ‘threshold’ that triggers anti-inflammatory responses [148].
Fig. 2. Expression of NF-κB transcripts in the DLPFC of controls vs. schizophrenia.

Two studies have measured NF-κB pathway transcripts in the DLPFC of people with schizophrenia compared to unaffected, age-matched controls. A Volk et al. [11] report up-regulations at multiple levels of the pathway in schizophrenia while Murphy et al. [12] replicated only one of these findings (TNFR1) and found several down-regulations of the same NF-κB pathway transcripts in schizophrenia. B When high inflammation controls (individuals with elevated levels of pro-inflammatory mRNAs) were omitted from analyses in Murphy et al. [12], many (13 of 18) of the diagnostic comparisons in NF-κB pathway transcripts were consistent with those reported by Volk et al. [11]. Red boxes indicate shared findings between both studies.
Contrary to the findings of Volk et al. we found that that RelB was downregulated in patients compared to controls overall [12]. RelB has been described as an ‘outlier’ in the NF-κB subunit family due to its several apparent anti-inflammatory roles (in addition to its dimer-forming, pro-inflammatory role) both in the nucleus and the cytoplasm [125], meaning less cortical RelB expression in people with schizophrenia could represent an additional failure to negatively regulate NF-κB. Overall, it appears that cortical inflammation in people with schizophrenia could be associated with a weaker-than-normal expression of the NF-κB subunits, such that levels may be sufficient to propagate, but not to ultimately shut off, the inflammatory response in brain.
Translocation of NF-κB dimers into the nucleus depends entirely on the status of upstream signals, since they cannot move from the cytoplasm without degradation of their bound inhibitors. Breakdown of NF-κB inhibitors is triggered by upstream signalling cascades. As such, without changes in NF-κB pathway regulators to facilitate dimer translocation, a higher number of NF-κB dimers in the cytoplasm may not necessarily mean higher NF-κB-mediated gene transcription. By working backwards from NF-κB in the pathway, we can examine which (if any) of the major NF-κB-activating cell-surface receptors and/or intracellular regulators may be changed in schizophrenia to add further support cytoplasmic release of NF-κB. This may provide clues as to which aspect(s) of the pathways are ultimately responsible for the drives NF-κB-related transcriptional increases in pro-inflammatory cytokines reported in schizophrenia.
The central mechanism of NF-κB inhibition may be inadequate in the PFC in schizophrenia
In the absence of pro-inflammatory stimuli, canonical NF-κB dimers are bound to inhibitor of κB (IκB), masking their nuclear localisation sequence and retaining them in the cytoplasm [149, 150]. IκB protein can enter the nucleus and actively remove NF-κB dimers from DNA [151, 152]. Thus, IκB alpha (IκBα), IκB beta (IκBβ) and IκB epsilon (IκBε) are the major negative regulators of the canonical pathway. Further, genes encoding IκBs are targets of NF-κB itself and are upregulated by NF-κB activation and resynthesised post-stimulation to provide negative feedback [153, 154]. The transcription rate of IκBα in particular is considered a critical parameter that modulates NF-κB dynamics post-stimulation [155]. As such, elevated expression of IκBα is considered to be a proxy for increased NF-κB activity, and it has been reported that IκBα mRNA is increased in the dorsolateral PFC of people with schizophrenia relative to controls [11]. Unsurprisingly, though, IκBα is actually only upregulated in the subset of patients who also have elevated brain cytokines, similar to the situation for RelA and NF-κB1 mRNAs [12]. Further, stratifying cohorts based on neuroinflammatory status also revealed that IκBα mRNA levels are lower in high inflammation patients than in high inflammation controls [12], again pointing to lower-than-expected expression and/or induction of NF-κB regulatory transcripts. This putative deficit in NF-κB signalling could mean failure to attenuate inflammation in people with schizophrenia, thus compounding existing neuropathology.
Further evidence of ‘blunted’ NF-κB activation in schizophrenia: altered expression of the regulatory transcript IKKβ
Once canonical NF-κB-inducing receptors become activated at the cell surface, IκBs are targeted for degradation by the intracellular kinase inhibitor-of-NF-κB kinase subunit β (IKKβ). IKKβ phosphorylates IκBs leading to their degradation, and this process liberates NF-κB for nuclear translocation (Fig. 3). Increased IKKβ mRNA in the dorsolateral PFC has been reported in schizophrenia relative to controls [11], but may be more related to inflammation than to diagnosis and not unique to ‘inflamed’ patients but also occur in ‘inflamed’ controls [12]. However, similar to RelA and NF-κB1 mRNAs, levels of IKKβ transcript are significantly lower in high inflammation patients than in high inflammation controls [12]. Given that IKKβ is a chief positive regulator of canonical NF-κB, these findings provide further support for the theory that NF-κB activation may be blunted in people with schizophrenia compared to what a ‘normal’ response to increased brain cytokines would be.
Fig. 3. Regulation of canonical NF-κB activation.
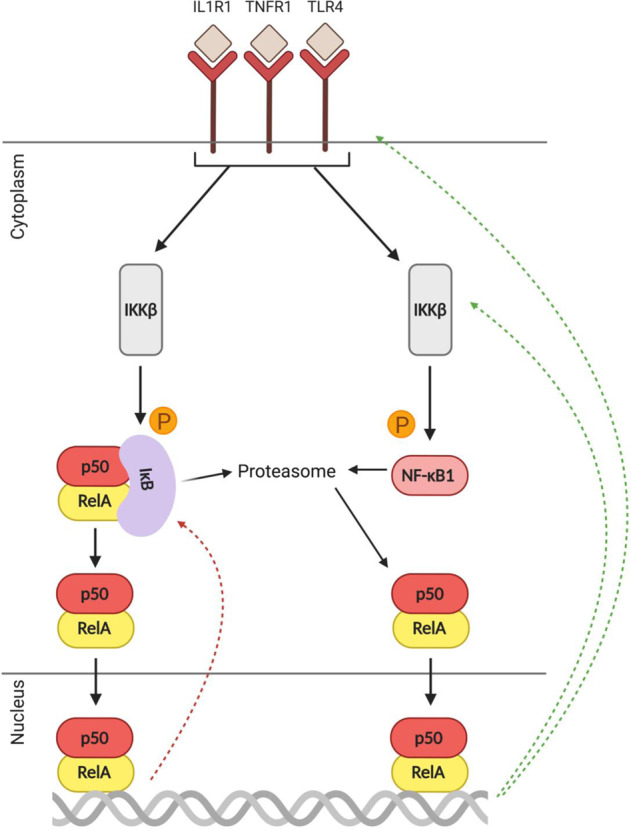
Cell-surface immunoreceptors activate IKKβ, which tags IκB for proteasomal degradation thereby freeing the p50-RelA/cRel dimer. Activation of canonical NF-κB receptors also enhances the partial processing of NF-κB1 into p50. As a result of NF-κB1 processing or IκBα degradation, the p50-RelA/cRel dimer moves into the nucleus where it initiates pro-inflammatory gene transcription. As such, IKKβ and IκB are considered the central regulators of the canonical pathway. Red dotted arrows indicate negative feedback via IκB transcription, green dotted arrows indicate positive feedback via receptor and IKKβ transcription.
Expected levels of the non-canonical NF-κB regulatory transcript NIK in the PFC in schizophrenia
Similar to canonical dimers sequestered by IκB, the NF-κB2-RelB dimer of the non-canonical NF-κB pathway is also retained in the cytoplasm in the absence of pro-inflammatory stimuli (Fig. 4). Following activation of cell-surface non-canonical NF-κB-inducing receptors, NF-κB2 is partially processed by the proteasome into its mature subunit. This process is mediated by the actions of NF-κB-inducing kinase (NIK) and IKKα, which generate the active non-canonical p52-RelB dimer that then translocates to the nucleus and initiates target gene transcription. As such, NIK is the central regulatory kinase of the non-canonical pathway. NIK activity is mainly controlled post-translationally [156] but also appears to be regulated at the transcriptional level [157] and is upregulated in response to non-canonical NF-κB activation [158, 159]. Interestingly, unlike the regulatory kinase of the canonical pathway (IKKβ), NIK mRNA is elevated in high inflammation schizophrenia to the same degree as in those with inflammation who do not have schizophrenia [12]. That is to say, NIK up-regulation in the dorsolateral PFC appears to reflect a heightened neuroinflammatory state in general and is not specific to schizophrenia, contrary to previous interpretations of disease-specific increases when controls are viewed as one group (~90% of whom are likely non-inflamed [11, 50, 52].
Fig. 4. Regulators of non-canonical NF-κB activation.
