

Abstract
Herein, we assessed the anti-inflammatory and intestinal barrier protective effects of butyrolactone-I (BTL-1), derived from the coral-derived endophytic fungus (Aspergillus terreus), using the LPS-induced IPEC-J2 inflammation model and the DSS-induced IBD model in mice. In IPEC-J2 cells, pretreatment with BTL-I significantly inhibited TLR4/NF-κB signaling pathway and JNK phosphorylation, resulting in the decrease of IL-1β and IL-6 expression. Interestingly, BTL-1 pretreatment activated the phosphorylation of ERK and P38, which significantly enhanced the expression of TNF-α. Meanwhile, BTL-1 pretreatment upregulated tight junction protein expression (ZO-1, occludin, and claudin-1) and maintained intestinal barrier and intestinal permeability integrity. In mice, BTL-1 significantly alleviated the intestinal inflammatory response induced by DSS, inhibited TLR4/NF-κB signaling pathway, and MAPK signaling pathway, thus reducing the production of IL-1, IL-6, and TNF-α. Further, the expression of tight junction proteins (ZO-1, occludin, and claudin-1) was upregulated in BTL-1 administrated mice. Therefore, it has been suggested that butyrolactone-I alleviates inflammatory responses in LPS-stimulated IPEC-J2 and DSS-induced murine colitis by TLR4/NF-κB and MAPK signal pathway. Thereby, BTL-1 might potentially be used as an ocean drug to prevent intestinal bowel disease.
Keywords: butyrolactone-I, anti-inflammatory, intestinal barrier, IBD, TLR4/NF-κB, MAPK
Graphical Abstract.
Butyrolactone-I alleviates inflammatory responses in LPS-stimulated IPEC-J2 and DSS-induced murine colitis by TLR4/NF-κB and MAPK signal pathway.

Introduction
Inflammatory bowel diseases (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), are a collection of chronically non-specific inflammatory disorders featured with chronic inflammation and abnormalities in the gastrointestinal epithelial barrier (1, 2). Although the cause of IBD has not yet been fully elucidated, current research assumed that it is mainly caused by the interaction of multiple factors, such as genetics, immunity, and gut microbes (3, 4). Presently, IBD has a high prevalence rate worldwide, and the incidence rate continues to increase. Its recurrence, persistence, and cancer risk significantly impact patients' lives. At present, there is no specific treatment for IBD. The ultimate goal of IBD treatment is to achieve clinical remission with sustained remission. IBD can be treated with glucocorticoids and immunosuppressant's; however, these drugs have substantial adverse effects (such as nephrotoxicity, hemolytic anemia, osteoporosis, insomnia, and pancreatitis) and poor clinical outcomes (5). Therefore, developing a new medicine with fewer side effects is warranted for treating IBD.
Previous studies have shown that TLR4 is the primary membrane signal receptor causing IBD inflammation. Several standard signaling' pathways, including the nuclear factor-κ B (NF-κB) and mitogen-activated protein kinases (MAPKs) pathways, play a potential role in this process (6). NF-κB is a ubiquitous transcriptional factor promoting the production of pro-inflammatory cytokines and chemokines and is essential in inflammation and immune response. IBD and other autoimmune diseases have also been linked to the NF-κB protein (7). MAPKs are composed of various serine and threonine kinases that induce inflammation (8). Other evidence suggests that inhibiting the NF-κB and MAPK signaling pathways is vital in IBD treatment (9).
Impaired epithelial barrier function is a typical symptom of IBD. Inflammation can damage the intestinal barrier integrity, resulting in the translocation of pathogens and infiltration of apical tight junction proteins (10). As a critical player in the epithelial barrier function of inflammatory bowel disease, tight junction proteins are essential for maintaining the epithelial barrier function and controlling the paracellular permeability (11). Thus, far, the relationship between the imbalance of tight junction (TJ) proteins and the occurrence of intestinal inflammation is still unclear. However, studies have pointed out that the imbalance of tight junction protein expression may precede intestinal inflammation (12), suggesting that the treatment of IBD by restoring the integrity of the epithelial barrier could be the future trend in treatment (13).
Butyrolactone-I (BTL-1) is derived from Aspergillus terreus C23-3, an endophytic fungus isolated from Puko Shore coral (Porites pukoensis) in Xuwen (Guangdong, China) (14). Previous studies have found that butyrolactone-I, as a cyclin CDC-2 kinase and CDK-2 kinase inhibitor (15), can prevent oocytes from decelerating division, inhibit tumor cell proliferation, induce cancer cell apoptosis, and prevent the apoptosis of central and peripheral neurons (16–21). Presently, researchers have discovered that it has antibacterial and anti-inflammatory activities and inhibits the secretion of NO and TNF-α in macrophages and microglia (22). However, whether BTL-1 can protect the intestinal barrier integrity and be used as a potential therapeutic drug for IBD is not fully elucidated. Herein, we established an LPS induced-inflammation model in IPEC cell and DSS-induced IBD model in mice and investigated the inhibitory effect of BTL-1 on intestinal inflammation and its protective effect on intestinal barrier integrity.
Materials and Methods
Chemicals and Reagents
Lipopolysaccharide (LPS) and Triton-X-100 were procured from Sigma-Aldrich (St. Louis, MO, USA). Cell Counting Kit-8 (CCK8) was secured from APExBIO Technology (Houston, USA). The commercial-specific complete Dulbecco's Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12), penicillin, and streptomycin were acquired from GIBCO (Carlsbad, CA, USA). The ECL kit was provided by Tanon (Shanghai, China). In addition, 40, 6-diamidino-2-phenylindole (DAPI) was purchased from Beyotime (Shanghai, China). Primary and secondary antibodies used for western blotting were brought from Cell Signaling Technology (Danvers, MA, USA). Biotin-conjugated anti-Rabbit IgG antibody and DyLight 594-conjugated Avidin were picked up from Jackson ImmunoResearch (Pennsylvania, USA).
Preparation of Butyrolactone-I
Butyrolactone-I (BTL-1) was prepared according to the method described elsewhere (23). The producer strain C23-3 was fermented at 28°C for 1 week in a 40 × 1 L flask on a rotary shaker at 150 rpm. The culture was filtered to isolate the broth and mycelium and then extracted with ethyl acetate and a combination of ethyl acetate and chloroform-methanol (1:1, v/v) after fermentation. The two extracts were mixed and concentrated in vacuo to make a crude extract. The extract was separated into nine fractions using a Sephadex LH-20 column treated with chloroform-methanol (1:1, v/v) (Fr 1 to Fr 9). Fr 7 was further purified using a Sephadex LH-20 column eluted with methanol and a preparative RP-18 HPLC eluted with a 7:3 methanol-water mixture, yielding pure compound BTL-1 (14).
Induction of Experimental Colitis
Male C57BL/6J mice weighing 6–8 weeks (19–21 g) were procured from the Guangzhou Dean Gene co., ltd (Guangzhou, China) and reared in a specific pathogen-free environment with controlled temperature (26 ± 1°C) and 12-h light/dark cycle in the animal hospital of Guangdong Ocean University. The animals were acclimatized for 1 week before experimental work and given ad libitum access to food and water. They were fed ad libitum DSS (MW 36,000–50,000, Dalian Meilun Biotechnology Co., Ltd, Dalian, China) in drinking water (3.5%, v/w) for 7 days to develop colitis (24). The animals were randomly allocated to one of the following five groups (n = 5): normal control (only given food and water without DSS), DSS model (administered DSS in drinking water), low-concentration BTL-1 (administered 1 mg/kg BTL-1, p.o), middle-concentration BTL-1 (taken 2 mg/kg BTL-1, p.o), and high-concentration BTL-1 group (given 5 mg/kg BTL-1, p.o) group. A schematic overview of the experiment design is shown in Figure 1. The body weight, feed intake, and disease activity index (DAI) were recorded 3 days before feeding BTL-1. The DAI, includes changes in the consistency of stool (0, none; 1 and 2, loose stool; 3 and 4, diarrhea), bleeding (0, none; 1, trace of fecal occult blood; 2, mild occult blood; 3, obvious occult blood; 4, gross bleeding), and weight loss (0, none; 1, 0–5%; 2, 5–10%; 3, 10–20%; 4, > 20%) (25). After the mice were euthanized, the spleen, thymus, liver, kidney, colon, and colon were collected for further analysis.
