

Abstract
Cancer vaccines that make use of tumor antigens represent a promising therapeutic strategy by stimulating immune responses against tumors to generate long-term anti-tumor immunity. However, vaccines have shown limited clinical efficacy due to inefficient delivery. In this study, we focus on vaccine delivery assisted by nanocomplexes for cancer immunotherapy. Nanocomplex-mediated vaccination can efficiently deliver nucleic acids encoding neoantigens to lymphoid tissues and antigen-presenting cells. Polyethylenimine (PEI) was conjugated with farnesylthiosalicylic acid (FTS) to form micelles. Subsequent interaction with nucleic acids led to formation of polymer/nucleic acid nanocomplexes of well-controlled structure. Tumor transfection via FTS-PEI was much more effective than that by PEI, other PEI derivatives, or naked DNA. Significant numbers of transfected cells were also observed in draining lymph nodes (LNs). In vivo delivery of ovalbumin (OVA; a model antigen) expression plasmid (pOVA) by FTS-PEI led to a significant growth inhibition of the OVA-expressing B16 tumor through presentation of OVA epitopes as well as other epitopes via epitope spreading. Moreover, in vivo delivery of an endogenous melanoma neoantigen tyrosinase-related protein 2 (Trp2) also led to substantial tumor growth inhibition. FTS-PEI represents a promising transfection agent for effective gene delivery to tumors and LNs to mediate effective neoantigen vaccination.
Keywords: farnesylthiosalicylic acid, polyethylenimine, nanocomplex, vaccination, neoantigen, immunotherapy
Graphical abstract
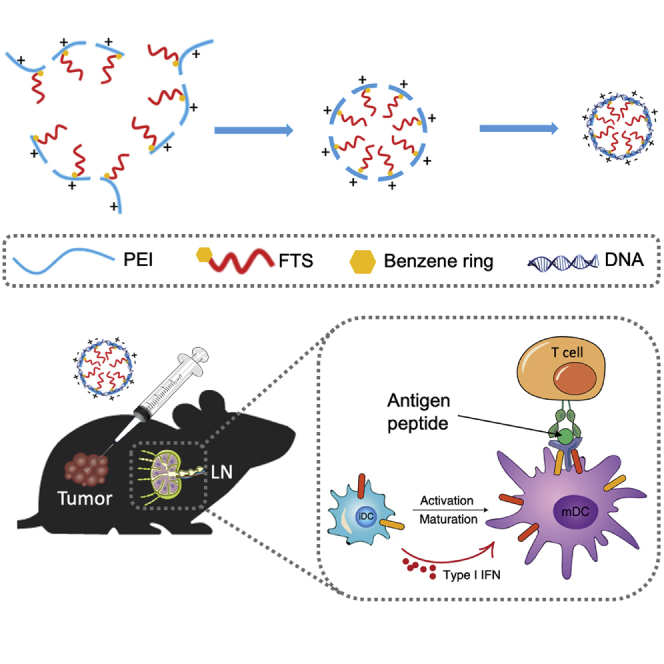
Nanocomplex-mediated vaccination can efficiently deliver nucleic acids encoding neoantigens to lymphoid tissues and antigen-presenting cells. Polyethylenimine (PEI) was conjugated with farnesylthiosalicylic acid (FTS) to form micelles. FTS-PEI represents a promising transfection agent for effective gene delivery to tumors and LNs to mediate effective neoantigen vaccination.
Introduction
Vaccines that activate our immune system for prevention and treatment of infections and other diseases play a significant role in human healthcare.1 Cancer neoantigens derived from somatic mutations in tumor tissues provide an attractive target for cancer immunotherapies such as cancer vaccine.2 Vaccination against tumor-specific neoantigens has the capability to decrease the induction of potential central and peripheral tolerance.3,4 It has been reported that neoantigen-based personalized vaccination shows remarkable therapeutic potential in preclinical and early-phase clinical studies.5, 6, 7 However, significant challenges remain in the efficient and safe delivery of the vaccine components to induce potent and generalized anti-cancer T cell responses.8
Currently, a variety of delivery systems have been developed for gene delivery including viral and non-viral carriers.9 Various non-viral systems have been reported including peptides, liposomes, and cationic polymers.10 Synthetic polymers have attracted increasing attention in nucleic acid delivery due to their versatility, favorable safety profiles, and the ease of production.9, 10, 11, 12 Nanocomplexes have been extensively investigated for vaccine delivery since they could protect vaccines from degradation, prolong retention time, and enhance lymphoid organ targeting.13
We herein developed a delivery system based on polyethylenimine (PEI), a cationic polymer that has been widely used in non-viral gene delivery including intratumor injection.14 PEI has a high charge density with proton sponge effect. Unprotonated secondary amines of PEI absorb protons upon internalization into the endosome, resulting in more protons being brought into the endosome and a concomitant increased influx of Cl– ions and water.15 This can lead to endosomal bursting and the subsequent release of endocytosed materials into cytosol.14 PEI is a water-soluble molecule that can randomly interact with nucleic acids in aqueous solutions. By anchoring PEI molecules on the surface of nanoparticles (NPs) of defined sizes, a more effective and controllable interaction with nucleic acids can be achieved.16 For example, PEI was modified with cholesterol, deoxycholic acid, or lipoic acid to improve the ability of the polymer to protect and deliver genes to cells. It is believed that a more lipophilic PEI derivative facilitates the interaction with cells.17,18 The lipid-derivatized PEI also self-assembles to form NPs that provide more controlled interaction with nucleic acids.