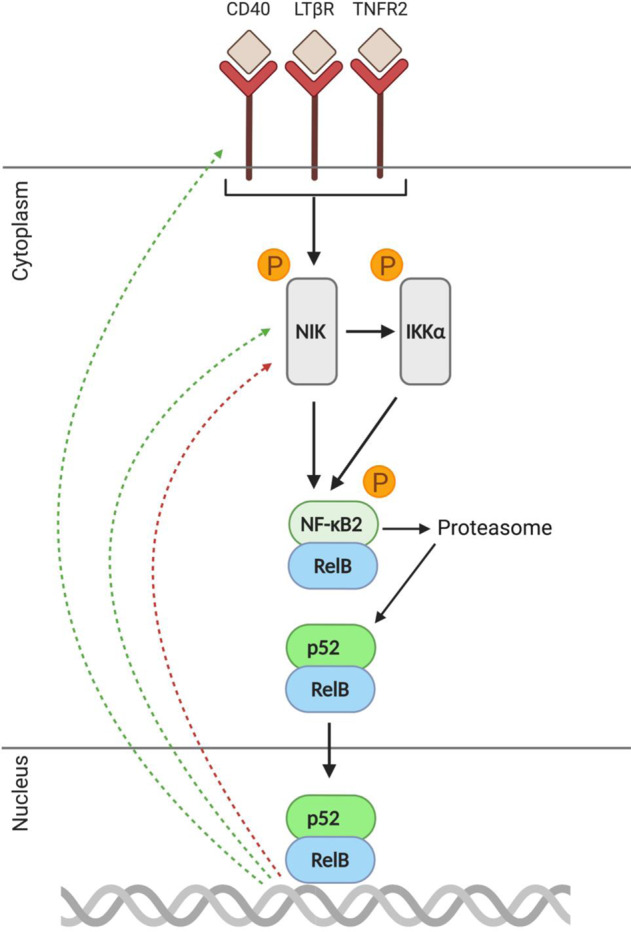
Cell-surface immunoreceptors activate NIK, which recruits IKKα to phosphorylate NF-κB2. NF-κB2 is then partially degraded by the proteasome into p52. As a result of NF-κB2 processing into p52, the p52-ReB dimer moves into the nucleus where it initiates pro-inflammatory gene transcription. As such, NIK is considered the central regulator of the non-canonical pathway. Red dotted arrow indicates negative feedback via NIK transcription, green dotted arrows indicate positive feedback via receptor and NIK transcription.
Focus on cell-surface receptors in schizophrenia: are changes in NF-κB-activating receptor mRNAs disease-specific?
Prior to kinases IKKβ and NIK liberating NF-κB dimers from their inhibitors, the first step of NF-κB induction is activation of cell-surface immunoreceptors. Several NF-κB -inducing receptor mRNAs are upregulated in the dorsolateral PFC in schizophrenia when examined by diagnosis [11], but stratification by inflammatory biotype indicates the majority of these changes are inflammation-specific and not disease-specific [12]. Therefore, changes in these cell-surface receptors tell us little about the specific pathways upstream of cytokine induction in schizophrenia, but instead suggest that it may be part of a more generalised brain reaction. The exception is the microglial-associated transcript TLR4, which is unchanged in high inflammation schizophrenia despite robust (approximately twofold) increases in high inflammation controls [12]. TLR4 mRNA is enriched in and highly functionally-relevant to microglia [160–163], as opposed to other canonical NF-κ-activating receptors such as IL1R1, the expression of which is unique to endothelial cells, astrocytes and some neurons but is not expressed by microglia at all under physiological conditions in mice [164]. Non-microglial cells have a limited TLR repertoire and TLR4 in particular appears largely unimportant for cells such as astrocytes, at least relative to its critical importance for the activation and effector functions of microglia [165]. The NF-κB subunits NF-κB1 and cRel also appear to be enriched in microglia [166] and as mentioned above, both of these transcripts were, like TLR4, lower in the cortex of high inflammation patients than high inflammation controls [12], further supporting a ‘blunting’ of canonical NF-κB activation in microglia in ‘inflamed’ patients. In light of conflicting reports regarding increased [11] and comparatively decreased [12] cortical TLR4 transcript levels in some patients, we posit that: (1) changes in TLR4 expression and microglial activation states in schizophrenia may be dynamic, and (2) neglecting to consider the neuroinflammatory status of individuals at their time of death when analysing immune-related transcripts in the brain may obscure the differences occurring in only a subset of patients.
Given that TLR4 is a potent activator of NF-κB and pro-inflammatory microglia [167], its putative suppression in the cortex in schizophrenia suggests that the increase in cytokines may not be microglia-derived and bolsters the theory of possible microglial suppression in a subset of patients. A putative lack of TLR4 expression by microglia also aligns with the above-mentioned blunting of transcriptional NF-κB1 in high inflammation patients since the abundance of NF-κB1 may coordinate the pro- to anti-inflammatory shift in microglia [168]. Together, these findings point to a lack of what may be physiologically appropriate pro-inflammatory microglia in patients with high levels of pro-inflammatory cytokines in the cortex.4
In contrast to TLR4, mRNA levels for one cytokine receptor that strongly induces the pro-inflammatory phenotype in astrocytes, IL1R1, are robustly increased in both high inflammation schizophrenia and high inflammation controls (see Fig. 5 for a summary of NF-κB pathway changes in high inflammation patients relative to high inflammation controls). Reactive astrocytes have been implicated in the inflamed patient subgroup previously [61], and are the major source of the inflammatory marker SERPINA3 that reliably identifies neuroinflammation across cohorts [50, 52, 66, 134, 170]. Together, these findings may indicate that normal microglial immune function via NF-κB is impaired in schizophrenia, and chronic NF-κB activation in non-microglial cells including astrocytes contribute to sustained, unimpeded cortical inflammation in some people with schizophrenia. It has even recently been proposed that reactive astrocytes release factors that keep microglia in a non-inflammatory state [171]; meaning inflammatory astrocytes and non-inflammatory microglia may mutually contribute to neuroinflammation in schizophrenia via their effects on each other. This is supported by studies finding evidence of reactive astrogliosis [53, 61, 131, 170, 172–175] and what appears to be paradoxical microglial suppression [63, 131, 176–179] in the PFC in at least some patients. However, it is important to note that some studies have failed to find evidence of astrogliosis [72–74, 180] while others have reported increased microgliosis in the PFC of people with schizophrenia [180, 181]. These conflicting results again highlight the neuropathological heterogeneity within the disease and the need for immune stratification to examine biologically distinct patient subgroups, but may also result from the use of different markers to identify glial pathology between studies. Future studies must consider the relative contributions of microglia, astrocytes and potentially even peripherally-derived immune cells to cortical inflammation in schizophrenia and to consider that these states are likely dynamic and not static.
Fig. 5. Expression of NF-κB transcripts in the postmortem DLPFC of schizophrenia patients with neuroinflammation relative to non-schizophrenic controls with neuroinflammation.

A Not all patients show evidence of neuroinflammation (red = proportion of ‘inflamed’ patients, grey = non-inflamed patients), and some individuals without schizophrenia do show evidence of neuroinflammation (blue = ‘inflamed’ controls, grey = non-inflamed controls). Comparing cortical mRNA levels of NF-κB pathway members between high inflammation patients and high inflammation controls allows for identification of schizophrenia-specific abnormalities in this critical immunomodulatory pathway. B Such comparisons have shown that high neuroinflammation patients may actually have ‘blunted’ NF-κB activation, which may represent cell-specific deficits and/or a failure to adequately induce NF-κB to the level required to initiate NF-κB-dependent anti-inflammatory processes in the brain.
Conclusions, future directions and implications for treatment
The findings discussed above suggest that impairment in NF-κB regulation in the brain may be pathogenic in some people with schizophrenia. While most research has focused on endpoint markers of inflammation such as cytokines and acute phase proteins, identifying upstream mechanisms that regulate cytokine synthesis may provide greater insight and efficacy for novel therapeutics. The papers discussed in this review support the contention that a fault in microglial responses to immune stress in the dorsolateral PFC may contribute to prolonged pro-inflammatory responses from other cells such as astrocytes in patients [182]. General up-regulation of several NF-κB pathway transcripts in the PFC in patients with neuroinflammation may therefore reflect NF-κB activation in astrocytes and other non-microglial cells as opposed to microglia.
One important consideration here is that that weak NF-κB induction is not likely to represent a failure of the immune response in all contexts. In fact, the degree of NF-κB activation would logically be proportional to the magnitude of the inflammatory insult, thus less activation could just mean less stimulation. However, the putative NF-κB ‘blunting’ in high inflammation schizophrenia relative to high inflammation controls is curious and somewhat contradictory since high inflammation controls and high inflammation patients have the same degree of elevated inflammatory signalling in the cortex [50, 52], yet do not have the same levels of NF-κB transcripts that induce, and are induced by, these pro-inflammatory signals. This is why it is plausible that NF-κB is ‘normally’ activated in some cells such as astrocytes yet inappropriately underactive in other cells such as microglia in people with schizophrenia, while NF-κB is ‘normally’ activated in both cell types in high inflammation controls.