Figure 1.

Mice treatment scheme.
Assessment of Disease Activity Index (DAI) Score
Stool consistency, body weight, and blood in the stool were determined according to a previously published grading system (26) to assess the severity of colitis. Briefly, blood in stool was scored as follows: score 0, normal; score 2, slight bleeding; and score 4, gross bleeding. Diarrhea was scored as follows: score 0, normal; score 2, loose stools; and score 4, watery diarrhea. Weight loss was scored as follows: score 0, none; score 1, 1–5%; score 2, 5–10%; score 3, 10–20%; and score 4, >20%.
Cell Culture and Viability Assay
The IPEC-J2 cells obtained from the Guoqiang Zhu of Yangzhou University (Yangzhou, China) were grown at 37°C in a humidified % CO2 incubator in DMEM/F12 supplemented with 10% FBS and 1% penicillin/streptomycin. An IPEC-J2 cell viability experiment was carried out using the Cell Counting Kit-8. Briefly, IPEC-J2 cells were scattered at density 1 × 104 cells/mL in 96-well plates and were treated with BTL-1 (10, 20, 50, and 100 μM) for 24 h. After that, 10 μL CCK-8 solution was added and incubated for another 1 h. The optical density at 450 nm was used to measure the cell viabilities using a microplate reader (BioTek, Vermont, USA).
Cytokine Assay
IPEC-J2 cells were seeded at a density of 5 × 106 cells/well in 6-well plates to investigate the inhibitory effect of BTL-1 on inflammatory cytokines. The cells were pretreated for 24 h with BTL-1 (10, 20, and 50 μM) and then activated for 1 h with LPS (50 μg/mL). The tissue waste (culture medium) was collected and centrifuged at 3,000 rpm for 10 min. Serum was treated the same as the culture medium. For cytokine detection, colon tissue was ground thoroughly and centrifuged at 3,000 rpm for 10 min. The level of IL-1β, TNF-α, IL-6, and IL-10 in the culture medium, serum, and colonic tissue were quantified using ELISA kits (Meimian, Shanghai, China).
Western Blotting Analysis
Protein levels were measured using a BCA reagent after IPEC-J2 cells, and colonic tissues were homogenized (Cwbio, Beijing, China). According to the manufacturer's recommendations, the cytoplasmic and nuclear extractions were carried out (keygen, Jiangsu, China). SDS-PAGE was used to load and electrophorese the samples, which were subsequently transferred through polyvinylidene fluoride (PVDF) membranes. After 30 min of blocking in QuickBlock™ Blocking Buffer (Beyotime, Shanghai, China), the membranes were incubated with primary antibodies. The following primary antibodies were used: TLR4, P65, phospho-P65, IκBα, phospho-IκBα, phospho-ERK1/2, ERK 1/2, phospho-P38, P38, phospho-JNK, JNK, occludin, ZO-1, claudin1, and β-actin at 1:1,000 dilution. Secondary antibodies include horseradish peroxidase (HRP)-conjugated goat anti-rabbit, and anti-mouse IgG at 1:1,000 dilution (Cell Signaling Technology, Danvers, MA, USA).
Total RNA Extraction and qPCR
RNA was extracted using TRIZOL followed by cDNA synthesis via a reverse transcriptase. Quantitative RT-PCR analyses were performed using SYBR Green (TransGen Biotech, Beijing, China). The results were evaluated using the 2–ΔΔCT method and normalized to β-actin expression. The following primers were used (Table 1).
Table 1.
Primer sequence.
| Gene ID | Primer | Sequence (5'-3') |
|---|---|---|
| 414396 | pig β-actin Forward | GCTGTCCCTGTATGCCTCT |
| pig β-actin Reverse | GATGTCACGCACGATTTCC | |
| 399500 | pig IL-6 Forward | ATAAGGGAAATGTCGAGGCTGTGC |
| pig IL-6 Reverse | GGGTGGTGGCTTTGTCTGGATTC | |
| 397122 | pig IL-1β Forward | CAAGCCAGAGAAGCAAGGTGTCC |
| pig IL-1β Reverse | GCCGTCCTCAGCAGCAAGAAG | |
| 397106 | pig IL-10 Forward | AGCCAGCATTAAGTCTGAGAACAGC |
| pig IL-10 Reverse | GGTCAGCAACAAGTCGCCCATC | |
| 397086 | pig TNF-α Forward | AAAGGACACCATGAGCACGGAAAG |
| pig TNF-α Reverse | CGCCACGAGCAGGAATGAGAAG | |
| 399541 | pig TLR4 Forward | GCCATCGCTGCTAACATCATC |
| pig TLR4 Reverse | CTCATACTCAAAGATACACCATCGG | |
| 100135665 | pig P65 Forward | CTGAGGCTATAACTCGCTTGGTGAC |
| pig P65 Reverse | CATGTCCGCAATGGAGGAGAAGTC | |
| 100157586 | pig IκB Forward | GGCAGTGACATGAGTGGCAGATAC |
| pig IκB Reverse | CTTCCTTGAGGCTGTGCTTCTTCC | |
| 445013 | pig ERK1 Forward | ACGTCATTGGCATCCGAGACATTC |
| pig ERK1 Reverse | GAGGAAGTAGCAGATGTGGTCGTTG | |
| 100626611 | pig P38 Forward | CGCCTGTGAAGACCTCCTTGAAC |
| pig P38 Reverse | TTCCTGTCCTCCACCTTCCGAAG | |
| 396610 | pig JNK Forward | ACTACAGAGCACCTGAGGTCATCC |
| pig JNK Reverse | ATTTCTCCCATAATGCACCCCACAG | |
| 397236 | pig Occludin Forward | CAGTGGTAACTTGGAGGCGTCTTC |
| pig Occludin Reverse | CGTCGTGTAGTCTGTCTCGTAATGG | |
| 100736682 | pig ZO-1 Forward | CCAGGGAGAGAAGTGCCAGTAGG |
| pig ZO-1 Reverse | TTTGGTGGGTTTGGTGGGTTGAC | |
| 100625166 | pig Claudin1 Forward | AGAAGATGCGGATGGCTGTCATTG |
| pig Claudin1 Reverse | ACCATACCATGCTGTGGCAACTAAG | |
| 11461 | mouse β-actin Forward | GTGACGTTGACATCCGTAAAGA |
| mouse β-actin Reverse | GCCGGACTCATCGTACTCC | |
| 16193 | mouse IL-6 Forward | CTCCCAACAGACCTGTCTATAC |
| mouse IL-6 Reverse | CCATTGCACAACTCTTTTCTCA | |
| 16176 | mouse IL-1β Forward | GAAATGCCACCTTTTGACAGTG |
| mouse IL-1β Reverse | TGGATGCTCATCAGGACAG | |
| 16153 | mouse IL-10 Forward | TTCTTTCAAACAAAGGACCAGC |
| mouse IL-10 Reverse | GCAACCCAAGTAACCCTTAAAG | |
| 21926 | mouse TNF-α Forward | ATGTCTCAGCCTCTTCTCATTC |
| mouse TNF-α Reverse | GCTTGTCACTCGAATTTTGAGA | |
| 21898 | mouse TLR4 Forward | GCCATCATTATGAGTGCCAATT |
| mouse TLR4 Reverse | AGGGATAAGAACGCTGAGAATT | |
| 19697 | mouse P65 Forward | TGCGATTCCGCTATAAATGCG |