In this study, we coupled farnesylthiosalicylic acid (FTS) with PEI to obtain an amphiphilic FTS-PEI conjugate that self-assembled to form micelles. Water-insoluble FTS served as the hydrophobic region of polymeric micelles, whereas PEI served as the cationic hydrophilic domain to bind nucleic acid and increase cellular uptake. FTS is a nontoxic RAS antagonist and can inhibit receptor-mediated RAS activation, resulting in the inhibition of RAS-dependent tumor growth.19 FTS-PEI was characterized with respect to the transfection efficiency in vitro and in vivo. In addition, ovalbumin (OVA) was used as a model antigen, and tyrosinase-related protein 2 (Trp2) was used as an endogenous melanoma neoantigen to examine the efficiency of FTS-PEI-mediated vaccination.20
Results
Synthesis and characterization of FTS-PEI polymer
The synthesis route of the FTS-PEI polymer is shown in Figure 1A. FTS was conjugated to PEI via condensation reaction. The input molar ratio of FTS and PEI units was 1:20. The conjugation efficiency was determined based on UV absorbance at the wavelength of 250 nm. By using a calibration curve made with FTS standards, the molar percentage of FTS in the final polymer was determined to be 3.2% with 72% of conjugation efficiency. The 1H nuclear magnetic resonance (NMR) spectra of the FTS-PEI showed the respective PEI peaks (2.5−2.9 ppm) and FTS lipid peaks (0.9−1.0 ppm) (Figures 1B and S1A−S1C). The matrix-assisted laser desorption/ionization (MALDI) spectra also showed that compared with free PEI, FTS-PEI had a peak with slightly larger molecular weight (MW), indicating FTS conjugation (Figure 1C).
Figure 1.

Synthesis scheme and chemical characterization of FTS-PEI polymers
(A) FTS-PEI was synthesized via a condensation reaction from linear PEI. PEI was reacted with FTS at different ratios in DMF at room temperature with DCC as a condensing reagent and DMAP as a catalyst. (B) 1H nuclear magnetic resonance (NMR) spectra of the FTS-PEI (5% FTS, PEI MW = 2.5 kDa). (C) The matrix-assisted laser desorption/ionization (MALDI) spectra of free PEI (PEI MW = 2.5 kDa, black line) and FTS-PEI (5% FTS, PEI MW = 2.5 kDa, blue line).
Characterization of micellar nanocomplexes
FTS-PEI polymer self-assembled to form micelles in aqueous solutions with a size of 164.2 nm and a zeta potential of 30.2 mV, as determined by a Zetasizer. FTS-PEI micelles were mixed with expression plasmid (p)GFP at various N/P ratios (the molar ratio of nitrogen residues [N] in the cationic polymer over phosphate [P] of nucleic acids). The hydrodynamic sizes of blank FTS/PEI micelles and pGFP/FTS-PEI nanocomplexes at N/P ratios from 0.5/1 to 10/1 were shown in Figures 2A and S2A−S2C. At N/P ratios below 3.8, the net charges of nanocomplexes were negative with the particle sizes around 100 nm. At the N/P ratio of 3.8, a significant increase in particle size was observed with particle charges close to neutral. Further increases in N/P ratios led to continuous increases in net positive charges and decreases in particle sizes, suggesting gradual condensation of nucleic acid by the FTS-PEI polymer. Both blank FTS-PEI micelles and pGFP/FTS-PEI nanocomplexes were spherical in morphology, as shown by transmission electron microscopy (TEM) (Figures S3A−S3C).
Figure 2.

In vitro and in vivo characterizations of DNA/FTS-PEI nanocomplexes
(A) The hydrodynamic sizes and zeta potentials of FTS-PEI micelles (5% FTS, PEI MW = 2.5 kDa) and expression plasmid (p)GFP/FTS-PEI nanocomplexes formed at various N/P ratios. (B) Gel retardation assay of pGFP/FTS-PEI nanocomplexes at different N/P ratios. Samples were incubated for 20 min at room temperature prior to electrophoresis on a 1.5% (w/v) agarose gel (120 V, 20 min). (C) Gel electrophoresis assay of DNA displacement from pGFP/FTS-PEI nanocomplexes (N/P = 1) by dextran sulfate at various S/P ratios. (D) Linear PEI (MW = 2.5 kDa or 25 kDa) conjugated with different percentages of FTS (1% or 5%) were complexed with pGFP at N/P ratios of 0.5/1, 1/1, and 2.5/1, respectively. A total of 12 nanocomplexes were obtained that vary in the FTS/PEI (m/m) as well as N/P ratios. Nanocomplexes were individually administered via intratumor injection into tumor-bearing mice. Tumor tissues were collected after 48 h, and transfection efficiency was evaluated by fluorescence microscopic examination of GFP expression.