If overactivity of astrocytic NF-κB and underactivity of microglial NF-κB contribute to schizophrenia pathology, this presents a challenging hurdle for the development or repurposing of therapeutics to target NF-kB dysregulation in the brain. Blocking NF-κB in all cell types may worsen the problem by further suppressing microglia, but could also shut-off inflammatory signalling from astrocytes, thereby alleviating neuroinflammation and associated symptoms. This might be achieved by boosting levels of the chief NF-κB inhibitor, IκB, by blocking the action of IKKβ in the PFC. Indeed, several such compounds have been identified and show pre-clinical efficacy in the treatment of other inflammatory conditions including arthritis, chronic obstructive pulmonary disease and acute organ injury [183]. However, further dampening NF-κB activity in microglia may also have deleterious non-immune effects given its purported roles in brain homoeostasis and neuronal support [139, 184], which has also been reported in oligodendrocytes [185]. Thus, selective inhibition of NF-κB in astrocytes might be the most viable option for treating neuroinflammation in the brain in people with schizophrenia, and has proven neuroprotective in many mouse models of neuroinflammation. Inhibition of NF-κB in murine astrocytes post-CNS injury lessens pro-inflammatory cytokine production and neurodegeneration [186, 187], improves functional recovery [186], and substantially limits the extent of leucocyte infiltration to the damaged region [188]. Similarly in experimental autoimmune encephalitis, NF-κB signalling specifically in astrocytes contributes to a large degree of tissue damage and the influx of inflammatory immune cells into the CNS [189], a pertinent consideration given recent reports of increased numbers of leucocytes in the brains of some patients [63, 64, 190]. NF-κB activation in astrocytes has even been linked to memory impairment in a mouse model of dementia [191], underscoring the potential for astrocytic NF-κB to interfere with normal cognition. Peripherally, constitutive activity of NF-κB in myeloid cells has also been shown to drive pathogenicity of monocytes and macrophages during autoimmune-type neuroinflammation in mice [192]. Activation of NF-κB in macrophages specifically is thought to cause disruption of the blood–brain barrier, subsequent immune cell infiltration to the brain and resultant cognitive impairment and sickness behaviour which can be attenuated with peripheral NF-κB inhibition [193–195]. Given the several reports of increased pro-inflammatory, macrophage-derived cytokines in patient blood [19, 82–84, 90], increased TLR4 mRNA in patient white blood cells [196, 197] and exaggerated inflammatory responses to LPS (which activates NF-κB in macrophages via TLR4) in patient white blood cells [198], NF-κB in circulating monocytes may be an additional therapeutic target in schizophrenia. Several over-the-counter and prescription anti-inflammatory medications such as aspirin, minocycline and celecoxib are known to inhibit NF-κB in white blood cells (including macrophages) in vitro [199–201] and some early studies investigating the utility of these drugs in schizophrenia have produced promising results across various symptom domains [13, 202–204]. However, assessing the therapeutic benefit of anti-inflammatory drugs in schizophrenia based on patients’ immune status at commencement of treatment is yet to become a mainstream practice, which may lead to underestimation of treatment efficacy in ‘inflamed’ individuals, as mentioned above. Overall, future treatments targeting NF-κB in the PFC of people with schizophrenia must consider its cell-specific roles, and the possibility that targeting astrocytic NF-κB in the brain may be least likely to cause off-target, undesired effects on cells where NF-κB serves homoeostatic or even cell protective functions.
At present, the relative contributions of microglia, astrocytes, and even peripherally-derived immune cells to neuroinflammation in people with schizophrenia remains speculative since pro-inflammatory cytokine transcripts in the dorsolateral PFC have not been localised to specific cells (though general assessment of microgliosis and astrogliosis in postmortem patient brain tissue has been reviewed comprehensively elsewhere [205–208]). Further, the more detailed phenotypes of microglia in schizophrenia have not been determined and are likely dynamic, as exemplified by conflicting findings regarding TLR4 expression in the PFC in patients. Answering these remaining questions is crucial to the development of anti-inflammatory drugs to treat neuroinflammation in a subset of patients with schizophrenia, since it is clear that the normal healthy brain relies on NF-κB activation in the right cells, at the right time and to the right extent. Further thought and research will be required to develop an optimal therapeutic strategy to bring NF-κB activation signalling back into cellular and temporal homoeostasis in human brain.
Author contributions
CEM, AKW and CSW were involved in conceptualisation and writing of the paper.
Funding
CSW is funded by the N.S.W. Ministry of Health, Office of Health and Medical Research. CSW is a recipient of a National Health and Medical Research Council (Australia) Principal Research Fellowship (PRF) (#1117079). CSW is in collaboration with Astellas Pharma Inc., Japan. AKW is a recipient of a National Breast Cancer Foundation (Australia) fellowship (PF-15-014), and Sydney Partnership for Health, Education, Research and Enterprise (SPHERE) grants, and is funded by Schizophrenia Research Laboratory and Neuroscience Research Australia (NeuRA).
Competing interests
The authors declare no competing interests.
Footnotes
The terms ‘high neuroinflammation’ and ‘high inflammation’ used hereafter are relative to low/normal expression of immune markers and reflects subclinical inflammation in the absence of infection. These terms do not necessarily imply levels of inflammation comparable to acute infection or injury.
Evidence from first episode psychosis and chronically ill patients supports that peripheral inflammation fluctuates throughout the course of the illness and is highest during acute psychosis [18, 121].
Though NF-κB encompasses five separate transcription factors, the term ‘NF-κB’ is also commonly used to refer to the dimer complex comprised of two transcription factors. Thus, these transcription factors are also often referred to as NF-κB subunits. In this review, the term NF-κB will be used to describe the collective family of transcription factors, and separate transcription factors will be referred to as NF-κB subunits. The term ‘NF-κB activation’ will be used to describe the induction of pathways resulting in NF-κB dimer-DNA binding in the nucleus.
It is also important to note that some evidence supports an additional, non-immune role for microglial NF-κB in neuronal homoeostasis and hippocampal-dependent learning [169].
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Messias E, Chen C, Eaton WW. Epidemiology of schizophrenia: review of findings and myths. Psychiatr Clin North Am. 2007;30:323–38. doi: 10.1016/j.psc.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Psychiatric Association. Schizophrenia spectrum and other psychotic disorders. In: Diagnostic and statistical manual of mental disorders. 5th ed. (Washington DC 2013). 10.1176/appi.books.9780890425596.dsm02.
- 3.World Health Organization. Schizophrenia or other primary psychotic disorders. in International statistical classification of diseases and related health problems. 11th ed. 2019. https://icd.who.int/browse10/2016/en#F20.
- 4.Sheffield JM, Karcher NR, Barch DM. Cognitive deficits in psychotic disorders: a lifespan perspective. Neuropsychol Rev. 2018;28:509–33. doi: 10.1007/s11065-018-9388-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickson H, Hedges EP, Ma SY, Cullen AE, MacCabe JH, Kempton MJ, et al. Academic achievement and schizophrenia: a systematic meta-analysis. Psychol Med. 2020;50:1949–65. doi: 10.1017/S0033291720002354. [DOI] [PubMed] [Google Scholar]
- 6.Holm M, Taipale H, Tanskanen A, Tiihonen J, Mitterdorfer-Rutz E. Employment among people with schizophrenia or bipolar disorder: a population-based study using nationwide registers. Acta Psychiatr Scand. 2021;143:61–71. doi: 10.1111/acps.13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin H, Mosweu I. The societal cost of schizophrenia: a systematic review. Pharmacoeconomics. 2017;35:25–42. doi: 10.1007/s40273-016-0444-6. [DOI] [PubMed] [Google Scholar]
- 8.Millier A, Schmidt U, Angermeyer MC, Chauhan D, Murthy V, Toumi M, et al. Humanistic burden in schizophrenia: a literature review. J Psychiatr Res. 2014;54:85–93. doi: 10.1016/j.jpsychires.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 9.Cho M, Lee TY, Kwak YB, Yoon YB, Kim M, Kwon JS. Adjunctive use of anti-inflammatory drugs for schizophrenia: A meta-analytic investigation of randomized controlled trials. Aust NZ J Psychiatry. 2019;53:742–59. doi: 10.1177/0004867419835028. [DOI] [PubMed] [Google Scholar]