| mouse P65 Reverse | ACAAGTTCATGTGGATGAGGC | |
| 12675 | mouse IκB Forward | TTGGGTTATGCCAAAGATGTTG |
| mouse IκB Reverse | GCTGTGTACGGCTTATTTTCAA | |
| 26417 | mouse ERK1 Forward | CAGCTCAACCACATTCTAGGTA |
| mouse ERK1 Reverse | TCAAGAGCTTTGGAGTCAGATT | |
| 26416 | mouse P38 Forward | AGGAATTCAATGACGTGTACCT |
| mouse P38 Reverse | AGGTCCCTGTGAATTATGTCAG | |
| 26419 | mouse JNK1 Forward | TTGAAAACAGGCCTAAATACGC |
| mouse JNK1 Reverse | GTTTGTTATGCTCTGAGTCAGC | |
| 18260 | mouse Occludin Forward | TGCTTCATCGCTTCCTTAGTAA |
| mouse Occludin Reverse | GGGTTCACTCCCATTATGTACA | |
| 21872 | mouse ZO-1 Forward | CTGGTGAAGTCTCGGAAAAATG |
| mouse ZO-1 Reverse | CATCTCTTGCTGCCAAACTATC | |
| 12737 | mouse Claudin1 Forward | AGATACAGTGCAAAGTCTTCGA |
| mouse Claudin1 Reverse | CAGGATGCCAATTACCATCAAG |
Immunofluorescence
IPEC-J2 cells were seeded on slide cover glass at 5 × 105 cells/mL, fixed in 4% paraformaldehyde at 4°C for 30 min, and then rinsed with phosphate-buffered saline (PBS). Cells were permeabilized with 0.2 % Triton-X-100, then rinsed with PBS before being blocked with 5% BSA. After that, the cells were incubated overnight with primary antibodies p-P65 and ZO-1 at 1:100 dilution and incubated with a biotin-conjugated anti-Rabbit IgG antibody for 1 h. Afterward, the cells were washed with PBS and treated for 1 h with DyLight 594-conjugated Avidin. Fluorescence microscopy was used to examine the nuclei stained with DAPI (Olympus, Tokyo, Japan).
In vitro Intestinal Paracellular Permeability Assay
The IPEC-J2 cells were seeded onto transwell at a density of 2 × 105 cells/mL and reached confluence. Cells were cultured for 16 days before use. Then, IPEC-J2 cells were washed three times with PBS to remove the antibiotic media. The treatment is the same with section Induction of Experimental Colitis. The Trans Epithelial Electric Resistance (TEER) was measured by a transmembrane resistance meter (MRRCK, NJ, USA). After TEER was measured, barrier function was determined by upper compartment to lower compartment flux of 4 kDa fluorescein isothiocyanate-labeled dextran (FITC-dextran). The upper compartment was filled with FITC-dextran (0.5 mL, 1 mg/mL) (27), and 1 h later, the concentrations of FITC-dextran in the lower compartment were determined using a fluorometer (Thermo, MA, USA; excitation, 427 nm; emission, 536 nm).
In vivo Intestinal Permeability Assay
This assay was based on the intestinal permeability toward 4 kDa FITC-dextran as described elsewhere (28). Briefly, rats fasted for 8 h were given 4 kDa FITC-dextran (MRRCK, NJ, USA) by gavage (40 mg/100 g body weight, 100 mg/mL). The blood was collected from the submandibular venous plexus 4 h later and centrifuged at 4°C at 4,000 rpm for 10 min. Plasma was diluted with an equal volume of PBS (pH 7.4) and analyzed for FITC-dextran concentration using a fluorometer (Thermo, MA, USA; excitation, 427 nm; emission, 536 nm).
H&E Staining
The colon was embedded in paraffin, sectioned, and placed on a microscope slide after being preserved in 4% phosphate-buffered paraformaldehyde. Hematoxylin and eosin (H & E) were used to stain the slides, and the histopathological changes were evaluated using an optical microscope (Olympus, Tokyo, Japan).
Molecular Docking
We used Chembiodraw Ultra 14.0 to draw the structure of BTL-1. After that, we used Chembio3d Ultra 14.0 to transform it into a 3D structure and optimized it with an mmff94 force field. 3D structure of Toll-like receptor (TLR4) (PDB ID: 2z64) was obtained from RCSB protein data bank (www.rcsb.org). Both TLR4 and BTL-1 were transformed into pdbqt format by autodock tools 1.5.6 (29, 30). In this project, autodock Vina 1.1.2 (31) was used for molecular docking. The coordinate of TLR4 active site is: Center_ x = −28.441, center_ y = −17.705, center_ z = −22.582, size_ x = 20, size_ y = 20, size_ z = 20. To increase the accuracy of the calculation, we set the parameter exhaust to 100. Except for special instructions, other parameters use default values. Finally, the conformation with the highest score was selected and analyzed with PyMOL 1.7.6.
Identification of BTL-1
Butyrolactone-I was obtained in the form of yellow oil after repeated column chromatography of the extract. Its NMR and specific optical rotation data listed below are identical to previous reports (14, 32).
1H NMR (CD3OD, 700 MHz): δH 7.59 (H-2'and H-6', d, 8.4), 6.87 (H-3' and H-5', d, 9.1), 6.54 (H-6”, dd, 8.1, 2.1), 6.50 (H-5”, d, 8.4), 6.41 (H-2”, d, 2.1), 5.07 (H-8”, m), 3.78 (H-7, s),3.43 (H-5, d, 14.0), 3.08 (H-7”, m), 1.67 (H-10”, s), 1.57 (H-11”, s). 13C-NMR NMR (CD3OD, 175 MHz):δC 171.41 (C-6), 170.11 (C-1), 159.13 (C-4'), 154.88 (C-4”), 139.46 (C-2), 132.78 (C-9”), 132.19 (C-2”),130.19 (C-2' and C-6'), 129.56 (C-3”), 129.01 (C-3), 128.23 (C-6”), 124.85 (C-1”), 123.36 (C-8”),122.94 (C-1'), 116.41 (C-3' and C-5'), 114.83 (C-5”), 86.59 (C-4), 53.64 (C-7), 39.41 (C-5), 28.48 (C-7”), 25.75 (C-10”), 17.56 (C-11”) (Figure 2). [α] 108.85°(c 0.2, MeOH).
Figure 2.
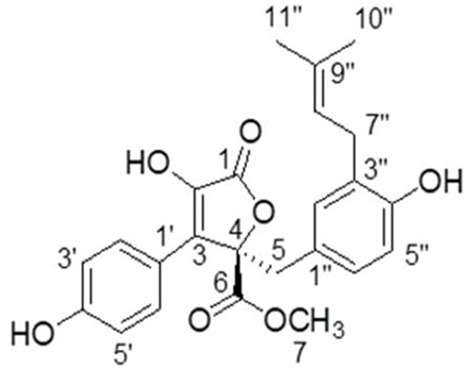
The structure of butyrolactone-I.
Statistical Analyses
All experimental results were obtained from at least three independent experiments. For statistical analyses, the values are expressed as mean ± SD (n = 3). One-way ANOVA and t-test determined the statistical significance. A value of P < 0.05 was considered statistically significant. IBM SPSS Statistics 26 was used for all statistical analyses, and GraphPad Prism 7.0 (GraphPad Software) was used for all column and line charts.