We then tested whether the cationic micelles could form stable complexes with pGFP via gel retardation assay. As shown in Figure 2B, complete complexation of pGFP by the FTS-PEI polymer was achieved at an N/P ratio of 5/1 or greater. To further investigate the interaction between FTS-PEI nanocarrier and pGFP, we examined the impact of dextran sulfate on the displacement of DNA from pGFP/FTS-PEI complexes and compared to pGFP/PEI complexes. As shown in Figure 2C, at an N/P ratio of 1/1, pGFP was only partially retarded by both PEI and FTS-PEI, but there appeared to have more DNA retardation with pGFP/FTS-PEI complexes. Addition of dextran sulfate to both DNA/polymer complexes led to gradual increases in the amounts of displaced DNA with the increases in the S/P ratios (molar ratio between the sulfur from dextran sulfate [S] and the phosphate from pGFP [P]) from 0.5 to 1. However, pGFP/FTS-PEI appeared to be more resistant to dextran sulfate-mediated DNA displacement compared to pGFP/PEI complexes. At an N/P ratio of 5/1, pGFP could not be replaced by dextran sulfate at an S/P ratio of 2.5 and 5, respectively. In contrast, an obvious release of pGFP was observed from the PEI carrier at the corresponding N/P ratios (Figure S4). These data suggest that FTS-PEI formed more stable complexes with DNA compared to PEI.
We then went on to evaluate the nucleic acid delivery efficacy of several polymer conjugates by loading pGFP into nanocomplexes and comparing GFP expression in tumor tissues. Linear PEIs of different MWs (2.5 kDa or 25 kDa) conjugated with different percentages of FTS (1% or 5%) were complexed with pGFP at N/P ratios of 0.5/1, 1/1, and 2.5/1, respectively. A total of 12 nanocomplexes were obtained that vary in the MW of PEI and FTS/PEI (m/m) as well as N/P ratios. They were individually administered via intratumor injection into B16F10 tumor-bearing mice, and tumor tissues were collected after 48 h. Tumor transfection from FTS-PEI nanocomplexes was much more effective than that by PEI or naked DNA alone (Figure 2D). Widespread transgene expression was observed in tumor tissues treated with nanocomplexes with a PEI of 2.5 kDa and 5% FTS derivatization at a N/P ratio of 1/1 (Figure 2D). This ratio was chosen for all subsequent studies.
FTS-PEI nanocomplex-mediated transfection of tumor cells and DCs
Effective nucleic acid-based vaccination requires efficient intracellular antigen expression and pursuant immune cell activation to generate an effective immune response. Therefore, we went to examine the other target tissues of the pGFP/FTS-PEI nanocomplexes besides tumor. Significant transfection was also observed in adjacent draining inguinal lymph nodes (LNs) (Figure 3A). We confirmed the expression of GFP via immunofluorescence using a GFP-specific antibody. We observed highly overlapping GFP green fluorescence and GFP-specific staining (red) signals in both tumors and draining LNs, ruling out the possibility of autofluorescence from the tissues (Figure 3B). We further confirmed and quantified the gene expression using luciferase as a reporter in tumors and LNs. Again, the highest level of gene expression was achieved with the nanocomplexes with a PEI of 2.5 kDa and 5% FTS derivatization at a 1/1 N/P ratio (Figure 3C).
Figure 3.

In vivo transfection of FTS-PEI
(A) GFP expression after 48 h in tumor tissues and LNs. (B) Immunofluorescence staining of GFP in tumors and LNs. (C) Luciferase expression after 48 h in tumors and LNs. Data are presented as mean ± SEM, n = 3. p values were generated by two-tailed Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (D) Fluorescence-activated cell sorting (FACS) analyses of GFP expression in fluorescence-labeled tumor cells (tdTomato+) in tumor tissues and DCs (CD11b+, MHC class II+) in LNs. (E) Comparison of pGFP transfection efficiency in tumors and LNs by PEI, OA-PEI, LA-PEI, and FTS-PEI, respectively.
Following demonstration of significant transfection of both tumors and adjacent LNs. We went on to quantify the percentages of transfected cells and examine whether DCs were effectively transfected by FTS-PEI, as DCs reside mostly in draining LNs and are believed to be one of the main target cells of vaccination due to their capability of antigen presentation and T cell activation. pGFP/FTS-PEI nanocomplexes were i.t. injected into 4T1.2-tdTomato-bearing mice. The fluorescence-labeled 4T1.2 tumor cells harbor a stable transfection and express tdTomato. Our results showed significant levels of GFP transfection in both tumor cells (tdTomato+) at around 12.9% and DCs (CD11b+, major histocompatibility complex [MHC] class II+) at around 4.8% (Figure 3D; p < 0.001, Student’s t test). Gating strategies for both tumor cells and DCs were shown in Figure S5. Moreover, compared to unmodified PEI, or PEI derivatized with other lipid motifs, like OA and LA, FTS-PEI was much more effective in transfecting both tumors and LNs (Figure 3E).
FTS-PEI nanocomplex-mediated anti-tumor immunity and antigen presentation
The above studies clearly showed the efficacy of our nanocomplexes in gene delivery to both tumor cells and antigen-presenting cells (APCs). As an initial step to test whether delivery of an antigen expression system via our nanocomplexes will lead to effective vaccination, OVA was used as a model antigen. We tested the adaptive immune response and anti-tumor efficacy of our vaccination strategy by establishing an OVA-expressing B16F10 mouse melanoma model and delivering a pOVA vaccine through intratumor administration. We found that pOVA-loaded nanocomplexes induced robust tumor suppression with only three doses of vaccination (once every 6 days), and tumor sizes were contained at less than 100 mm3 for over 3 weeks. Free pOVA showed a modest antitumor activity, whereas empty plasmid vector-loaded nanocomplexes showed almost no anti-tumor efficacy (Figure 4A).
Figure 4.