- 10.Volk DW, Chitrapu A, Edelson JR, Roman KM, Moroco AE, Lewis DA. Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry. 2015;172:1112–21. doi: 10.1176/appi.ajp.2015.15010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volk DW, Moroco AE, Roman KM, Edelson JR, Lewis DA. The role of the Nuclear Factor-κB transcriptional complex in cortical immune activation in schizophrenia. Biol Psychiatry. 2019;85:25–34. doi: 10.1016/j.biopsych.2018.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy CE et al. Nuclear factor kappa B activation appears weaker in schizophrenia patients with high brain cytokines than in non-schizophreni ccontrols with high brain cytokines. J Neuroinflammation. 2020;215. [DOI] [PMC free article] [PubMed]
- 13.Laan W, Grobbee DE, Selten JP, Heijnen CJ, Kahn RS, Burger H. Adjuvant aspirin therapy reduces symptoms of schizophrenia spectrum disorders: results from a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2010;71:520–7. doi: 10.4088/JCP.09m05117yel. [DOI] [PubMed] [Google Scholar]
- 14.Müller N, Ulmschneider M, Scheppach C, Schwarz MJ, Ackenheil M, Möller HJ, et al. COX-2 inhibition as a treatment approach in schizophrenia: Immunological considerations and clinical effects of celecoxib add-on therapy. Eur Arch Psychiatry Clin Neurosci. 2004;254:14–22. doi: 10.1007/s00406-004-0478-1. [DOI] [PubMed] [Google Scholar]
- 15.Sommer IE, van Westrhenen R, Begemann MJ, de Witte LD, Leucht S, Kahn RS. Efficacy of anti-inflammatory agents to improve symptoms in patients with schizophrenia: an update. Schizophrenia Bull. 2014;40:181–91. doi: 10.1093/schbul/sbt139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan X, Pristach C, Liu EY, Freudenreich O, Henderson DC, Goff DC. Elevated serum levels of C-reactive protein are associated with more severe 182 psychopathology in a subgroup of patients with schizophrenia. Psychiatry Res. 2007;149:267–71. doi: 10.1016/j.psychres.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 17.Dickerson F, Stallings C, Origoni A, Boronow J, Yolken R. C-reactive protein is associated with the severity of cognitive impairment but not of psychiatric symptoms in individuals with schizophrenia. Schizophrenia Res. 2007;93:261–5. doi: 10.1016/j.schres.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 18.Jacomb I, Stanton C, Vasudevan R, Powell H, O'Donnell M, Lenroot R, et al. C-reactive protein: higher during acute psychotic episodes and related to cortical thickness in schizophrenia and healthy controls. Front Immunol. 2018;9:2230. doi: 10.3389/fimmu.2018.02230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fillman SG, Weickert TW, Lenroot RK, Catts SV, Bruggemann JM, Catts VS, et al. Elevated peripheral cytokines characterize a subgroup of people with schizophrenia displaying poor verbal fluency and reduced Broca’s area volume. Mol Psychiatry. 2016;21:1090–8. doi: 10.1038/mp.2015.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenberg SD, Lu W, Mueser KT, Jankowski MK, Cournos F. Correlates of adverse childhood events among adults with schizophrenia spectrum disorders. Psychiatr Serv. 2007;58:245–53. doi: 10.1176/ps.2007.58.2.245. [DOI] [PubMed] [Google Scholar]
- 21.Trotman HD, Holtzman CW, Walker EF, Addington JM, Bearden CE, Cadenhead KS, et al. Stress exposure and sensitivity in the clinical high risk syndrome: initial findings from the North American Prodrome Longitudinal Study (NAPLS) Schizophrenia Res. 2014;160:104–9. doi: 10.1016/j.schres.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allebeck P, Adamsson C, Engström A. Cannabis and schizophrenia: a longitudinal study of cases treated in Stockholm County. Acta Psychiatr Scand. 1993;88:21–4. doi: 10.1111/j.1600-0447.1993.tb03408.x. [DOI] [PubMed] [Google Scholar]
- 23.Hambrecht M, Häfner H. Substance abuse and the onset of schizophrenia. Biol Psychiatry. 1996;40:1155–63. doi: 10.1016/S0006-3223(95)00609-5. [DOI] [PubMed] [Google Scholar]
- 24.Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–7. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe'er I, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–7. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) C. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–76. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kähler AK, Akterin S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–9. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matzaraki V, Kumar V, Wijmenga C, Zhernakova A. The MHC locus and genetic susceptibility to autoimmune infectious diseases. Genome Biol. 2017;18:76. doi: 10.1186/s13059-017-1207-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-κB transcription factor. Mol Cell Biol. 1990;10:2327–34. doi: 10.1128/mcb.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Broser M, Rom WM. Activation of the interleukin 6 gene by myobacterium tuberculosis or lipopolysaccharide is mediated by nuclear factors NF-IL6 and NF-κB. PNAS. 1994;91:2225–9. doi: 10.1073/pnas.91.6.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiscott J, Marois J, Garoufalis J, D'Addario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993;13:6231–40. doi: 10.1128/mcb.13.10.6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsusaka T, Fujikawa K, Nishio Y, Mukaida N, Matsushima K, Kishimoto T, et al. Transcription factors NF-IL6 and NF-κB synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. PNAS. 1993;90:10193–7. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kunsch C, Rosen CA. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993;13:6137–46. doi: 10.1128/mcb.13.10.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collart MA, Baeuerle P, Vassali P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four κB-Like motifs and of constitutive and inducible forms of NF-κB. Mol Cell Biol. 1990;10:1498–506. doi: 10.1128/mcb.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drouet C, Shakhov AN, Jongeneel CV. Enhancers and transcription factors controlling the inducibility of the tumor necrosis factor-alpha promoter in primary macrophages. J Immunol. 1991;147:1694–700. [PubMed] [Google Scholar]
- 38.Sasayama D, Hori H, Teraishi T, Hattori K, Ota M, Iijima Y, et al. Possible association between Interleukin-1beta gene and schizophrenia in a Japanese population. Behav Brain Funct. 2011;7:35. doi: 10.1186/1744-9081-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papiol S, Rosa A, Gutiérrez B, Martín B, Salgado P, Catalán R, et al. Interleukin-1 cluster is associated with genetic risk for schizophrenia and bipolar disorder. J Med Genet. 2004;41:219–23. doi: 10.1136/jmg.2003.012914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katila H, Hänninen K, Hurme M. Polymorphisms of the interleukin-1 gene complex in schizophrenia. Mol Psychiatry. 1999;4:179–81. doi: 10.1038/sj.mp.4000483. [DOI] [PubMed] [Google Scholar]
- 41.Xu M, He L. Convergent evidence shows a positive association of interleukin-1 gene complex locus with susceptibility to schizophrenia in the Caucasian population. Schizophrenia Res. 2010;120:131–42. doi: 10.1016/j.schres.2010.02.1031. [DOI] [PubMed] [Google Scholar]
- 42.Kapelski P, Skibinska M, Maciukiewicz M, Wilkosc M, Frydecka D, Groszewska A, et al. Association study of functional polymorphisms in interleukins and interleukin receptors genes: IL1A, IL1B, IL1RN, IL6, IL6R, IL10, IL10RA and TGFB1 in schizophrenia in Polish population. Schizophrenia Res. 2015;169:1–9. doi: 10.1016/j.schres.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 43.Borkowska P, Kucia K, Rzezniczek S, Paul-Samojedny M, Suchanek R, Owczarek A, et al. Interleukin-1B promoter (-31T/C and -511C/T) polymorphisms in paranoid schizophrenia. Psychiatr Genet. 2012;22:311. doi: 10.1097/YPG.0b013e3283586274. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida M, Shiroiwa K, Mouri K, Ishiguro H, Supriyanto I, Ratta-Apha W, et al. Haplotypes in the expression quantitative trait locus of interleukin-1β gene are associated with schizophrenia. Schizophrenia Res. 2012;140:185–91. doi: 10.1016/j.schres.2012.06.031. [DOI] [PubMed] [Google Scholar]
- 45.Laurent C, Thibaut F, Ravassard P, Campion D, Samolyk D, Lafargue C, et al. Detection of two new polymorphic sites in the human interleukin-1 beta gene: lack of association with schizophrenia in a French population. Psychiatr Genet. 1997;7:103–5. doi: 10.1097/00041444-199723000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Tatsumi M, Sasaki T, Sakai T, Kamijima K, Fukuda R, Kunugi H, et al. Genes for interleukin-2 receptor beta chain, interleukin-1 beta, and schizophrenia: no evidence for the association or linkage. Am J Med Genet. 1997;74:338–41. doi: 10.1002/(SICI)1096-8628(19970531)74:3<338::AID-AJMG17>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 47.Shibuya M, Watanabe Y, Nunokawa A, Egawa J, Kaneko N, Igeta H, et al. Interleukin 1 beta gene and risk of schizophrenia: detailed case-control family-based studies and an updated meta-analysis. Hum Psychopharmacol. 2014;29:31–7. doi: 10.1002/hup.2365. [DOI] [PubMed] [Google Scholar]
- 48.Yang J, Si T, Ling Y, Ruan Y, Han Y, Wang X, et al. Association study between interleukin-1beta gene (IL-1beta) and schizophrenia. Life Sci. 2003;72:3017–21. doi: 10.1016/S0024-3205(03)00248-0. [DOI] [PubMed] [Google Scholar]
- 49.Saiz PA, Garcia-Portilla MP, Arango C, Morales B, Martinez-Barrondo S, Alvarez V, et al. Interleukin-1 gene complex in schizophrenia: an association study. Neuropsychiatr Genet. 2006;141B:678–80. doi: 10.1002/ajmg.b.30394. [DOI] [PubMed] [Google Scholar]
- 50.Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, et al. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry. 2013;18:206–14. doi: 10.1038/mp.2012.110. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Catts VS, Sheedy D, McCrossin T, Kril JJ, Shannon Weickert C. Cortical grey matter volume reduction in people with schizophrenia is associated with neuro-inflammation. Transl Psychiatry. 2016;6:e982. [DOI] [PMC free article] [PubMed]