Results
BTL-1 Decreases IL-6 and IL-1β and Increases TNF-α in IPEC-J2 Exposed to LPS
The toxicity of BTL-1 on IPEC-J2 cells was first examined by CCK8 assay. After treatment with several concentrations of BTL-1 for 24 h, the cell proliferation rate was dramatically decreased when exposed to 100 μM BTL-1; thereby, the highest concentration for in vivo study was set at 50 μM (Figure 3A). Three concentrations (10, 20, and 50 μM) were used in subsequent experiments. As shown in Figure 3B, LPS considerably decreased the cell proliferation rate of IPEC-J2, whereas BTL-1 increased that in a dose-dependent manner. Further, LPS significantly upregulates the expression of IL-6, IL-10, IL-1β, and TNF-α at mRNA and protein levels, whereas BTL-1 decreased IL-10 and IL-1β in a dose-dependent way (Figures 3C–J). Interestedly, BTL-1 significantly increased the expression of TNF-α. In short, these findings suggest that BTL-1 may have a protective impact on the IPEC-J2 cell model of LPS-induced inflammation.
Figure 3.

Impact of BTL-1 on inflammatory cytokines production in LPS-induced IPEC-J2 cell. The outcome of BTL-1 on IPEC-J2 cell viability (A,B). The consequences of BTL-1 on the expression of cytokines were determined by ELISA (C–F) and qPCR (G–J). The consequences of BTL-1 on the expression of cytokines were determined by ELISA. The cells were treated with BTL-1 (10, 20, and 50 μM) for 24 h and then treated with LPS (50 μg/mL) for another 1 h. The data shown are representative of three independent experiments. The results were expressed as the means ± SEM. #P < 0.05, ##P < 0.01, compared to non-treated group, *P < 0.05, **P < 0.01 compared to LPS group.
BTL-1 Inhibits TLR4/NF-κB Signaling Pathway in IPEC-J2 Cells Exposed to LPS
TLR4, IκB, P65, and the phosphorylated IκB and P65 were measured to evaluate the sequel of BTL-1 on the TLR4/NF-κB signaling pathway. As shown in Figures 4A–C, the mRNA expressions of TLR4, p65, and IκB were significantly upregulated after LPS treatment. However, they have downregulated in a dose-dependent manner after BTL-1 pretreatment. Meanwhile, when the cells were exposed to LPS, TLR4 and the phosphorylated P65 and IκB were greatly enhanced; however, they were significantly decreased when the cells were pretreated with BTL-1 (Figures 4D–G).
Figure 4.

Inhibitory effects of BTL-1 on TLR4/NF-κB signaling pathway. BTL-1 inhibits TLR4/NF-κB signaling pathway as determined by qPCR (A–C) and western blotting (D–G). BTL-1 inhibits the translocation of phosphorylated P65 as determined by western blotting (D,H) and IF (I). The data are representative of three independent experiments. The results were expressed as the means ± SEM. #P < 0.05, ##P < 0.01, compared to non-treated group, *P < 0.05, **P < 0.01 compared to LPS group.
Western blotting and IF measured translocation of phosphorylated P65 from the cytoplasm into the nucleus. As shown in Figures 4D,H, LPS significantly increased the translocation of phosphorylated P65 from the cytoplasm to the nucleus; however, the translocation was inhibited when the cells were pretreated with BTL-1. In IPEC-J2 cells, immunofluorescence was used to observe the site of phosphorylated P65. In the blue channel, the nucleus was stained with DAPI, and the phosphorylated P65 was stained red. The phosphorylated P65 was found outside the nucleus with weak immunofluorescence in the control group. Still, the immunofluorescence became brighter and approached the nucleus when the cells were exposed to LPS, suggesting that LPS enhances translocation of phosphorylated P65 from the cytoplasm to the nucleus. When cells were pretreated with BTL-1, the translocation of phosphorylated P65 was inhibited. The above results denote that BTL-1 can inhibit the TLR4/NF-κB signaling pathway by downregulating the expression of TLR4, the phosphorylation of IκB and P65, and reduces the translocation of phosphorylated P65 (Figure 4I).
BTL-1 Upregulates TNF-α via MAPK Signaling Pathway in IPEC-J2 Exposed to LPS
P38, ERK1/2, JNK, and the phosphorylation of P38, ERK1/2, and JNK were tested to assess the impact of BTL-1 on the MAPK signaling pathway. As shown in Figures 5A–C, the mRNA expressions of P38, ERK1, and JNK were significantly upregulated after LPS treatment. However, after treatment with BTL-1, the mRNA expression levels of ERK1 and P38 were further upregulated. In contrast, JNK was significantly downregulated after BTL-1 treatment. Additionally, the western blotting analysis showed that the phosphorylation of P38, ERK1/2, and JNK was dramatically increased when the cells were exposed to LPS. In contrast, phosphorylation of ERK1 and P38 were significantly decreased following BTL-1 treatment (Figures 5D–G). On the contrary, the phosphorylated JNK was downregulated considerably after BTL-1 treatment.
Figure 5.

Effect of BTL-1 on MAPK signaling pathway. The impact of BTL-1 on the MAPK signaling pathway determined by qPCR (A–C). The outcome of BTL-1 on the MAPK signaling pathway determined by western blotting (D–G). Effect of P38 inhibitor (SB203580, 10 μM, 1 h) on the phosphorylation of P38. (H,I) determined by western blotting. Impact of P38 inhibitor on the mRNA and protein of TNF-α determined by qPCR and ELISA. (J,K). The outcome of ERK1/2 inhibitor (PD98095,50 μM, 1 h) on the phosphorylation of ERK1/2 (L,M) determined by western blotting analysis. Inhibitory effect of ERK1/2 inhibitor on the mRNA and protein of TNF-α determined by qPCR and ELISA (N,O). The data shown are representative of three independent experiments. The inhibitors of ERK1/2 and P38 were added 1 h before LPS and BTL-1 treatment, and the cells were washed with PBS three times before adding LPS and BTL-1. The results were expressed as the means ± SEM. #P < 0.05, ##P < 0.01, compared to non-treated group, *P < 0.05, **P < 0.01 compared to LPS group.
ERK1/2 and P38 inhibitors were added to the IPEC-J2 cell to investigate the mechanism contributing to increased TNF-α after BTL-1 treatment. As shown in Figures 5H,I, the phosphorylation of P38 was significantly inhibited by adding a P38 inhibitor (SB203580,10 μM). Meanwhile, the mRNA and protein expression of TNF-α was decreased when SB203580 was added (Figures 5J,K). Same as phosphorylated P38, the activity of phosphorylated ERK1/2 was reduced after adding inhibitor PD98095 (Figures 5L–O). Moreover, the mRNA and protein expression of TNF-α was significantly decreased after inhibition of ERK1/2 phosphorylation. The above findings indicate that the expression of TNF-α was closely associated with the phosphorylation of P38 and ERK1/2.
BTL-1 Protects Intestinal Paracellular Permeability via Upregulating Tight Junction Proteins
From the finding mentioned above, BTL-1 could exert an anti-inflammatory effect. Subsequently, we evaluated the intestinal barrier protection function of BTL-1 in vitro. The transepithelial resistance was stable on 14–16th day after cell culture (Figure 6A), followed by BTL-1 treatment and LPS stimulation. As shown in Figures 5B,C, exposure to LPS significantly decreased transmembrane resistance (TEER), 1 h post-exposure (Figure 6B). In addition, it increased the passage of FITC-dextran (FITC-D) (Figure 6C), indicating that LPS significantly impaired epithelial barrier integrity. Treatment of IPEC-J2 cells with BTL-1 significantly increased TEER and decreased the paracellular flux of FITC-D in a dose-dependent manner.