FTS-PEI nanocomplex-mediated anti-tumor immunity, antigen presentation, and epitope spreading
(A) PBS, pcDNA/FTS-PEI, free p-ovalbumin (pOVA), and pOVA/FTS-PEI were injected locally (intratumor) to B16F10-OVA (local tumors)-bearing mice once every 6 days for three times. Tumor growth was monitored once every 2 days. (B) Re-challenged tumors (distant tumors) were established via inoculation of tumor cells at the contralateral side after the 3rd vaccination of the primary tumors, and tumor growth in both groups were monitored. Values reported are the mean ± SEM, n = 5. p values were generated by one-way ANOVA using the Tukey test for multiple comparisons. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (C) SIINFEKL-H2K(b) presentation by B16F10-OVA tumors treated with pOVA/FTS-PEI or PBS. Cells were stimulated with recombinant mouse IFN-γ (10 ng/mL) overnight to induce OVA surface expression. MFI, mean fluorescence intensity. Data are presented as the mean ± SEM, n = 5. p values were generated by two-tailed Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (D) CD8+ T cells were isolated from B16F10-OVA tumors treated with PBS, pcDNA/FTS-PEI, free pOVA, or pOVA/FTS-PEI and co-cultured with B16F10 or MC38 for 6 h. Tumor cells were collected afterward for flow cytometry analysis for CFSE intensity. Data are presented as the mean ± SEM, n = 3. p values were generated by two-tailed Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (E) CD8+ T cells were isolated from B16F10-OVA tumors treated with PBS, pcDNA/FTS-PEI, free pOVA, or pOVA/FTS-PEI and co-cultured with B16F10 or MC38 for 6 h. Culture medium was collected, and the level of IFN-γ in the supernatant was measured by ELISA.
Given the importance of adaptive immunity in driving antitumor responses, we examined whether intratumoral delivery of pOVA/FTS-PEI could elicit systemic immunity that affects distant metastasis. Following the 3rd vaccination, “metastatic” tumors were established at the contralateral site by inoculating 1/10 of the number of cells used to establish primary tumors. Figure 4B shows that the growth of the tumors at the distal site was significantly inhibited in mice that received administration of pOVA/FTS-PEI only to primary tumors. We reasoned that the increased antitumor activity could be attributed to improved tumor cell recognition through increased antigen presentation. As shown in Figure 4C, the level of staining of the SIINFEKL epitope from OVA in the context of H2K(b) was significantly higher in the group treated with pOVA/FTS-PEI, suggesting that the vaccination increased the levels of antigen-loaded MHC class I on the surface of tumor cells. The observation of an abscopal effect indicates that vaccination with pOVA/FTS-PEI led to generation of systemic antitumor immunity.
Epitope spreading has been reported to be implicated in the induction of antitumor cytotoxic T lymphocyte (CTL) responses following vaccination. To examine whether epitope spreading is also involved in the antitumor immune response following vaccination with pOVA/FTS-PEI, we isolated CD8+ T cells from mice inoculated with B16F10-OVA tumor cells and treated with PBS, pcDNA/FTS-PEI, free pOVA, or pOVA/FTS-PEI and examined the cytotoxic effect on wild-type B16F10 cells. As shown in Figure 4D, CD8+ T cells isolated from mice that were challenged with B16F10-OVA tumor cells and treated with pOVA/FTS-PEI showed significant toxicity toward wild-type B16F10 cells. These CTL responses are B16F10 specific, as they showed no effect on MC38 tumor cells. Figure 4E shows that the medium interferon (IFN)-γ level in the co-culture was the highest in the group of pOVA/FTS-PEI, suggesting that FTS-PEI nanocomplexes can induce anti-tumor activity across epitopes within a specific tumor cell line. These data suggest that vaccination with pOVA/FTS-PEI induced strong epitope spreading, which may contribute to the overall antitumor activity.
The above study clearly shows the therapeutic efficacy of FTS-PEI-mediated cancer vaccination using OVA as a model antigen. To further investigate its potential in clinical translation, we went on to examine the therapeutic benefit of vaccination with Trp2, an endogenous neoantigen. Trp2 is an enzyme involved in melanin synthesis that undergoes N-glycosylation and translocation into the melanosome in melanocytes. It has been reported to be a tumor-associated neoantigen present in both melanocytes and melanoma, and as such, Trp2 has been intensely studied as a viable therapeutic and prophylactic vaccine candidate for melanoma. By using the same experimental design as in the delivery of pOVA, we delivered a pTrp2 to B16F10-bearing mice through intratumor administration. As shown in Figure 5A, intratumor delivery of pTrp2 via FTS-PEI led to significant inhibition of tumor growth. In addition, treatment of the primary tumors led to a strong abscopal effect in inhibiting the growth of untreated distal tumors (Figure 5B).
Figure 5.

FTS-PEI nanocomplex-mediated neoantigen vaccination induces tumor inhibition
(A) PBS, pcDNA/FTS-PEI, free pTrp2, and pTrp2/FTS-PEI were injected locally (intratumor) to B16F10 (local tumors)-bearing mice, respectively, once every 6 days for three times. (B) Re-challenged tumors (distant tumors) were established via inoculation of B16F10 cells at the contralateral side, 2 days after the final vaccination, and tumor growth in all groups was monitored. Values reported are the mean ± SEM, n = 5. p values were generated by one-way ANOVA using the Tukey test for multiple comparisons. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Effect of FTS treatment on PEI-mediated in vitro transfection
The significantly improved transfection efficiency of FTS-PEI over PEI or other PEI derivatives prompted us to examine whether FTS treatment will have an impact on transfection efficiency. Cultured B16F10 cells were pre-treated with different concentrations of FTS ranging from 0.625 μM to 5 μM for 24 h, followed by PEI-mediated pGFP transfection. FTS treatment led to a significant increase in GFP transgene expression in tumor cells (Figure 6A). We then similarly examined the effect of doxorubicin (Dox) and paclitaxel (PTX), two commonly used anticancer agents on transfection, and the transfection efficiency was quantitatively assessed by flow cytometry. As shown in Figure 6B, Dox and PTX had no significant impact on transfection, whereas FTS pretreatment led to significant increases in the numbers of GFP-positive cells in a dose-dependent manner.