- 52.Fillman SG, Sinclair D, Fung SJ, Webster MJ, Shannon Weickert C. Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Transl Psychiatry. 2014;4:e365. doi: 10.1038/tp.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purves-Tyson TD, Weber-Stadlbauer U, Richetto J, Rothmond DA, Labouesse MA, Polesel M, et al. Increased levels of midbrain immune-related transcripts in schizophrenia and in murine offspring after maternal immune activation. Mol Psychiatry. 2019. 10.1038/s41380-019-0434-0. [DOI] [PMC free article] [PubMed]
- 54.Weinberger DR, Berman KF, Illowsky BP. Physiological dysfunction of dorsolateral prefrontal cortex in schizophrenia. Arch Gen Psychiatry. 1988;45:609–15. doi: 10.1001/archpsyc.1988.01800310013001. [DOI] [PubMed] [Google Scholar]
- 55.Zhou Y, Liang M, Jiang T, Tian L, Liu Y, Liu Z, et al. Functional dysconnectivity of the dorsolateral prefrontal cortex in first-episode schizophrenia using resting-state fMRI. Neurosci Lett. 2007;417:297–302. doi: 10.1016/j.neulet.2007.02.081. [DOI] [PubMed] [Google Scholar]
- 56.Potkin SG, Turner JA, Brown GG, McCarthy G, Greve DN, Glover GH, et al. Working memory and DLPFC inefficiency in schizophrenia: the FBIRN study. Schizophrenia Bull. 2009;35:19–31. doi: 10.1093/schbul/sbn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Callicott JH, Ramsey NF, Tallent K, Bertolino A, Knable MB, Coppola R, et al. Functional Magnetic Resonance Imaging Brain Mapping in Psychiatry: Methodological Issues Illustrated in a Study of Working Memory in Schizophrenia. Neuropsychopharmacology. 1998;18:186–96. doi: 10.1016/S0893-133X(97)00096-1. [DOI] [PubMed] [Google Scholar]
- 58.Hill K, Mann L, Laws KR, Stephenson CM, Nimmo-Smith I, McKenna PJ. Hypofrontality in schizophrenia: a meta-analysis of functional imaging studies. Acta Psychiatr Scand. 2004;110:243–56. doi: 10.1111/j.1600-0447.2004.00376.x. [DOI] [PubMed] [Google Scholar]
- 59.Van Snellenberg JX, Girgis RR, Horga G, van de Giessen E, Slifstein M, Ojeil N, et al. Mechanisms of working memory impairment in schizophrenia. Biol Psychiatry. 2016;80:617–26. doi: 10.1016/j.biopsych.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kikinis Z, Fallon JH, Niznikiewicz M, Nestor P, Davidson C, Bobrow L, et al. Gray matter volume reduction in rostral middle frontal gyrus in patients with chronic schizophrenia. Schizophrenia Res. 2010;123:153–9. doi: 10.1016/j.schres.2010.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Catts VS, Wong J, Fillman SG, Fung SJ, Shannon Weickert C. Increased expression of astrocyte markers in schizophrenia: association with neuroinflammation. Aust NZ J Psychiatry. 2014;48:722–34. doi: 10.1177/0004867414531078. [DOI] [PubMed] [Google Scholar]
- 62.Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, et al. Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci. 2003;23:6315–26. doi: 10.1523/JNEUROSCI.23-15-06315.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cai HQ, Catts VS, Webster MJ, Galletly C, Liu D, O'Donnell M, et al. Increased macrophages and changed brain endothelial cell gene expression in the frontal cortex of people with schizophrenia displaying inflammation. Mol Psychiatry. 2018;25:761–75. doi: 10.1038/s41380-018-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Purves-Tyson T, et al. Increased macrophages and C1qA, C3, C4 transcripts in the midbrain of people with schizophrenia. Frontiers in Immunology, 2020;11:2002. [DOI] [PMC free article] [PubMed]
- 65.Pandey GN, Rizavi HS, Zhang H, Ren X. Abnormal gene expression and protein expression of inflammatory cytokines in the postmortem brain of schizophrenia patients. Schizophrenia Res. 2018;192:247–54. doi: 10.1016/j.schres.2017.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007;62:711–21. doi: 10.1016/j.biopsych.2006.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lanz TA, Reinhart V, Sheehan MJ, Rizzo S, Bove SE, James LC, et al. Postmortem transcriptional profiling reveals widespread increase in inflammation in schizophrenia: a comparison of prefrontal cortex, striatum, and hippocampus among matched tetrads of controls with subjects diagnosed with schizophrenia, bipolar or major depressive disorder. Transl Psychiatry. 2019;9:151. doi: 10.1038/s41398-019-0492-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Collado-Torres L, Burke EE, Peterson A, Shin J, Straub RE, Rajpurohit A, et al. Regional heterogeneity in gene expression, regulation, and coherence in the frontal cortex and hippocampus across development and schizophrenia. Neuron. 2019;103:203–16.e8. doi: 10.1016/j.neuron.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Toyooka K, Watanabe Y, Iritani S, Shimizu E, Iyo M, Nakamura R, et al. A decrease in interleukin-1 receptor antagonist expression in the prefrontal cortex of schizophrenic patients. Neurosci Res. 2003;46:299–307. doi: 10.1016/S0168-0102(03)00093-2. [DOI] [PubMed] [Google Scholar]
- 70.Dean B, Gibbons AS, Tawadros N, Brooks L, Everall IP, Scarr E. Different changes in cortical tumor necrosis factor-α-rated pathways in schizophrenia and mood disorders. Mol Psychiatry. 2013;18:767–73. doi: 10.1038/mp.2012.95. [DOI] [PubMed] [Google Scholar]
- 71.Arnold SE, et al. Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. JAMA Psychiatry. 1998;55:225–323. doi: 10.1001/archpsyc.55.3.225. [DOI] [PubMed] [Google Scholar]
- 72.Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, et al. Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet. 2003;362:798–805. doi: 10.1016/S0140-6736(03)14289-4. [DOI] [PubMed] [Google Scholar]
- 73.Dean B, Gray L, Scarr E. Regionally specific changes in levels of cortical S100beta in bipolar 1 disorder but not schizophrenia. Aust NZ J Psychiatry. 2006;40:217–24. doi: 10.1080/j.1440-1614.2006.01777.x. [DOI] [PubMed] [Google Scholar]
- 74.Steffek AE, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH. Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia. Schizophrenia Res. 2008;103:71–82. doi: 10.1016/j.schres.2008.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Birnbaum R, Jaffe AE, Chen Q, Shin JH, BrainSeq C, Kleinman JE, et al. Investigating the neuroimmunogenic architecture of schizophrenia. Mol Psychiatry. 2018;23:1251–60. doi: 10.1038/mp.2017.89. [DOI] [PubMed] [Google Scholar]
- 76.Wrona D. Neural-immune interactions: an integrative view of the bidirectional relationship between the brain and immune systems. Journal of Neuroimmunology 2006;172:38–58. [DOI] [PubMed]
- 77.Kuznetsova NI, Semenov SF. Detection of anti-brain antibodies in the blood serum of patients with neuropsychiatric diseases. Zh Nevrologii i Psikhiatrii Im S S Korsakova. 1961;61:869–74. [PubMed] [Google Scholar]
- 78.De Lisi LE, King AC, Targum S. Serum immunoglobulin concentrations in patients admitted to an acute psychiatric in-patient service. Br J Psychiatry. 1984;145:661–5. doi: 10.1192/bjp.145.6.661. [DOI] [PubMed] [Google Scholar]
- 79.Prebel OT, Torrey EF. Serum interferon in patients with psychosis. Am J Psychiatry. 1985;142:1184–6. doi: 10.1176/ajp.142.10.1184. [DOI] [PubMed] [Google Scholar]
- 80.Ganguli R, Rabin BS, Kelly RH, Lyte M, Ragu U. Clinical and laboratory evidence of autoimmunity in acute schizophrenia. Ann NY Acad Sci. 1987;496:676–85. doi: 10.1111/j.1749-6632.1987.tb35829.x. [DOI] [PubMed] [Google Scholar]
- 81.Rapaport MH, Nelson DL, Paul SM. Elevated levels of soluble interleukin 2 receptors in schizophrenia. Arch Gen Psychiatry. 1989;46:291–2. doi: 10.1001/archpsyc.1989.01810030097017. [DOI] [PubMed] [Google Scholar]
- 82.Di Nicola M, Cattaneo A, Hepgul N, Di Forti M, Aitchison KJ, Janiri L, et al. Serum and gene expression profile of cytokines in first-episode psychosis. Brain Behav Immun. 2013;31:90–5. doi: 10.1016/j.bbi.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pandey GN, Ren X, Rizavi HS, Zhang H. Proinflammatory cytokines and their membrane-bound receptors are altered in the lymphocytes of schizophrenia patients. Schizophrenia Res. 2015;164:193–8. doi: 10.1016/j.schres.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chase KA, Cone JJ, Rosen C, Sharma RP. The value of interleukin 6 as a peripheral diagnostic marker in schizophrenia. BMC Psychiatry. 2016;16:152. doi: 10.1186/s12888-016-0866-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Song X, Lv L, Li W, Zhao J. The interaction of nuclear factor-kappa B and cytokines is associated with schizophrenia. Biol Psychiatry. 2009;65:481–8. doi: 10.1016/j.biopsych.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 86.Al-Hakeim HK, Al-Rammahi DA, Al-Dujaili AH. IL-6, IL-18, sIL-2R and TNFαproinflammatory markers in depression and schizophrenia patients who are free of overt inflammation. J Affect Disord. 2015;182:106–14. doi: 10.1016/j.jad.2015.04.044. [DOI] [PubMed] [Google Scholar]
- 87.Frydecka D, Misiak B, Pawlak-Adamska E, Karabon L, Tomkiewicz A, Sedlaczek P, et al. Interleukin-6: the missing element of the neurocognitive deterioration in schizophrenia? The focus on genetic underpinnings, cognitive impairment and clinical manifestation. Eur Arch Psychiatry Clin Neurosci. 2015;265:449–59. doi: 10.1007/s00406-014-0533-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lesh TA, Careaga M, Rose DR, McAllister AK, Van de Water J, Carter CS, et al. Cytokine alterations in first-episode schizophrenia and bipolar disorder: relationships to brain structure and symptoms. J Neuroinflammation. 2018;15:165. doi: 10.1186/s12974-018-1197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kalmady SV, Shivakumar V, Jose D, Ravi V, Keshavan MS, Gangadhar BN, et al. Plasma cytokines in minimally treated schizophrenia. Schizophrenia Res. 2018;199:292–6. doi: 10.1016/j.schres.2018.04.022. [DOI] [PubMed] [Google Scholar]