Figure 6.

In vitro effect of BTL-1 on intestinal paracellular permeability. Transmembrane resistance of IPEC-J2 monolayer measured by IPEC-J2 cell resistance voltmeter (A). The effect of BTL-1 on transmembrane resistance in vitro intestinal paracellular permeability was measured by a fluorometer (B). The effect of BTL-1 on FITC-D fluorescence intensity in the lower compartment measured by a fluorescence microplate reader (C). The effect of BTL-1 on TJ protein determined by qPCR (D–F), IF (G), and western blotting (H–K). The data are representative of three independent experiments. The results were expressed as the means ± SEM. #P < 0.05, ##P < 0.01, compared to non-treated group, *P < 0.05, **P < 0.01 compared to LPS group.
Subsequently, the expression of TJ proteins was determined. As shown in Figures 6D–F, the mRNA expressions of ZO-1, occludin, and claudin1 were significantly downregulated after LPS treatment; however, they were upregulated following BTL-1 treatment. Meanwhile, when the cells were exposed to LPS, ZO-1, occludin, and claudin1 were significantly reduced; nevertheless, they significantly increased when treated with BTL-1 (Figures 6H–K) protein level. The immunofluorescence analysis showed that ZO-1 expression was decreased after LPS treatment (Figure 6G). However, when the cells were treated with BTL-1, ZO-1 expression was significantly enhanced. These results suggest that LPS may inhibit the expression of tight junction proteins, thereby increasing intestinal paracellular permeability. In contrast, BTL-1 can increase the expression of TJ proteins in a dose-dependent manner to protect the integrity of the intestinal barrier.
BTL-1 Protects DSS-Induced IBD in Mice
Previous experiments confirmed that BTL-1 has a protective effect on the LPS-induced inflammation model in IPEC-J2 cells. Subsequently, we examined the impact of BTL-1 on the DSS-induced IBD model in mice. Before DSS treatment, intragastric administration of BTL-1 reduced the average daily feed intake (Figure 7A) of mice; however, it did not affect the body weight (Figure 7B) and disease activity index (DAI) (Figure 7C). After DSS treatment, the average daily gain of mice decreased significantly. Further, the symptoms of wilting, loose stool, bloody stool, and anemia (Figure 7J) were appeared, leading to the increase in the DAI. At the same time, DSS significantly increased the immune organ index (thymus index and spleen index) (Figures 7D–G) and the total number of white blood cells (Figure 7K), decreased the length of the colon (Figures 7H,I). Thus, BTL-1 could rescue the symptoms and pathological changes caused by DSS in a dose-dependent manner compared with the DSS group. These results denote that BTL-1 has a potential protective effect on DSS-Induced IBD of mice.
Figure 7.

The effect of BTL-1 on DSS-induced IBD model in mice. Mice were given DSS for 7 days with or without BTL-1, the PBS group was given only PBS, and the control group was fed normally. The effect of BTL-1 on average daily feed intake (A), average daily gain (B), DAI (C), organ index (D–G), and colon (H,I) of IBD mouse model induced by DSS. The effects of BTL-1 on blood routine examination in DSS-induced IBD mice (J,K). The results were expressed as the means ± SEM of n = 5. #P < 0.05, ##P < 0.01, compared to control group, *P < 0.05, **P < 0.01 compared to DSS group.
BTL-1 Inhibits the Expression of Inflammatory Factors in DSS Induced IBD of Mice
The expression of inflammatory factors in the blood and intestinal tissues were measured to investigate the impacts of BTL-1 on blood indexes and intestinal inflammatory factors in the DSS-induced IBD mice model. Consistent with blood routine results, the ELISA results of serum inflammatory factors (Figures 8A–D) showed that DSS treatment results in a measurable increase in serum inflammatory factors (IL-10, IL-6, TNF-α, and IL-1β), and BTL-1 would significantly inhibit such increases. Furthermore, the changes in the mRNA and concentration of inflammatory factors in the intestinal tissue also showed the same trend (Figures 8E–L). These results suggest that DSS treatment can upregulate the inflammatory factors in mice and the occurrence of intestinal inflammation. At the same time, BTL-1 can effectively inhibit the expression of inflammatory factors, in turn, inflammation.
Figure 8.

The effects of BTL-1 on blood routine examination and expression of inflammatory factors in DSS-induced IBD mice. The effects of BTL-1 on inflammatory factors in serum (A–D) and intestinal tissue as measured by qPCR (D–H) and ELIS A (I–L). The results were expressed as the means ± SEM of n = 3. #P < 0.05, ##P < 0.01, compared to control group, *P < 0.05, **P < 0.01 compared to DSS group.
BTL-1 Inhibits the Activity of TLR4/NF-κB and MAPK Signaling Pathways in the DSS-Induced IBD Model
The critical proteins of NF-κB and MAPK signaling pathways were measured by qPCR and WB to investigate the mechanism of BTL-1 inhibiting inflammation in the DSS-induced IBD mouse model. As shown in Figures 9A,B, DSS treatment increased TLR4, IκB, and P65's mRNA expression and upregulated the protein expression of TLR4 and the phosphorylation of IκB and P65. However, the expression and phosphorylation of key proteins (TLR4, IκB, and P65) of the NF-κB pathway were inhibited after BTL-1 was administered intragastrically. Furthermore, unlike the IPEC-J2 cell experiment, BTL-1 also significantly inhibited the phosphorylation of ERK, P38, and JNK in mice induced by DSS. In sum, the TNF-α in mice was downregulated considerably, indicating that BTL-1 regulates the expression of TNF-α through phosphorylation of ERK and P38 (Figures 9C,D).
Figure 9.

The effect of BTL-1 on the intestinal NF-κB and MAPK signaling pathways of IBD mouse model induced by DSS. The inhibitory effect of BTL-1 on TLR4/NF-κB signaling pathway determined by qPCR (A) and western blotting (B). The inhibitory effect of BTL-1 on the MAPK signaling pathway determined by qPCR (C) and western blotting (D). The results were expressed as the means ± SEM of n = 3. #P < 0.05, ##P < 0.01, compared to control group, *P < 0.05, **P < 0.01 compared to DSS group.
BTL-1 Alleviates DSS-Induced Intestinal Injury in Mice by Upregulating TJ Proteins
Intestinal permeability, intestinal structure, and tight junction proteins were measured to investigate the effect of BTL-1 on intestinal injury in the DSS-induced IBD mice model. As shown in Figure 10B, the intestinal permeability was increased after DSS treatment, and the serum FITC-D fluorescence intensity was significantly increased. Although the permeability of the intestine was not restored to normal levels following treatment with BTL-1, it is greatly improved compared with the DSS group. As shown in Figure 10A, there was no apparent colon injury in the control and PBS groups. On the contrary, the mucosa structure in the DSS group was not clear, and many inflammatory cells infiltrated in lamina propria and submucosa, accompanied by submucosal edema. Compared with the DSS group, the structural damage, inflammatory cell infiltration, and submucosal edema of each dose of the BTL-1 group were all improved. We also measured the expression of TJ proteins, as shown in Figures 10C–I; DSS treatment decreased the mRNA and proteins expression of ZO-1, occludin, and claudin1. However, the expression of TJ proteins (ZO-1, occludin, and claudin1) was upregulated after BTL-1 was administered intragastrically.
Figure 10.