Figure 6.

Effect of FTS treatment on PEI-mediated in vitro transfection
(A) GFP expression of cultured B16F10 tumor cells without transfection (negative control [NC]), cells transfected with pGFP/PEI without FTS pre-treatment, and cells transfected with pGFP/PEI and FTS pre-treatment (0.625 μM, 1.25 μM, 2.5 μM, and 5 μM). (B) Flow cytometry analysis of GFP+ cells transfected with pGFP/PEI, with or without pre-treatment of FTS (0.625−5 μM), doxorubicin (0.3−2.4 μM), and paclitaxel (0.5−4 μM). Values reported are the mean ± SEM, n = 5. p values were generated by one-way ANOVA using the Tukey test for multiple comparisons. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
In general, locally delivered soluble antigens or molecular adjuvants rapidly diffuse into systemic circulation due to their small molecular sizes.21 They are disseminated systemically and are of limited efficiency in targeting and accumulating in LNs, resulting in a poor immune response.14 Intratumor injection of naked nucleic acids has been shown to be capable of transfecting various types of tumors, but the efficiency is relatively low and varies greatly among different types of tumors.22,23 In addition, it has limited efficiency in targeting APCs due to inefficient delivery to lymphoid organs.24
Compared to soluble antigens, antigens in particulate form are more effectively taken up by APCs through phagocytosis.25 Various PEI-based nanocomplexes have been developed for tumor vaccination via intratumor administration, including cholesterol, deoxycholic acid, and lipoic acid derivatives of PEI. Our preliminary data showed that FTS-PEI was more effective in mediating in vivo transfection via intratumor injection compared to several other lipid-derivatized PEIs such as LA-PEI and OA-PEI. One unique feature of FTS compared to other lipid motifs (cholesterol or lipoic acid) lies in the presence of a benzene ring in addition to a hydrophobic alkyl chain. The benzyl rings in FTS-PEI can interact with the base π systems of nucleic acids through hydrophobic interaction and π–π stacking.26 The multiple modes of interactions between FTS-PEI and nucleic acids may offer better protection against nuclease-mediated degradation and at the same time facilitate the release of nucleic acids from endosomes into cytosol, resulting in improved transfection.27 This is supported by our data showing that pGFP formed more stable nanocomplexes with FTS-PEI and was more resistant to dextran sulfate-mediated displacement (Figures 2B and 2C). It is also interesting to notice that treatment of cultured tumor cells with FTS led to enhanced transfection in a dose-dependent manner (Figures 6A and 6B), whereas Dox and PTX had no effect on transfection. FTS is a known inhibitor of Ras signaling.19 More studies are needed to define whether Ras activity negatively affects the transfection efficiency. It also remains to be examined whether FTS can be effectively released in the tumor tissues to similarly affect the in vivo transfection as noted in vitro.
Intratumor gene delivery via FTS-PEI led to efficient transfection of both tumor cells and DCs in the draining LNs. This was translated into effective control of tumor growth in a B16F10-OVA tumor model. In addition to effective inhibition of the growth of vaccinated tumors, we observed a significant antitumor effect on the distal non-vaccinated metastatic tumors, suggesting development of a systemic antitumor immunity following treatment of the primary tumor with pOVA/FTS-PEI. Furthermore, vaccination of the B16F10-OVA tumor with pOVA/FTS-PEI triggered a specific CTL response against control B16F10 tumor cells, suggesting a possible involvement of epitope spreading in the overall antitumor activity. It is well known that there is significant heterogeneity both among tumors from different patients and within the same tumors.28 The epitope spreading subsequent to tumor vaccination opens the possibility of eradicating both antigen-positive and antigen-negative tumor cells.
In summary, we have developed a simple and effective gene-delivery nanocomplex (FTS-PEI) for tumor vaccination. Both tumor cells and DCs in draining LNs were effectively transfected. Delivery of a model antigen (OVA) led to the development of a strong systemic antitumor immunity in a B16F10-OVA tumor model. We have further demonstrated significant antitumor activity following vaccination with Trp2, an endogenous melanoma neoantigen.
Materials and methods
Reagents
FTS was synthesized and purified following published literature.9 PEI was purchased from Polysciences (Warrington, PA, USA). Dulbecco’s modified Eagle’s medium (DMEM) and trypsin-EDTA solution were purchased from Sigma-Aldrich (St. Louis, MO, USA). Pierce Firefly Luciferase Glow Assay Kit was purchased from Thermo Scientific (Waltham, MA, USA). OVA polyclonal antibody, fetal bovine serum (FBS), and penicillin-streptomycin solution were purchased from Invitrogen (Grand Island, NY, USA).
Cell culture
Murine melanoma cell line B16F10, murine triple-negative breast carcinoma cell line 4T1.2, and murine colon cancer cell line MC38 were obtained from ATCC (Manassas, VA, USA). 4T1.2-tdTomato subline was kindly provided by Dr. Da Yang. OVA-expressing B16F10 cells were generated by transfection of B16F10 cells with pOVA followed by G418 selection. All cell lines used in this work were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C in a humidified atmosphere with 5% CO2. All cell lines were subject to periodic testing for mycoplasma using the LookOut Mycoplasma PCR Detection Kit (Sigma).