- 90.Boerrigter D, Weickert TW, Lenroot R, O'Donnell M, Galletly C, Liu D, et al. Using blood cytokine measures to define high inflammatory biotype of schizophrenia and schizoaffective disorder. J Neuroinflammation. 2017;14:188. doi: 10.1186/s12974-017-0962-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Akanji AO, Ohaeri JU, Al-Shammri S, Fatania HR. Association of blood levels of C-reactive protein with clinical phenotypes in Arab schizophrenic patients. Psychiatry Res. 2009;169:56–61. doi: 10.1016/j.psychres.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 92.Fawzi MH, Fawzi MM, Fawzi MM, Said NS. C-reactive protein serum level in drug-free male Egyptian patients with schizophrenia. Psychiatry Res. 2011;190:91–7. doi: 10.1016/j.psychres.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 93.Lin C, Chang C, Liu C, Huang T. Increased high-sensitivity C-reactive protein levels in Taiwanese schizophrenic patients. Asia-Pac Psychiatry. 2013;5:E58–63. doi: 10.1111/appy.12078. [DOI] [PubMed] [Google Scholar]
- 94.Joseph J, Depp C, Martin AS, Daly RE, Glorioso DK, Palmer BW, et al. Associations of high sensitivity C-reactive protein levels in schizophrenia and comparison groups. Schizophrenia Res. 2015;168:456–60. doi: 10.1016/j.schres.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pan S, Tan Y, Yao S, Zhao X, Xiong J. Serum high-sensitivity C-reactive protein: a delicate sentinel elevated in drug-free acutely agitated patients with schizophrenia. Psychiatry Res. 2016;246:89–94. doi: 10.1016/j.psychres.2016.09.033. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Q, Hong W, Li H, Peng F, Wang F, Li N, et al. Increased ratio of high sensitivity C-reactive protein to interleukin-10 as a potential peripheral biomarker of schizophrenia and aggression. Int J Psychophysiol. 2017;114:9–15. doi: 10.1016/j.ijpsycho.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 97.North NF, Bruggemann J, Cropley V, Swaminathan V, Sundram S, Lenroot R, et al. Increased peripheral inflammation in schizophrenia is associated with worse cognitive performance and related cortical thickness reductions. European Arch Psychiatry Clin Neurosci. 2021. 10.1007/s00406-021-01237-z. [DOI] [PubMed]
- 98.Erbagci AB, Herken H, Köylüoglu O, Yilmaz N, Tarakçioglu M. Serum IL-1β, sIL-2R, IL-6, IL-8 and TNF-α in schizophrenic patients, relation with symptomatology and responsiveness to risperidone treatment. Mediators Inflamm. 2001;10:109–15. doi: 10.1080/09629350123895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hope S, Melle I, Aukrust P, Steen NE, Birkenaes AB, Lorentzen S, et al. Similar immune profile in bipolar disorder and schizophrenia: selective increase in soluble tumor necrosis factor receptor I and von Willebrand factor. Bipolar Disord. 2009;11:726–34. doi: 10.1111/j.1399-5618.2009.00757.x. [DOI] [PubMed] [Google Scholar]
- 100.Sarandol A, Kirli S, Akkaya C, Ocak N, Eroz E, Sarandol E. Coronary artery disease risk factors in patients with schizophrenia: effects of short term antipsychotic treatment. J Psychopharmacol. 2007;21:857–63. doi: 10.1177/0269881107077609. [DOI] [PubMed] [Google Scholar]
- 101.Gattaz WF, Dalgalarrondo P, Schröder HC. Abnormalities in serum concentrations of interleukin-2, interferon-α and interferon-γ in schizophrenia not detected. Schizophrenia Res. 1992;6:237–41. doi: 10.1016/0920-9964(92)90006-Q. [DOI] [PubMed] [Google Scholar]
- 102.Haack M, Hinze-Selch D, Fenzel T, Kraus T, Kühn M, Schuld A, et al. Plasma levels of cytokines and soluble cytokine receptors in psychiatric patients 186 upon hospital admission: effects of confounding factors and diagnosis. J Psychiatr Res. 1993;33:407–18. doi: 10.1016/S0022-3956(99)00021-7. [DOI] [PubMed] [Google Scholar]
- 103.Dimitrov DH, Lee S, Yantis J, Valdez C, Paredes RM, Braida N, et al. Differential correlations between inflammatory cytokines and psychopathology in veterans with schizophrenia: potential role for IL-17 pathway. Schizophrenia Res. 2013;151:29–35. doi: 10.1016/j.schres.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 104.van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, et al. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64:820–2. doi: 10.1016/j.biopsych.2008.04.025. [DOI] [PubMed] [Google Scholar]
- 105.Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med. 2009;50:1801–7. doi: 10.2967/jnumed.109.066647. [DOI] [PubMed] [Google Scholar]
- 106.Bloomfield PS, Selvaraj S, Veronese M, Rizzo G, Bertoldo A, Owen DR, et al. Microglial activity in people at ultra high risk of psychosis and in schizophrenia; an [11C]PBR28 PET brain imaging study. Am J Psychiatry. 2016;173:44–52. doi: 10.1176/appi.ajp.2015.14101358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Conen S, Gregory CJ, Hinz R, Smallman R, Corsi-Zuelli F, Deakin B, et al. Neuroinflammation as measured by positron emission tomography in patients with recent onset and established schizophrenia: implications for immune pathogenesis. Mol Psychiatry. 2020. 10.1038/s41380-020-0829-y. [DOI] [PMC free article] [PubMed]
- 108.Di Biase MA, Zalesky A, O'keefe G, Laskaris L, Baune BT, Weickert CS, et al. PET imaging of putative microglial activation in individuals at ultra-high risk for psychosis, recently diagnosed and chronically ill with schizophrenia. Transl Psychiatry. 2017;7:e1225. doi: 10.1038/tp.2017.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cosenza-Nashat M, Zhao ML, Suh HS, Morgan J, Natividad R, Morgello S, et al. Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol. 2009;35:306–28. doi: 10.1111/j.1365-2990.2008.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Betlazar C, Harrison-Brown M, Middleton RJ, Banati R, Liu G. Cellular sources and regional variations in the expression of the neuroinflammatory markers translocator protein (TSPO) in the normal brain. Int J Mol Sci. 2018;19:2707. doi: 10.3390/ijms19092707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Notter T, et al. Neuronal activity increases translocator protein (TSPO) levels. Mol Psychiatry. 2020. 10.1038/s41380-020-0745-1. [DOI] [PMC free article] [PubMed]
- 112.Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, et al. Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. 2011;52:24–32. doi: 10.2967/jnumed.110.079459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Albrecht P, Torrey EF, Boone E, Hicks JT, Daniel N. Raised cytomegalovirus-antibody level in cerebrospinal fluid of schizophrenic patients. Lancet. 1980;2:769–72. doi: 10.1016/S0140-6736(80)90386-4. [DOI] [PubMed] [Google Scholar]
- 114.Müller N, Ackenheil M. Immunoglobulin and albumin content of cerebrospinal fluid in schizophrenic patients: Relationship to negative symptomatology. Schizophrenia Res. 1995;14:223–8. doi: 10.1016/0920-9964(94)00045-A. [DOI] [PubMed] [Google Scholar]
- 115.Müller N, Dobmeier P, Empl M, Riedel M, Schwarz M, Ackenheil M. Soluble IL-6 receptors in the serum and cerebrospinal fluid of paranoid schizophrenic patients. Eur Psychiatry. 1997;12:294–9. doi: 10.1016/S0924-9338(97)84789-X. [DOI] [PubMed] [Google Scholar]
- 116.Nikkilä HV, Müller K, Ahokas A, Rimón R, Andersson LC. Increased frequency of activated lymphocytes in the cerebrospinal fluid of patients with acute schizophrenia. Schizophrenia Res. 2001;49:99–105. doi: 10.1016/S0920-9964(99)00218-2. [DOI] [PubMed] [Google Scholar]
- 117.Erhardt S, Blennow K, Nordin C, Skogh E, Lindström LH, Engberg G. Kynurenic acid levels are elevated in the cerebrospinal fluid of patients with schizophrenia. Neurosci Lett. 2001;313:96–8. doi: 10.1016/S0304-3940(01)02242-X. [DOI] [PubMed] [Google Scholar]
- 118.Endres D, Perlov E, Baumgartner A, Hottenrott T, Dersch R, Stich O, et al. Immunological findings in psychotic syndromes: a tertiary care hospital’s CSF sample of 180 patients. Front Hum Neurosci. 2015;9:476. doi: 10.3389/fnhum.2015.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gallego JA, Blanco EA, Husain-Krautter S, Madeline Fagen E, Moreno-Merino P, del Ojo-Jiménez JA, et al. Cytokines in cerebrospinal fluid of patients with schizophrenia spectrum disorders: new data and an updated meta-analysis. Schizophrenia Res. 2018;202:64–71. doi: 10.1016/j.schres.2018.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kirkpatrick B, Miller BJ. Inflammation and Schizophrenia. Schizophrenia Bull. 2013;39:1174–9. doi: 10.1093/schbul/sbt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Perkins DO, Jeffries CD, Addington J, Bearden CE, Cadenhead KS, Cannon TD, et al. Towards a psychosis risk blood diagnostic for persons experiencing high-risk symptoms: preliminary results from the NAPLS project. Schizophrenia Bull. 2015;41:419–28. doi: 10.1093/schbul/sbu099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Föcking M, Dicker P, Lopez LM, Cannon M, Schäfer MR, McGorry PD, et al. Differential expression of the inflammation marker IL12p40 in the at-risk mental state for psychosis: a predictor of transition to psychotic disorder? BMC Psychiatry. 2016;16:326. doi: 10.1186/s12888-016-1039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Marcinowicz P, Więdłocha M, Zborowska N, Dębowska W, Podwalski P, Misiak B, et al. A meta-analysis of the influence of antipsychotics on cytokines levels in first episode psychosis. J Clin Med. 2021;10:2488. doi: 10.3390/jcm10112488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vuksan-Ćusa B, Šagud M, Jakovljević M. C-reactive protein and metabolic syndrome in patients with bipolar disorder compared to patients with schizophrenia. Psychiatr Danubina. 2010;22:275–7. [PubMed] [Google Scholar]