The effect of BTL-1 on the intestinal injury of IBD mouse model induced by DSS. Histopathological changes after DSS stimulation in the colon (A). The in vitro intestinal barrier function evaluated by FITC-D fluorescence intensity (B). Upregulation of BTL-1 on tight junction proteins as determined by qPCR (C–E) and western blotting (F–I). The results were expressed as the means ± SEM of n = 3. #P < 0.05, ##P < 0.01, compared to control group, *P < 0.05, **P < 0.01 compared to DSS group.
Docking of BTL-1 With TLR4
To elucidate the interaction mode between TLR4 and compound BTL-1 at the molecular level, we docking compound BTL-1 to the active pocket of TLR4 with an affinity of −8.8 kcal mol−1. The 3D structure of BTL-1 is shown in Figure 11A. The theoretical binding mode is shown in Figures 11B,C. It can be seen from Figure 11B that the active pocket of compound butyrolactone-I presents a compact binding mode. It can be seen from Figures 11B,C that BTL-1 is in a cavity composed of amino acids Ile-32, Ile-46, Ser-47, Ile-52, Val-61, Leu-78, Phe-121, Cys-133, Phe-151, and Ile-153, forming a robust hydrophobic interaction. It is crucial that one hydroxyl group of BTL-1 can form a hydrogen bond with amino acid Ser-47 to 3.6 Å (Figure 11D) and form a Pi-Pi bond with amino acid Phe-151, which is the most critical force between butyrolactone I and TLR4. All these interactions make BTL-1 and TLR4 form stable complexes. In conclusion, the above docking studies provide a reasonable explanation for the interaction between BTL-1 and TLR4.
Figure 11.

Binding mode of BTL-1 into the TLR4. 3D structure of BTL-1 (A). Docking of BTL-1 to the active pocket of TLR4 (overall) (B). Docking of BTL-1 to the active pocket of TLR4 (detail) (C). Docking of BTL-1 to the active pocket of TLR4 (2D) (D).
Discussion
The incidence rate of IBD has been increasing worldwide, which seriously threatens human health. Even though the pathogenic mechanism of IBD is still unknown, current data suggests that IBD symptoms are caused by abnormal immune responses and damage to the intestinal barrier. Therefore, the development of effective anti-inflammatory and intestinal barrier protective function agents may be a future treatment strategy for IBD (33). This study first reported that butyrolactone-I alleviates LPS induced inflammatory response in IPEC-J2 via TLR4/NF-κB and MAPK signaling pathways. Therefore, BTL-1 can be used as an ocean drug to prevent intestinal bowel disease.
Butyrolactone-I was first reported in 1977 (34). Butyrolactone-I has a furan ring system and two phenolic groups in terms of structure. Many studies have shown that phenolic compounds can inactivate toll-like receptors, NF-κB, MAPK, kappa kinase/c-Jun amino-terminal kinases (IKK/JNK), inhibiting pro-inflammatory genes expression (35, 36). Similarly, natural furan derivatives have many pharmacological activities, such as antibacterial, antioxidant, antiviral, anti-inflammatory, anti-tumor, analgesic, anti-convulsant, and anti-fungal effects (32, 37). Furthermore, it has been proved that butyrolactone-I has the functions of inhibiting mammalian oocyte division, inhibiting tumor cell proliferation, inducing cancer cell apoptosis, preventing neuron apoptosis, has antibacterial and anti-inflammatory effects on microglia (14, 16, 18, 21, 22, 38). However, there is still a gap in demonstrating its ability to inhibit intestinal inflammation and protect intestinal function at the cellular and animal levels.
Given the LPS-induced IPEC, the cell inflammation and DSS-induced IBD mouse models are widely accepted as experimental models for assessing intestinal inflammation and IBD (39). We used IPEC-J2 cells to create an LPS-induced inflammatory model in vitro and found IL-1, IL-6, and TNF- elevated. Meanwhile, after being exposed to LPS, cell viability was reduced. In in vivo study, DSS administration significantly reduced the body weight gain and increased the disease activity index in mice, denoting that the experimental model was successfully established.
Toll-like receptors play an essential role in innate immunity. It can recognize characteristics that are shared by many viruses and their pathogenic components (PAMP) and trigger a natural immune response. LPS first activates TLR4 to trigger the downstream of the MAPK and NF-κB signaling pathways, thereby regulating cytokine production and the expression of many inflammatory genes. In IPEC-J2, we found that BTL-1 could inhibit the phosphorylation and nuclear entry of P65 via inhibiting TLR4, thereby decreasing the expression of IL-1β and IL-6. Studies have pointed out that BTL-1 has an active site that binds to P65 (14), so it is not ruled out that BTL-1 can also directly bind to P65, inhibiting the expression of downstream inflammatory factors. At the same time, BTL-1 can regulate the expression of TNF-α by upregulating the phosphorylation of ERK1/2 and P38 when TLR4 was inhibited. In line with our results, studies have pointed out that ERK and P38 can regulate the expression of TNF-α (40). Unfortunately, we cannot explore how BTL-1 upregulates the phosphorylation of ERK1/2 and P38 in IPEC-J2. Although TNF-α was upregulated, the downregulation of anti-inflammatory factor IL-10 (41) and increased cell viability denote that BTL-1 had an anti-inflammatory effect.
In general, the improvement of barrier function was correlated with the increase of TEER and the decrease of paracellular permeability (42, 43). The three proteins ZO-1, occludin, and claudin-1, are crucial for the TJ of the intestine to maintain intestinal permeability (44, 45). We found that BTL-1 can increase TEER and decrease paracellular permeability by upregulating tight junction protein. Furthermore, it shows that BTL-1 can enhance the intestinal barrier function in vitro.
To explore the role of BTL-1 in preventing IBD, we also used a DSS-induced IBD mouse model. We found that BTL-1 can effectively alleviate DSS-induced decreased food intake, increased disease activity index, immune organ index, anemia, and total white blood cell count. Further, we found that BTL-1 effectively inhibited the NF-κB and the MAPK signaling pathways, indicating that BTL-1 can downregulate the expression of pro-inflammatory factors (IL-6, IL-1β, and TNF-α). On the other hand, BTL-1 can promote the phosphorylation of P38 and ERK in IPEC-J2 cells, thus regulating the expression of TNF-α. However, BTL-1 has different activities in the IBD mouse model, which inhibits the phosphorylation of ERK and P38, thus downregulating the expression of TNF-α. Our results are consistent with the results of related literature in inhibiting the expression of TNF-α in mice (46).
In terms of protecting the intestinal barrier, BTL-1 significantly increased the expression of TJ proteins and reduced the permeability of the intestinal tract. In addition, the histological changes of the intestinal mucosa were close to those of the control group. This finding denotes that BTL-1 can sustain the integrity of the mouse intestinal barrier by increasing the expression of TJ proteins.
Herein, BTL-1 showed different activities on the MAPK signaling pathway in IPEC-J2 and mouse models. In IPEC, BTL-1 upregulates TNF-α by activating the phosphorylation of ERK1/2 and P38. Whereas, in mice, BTL-1 downregulates TNF-α by inhibiting the phosphorylation of ERK1/2 and P38. Due to different animal species and cell types, the same component often shows various activities. In this context, Gong et al. found that cortisol inhibition of oocyte maturation could not be relieved by NR3C1 inhibitor RU486 in mice, but RU486 overcame it in pigs (47). Further, Simon et al. found that Ascaris suum adult body fluid (ABF) did not affect cytokine production in IPEC-J2 cells; however, it suppressed LPS-induced secretion of IL-6 and TNF-α in PBMC (41). Therefore, it is reasonable that BTL-1 has different activities on the MAPK signaling pathway in IPEC and mice. However, we did not explore the mechanism of this difference that necessitated further experimental works.