Animals
Female BALB/c and C57BL/6 mice (4−6 weeks) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). All animals were housed under pathogen-free conditions according to the AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) guidelines. All animal-related experiments were performed in full compliance with institutional guidelines and approved by the Animal Use and Care Administrative Advisory Committee at the University of Pittsburgh.
Synthesis of polymers
FTS-PEI was synthesized via condensation reaction using linear PEI (MW = 2,500 Da or 25,000 Da). PEI (129 mg, 3 mmol NH−) was reacted with FTS at different ratios (10.74 mg, 0.03 mmol; 53.7 mg, 0.15 mmol) in DMF (5 mL) at room temperature (RT) under stirring with N,N′-dicyclohexylcarbodiimide (DCC; same equivalent to FTS) as a condensing reagent and DMAP (5 mg) as a catalyst. After 16 h, the reaction mixture was filtered by cotton, followed by addition of 50 mL diethyl ether. The mixture was centrifuged at 4,500 rpm for 10 min, and the precipitate was collected. Finally, the FTS-PEI polymer was obtained by precipitation in ether for 3 times. 1H NMR spectra were examined on a 600.0 MHz Bruker spectrometer using DMSO-d6 as the solvent. MALDI analysis of FTS-PEI and PEI was conducted by Scripps Research as a paid service.
Preparation and physicochemical characterization of nucleic acid/FTS-PEI nanocomplexes
Plasmid DNA (0.5 mg/mL in distilled deionized [DD] water) was mixed with FTS-PEI micelles (2 mg/mL in DD water) at desired N/P ratios. DNA/FTS-PEI nanocomplexes were allowed to incubate at RT for 20 min prior to further characterization. Both in vitro and in vivo studies were performed with freshly prepared nanocomplexes. The particle size and zeta potential of nucleic acid/FTS-PEI nanocomplexes at different N/P ratios were assessed by a Zetasizer (Nano ZS instrument; Malvern, Worcestershire, UK). The morphology of FTS-PEI blank micelles and DNA/FTS-PEI nanocomplexes was examined by TEM. The micelles and DNA/polymer nanocomplexes were mounted on a copper grid with nitrocellulose-supporting film. The samples were negatively stained with uranyl acetate and dried at RT before examination.
Gel retardation assay and DNA replacement assay
pGFP/FTS-PEI complexes were prepared at different N/P ratios, ranging from 0.5 to 10. (pGFP concentration was fixed at 0.02 mg/mL.) The resulting nanocomplexes were then electrophoresed on a 1.5% agarose gel. The gels were prepared with 1.5% agarose in Tris-acetate-EDTA (TAE) buffer containing 0.5 μg/mL ethidium bromide (Biotium, Fremont, CA, USA). Samples were incubated at RT for 20 min, and BlueJuice Gel Loading Buffer (10×; Invitrogen, Grand Island, NY, USA) was added to the samples. Gel electrophoresis was carried out at 120 V for 20 min, and the gel was subsequently visualized using a UV illuminator. Free pGFP was served as a control.
For DNA replacement assay, pGFP/FTS-PEI complexes of various N/P ratios were first prepared. Dextran sulfate was then added to the samples at various S/P ratios (molar ratio between the sulfur from dextran sulfate and the phosphate from nucleic acid). The samples were then similarly electrophoresed as described above.
In vivo transfection of DNA/FTS-PEI nanocomplexes
For in vivo transfection of pGFP/FTS-PEI, C57BL/6J mice aged 4−6 weeks were inoculated subcutaneously (s.c.) with B16F10 cells (1 × 105) into the right flank. Mice were given intratumor injection of blank FTS-PEI polymer or pGFP/FTS-PEI nanocomplexes of different N/P ratios on day 6 after tumor inoculation (50 μg pGFP/mouse). Mice were sacrificed at 48 h post-injection. Tumors and LNs were collected, sectioned, and observed under a fluorescence microscope (BZ-X710; KEYENCE, Osaka, Japan). In addition, the sections were stained with Cy5-labeled anti-GFP monoclonal antibody to further confirm GFP transgene expression. Assessment of in vivo gene transfer using p-luciferase (pLuc) was similarly performed as described above. Mice were sacrificed at 48 h post-injection. Tumors and LNs were collected and subjected to luciferase assay.