- 125.Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yang Y, Wu J, Wang J. A database and functional annotation of NF-κB target genes. Int J Clin Exp Med. 2016;9:7986–95. [Google Scholar]
- 127.Ten RM, Paya CV, Israël N, Le Bail O, Mattei MG, Virelizier JL, et al. The characterization of the promoter of the gene encoding the p50 subunit of NF-κB participates in its own regulation. EMBO J. 1992;11:195–203. doi: 10.1002/j.1460-2075.1992.tb05042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liptay S, Schmid RM, Nabel EG, Nabel GJ. Transcriptional regulation of NF-κB2: evidence for κB-mediated positive and negative autoregulation. Mol Cell Biol. 1994;14:7695–703. doi: 10.1128/mcb.14.12.7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Renner F, Schmitz ML. Autoregulatory feedback loops terminating the NF-kB response. Trends Biochem Sci. 2009;34:128–35. doi: 10.1016/j.tibs.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 130.Valiño-Rivas L, Vaquero JJ, Sucunza D, Gutierrez S, Sanz AB, Fresno M, et al. NIK as a druggable mediator of tissue injury. Trends Mol Med. 2019;25:341–60. doi: 10.1016/j.molmed.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 131.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018;362:eaat8127. doi: 10.1126/science.aat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Roussos P, Katsel P, Davis KL, Giakoumaki SG, Lencz T, Malhotra AK, et al. Convergent findings for abnormalities of the NF-κB signaling pathway in schizophrenia. Neuropsychopharmacology. 2013;38:533–9. doi: 10.1038/npp.2012.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kordula T, Bugno M, Rydel RE, Travis J. Mechanism of Interleukin-1- and tumor necrosis factor α-dependent regulation of the α1-antichymotrypsin gene in human astrocytes. J Neurosci. 2000;20:7510–6. doi: 10.1523/JNEUROSCI.20-20-07510.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E. Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry. 2007;7:46. doi: 10.1186/1471-244X-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 136.Shih VF, Tsui R, Caldwell A, Hoffman A. A single NF-κB system for both canonical and non-canonical signaling. Cell Res. 2011;21:86–102. doi: 10.1038/cr.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17:545–58. doi: 10.1038/nri.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cildir G, Low KC, Tergaonkar V. Noncanonical NF-κB signaling in health and disease. Trends Mol Med. 2016;22:414–29. doi: 10.1016/j.molmed.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 139.Dresselhaus EC, Meffert MK. Cellular specificity of NF-κB function in the nervous system. Front Immunol. 2019;10:1043. doi: 10.3389/fimmu.2019.01043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Brain synapses contain inducible forms of the transcription factor NF-κB. Mech Dev. 1993;43:135–47. doi: 10.1016/0925-4773(93)90031-R. [DOI] [PubMed] [Google Scholar]
- 141.Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Stimulation of ionotropic glutamate receptors activates transcription factor NF-κB in primary neurons. PNAS. 1995;92:9618–22. doi: 10.1073/pnas.92.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kaltschmidt C, Kaltschmidt B, Neumann H, Wekerle H, Baeuerle PA. Constitutive NF-κB activity in neurons. Mol Cell Biol. 1994;14:3981–92. doi: 10.1128/mcb.14.6.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Meberg PJ, Kinney WR, Valcourt EG, Routtenberg A. Gene expression of the transcription factor NF-κB in hippocampus: regulation by synaptic activity. Mol Brain Res. 1996;38:179–90. doi: 10.1016/0169-328X(95)00229-L. [DOI] [PubMed] [Google Scholar]
- 144.Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappaB functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–8. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- 145.Mikenberg I, Widera D, Kaus A, Kaltschmidt B, Kaltschmidt C. Transcription factor NF-κB is transported to the nucleus via cytoplasmic dynein/dynactin motor complex in hippocampal neurons. PLoS ONE. 2007;7:e589. doi: 10.1371/journal.pone.0000589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jarosinski KW, Whitney LW, Massa PT. Specific deficiency in nuclear factor-κB activation in neurons of the central nervous system. Lab Investig. 2001;81:1275–88. doi: 10.1038/labinvest.3780341. [DOI] [PubMed] [Google Scholar]
- 147.Listwak SJ, Rathore P, Herkenham M. Minimal NF-κB activity in neurons. Neuroscience. 2013;250:282–99. doi: 10.1016/j.neuroscience.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nam J, Aguda BD, Rath B, Agarwal S. Biomechanical thresholds regulate inflammation through the NF-κB pathway: experiments and modeling. PLoS ONE. 2009;4:e5262. doi: 10.1371/journal.pone.0005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Malek S, Chen Y, Huxford T, Ghosh G. IκBβ, but not IκBα, functions as a classical cytoplasmic inhibitor of NF-κB dimers by masking both NF-κB nuclear localization sequences in resting cells. Journal of Biological Chemistry 2001;276:45225–35. [DOI] [PubMed]
- 150.Oeckinghaus A, Ghosh S. The NF-kB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, et al. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J Cell Sci. 1997;110:369–78. doi: 10.1242/jcs.110.3.369. [DOI] [PubMed] [Google Scholar]
- 152.Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NF-κB•DNA dissociation by IκBα. PNAS. 2009;106:19328–33. doi: 10.1073/pnas.0908797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Scott ML, Fujita T, Liou H, Nolan GP, Baltimore D. The p65 subunit of NF-κB regulates IκB by two distinct mechanisms. Genes Dev. 1993;7:1266–76. doi: 10.1101/gad.7.7a.1266. [DOI] [PubMed] [Google Scholar]
- 154.Kearns JD, Basak S, Werner SL, Huang CS, Hoffman A. IκBε provides negative feedback to control NF-κB oscillations, signaling dynamics, and inflammatory gene expression. J Cell Biol. 2006;173:659–64. doi: 10.1083/jcb.200510155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mothes J, Busse D, Kofahl B, Wolf J. Sources of dynamic variability in NF-κB signal transduction: a mechanistic model. Bioessays. 2015;37:452–62. doi: 10.1002/bies.201400113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liao G, Zhang M, Harhaj EW, Sun S. Regulation of the NF-κB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem. 2004;25:26243–50. doi: 10.1074/jbc.M403286200. [DOI] [PubMed] [Google Scholar]
- 157.Lu J, Plank TD, Su F, Shi X, Liu C, Ji Y, et al. The nonsense-mediated RNA decay pathway is disrupted in inflammatory myofibroblastic tumors. J Clin Investig. 2016;126:3058–62. doi: 10.1172/JCI86508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bista P, Zeng W, Ryan S, Bailly V, Browning JL, Lukashev ME. TRAF3 controls activation of the canonical and alternative NFκB by the lymphotoxin beta receptor. J Biol Chem. 2010;285:12971–8. doi: 10.1074/jbc.M109.076091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Cherry EM, Lee DW, Jung J, Sitcheran R. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) promotes glioma cell invasion through induction of NF-κB-inducing kinase (NIK) and noncanonical NF-κB signaling. Mol Cancer. 2015;14:9. doi: 10.1186/s12943-014-0273-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, et al. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–86. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fernandez-Lizarbe S, Pascual M, Guerri C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J Immunol. 2009;183:4733. doi: 10.4049/jimmunol.0803590. [DOI] [PubMed] [Google Scholar]
- 162.Yao L, et al. Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia. J Neuroinflammation. 2014;10:23. doi: 10.1186/1742-2094-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cui W, Sun C, Ma Y, Wang S, Wang X, Zhang Y. Inhibition of TLR4 induces M2 microglial polarization and provides neuroprotection via the NLPR3 inflammasome in Alzheimer’s disease. Front Neurosci. 2020;14:444. doi: 10.3389/fnins.2020.00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liu X, Nemeth DP, McKim DB, Zhu L, DiSabato DJ, Berdysz O, et al. Cell-type-specific interleukin 1 receptor 1 signaling in the brain regulates distinct neuroimmune activities. Immunity. 2019;50:317–33. doi: 10.1016/j.immuni.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci. 2011;121:367–87. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2016;89:37–53. 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed]
- 167.Kielian T. Toll-Like receptors in central nervous system glial inflammation and homeostasis. J Neurosci Res. 2006;83:711–30. doi: 10.1002/jnr.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Taetzsch T, Levesque S, McGraw C, Brookins S, Luqa R, Bonini MG, et al. Redox regulation of NF-κB p50 and M1 polarization in microglia. Glia. 2015;63:423–40. doi: 10.1002/glia.22762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kyrargyri V, Vega-Flores G, Gruart A, Delgado-García JM, Probert L. Differential contributions of microglial and neuronal IKKβ to synaptic plasticity and associative learning in alert behaving mice. Glia. 2015;63:549–66. doi: 10.1002/glia.22756. [DOI] [PubMed] [Google Scholar]