Conclusion
In conclusion, BTL-1 showed an anti-inflammatory effect in LPS-induced IPEC-J2 inflammation and DSS-induced IBD mouse models via TLR4/NF-κB and MAPK signaling pathways, resulting in the inhibition of pro-inflammatory cytokines expression. Meanwhile, by upregulating tight junction proteins, BTL-1 helps maintain the integrity of the intestinal barrier. These beneficial effects of BTL-1 in colitis may suggest the applicability of BTL-1 as a functional food or a new treatment strategy for human IBD. Identifying a novel function for BTL-1 may provide a basis for developing L-arabinose and new approaches to promote or protect gut health.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by the protocol was approved by the Committee on the Ethics of Animal Experiments at Guangdong Ocean University (Permit No.: 201–1231).
Author Contributions
SC: conceptualization, methodology, validation, and writing. XN, SM, JW, MB, TY, LW, CH, YZ, YY, and XL: resources and supervision. AA: review and editing. XJ: funding acquisition, project administration, and writing—review and editing. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the National Natural Science Foundation of China [grant numbers: 31472243, 31902314]; Natural Science Foundation of Guangdong Province, China [grant numbers: 2019A1515011142, 2018A030307046]; the Project of Enhancing School with Innovation of Guangdong Ocean University [grant number: GDOU230419057] and the Program for Scientific Research Start-Fund of Guangdong Ocean University [grant number: 101402/R17088]; the Basic Research Project of Shenzhen Science and Technology Innovation Commission [JCYJ20190813105005619, JCYJ20190813142005766]; Shenzhen Dapeng New District Industrial Development Fund [No. KY20180203]; Shenzhen Dapeng New District Scientific and Technological Research and Development Fund [No. KJYF202001-07]; The Special Project in Key Fields of Guangdong Provincial Higher Education Institutions (Biomedicine and Health Care) [2021ZDZX2064]; and The Innovation and Development Project about Marine Economy Demonstration of Zhanjiang City [XM-202008-01B1].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2021.748118/full#supplementary-material
References
- 1.Cui J, Li Y, Jiao C, Gao J, He Y, Nie B, et al. Improvement of magnesium isoglycyrrhizinate on DSS-induced acute and chronic colitis. Int Immunopharmacol. (2021) 90:107194. 10.1016/j.intimp.2020.107194 [DOI] [PubMed] [Google Scholar]
- 2.Ott C, Scholmerich J. Extraintestinal manifestations and complications in IBD. Nat Rev Gastroenterol Hepatol. (2013) 10:585–95. 10.1038/nrgastro.2013.117 [DOI] [PubMed] [Google Scholar]
- 3.Claudia O, Jürgen S. Extraintestinal manifestations and complications in IBD. Nat Rev Gastroenterol Hepatol. (2013) 10:585–95. [DOI] [PubMed] [Google Scholar]
- 4.Khatri V, Amdare N, Tarnekar A, Goswami K, Reddy M. Brugia malayi cystatin therapeutically ameliorates dextran sulfate sodium-induced colitis in mice. J Dig Dis.(2015) 16:585–94. 10.1111/1751-2980.12290 [DOI] [PubMed] [Google Scholar]
- 5.Khatri V, Kalyanasundaram R. Therapeutic implications of inflammasome in inflammatory bowel disease. FASEB J. (2021) 35:e21439. 10.1096/fj.202002622R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lai JL, Liu YH, Liu C, Qi MP, Liu RN, Zhu XF, et al. Indirubin inhibits LPS-induced inflammation via TLR4 abrogation mediated by the NF-kB and MAPK signaling pathways. Inflammation. (2017) 40:1–12. 10.1007/s10753-016-0447-7 [DOI] [PubMed] [Google Scholar]
- 7.Goudarzi R, Partoazar A, Mumtaz F, Yousefi-Manesh H, Abdollahi A, Dehpour A, et al. Arthrocen, an avocado-soy unsaponifiable agent, improves acetic acid-induced colitis in rat by inhibition of NF-kB signaling pathway. J Food Biochem. (2020) 44:e13244. 10.1111/jfbc.13244 [DOI] [PubMed] [Google Scholar]
- 8.Zobeiri M, Momtaz S, Parvizi F, Tewari D, Farzaei MH, Nabavi SM. Targeting mitogen-activated protein kinases by natural products: a novel therapeutic approach for inflammatory bowel diseases. Curr Pharm Biotechnol. (2020) 21:1342–53. 10.2174/1389201021666191216122555 [DOI] [PubMed] [Google Scholar]
- 9.Chen S, Zhang CL, Shen HQ, Zhou XF, Li JH, Yu JL, et al. Sesamin protects against DSS-induced colitis in mice by inhibiting NF-kappaB and MAPK signaling pathways. Food Funct. (2021) 12:1688–94. 10.1039/D0FO00950D [DOI] [PubMed] [Google Scholar]
- 10.Groschwitz KR, Hogan SP. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. (2009) 124:3–20. 10.1016/j.jaci.2009.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee SH. Intestinal permeability regulation by tight junction: implication on inflammatory bowel diseases. Intest Res. (2015) 13:11–8. 10.5217/ir.2015.13.1.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka H, Takechi M, Kiyonari H, Shioi G, Tamura A, Tsukita S. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut. (2015) 64:1529–38. 10.1136/gutjnl-2014-308419 [DOI] [PubMed] [Google Scholar]
- 13.Sugita K, Kabashima K. Tight junctions in the development of asthma, chronic rhinosinusitis, atopic dermatitis, eosinophilic esophagitis, and inflammatory bowel diseases. J Leukoc Biol. (2020) 107:749–62. 10.1002/JLB.5MR0120-230R [DOI] [PubMed] [Google Scholar]
- 14.Yuan ZY, Yi Z, Yuan-Bei Y, Xiao-Ling L, Zhong-Ji Q. Butyrolactone-I from coral-derived fungus aspergillus terreus attenuates neuro-inflammatory response via suppression of NF-κB pathway in BV-2 Cells. Mar Drugs. (2018) 16:202. 10.3390/md16060202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atkinson DA, Hill HH, Shultz TD. Quantification of mammalian lignans in biological fluids using gas chromatography with ion mobility detection. J Chromatogr. (1993) 617:173–9. 10.1016/0378-4347(93)80485-M [DOI] [PubMed] [Google Scholar]
- 16.Maas JW, Horstmann S, Borasio GD, Anneser JM, Shooter EM, Kahle PJ. Apoptosis of central and peripheral neurons can be prevented with cyclin-dependent kinase/mitogen-activated protein kinase inhibitors. J Neurochem. (1998) 70:1401–10. 10.1046/j.1471-4159.1998.70041401.x [DOI] [PubMed] [Google Scholar]
- 17.Wada M, Hosotani R, Lee JU, Doi R, Koshiba T, Fujimoto K, et al. An exogenous cdk inhibitor, butyrolactone-I, induces apoptosis with increased Bax/Bcl-2 ratio in p53-mutated pancreatic cancer cells. Anticancer Res. (1998) 18:2559–66. [PubMed] [Google Scholar]
- 18.Nishio K, Ishida T, Arioka H, Kurokawa H, Fukuoka K, Nomoto T, et al. Antitumor effects of butyrolactone I, a selective CDC2 kinase inhibitor, on human lung cancer cell lines. Anticancer Res. (1996) 16:3387–95. [PubMed] [Google Scholar]
- 19.Maziero RRD, Guaitolini CRdF, Paschoal DM, Crespilho AM, Monteiro BA, Lima JSd, et al. Treatment with roscovitine and butyrolactone I prior to in vitro maturation alters blastocyst production. Zygote. (2020) 28:24–31. 10.1017/S0967199419000571 [DOI] [PubMed] [Google Scholar]