Transfection of tumor cells and DCs by pGFP/FTS-PEI nanocomplexes was further assessed by flow cytometry. In this experiment, 4T1.2-tdTomato cells (1 × 105) were s.c. inoculated into the right flank of female BALB/c mice to establish a tumor model. Mice similarly received intratumor injection of pGFP/FTS-PEI nanocomplexes, as described above, and tumors and LNs were collected 48 h later. Tumors and LNs were cut mechanically with scissors and digested with Liberase TL and DNase I. Tissues were further grinded and filtered through a 70-μm cell strainer with red blood cells lysed by ACK lysis buffer. Tumor cells were first gated under the Zombie NIR− and CD45− cell population and further characterized by tdTomato+ expression. DCs were first gated under Zombie NIR− and CD45+ cells as a myeloid cell population and then further characterized by using Gr-1−, CD11b+, and MHC class II+ gating. The graphic representation of gating strategy was detailed in Figure S5. Data are presented as the mean ± standard error of mean (SEM), n = 3. p values were generated by two-tailed Student’s t test; ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Immunization and anti-tumor efficacy
Female C57BL/6J mice aged 4–6 weeks were inoculated s.c. with 1 × 105 B16F10-OVA or B16F10 into the right flank. Vaccination began when tumor sizes were ∼50 mm3. Mice were immunized by intratumor injection of FTS-PEI nanocomplexes loaded with 50 μg pOVA (pOVA/FTS-PEI) or pTrp2 (pTrp2/FTS-PEI). For FTS-PEI nanocomplexes loaded with 50 μg empty backbone plasmid pcDNA (pcDNA/FTS-PEI), free DNA and PBS were used as controls. Three doses were given every 6 days. For the re-challenge model, tumor-bearing mice with or without prior vaccinations were re-challenged by s.c. inoculation of 1 × 104 B16F10-OVA or B16F10 cells per mouse into the contralateral side, the day after the final vaccination. The growth of rechallenged tumor cells was compared to that of the amounts of tumor cells inoculated into naive mice. Tumors were measured every 3 days and calculated following the formula: (L × W2)/2 in which L is the longest dimension (length), and W is the longest perpendicular dimension (width). CO2 inhalation was used to euthanize mice on the day of euthanasia.
Cytotoxicity assay
1 × 104 B16F10 or MC38 cells were seeded into flat-bottom 96-well plates and cultured at 37°C for 6 h. B16F10 and MC38 cells were stained with different concentrations of CFSE (B16F10, 1:20,000; MC38, 1:5,000) prior to being seeded into the plates. CD8+ T cells were isolated from mice immunized with PBS, pcDNA/FTS-PEI, free pOVA, or pOVA/FTS-PEI. 1 × 105 T cells from each group were co-cultured with both B16F10 and MC38 for 6 h. Tumor cells were collected after co-culture for flow cytometry analysis for CFSE intensity. Supernatants were harvested and assayed for IFN-γ by a murine IFN-γ ELISA Kit (BD Biosciences, Pharmingen, San Diego, CA, USA).
PEI-mediated in vitro transfection with cells pre-treated with FTS, Dox, or PTX
B16F10 cells were seeded in 24-well plates at a density of 1 × 104 cells/well followed by overnight incubation in DMEM containing 10% FBS and 1% streptomycin/penicillin. The cells were pre-treated with FTS (0.625−5 μM), Dox (0.3−2.4 μM), or PTX (0.5−4 μM) for 24 h followed by pGFP (1 μg)/PEI transfection in Opti-MEM medium. Cells transfected with pGFP/PEI without drug pre-treatment were used as controls. 24 h later, GFP expression was examined under a fluorescence microscope (BZ-X710; KEYENCE, Osaka, Japan) or quantitatively analyzed by flow cytometry (BD Biosciences LSR II).
Statistical analysis
All values were presented as mean ± SEM. Statistical analysis was performed with two-tailed Student’s t test for comparison between two groups and one-way analysis of variance (ANOVA) for comparison among multiple groups. Results were considered statistically significant if p < 0.05.
Acknowledgments
This work was supported by National Institutes of Health grants R01CA219399, RO1239716, and R01CA223788.
Author contributions
Conceptualization, Y.C., Y.H., and S.L.; methodology, Y.C. and Y.H.; validation, Y.C., Y.H., H.H., Z.L., Z.Z., R.S., and Z.W.; formal analysis, Y.C.; investigation, Y.C., Y.H., H.H., Z.L., Z.Z., R.S., Z.W., J.S., and B.L.; visualization, Y.C. and Y.H.; writing – original draft, Y.C.; writing – review & editing, Y.C. and S.L.; project administration, Y.C. and S.L.; funding acquisition, S.L.; resources, S.L.; supervision, S.L.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.09.006.
Supplemental information
References
- 1.van der Burg S.H., Arens R., Ossendorp F., van Hall T., Melief C.J. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat. Rev. Cancer. 2016;16:219–233. doi: 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- 2.Aldous A.R., Dong J.Z. Personalized neoantigen vaccines: A new approach to cancer immunotherapy. Bioorg. Med. Chem. 2018;26:2842–2849. doi: 10.1016/j.bmc.2017.10.021. [DOI] [PubMed] [Google Scholar]