- 170.Murphy CE, et al. Regional, cellular and species difference of two key neuroin- flammatory genes implicated in schizophrenia. Brain, Behav Immunol. 2020;88:826–39. doi: 10.1016/j.bbi.2020.05.055. [DOI] [PubMed] [Google Scholar]
- 171.Corsi-Zuelli F, Deakin B. Impaired regulatory T cell control of astroglial overdrive and microglial pruning in schizophrenia. Neurosci Biobehav Rev. 2021. 10.1016/j.neubiorev.2021.03.004. [DOI] [PubMed]
- 172.Feresten AH, Barakauskas V, Ypsilanti A, Barr AM, Beasley CL. Increased expression of glial fibrillary acidic protein in prefrontal cortex in psychotic illness. Schizophrenia Res. 2013;150:252–7. doi: 10.1016/j.schres.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 173.Martins-de-Souza D, Gattaz WF, Schmitt A, Novello JC, Marangoni S, Turck CW, et al. Proteome analysis of schizophrenia patients Wernicke’s area reveals an energy metabolism dysregulation. BMC Psychiatry. 2009;9:17. doi: 10.1186/1471-244X-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, Meltzer H, Overholser J, Stockmeier C. Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophrenia Res. 2002;57:127–38. doi: 10.1016/S0920-9964(02)00339-0. [DOI] [PubMed] [Google Scholar]
- 175.Toro CT, Hallak JE, Dunham JS, Deakin JF. Glial fibrillary acidic protein and glutamine synthetase in subregions of prefrontal cortex. Neurosci Lett. 2006;404:276–81. doi: 10.1016/j.neulet.2006.05.067. [DOI] [PubMed] [Google Scholar]
- 176.Hercher C, Chopra V, Beasley CL. Evidence for morphological alterations in prefrontal white matter glia in schizophrenia and bipolar disorder. J Psychiatry Neurosci. 2014;39:376–85. doi: 10.1503/jpn.130277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kano S, Nwulia E, Niwa M, Chen Y, Sawa A, Cascella N. Altered MHC class I expression in dorsolateral prefrontal cortex of nonsmoker patients with schizophrenia. Neurosci Res. 2011;71:289–93. doi: 10.1016/j.neures.2011.07.1818. [DOI] [PubMed] [Google Scholar]
- 178.Nakatani N, Hattori E, Ohnishi T, Dean B, Iwayama Y, Matsumoto I, et al. Genome-wide expression analysis detects eight genes with robust alterations specific to bipolar I disorder: relevance to neuronal network perturbation. Hum Mol Genet. 2006;15:1949–62. doi: 10.1093/hmg/ddl118. [DOI] [PubMed] [Google Scholar]
- 179.Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein HG, et al. Distribution of HLA-DR-positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol. 2006;112:305–16. doi: 10.1007/s00401-006-0090-8. [DOI] [PubMed] [Google Scholar]
- 180.Radewicz K, Garey LJ, Gentleman SM, Reynolds R. Increase in HLADR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J Neuropathol Exp Neurol. 2000;59:137–50. doi: 10.1093/jnen/59.2.137. [DOI] [PubMed] [Google Scholar]
- 181.Bayer TA, Buslei R, Havas L, Falkai P. Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett. 1999;271:126–8. doi: 10.1016/S0304-3940(99)00545-5. [DOI] [PubMed] [Google Scholar]
- 182.Jha MK, Jo M, Kim J, Suk K. Microglia-astrocyte crosstalk: an intimate molecular conversation. Neuroscientist. 2018;25:227–40. doi: 10.1177/1073858418783959. [DOI] [PubMed] [Google Scholar]
- 183.Prescott JA, Cook SJ. Targeting IKKβ in cancer: challenges and opportunities for the therapeutic utilisation of IKKβ inhibitors. Cells. 2018;7:115. doi: 10.3390/cells7090115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7. 10.3389/fncel.2013.00006. [DOI] [PMC free article] [PubMed]
- 185.Shao X. Protective role of NF-κB in inflammatory demyelination. J Neurosci. 2018;38:2416–7. doi: 10.1523/JNEUROSCI.3286-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–56. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li YX, Sibon OC, Dijkers PF. Inhibition of NF-κB in astrocytes is sufficient to delay neurodegeneration induced by proteotoxicity in neurons. J Neuroinflammation. 2018;15:261. doi: 10.1186/s12974-018-1278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Khorooshi R, Babcock AA, Owens T. NF-kappaB-driven STAT2 and CCL2 expression in astrocytes in response to brain injury. J Immunol. 2008;181:7284–91. doi: 10.4049/jimmunol.181.10.7284. [DOI] [PubMed] [Google Scholar]
- 189.van Loo G, De Lorenzi R, Schmidt H, Huth M, Mildner A, Schmidt-Supprian M, et al. Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol. 2006;7:954–61. doi: 10.1038/ni1372. [DOI] [PubMed] [Google Scholar]
- 190.Schlaaff K, Dobrowolny H, Frodl T, Mawrin C, Gos T, Steiner J, et al. Increased densities of T and B lymphocytes indicate neuroinflammation in subgroups of schizophrenia and mood disorder patients. Brain Behav Immun. 2020;88:497–506. doi: 10.1016/j.bbi.2020.04.021. [DOI] [PubMed] [Google Scholar]
- 191.Saggu R, Schumacher T, Gerich F, Rakers C, Tai K, Delekate A, et al. Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol Commun. 2016;4:76. doi: 10.1186/s40478-016-0350-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sehnert B, Burkhardt H, Dübel S, Voll RE. Cell-type targeted NF-kappaB inhibition for the treatment of inflammatory diseases. Cells. 2020;9:1627. doi: 10.3390/cells9071627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Terrando N, Eriksson LI, Kyu Ryu J, Yang T, Monaco C, Feldmann M, et al. Resolving postoperative neuroinflammation and cognitive decline. Ann Neurol. 2011;70:986–95. doi: 10.1002/ana.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhang J, Jiang W, Zuo Z. Pyrrolidine dithiocarbamate attenuates surgery-induced neuroinflammation and cognitive dysfunction possibly via inhibition of nuclear factor κB. Neuroscience. 2014;261:1–10. doi: 10.1016/j.neuroscience.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Badshah H, Ali T, Kim MO. Osmotin attenuates LPS-induced neuroinflammation and memory impairments via the TLR4/ NFκB signaling pathway. Scientific Reports 2016;6:24493. [DOI] [PMC free article] [PubMed]
- 196.Kéri S, Szabó C, Kelemen O. Uniting the neurodevelopmental and immunological hypotheses: Neuregulin 1 receptor ErbB and Toll-like receptor activation in first-episode schizophrenia. Sci Rep. 2017;7:4147. doi: 10.1038/s41598-017-03736-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Balaji R, Subbanna M, Shivakumar V, Abdul F, Venkatasubramanian G, Debnath M. Pattern of expression of Toll like receptor (TLR)-3 and -4 genes in drug-naïve and antipsychotic treated patients diagnosed with schizophrenia. Psychiatry Res. 2020;285:112727. doi: 10.1016/j.psychres.2019.112727. [DOI] [PubMed] [Google Scholar]
- 198.McKernan DP, Dennison U, Gaszner G, Cryan JF, Dinan TG. Enhanced peripheral toll-like receptor responses in psychosis: further evidence of a pro- inflammatory phenotype. Transl Psychiatry. 2011;1:e36. doi: 10.1038/tp.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Liu Y, Fang S, Li X, Feng J, Du J, Guo L, et al. Aspirin inhibits LPS- induced macrophage activation via the NF-κB pathway. Sci Rep. 2017;7:11549. doi: 10.1038/s41598-017-10720-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Funakoshi-Tago M, Shimizu T, Tago K, Nakamura M, Itoh H, Sonoda Y, et al. Celecoxib potently inhibits TNFα-induced nuclear translocation and activation of NF-κB. Biochem Pharmacol. 2008;76:662–71. doi: 10.1016/j.bcp.2008.06.015. [DOI] [PubMed] [Google Scholar]
- 201.Sun J, Shigemi H, Tanaka Y, Yamauchi T, Ueda T, Iwasaki H. Tetracyclines downregulate the production of LPS-induced cytokines and chemokines in THP-1 cells via ERK, p38, and nuclear factor-κB signaling pathways. Biochem Biophys Rep. 2015;4:397–404. doi: 10.1016/j.bbrep.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Attari A, Mojdeh A, Soltani FA, Reza M. Aspirin inclusion in antipsychotic treatment on severity of symptoms in schizophrenia: a randomized clinical trial. Iran J Psychiatry Behav Sci. 2017;11:e5848. [Google Scholar]
- 203.Akhondzadeh S, Nejatisafa AA, Amini H, Mohammadi MR, Larijani B, Kashani L, et al. Adjunctive estrogen treatment in women with chronic schizophrenia: a double-blind, randomized, and placebo-controlled trial. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:1007–12. doi: 10.1016/S0278-5846(03)00161-1. [DOI] [PubMed] [Google Scholar]
- 204.Liu F, Guo X, Wu R, Ou J, Zheng Y, Zhang B, et al. Minocycline supplementation for treatment of negative symptoms in early-phase schizophrenia: a double-blind, randomized, controlled trial. Schizophrenia Res. 2014;153:169–76. doi: 10.1016/j.schres.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 205.Schnieder TP, Dwork AJ. Searching for neuropathology: gliosis in schizophrenia. Biol Psychiatry. 2011;69:134–9. doi: 10.1016/j.biopsych.2010.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wang C, Aleksic B, Ozaki N. Glia-related genes and their contribution to schizophrenia. Psychiatry Clin Neurosci. 2015;69:448–61. doi: 10.1111/pcn.12290. [DOI] [PubMed] [Google Scholar]
- 207.Dietz AG, Goldman SA, Nedergaard M. Glial cells in schizophrenia: a unified hypothesis. Lancet Psychiatry. 2020;7:272–81. doi: 10.1016/S2215-0366(19)30302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Snijders GJLJ, Zuiden W, Sneeboer MAM, Berdenis van Berlekom A, Geest AT, Schnieder T, et al. A loss of mature microglial markers without immune activation in schizophrenia. Glia. 2020;69:1251–67. doi: 10.1002/glia.23962. [DOI] [PMC free article] [PubMed] [Google Scholar]