- 20.Ferreira EM, Vireque AA, Adona PR, Ferriani RA, Navarro PA. Prematuration of bovine oocytes with butyrolactone I reversibly arrests meiosis without increasing meiotic abnormalities after in vitro maturation. Eur J Obstet Gynecol Reprod Biol. (2009) 145:76–80. 10.1016/j.ejogrb.2009.03.016 [DOI] [PubMed] [Google Scholar]
- 21.Lonergan P, Dinnyes A, Fair T, Yang X, Boland M. Bovine oocyte and embryo development following meiotic inhibition with butyrolactone I. Mol Reprod Dev. (2000) 57:204–9. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Zhong M, Lei X, Xiao S, Li Z. Diversity and antibacterial activities of culturable fungi associated with coral Porites pukoensis. World J Microbiol Biotechnol. (2014) 30:2551–8. 10.1007/s11274-014-1701-5 [DOI] [PubMed] [Google Scholar]
- 23.Yang JM, Liu YY, Yang WC, Ma XX, Nie YY, Glukhov E, et al. An anti-inflammatory isoflavone from soybean inoculated with a marine fungus Aspergillus terreus C23-3. Biosci Biotechnol Biochem. (2020) 84:1546–53. 10.1080/09168451.2020.1764838 [DOI] [PubMed] [Google Scholar]
- 24.Yuan Z, Yang L, Zhang X, Ji P, Hua Y, Wei Y. Huang-Lian-Jie-Du decoction ameliorates acute ulcerative colitis in mice via regulating NF-κB and Nrf2 signaling pathways and enhancing intestinal barrier function. Front Pharmacol. (2019) 10:1354. 10.3389/fphar.2019.01354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peeraphong L, Yiwen J, Nami K, Wenwei F, Changwu Z, Hongsheng T, et al. Safranal alleviates dextran sulfate sodium-induced colitis and suppresses macrophage-mediated inflammation. Front Pharmaco. (2019) 10:1281. 10.3389/fphar.2019.01281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Liu G, Yuan Y, Wu G, Yuan L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. (2019) 10:906. 10.1038/s41419-019-2157-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valdez JC, Cho J, Bolling BW. Aronia berry inhibits disruption of Caco-2 intestinal barrier function. Arch Biochem Biophys. (2020) 688:108409. 10.1016/j.abb.2020.108409 [DOI] [PubMed] [Google Scholar]
- 28.Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. (2008) 57:1470–81. 10.2337/db07-1403 [DOI] [PubMed] [Google Scholar]
- 29.Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. (1999) 17:57-61. [PubMed] [Google Scholar]
- 30.Garrett MM, Ruth H, William L, Michel FS, Richard KB, David SG, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. (2009) 30:2785–91. 10.1002/jcc.21256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. (2009) 30:2785–91. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhatiya R, Vaidya A, Kashaw SK, Jain AK, Agrawal RK. QSAR analysis of furanone derivatives as potential COX-2 inhibitors: kNN MFA approach. J. Saudi Chem. Soc. (2014) 18:977–84. 10.1016/j.jscs.2011.12.002 [DOI] [Google Scholar]
- 33.Mehandru S, Colombel JF. The intestinal barrier, an arbitrator turned provocateur in IBD. Nat Rev Gastroenterol Hepatol. (2021) 18:83–4. 10.1038/s41575-020-00399-w [DOI] [PubMed] [Google Scholar]
- 34.Kiriyama N, Nitta K, Sakaguchi Y, Taguchi Y, Yamamoto Y. Studies on the metabolic products of Aspergillus terreus. III. Metabolites of the stain IFO 8835 (1). Chem Pharm Bull. (1977) 25:2593–601. 10.1248/cpb.25.2593 [DOI] [Google Scholar]
- 35.Toma L, Sanda GM, Niculescu LS, Deleanu M, Sima AV, Stancu CS. Phenolic compounds exerting lipid-regulatory, anti-inflammatory and epigenetic effects as complementary treatments in cardiovascular diseases. Biomolecules. (2020) 10:641. 10.3390/biom10040641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biluca FC, da Silva B, Caon T, Mohr ETB, Vieira GN, Gonzaga LV, et al. Costa, Investigation of phenolic compounds, antioxidant and anti-inflammatory activities in stingless bee honey (Meliponinae). Food Res Int. (2020) 129:108756. 10.1016/j.foodres.2019.108756 [DOI] [PubMed] [Google Scholar]
- 37.Mohammad J, Moludi H, Khodaei S, Amir K, Sorayya PFT, Fatemeh P, et al. Recent updates on anti-inflammatory and antimicrobial effects of furan natural derivatives. J Inflamm Res. (2020) 13:451–63. 10.2147/JIR.S262132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu M, Zhou Q, Wang J, Liu J, Qi C, Lai Y, et al. Anti-inflammatory butenolide derivatives from the coral-derived fungus Aspergillus terreus and structure revisions of aspernolides D and G, butyrolactone VI and 4,8-diacetoxy butyrolactone VI. RSC Adv. (2018) 8:13040–7. 10.1039/C8RA01840E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng Z, Cui C, Wang Y, Ni J, Zheng L, Wei HK, et al. FSGHF3 and peptides, prepared from fish skin gelatin, exert a protective effect on DSS-induced colitis via the Nrf2 pathway. Food Funct. (2020) 11:414-23. 10.1039/C9FO02165E [DOI] [PubMed] [Google Scholar]
- 40.Dong N, Xu X, Xue C, Wang C, Li X, Bi C, et al. Ethyl pyruvate inhibits LPS induced IPEC-J2 inflammation and apoptosis through p38 and ERK1/2 pathways. Cell Cycle. (2019) 18:2614–28. 10.1080/15384101.2019.1653106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakobsen SR, Myhill LJ, Williams AR. Effects of Ascaris and Trichuris antigens on cytokine production in porcine blood mononuclear and epithelial cells. Vet Immunol Immunopathol. (2019) 12:2116–9. 10.1016/j.vetimm.2019.03.012 [DOI] [PubMed] [Google Scholar]
- 42.Balaji S, Aditya RK, Mandy BE, Hasan EA, Michael LS, James J. TEER measurement techniques for in vitro barrier model systems. J Lab Autom. (2015) 20:107–26. 10.1177/2211068214561025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Putt KK, Pei R, White HM, Bolling BW. Yogurt inhibits intestinal barrier dysfunction in Caco-2 cells by increasing tight junctions. Food Funct. (2017) 8:406–14. 10.1039/C6FO01592A [DOI] [PubMed] [Google Scholar]
- 44.Anderson JM, Itallie C. Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol Cell Physiol. (1995) 269:G467–75. 10.1152/ajpgi.1995.269.4.G467 [DOI] [PubMed] [Google Scholar]
- 45.Caimei H, Jun D, Xin H, Sichun Z, Jingtao W, Di X, et al. Xiaoping, Vitamin A inhibits the action of LPS on the intestinal epithelial barrier function and tight junction proteins. Food Funct. (2019) 10:1235–42. 10.1039/c8fo01123k [DOI] [PubMed] [Google Scholar]
- 46.Wu W, Liu L, Zhu H, Sun Y, Wu Y. Butyrolactone-I, an efficient α-glucosidase inhibitor, improves type 2 diabetes with potent TNF-α-lowering properties through modulating gut microbiota in db/db mice. FASEB J. (2019) 33:12616–29. 10.1096/fj.201901061R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shuai G, Guang-Yi S, Min Z, Hong-Jie Y, Shuai Z, Guang-Zhong J, et al. Mechanisms for the species difference between mouse and pig oocytes in their sensitivity to glucorticoids. Biol Reprod. (2017) 96 :1019–30. 10.1093/biolre/iox026 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.