- 3.Melero I., Gaudernack G., Gerritsen W., Huber C., Parmiani G., Scholl S., Thatcher N., Wagstaff J., Zielinski C., Faulkner I., Mellstedt H. Therapeutic vaccines for cancer: an overview of clinical trials. Nat. Rev. Clin. Oncol. 2014;11:509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 4.Efremova M., Finotello F., Rieder D., Trajanoski Z. Neoantigens Generated by Individual Mutations and Their Role in Cancer Immunity and Immunotherapy. Front. Immunol. 2017;8:1679. doi: 10.3389/fimmu.2017.01679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ott P.A., Hu Z., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sahin U., Derhovanessian E., Miller M., Kloke B.P., Simon P., Löwer M., Bukur V., Tadmor A.D., Luxemburger U., Schrörs B. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547:222–226. doi: 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- 7.Hellmann M.D., Snyder A. Making It Personal: Neoantigen Vaccines in Metastatic Melanoma. Immunity. 2017;47:221–223. doi: 10.1016/j.immuni.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Matsushita H., Vesely M.D., Koboldt D.C., Rickert C.G., Uppaluri R., Magrini V.J., Arthur C.D., White J.M., Chen Y.S., Shea L.K. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–404. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X., Huang Y., Zhao W., Chen Y., Zhang P., Li J., Venkataramanan R., Li S. PEG-farnesyl thiosalicylic acid telodendrimer micelles as an improved formulation for targeted delivery of paclitaxel. Mol. Pharm. 2014;11:2807–2814. doi: 10.1021/mp500181x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miao L., Li L., Huang Y., Delcassian D., Chahal J., Han J., Shi Y., Sadtler K., Gao W., Lin J. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 2019;37:1174–1185. doi: 10.1038/s41587-019-0247-3. [DOI] [PubMed] [Google Scholar]
- 11.Zhu G., Zhang F., Ni Q., Niu G., Chen X. Efficient Nanovaccine Delivery in Cancer Immunotherapy. ACS Nano. 2017;11:2387–2392. doi: 10.1021/acsnano.7b00978. [DOI] [PubMed] [Google Scholar]
- 12.Wilson D.S., Hirosue S., Raczy M.M., Bonilla-Ramirez L., Jeanbart L., Wang R., Kwissa M., Franetich J.F., Broggi M.A.S., Diaceri G. Antigens reversibly conjugated to a polymeric glyco-adjuvant induce protective humoral and cellular immunity. Nat. Mater. 2019;18:175–185. doi: 10.1038/s41563-018-0256-5. [DOI] [PubMed] [Google Scholar]
- 13.Xu J., Sun J., Ho P.Y., Luo Z., Ma W., Zhao W., Rathod S.B., Fernandez C.A., Venkataramanan R., Xie W. Creatine based polymer for codelivery of bioengineered MicroRNA and chemodrugs against breast cancer lung metastasis. Biomaterials. 2019;210:25–40. doi: 10.1016/j.biomaterials.2019.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H., Moynihan K.D., Zheng Y., Szeto G.L., Li A.V., Huang B., Van Egeren D.S., Park C., Irvine D.J. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519–522. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjaminsen R.V., Mattebjerg M.A., Henriksen J.R., Moghimi S.M., Andresen T.L. The possible “proton sponge ” effect of polyethylenimine (PEI) does not include change in lysosomal pH. Mol. Ther. 2013;21:149–157. doi: 10.1038/mt.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W., Joshi M.D., Singhania S., Ramsey K.H., Murthy A.K. Peptide Vaccine: Progress and Challenges. Vaccines (Basel) 2014;2:515–536. doi: 10.3390/vaccines2030515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Appelbe O.K., Kim B.K., Rymut N., Wang J., Kron S.J., Yeo Y. Radiation-enhanced delivery of plasmid DNA to tumors utilizing a novel PEI polyplex. Cancer Gene Ther. 2018;25:196–206. doi: 10.1038/s41417-017-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J., Meng F., Kim B.K., Ke X., Yeo Y. In-vitro and in-vivo difference in gene delivery by lithocholic acid-polyethyleneimine conjugate. Biomaterials. 2019;217:119296. doi: 10.1016/j.biomaterials.2019.119296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X., Lu J., Huang Y., Zhao W., Chen Y., Li J., Gao X., Venkataramanan R., Sun M., Stolz D.B. PEG-farnesylthiosalicylate conjugate as a nanomicellar carrier for delivery of paclitaxel. Bioconjug. Chem. 2013;24:464–472. doi: 10.1021/bc300608h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manguso R.T., Pope H.W., Zimmer M.D., Brown F.D., Yates K.B., Miller B.C., Collins N.B., Bi K., LaFleur M.W., Juneja V.R. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–418. doi: 10.1038/nature23270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fifis T., Gamvrellis A., Crimeen-Irwin B., Pietersz G.A., Li J., Mottram P.L., McKenzie I.F., Plebanski M. Size-dependent immunogenicity: therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004;173:3148–3154. doi: 10.4049/jimmunol.173.5.3148. [DOI] [PubMed] [Google Scholar]
- 22.Szeto G.L., Van Egeren D., Worku H., Sharei A., Alejandro B., Park C., Frew K., Brefo M., Mao S., Heimann M. Microfluidic squeezing for intracellular antigen loading in polyclonal B-cells as cellular vaccines. Sci. Rep. 2015;5:10276. doi: 10.1038/srep10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ali O.A., Huebsch N., Cao L., Dranoff G., Mooney D.J. Infection-mimicking materials to program dendritic cells in situ. Nat. Mater. 2009;8:151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alspach E., Lussier D.M., Miceli A.P., Kizhvatov I., DuPage M., Luoma A.M., Meng W., Lichti C.F., Esaulova E., Vomund A.N. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696–701. doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snapper C.M. Distinct Immunologic Properties of Soluble Versus Particulate Antigens. Front. Immunol. 2018;9:598. doi: 10.3389/fimmu.2018.00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson K.A., Holland D.J., Wetmore S.D. Topology of RNA-protein nucleobase-amino acid π-π interactions and comparison to analogous DNA-protein π-π contacts. RNA. 2016;22:696–708. doi: 10.1261/rna.054924.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y., Sun J., Huang Y., Liu Y., Liang L., Yang D., Lu B., Li S. Targeted codelivery of doxorubicin and IL-36γ expression plasmid for an optimal chemo-gene combination therapy against cancer lung metastasis. Nanomedicine (Lond.) 2019;15:129–141. doi: 10.1016/j.nano.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marusyk A., Polyak K. Tumor heterogeneity: causes and consequences. Biochim. Biophys. Acta. 2010;1805:105–117. doi: 10.1016/j.bbcan.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.